

Abstract
Oncolytic viruses and active immunotherapeutics have complementary mechanisms of action (MOA) that are both self amplifying in tumors, yet the impact of dose on subject outcome is unclear. JX-594 (Pexa-Vec) is an oncolytic and immunotherapeutic vaccinia virus. To determine the optimal JX-594 dose in subjects with advanced hepatocellular carcinoma (HCC), we conducted a randomized phase 2 dose-finding trial (n = 30). Radiologists infused low-or high-dose JX-594 into liver tumors (days 1, 15 and 29); infusions resulted in acute detectable intravascular JX-594 genomes. Objective intrahepatic Modified Response Evaluation Criteria in Solid Tumors (mRECIST) (15%) and Choi (62%) response rates and intrahepatic disease control (50%) were equivalent in injected and distant noninjected tumors at both doses. JX-594 replication and granulocyte-macrophage colony-stimulating factor (GM-CSF) expression preceded the induction of anticancer immunity. In contrast to tumor response rate and immune endpoints, subject survival duration was significantly related to dose (median survival of 14.1 months compared to 6.7 months on the high and low dose, respectively; hazard ratio 0.39; P = 0.020). JX-594 demonstrated oncolytic and immunotherapy MOA, tumor responses and dose-related survival in individuals with HCC.
Despite advances in cancer treatment over the past 30 years with chemotherapy and biologics, the majority of solid tumors remain incurable once they are metastatic. Truly new agents with multiple complementary MOA are required to move beyond the modest benefits achieved so far. Extensive study in the field of active immunotherapy has recently culminated in regulatory approvals of sipuleucelt (Provenge; Dendreon) and ipilimumab (Yervoy; Bristol-Myers Squibb). Although these agents comprise the first approvals for a new therapeutic class, their limited long-term benefit warrants development of more potent active immunotherapies. The oncolytic and immunotherapeutic herpes simplex virus T-Vec (Amgen), which expresses GM-CSF after local intratumoral injection, recently demon-strated anticancer immune induction and durable objective responses in an intratumoral phase 2 melanoma study1.
Oncolytic immunotherapies are designed to selectively replicate within, and subsequently lyse, cancer cells2–5 while inducing tumor-specific immunity. JX-594 (also called PexaVec; Jennerex Inc.) is a vaccinia virus (Wyeth vaccine strain) with disruption of the viral thymidine kinase gene (TK) for cancer selectivity and insertion of human granulocyte-macrophage colony-stimulating factor (hGM-CSF) and β-galactosidase transgenes for immune stimulation and replication assessment, respectively6–8. JX-594 is designed to induce both virus replication–dependent oncolysis and tumor-specific immunity6,9,10.
The advantages of a vaccinia virus include intravenous (i.v.) stability and delivery11, enhanced potency12, extensive safety experience as a live vaccine, demonstrated ability to induce efficient immune responses and a large transgene-encoding capacity6. The oncolytic immunotherapies JX-594 (Pexa-Vec), T-VEC (Amgen)13 and an adenoviral construct expressing GM-CSF (Ad-GM-CSF)3 comprise a new platform that has advantages over the currently approved immunotherapeutics given their direct tumor lysis, induction of subject tumor-specific immunity and use as off-the-shelf products.
In contrast to other agents in this class, JX-594 showed both complete responses of bulky tumors and systemic efficacy in phase 1 studies14,15. In a phase 1 clinical trial of intratumoral JX-594 (ref. 14), as can be expected with a self-amplifying agent, injection-site responses were seen at all doses; however, systemic tumor responses and delivery through the blood to distant tumors required a high dose. High-dose JX-594 was also required for i.v. delivery and efficacy in a dose-escalation i.v. phase 1 trial15. Thus far, however, no randomized dose-finding trial has been reported with these self-amplifying active immunotherapies. In addition, proof of active immunotherapy induction in subjects with these agents was lacking. The objectives of this randomized trial were to compare outcomes with low-dose (108 PFU) and high-dose (109 PFU) JX-594 in a uniform advanced solid tumor population (HCC), including safety, intrahepatic tumor response, induction of immunity to both cancer cells and vaccinia, and overall survival.
RESULTS
Subject enrollment and baseline and treatment characteristics
Between December 2008 and May 2011, we screened 49 subjects for enrollment. Study enrollment was halted early by an independent data safety monitoring board because of a significant survival benefit favoring the high-dose group. We enrolled 30 subjects and stratified (viral or nonviral tumor etiology) and randomized them (16 high-dose arm and 14 low-dose arm; the trial profile is shown in Supplementary Fig. 1). Baseline subject characteristics and prognostic factors were well balanced between the two groups (Table 1). We noted no statistically significant differences for prognostic factors between groups. However, the high-dose subjects were more likely to have failed previous systemic therapy, which is a negative prognostic factor (with six high-dose subjects compared to one low-dose subject failing previous systemic therapy; P = 0.09, Fisher’s exact test), including previous sorafenib treatment (all four subjects with previous sorafenib treatment in the high-dose group had tumor progression while on this therapy).
Table 1.
Demographic and baseline characteristics of the subjects according to treatment group (safety population)
| High dose (n = 16) |
Low dose (n = 14) |
|
|---|---|---|
| DEMOGRAPHICS | ||
| Age (years)a | 62.9 ± 12.7 | 67.1 ±11.5 |
| Sex (n (%)) | ||
| Male | 13 (81) | 10 (71) |
| Female | 3 (19) | 4 (29) |
| Region (n (%)) | ||
| North America | 9 (56) | 8 (57) |
| Asia | 7 (44) | 6 (43) |
| Cause of disease (n (%)) | ||
| Virally associated | 9 (56) | 9 (64) |
| HCV | 3 (19) | 3 (21) |
| HBV | 6 (37) | 6 (43) |
| Alcohol only | 0 (0) | 2 (14) |
| Other | 7 (44) | 3 (21) |
| Karnofsky performance status (KPS)of 70–100 (n (%)) | 16 (100) | 14 (100) |
| Baseline neutralizing antibodies to vaccinia (n (%)) | ||
| Positive | 9 (56) | 6 (43) |
| Negative | 7 (44) | 8 (57) |
| TUMOR BURDEn | ||
| BCLC stage (n (%)) | ||
| B (intermediate) | 2 (12) | 1 (7) |
| C (advanced) | 14 (88) | 13 (93) |
| Baseline tumors (n (%)) | ||
| Single | 6 (37) | 4 (29) |
| Multiple | 10 (63) | 10 (71) |
| At least four | 10 (63) | 6 (43) |
| Baseline tumors (longest diameter (sum))a | 9.6 ±3.9 | 10.6 ±5.1 |
| Baseline tumors (hepatic or portal vein invasion) (n (%)) | 3 (19) | 3 (21) |
| Extrahepatic spread (n (%)) | ||
| Noted | 3 (19) | 1 (7) |
| Unknown | 13 (81) | 13 (93) |
| Previous therapy (n patients (%))b | ||
| Locoregional therapy | 11 (69) | 11 (79) |
| Number of previous locoregional interventions (per patient)a | 3.4 ±3.2 | 2.9 ±2.3 |
| Progressed on previous systemic therapyc | 6 (38) | 1 (7) |
| Progressed on previous sorafenib therapyc | 4 (25) | 0 (0) |
| BASELInE LIVER FUnCTIOn | ||
| Child-Pugh classification (n (%)) | ||
| A (5–6) | 16 (100) | 12 (86) |
| B (7–9) | 0 (0) | 2 (14) |
| Baseline albumin (gm d−l | ||
| Mean ±s.d. | 4.2 ±0.3 | 3.8 ±0.6 |
| Median | 4.2 | 3.8 |
| Range | 3.7–4.7 | 2.9–4.8 |
| Baseline prothrombin time (s) | ||
| Mean ±s.d. | 12.6 ±1.2 | 13.3 ±2.4 |
| Median | 12.8 | 12.7 |
| Range | 10.1–14.2 | 10.5–16.8 |
| Baseline aPTT (s) | ||
| Mean ±s.d. | 32.6 ±3.9 | 37.9 ±9.0 |
| Median | 31.8 | 37.9 |
| Range | 26.0–39.6 | 28.1–59.6 |
| Baseline bilirubin (mg/dl) | ||
| Mean ±s.d. | 0.8 ±0.3 | 1.3 ±1.1 |
| Median | 0.7 | 0.8 |
| Range | 0.2–1.4 | 0.4–3.8 |
Data are shown as the mean ± s.d. None of the differences between the two study groups was significant (P≥ 0.05).
Patients may have received more than one type of therapy.
Patients receiving systemic therapies had tumor progression while on therapy (refractory disease). BCLC, Barcelona Clinic Liver Cancer staging system; aPTT, activated partial thromboplastin time.
Twenty-nine subjects received all three doses; one subject received only two doses because of an unrelated adverse event. All subjects were evaluable for safety (as they all received at least one dose) and all but one (because of a major undocumented protocol deviation, biopsy-confirmed cholangiocarcinoma) were evaluable for survival. Twenty-eight subjects were considered evaluable for radiographic endpoints; this included two subjects who had clinical disease progression and died without a scan at week 8 (and were considered to have had progression). Therapies given after JX-594 treatment and off protocol, as reported by the principal investigators, were similar in the two groups; these included sorafenib treatment (full-dose sorafenib for ≥8 weeks in two high-dose subjects and one low-dose subject) and local-regional palliative therapies (two high-dose subjects and one low-dose subject).
Safety and toxicity
JX-594 was generally well tolerated at both doses. No treatment-related deaths were reported. One treatment-related serious adverse event was reported in the high-dose group (nausea and vomiting requiring prolonged hospitalization). Ten non–treatment-related serious adverse events were reported (in eight subjects, four high-dose and four low-dose). Treatment-related adverse events are summarized by grade and treatment arm in Supplementary Table 1. The frequency and grade (according to the National Cancer Institute Common Terminology Criteria for Adverse Events) of the adverse events were similar between the two dose groups. Flu-like symptoms (grade 1–2) occurred in all subjects over the first 12–24 h after treatment, including fever, rigors, nausea or vomiting. Subjects treated at the high dose showed a larger temperature increase after the first JX-594 treatment compared with low-dose subjects (P = 0.0023, t test) and had a higher incidence of anorexia (31% compared to 0%; P = 0.04). One possibly related grade 4 event of lymphopenia (2 week duration) was reported in a high-dose subject. Increases in serum transaminase concentrations were reported in six subjects (four low-dose and two high-dose).
A single high-dose subject developed eight to ten grade 1 skin pustules measuring <1 cm diameter each on the extremities, forehead and trunk. The lesions developed approximately 4 d after treatment and resolved completely without scar formation within approximately 6 weeks. The subject received two subsequent doses of JX-594 without delays.
Intrahepatic disease control and mRECIST and Choi responses
We performed serial dynamic magnetic resonance imaging (MRI) scans of the liver and abdomen, and these were subsequently read by expert independent central readers who were blinded to treatment arm. We applied the mRECIST response criteria developed for individuals with HCC16 to assess the effects of JX-594 treatment in the liver. In addition, as JX-594 has been shown to disrupt tumor blood flow and induce tumor necrosis17, we performed tumor contrast enhancement measurements according to the modified Choi criteria18 to assess effects on perfusion and the development of tumor necrosis.
As demonstrated in a previous study14, both doses were associated with intrahepatic antitumor activity. The intrahepatic mRECIST disease control rate at week 8 was 46% overall (28 evaluable rates of 47% and 46% for the high-dose and low-dose groups, respectively) and was 50% at any time point (meaning at week 8 or any other time, whichever was better). As the primary radiographic endpoint was at week 8, we did not consistently perform subsequent scans and as a result did not assess time to tumor progression.
We found objective mRECIST responses and decreased tumor perfusion and contrast enhancement in both injected and noninjected tumors within both dose groups (Fig. 1a–d and SupplementaryFig. 2). In some cases, tumors with decreased contrast enhancement showed swelling and edema (Fig. 1e). The modified Choi response rate was 62% overall (26 evaluable subjects, with response rates of 57% and 67% for the high-dose and low-dose groups, respectively). The mean changes in the Choi parameter were −35.6% and −28.8% in the high-dose and low-dose groups, respectively (P = 0.73). Four objective mRECIST responses (one complete and three partial; Fig. 1f) and ten cases of stable disease were reported; dose did not correlate with intrahepatic response (Fig. 1a–d). The mean change in longest tumor diameter (mRECIST) may have reflected dose-related edema formation (changes of 23.8% and −8.7% in the high-dose and low-dose groups, respectively; P = 0.11). We found similar effects on tumor vascularity in non-injected tumors after high-dose treatment (Fig. 1g), including small masses (<1 cm) that were poorly visualized at baseline.
Figure 1.
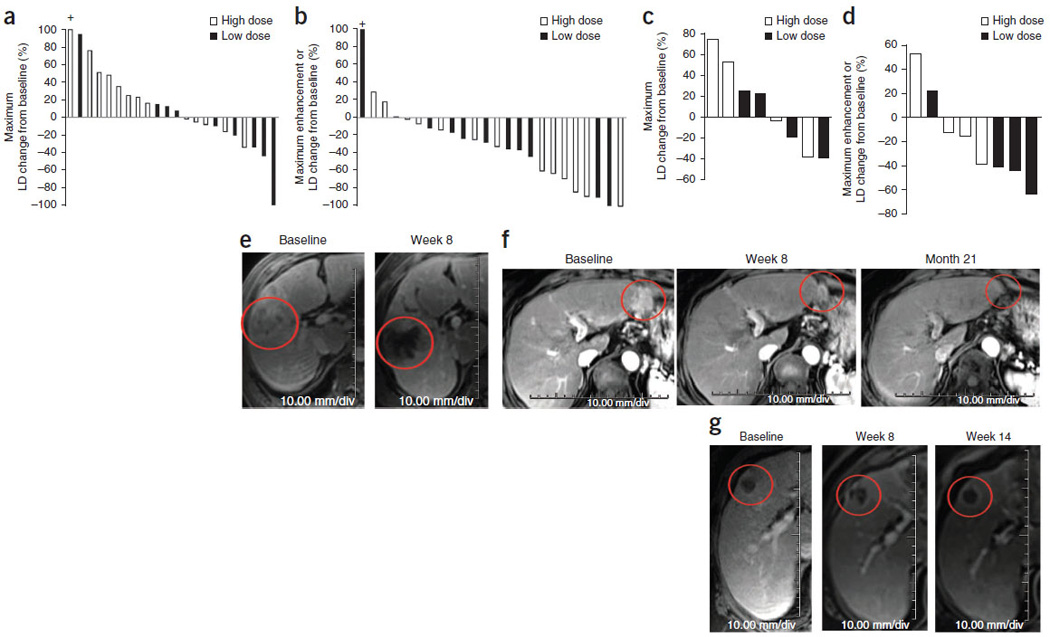
Radiographic antitumor activity after JX-594 therapy determined by dynamic MRI with a central radiographic reader blinded to treatment group. (a) Greatest decrease in the sum of the longest diameter (LD) of target tumors from baseline (mRECIST criteria) in livers of individual patients after JX-594 treatment. n = 22 patients who had measurable and evaluable tumors at baseline and at least one follow-up time point. A tumor with an increase >100% is indicated with a + above the bar. (b) Greatest decrease in tumor contrast enhancement or longest diameter from baseline (Choi criteria) in target tumors in livers of individual patients after JX-594 treatment. n = 24 patients who had measurable and evaluable tumors at baseline and at least one follow-up time point. A tumor with an increase >100% is indicated with a + above the bar. (c) Greatest decrease in the sum of the longest tumor diameter from baseline (RECIST criteria) in noninjected liver tumors of individual patients after JX-594 treatment. n = 8 evaluable. (d) Greatest decrease in tumor contrast enhancement or longest diameter from baseline (Choi criteria) in noninjected liver tumors of individual patients after JX-594 treatment. n = 8 evaluable. (e) Example of the effects of JX-594 on contrast enhancement and perfusion in an injected tumor (JX7-1401; high dose). (f) Example of the effects of JX-594 on the longest diameter of an injected tumor (complete mRECIST response) (JX7-1715; low dose). (g) Example of the effects of JX-594 on contrast enhancement and perfusion in a noninjected (distant) tumor (JX7-1403; high dose). The red circles (e–g) indicate the same (responding) tumors over time.
Pharmacokinetics, replication and transgene expression
JX-594 injections into tumors resulted in acute diffusion of JX-594 genomes into the blood, thus resulting in systemic distribution. We monitored the concentrations of JX-594 in the blood over time at 15 min, 3 h, 5–7 d and 14 d after completion of each injection procedure. The pharmacokinetic profiles were similar for all three injections per subject (Fig. 2a). Concentrations were highest at 15 min after injection; genomes were detectable in blood at this time point in all subjects on both arms. The peak concentrations of JX-594 were significantly greater for subjects in the high-dose arm (Fig. 2a); the mean concentration (± s.e.m.) for all cycles was 273,700 ± 55,650 genomes ml−1 for the high-dose group and 31,650 ± 5,317 genomes ml−1 for the low-dose group (P = 0.0002, t test). Acute blood concentrations at 15 min after injection were consistent with a substantial fraction of the input dose acutely entering the blood during or immediately after the injection procedure (half-life of ~60 min). The peak concentrations in these subjects were similar to peak concentrations in subjects after i.v. infusion at similar doses in a phase 1 trial15; the peak concentrations in the high-dose group in this trial were above the threshold for i.v. delivery to tumors (as defined in the i.v. phase 1 trial), whereas the concentrations in the low-dose group were not. The frequency and concentrations of detectable JX-594 genomes in the blood were also significantly greater in the high-dose subjects at 3 h after injection (78% of subjects had detectable JX-594 compared to 45% for the high-dose and low-dose groups, respectively, for all treatment cycles; mean concentrations of 10,450 ± 2,090 genomes ml−1 and 2,098 ± 488 genomes ml−1 for the high-dose and low-dose groups, respectively, for all treatment cycles; P = 0.0006, t test).
Figure 2.
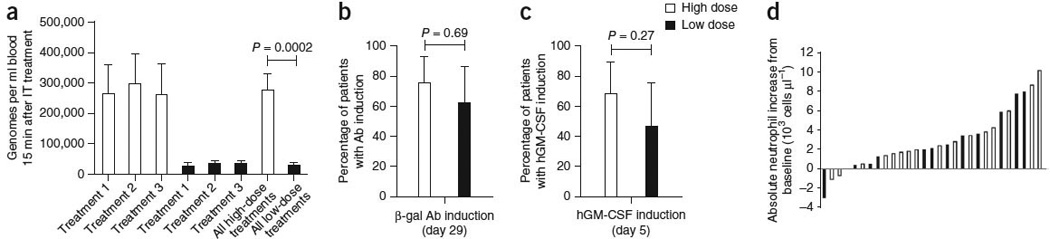
Laboratory evidence for JX-594 replication, transgene expression and GM-CSF protein function. (a) The mean (± s.e.m.) peak concentration of JX-594 (genomes measured by quantitative PCR (qPCR)) in blood after each treatment cycle (using blood obtained 15 min after the completion of treatment) by dose group (t test). IT, intratumoral. (b) The percentage of patients with evidence of β-gal transgene expression (+95% confidence interval (CI)) after JX-594 treatment (generation of antibodies (Ab) to the β-gal transgene product within 29 d of treatment is indicative of JX-594 replication, as β-gal protein expression is associated with virus replication) (Fisher’s exact test). (c) The percentage of patients with evidence of hGM-CSF transgene expression (+95% CI) on day 5 after JX-594 treatment (Fisher’s exact test). (d) Maximum induction of neutrophil concentration in blood after treatment cycle 1 by dose group (using blood obtained on days 5 and 15 after treatment). Black bars, low-dose JX-594; white bars, high-dose JX-594.
We assessed JX-594 replication and dual transgene expression using three methods. We first assessed antibody development to the β-galactosidase (β-gal) protein (the lacZ transgene product), which is dependent on viral replication and associated protein expression. Second, JX-594 expresses the hGM-CSF transgene under control of the synthetic early-late promoter such that high-level expression would occur during replication of the product in cancer cells6–8. Whereas i.v. JX-594 can acutely stimulate production of multiple cytokines, delayed hGM-CSF expression 5 or more days after infusion (when other cytokine concentrations have returned to baseline levels) was indicative of hGM-CSF expression from JX-594 in the context of replication14,15.
Third, we evaluated the delayed re-emergence of JX-594 genomes in the blood on days 5, 15, 22, 29, 36, 43 and 57. The detection of JX-594 genomes in blood at any of these time points after JX-594 injection would suggest intratumoral replication and subsequent leakage into the systemic circulation. We were able to detect JX-594 genomes in the blood of three subjects (two high dose and one low dose) at the late time points (days 15–36).
As can be expected because of replication of JX-594 at both doses, β-gal–specific antibodies developed in most of the subjects during the treatment phase: 75% of high-dose and 62% of low-dose subjects developed these antibodies (Fig. 2b). Neutralizing antibody titers to vaccinia (and, hence, JX-594) were detectable at baseline (from childhood vaccination) in 50% of subjects; all subjects developed detectable titers by day 29.
hGM-CSF protein was quantifiable in the plasma on day 5 in 69% of the high-dose and 46% of the low-dose subjects (P = 0.27, Fisher’s exact test; all baseline samples were negative for hGM-CSF) (Fig. 2c). Exploratory data from a subset of subjects treated with the same manufacturing lot of clinical trial material showed that hGM-CSF concentration (day 5) was higher in the high-dose subjects than in the low-dose subjects (median for the high-dose subjects was 47.6 pg ml−1 compared to 1.5 pg ml−1 for the low-dose subjects; P< 0.001, t test). Neutrophils and eosinophils are GM-CSF–responsive subsets of white blood cells. The absolute neutrophil and eosinophil concentrations increased substantially between 5–14 d after treatment in 65% of subjects with quantifiable hGM-CSF in the blood (Fig. 2d).
Induction of humoral and cellular anticancer immunity
We analyzed subject blood, tumor tissue and radiographic imaging to assess the induction of anticancer immunity (Fig. 3). As a measure of induction of antitumoral immunity, we assessed antibody-mediated complement-dependent cytotoxicity (CDC) in subjects’ serum over time after treatment. Eleven of 16 (69%) subjects evaluated developed CDC against at least one of four HCC cell lines (six high-dose and five low-dose subjects) (Fig. 3a,b); CDC was evident even after serum dilution down to 5%. All HCC cell lines tested contained hepatitis B virus DNA except HepG2 (which is not virally associated); six subjects with CDC induction had hepatitis B–associated HCC, two had hepatitis C–associated HCC and three had non-viral HCC.
Figure 3.
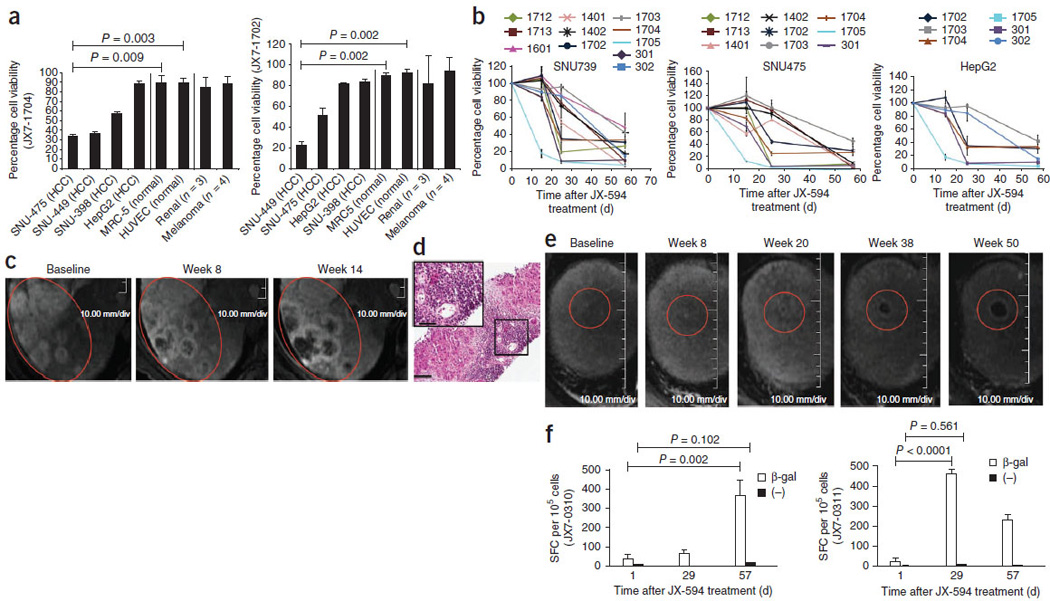
Laboratory, radiographic and biopsy evidence for JX-594– associated induction of anticancer immunity. (a) Antibody-mediated complement-dependent cytotoxicity induction after JX-594 therapy in HCC (n = 4), normal (n = 2; HUVEC and MRC-5) and non-HCC (RCC, n = 3; melanoma, n = 4) cell lines. Each graph shows the mean percentage cell viability (+s.d.) after incubation with each individual patient’s serum (diluted to 5%) collected on day 43 after the initiation of treatment compared to baseline. JX7-1704 and JX7-1702, two high-dose patients. (b) Antibody-mediated CDC induction against HCC cell lines in individual patients over time after JX-594 therapy (serum diluted to 5%; mean ± s.d.). The data shown are from serum of patients that induced ≤50% cell viability (>50% cell killing) on at least one follow-up time point. (c) Radiographic evidence of progressive necrosis and peripheral enhancement over time in noninjected tumors (JX7-0307; low dose). (d) H&E staining of a biopsy sample from a tumor collected from patient JX7-0301 (low dose) 1.5 years after the initiation of JX-594 treatment. Scale bars, 100 αm (low magnification); 50 αm (high-magnification inset). (e) Radiographic evidence of progressive necrosis and peripheral enhancement over time in a noninjected tumor (JX7-0301; low dose). Red circles (c,e) indicate the same (responding) tumors over time. (f) ELISPOT analysis detecting T cells producing interferon-γ in response to stimulation with β-gal peptides at baseline and after JX-594 treatment; data are expressed as the mean number of spot-forming cells (SFC) per 105 cells (+s.d.) (JX7-0310, low dose; JX7-0311, high dose). (−), negative control peptide. The P values in a and f were calculated by t test.
Radiographic evidence included progressive near-complete tumor hypovascularity and necrosis associated with a markedly enhancing rim in noninjected tumors developing over 3–4 months in four subjects (Fig. 3); these findings are considered highly unusual for tumor progression in HCC, but we could not completely rule out progression. We biopsied one of these masses 1.5 years after the last JX-594 treatment (Fig. 3e); the biopsy showed diffuse lymphocytic infiltration (Fig. 3d).
We also assessed cellular immunity. Cytotoxic T cells were induced to vaccinia peptides (data not shown) and the JX-594 transgene product β-gal (enzyme-linked immunosorbent spot (ELISPOT) analysis) (Fig. 3f); β-gal cytotoxic T cell activity was present in a high-dose subject 1.5 years after treatment initiation (Supplementary Fig. 3). In contrast, T cells collected from healthy donors not exposed to JX-594 did not have reactivity to β-gal (Supplementary Fig. 3)
Overall survival
The median overall survival was 9.0 months for the entire study population. We assessed the correlation between various baseline variables and overall survival (Table 2). Only dose cohort and peak JX-594 blood concentration correlated significantly with overall survival. Overall survival was significantly longer in the high-dose arm compared to in the low-dose arm (hazard ratio 0.39, P = 0.020, Gehan-Breslow-Wilcoxon test, one-sided test for superiority of the high dose). The median overall survival was 14.1 months for the high-dose group compared to 6.7 months for the low-dose group (Fig. 4a). Kaplan-Meier survival estimates for the high-dose and low-dose groups at 1 year were 66% and 23%, respectively, and at 18 months were 35% and 11%, respectively. Survival did not correlate with tumor etiology (viral or nonviral). In subjects with multiple tumors at base-line (ten high-dose and nine low-dose subjects), high-dose JX-594 was associated with a significant survival benefit; the median over-all survival was 13.6 months in the high-dose group compared to 4.3 months in the low-dose group (hazard ratio 0.19, P = 0.018, Gehan-Breslow-Wilcoxon test, one-sided test for superiority of the high dose) Fig. 4b). Subjects with multiple tumors (n = 19) had a median survival that was half that of subjects with single tumors (n = 10) in this trial (8.8 months compared to 16.6 months, respectively). The presence or absence of detectable neutralizing antibodies to vaccinia at baseline did not correlate with survival duration (hazard ratio of 0.68 in favor of baseline antibody-positive compared to antibody-negative subjects; P = 0.24, Gehan-Breslow-Wilcoxon test) (Fig. 4c). We performed an exploratory multivariate stepwise regression analysis to evaluate dose and peak genome concentration, neutrophil induction and antibody induction to the JX-594 β-gal transgene; as only 24 subjects were evaluable for all variables, this was only a hypothesis-generating analysis. This multivariate analysis confirmed that dose group was the best predictor of overall survival (χ2 = 2.0085, hazard ratio of 0.358); other variables did not add additional overall survival predictive value after dose.
Table 2.
Correlations by dose
| High dose | Low dose | P | Statistical test | |
|---|---|---|---|---|
| Mean peak acute pharmacokinetics (genomes ml−1 blood)a | 273,700 ± 55,650 | 31,650 ± 5,317 | 0.0002 | t test |
| Median overall survival in patients with multiple tumors (months) | 13.6 | 4.3 | 0.018 | Gehan-Breslow |
| Median overall survival (months) | 14.1 | 6.7 | 0.02 | Gehan-Breslow |
| GM-CSF induction (yes/no) | 11/16 (69%) | 6/13 (46%) | 0.27 | Fisher’s exact |
| Peak GM-CSF concentration (day 5) (pg ml−1)a | 83.8 ± 38.1 | 38.9 ± 18.5 | 0.33 | t test |
| Best mRECIST response (yes/no) | 1/15 (7%) | 3/13 (23%) | 0.31 | Fisher’s exact |
| β-gal–specific antibody induction (day 29) (yes/no) | 12/16 (75%) | 8/13 (62%) | 0.69 | Fisher’s exact |
| Best Choi response (yes/no) | 8/14 (57%) | 8/12 (67%) | 0.70 | Fisher’s exact |
| Peak neutrophil induction (day 5–15) (αl−1)a | 3.0 × 103 ± 0.76 × 103 | 2.7 × 103 ± 0.87 × 103 | 0.75 | t test |
| mRECIST disease control (week 8) (yes/no) | 7/15 (47%) | 6/13 (46%) | 1.0 | Fisher’s exact |
| CDC induction versus SNU739 cell line (≥50% cell viability (yes/no)) | 6/8 (75%) | 5/8 (63%) | 1.0 | Fisher’s exact |
| CDC induction versus HepG2 cell line (≥50% cell viability (yes/no)) | 3/8 (38%) | 3/8 (38%) | 1.0 | Fisher’s exact |
| CDC induction versus SNU475 cell line (≥50% cell viability (yes/no)) | 5/8 (63%) | 4/8 (50%) | 1.0 | Fisher’s exact |
Data are shown as the mean ± s.e.m.
Figure 4.
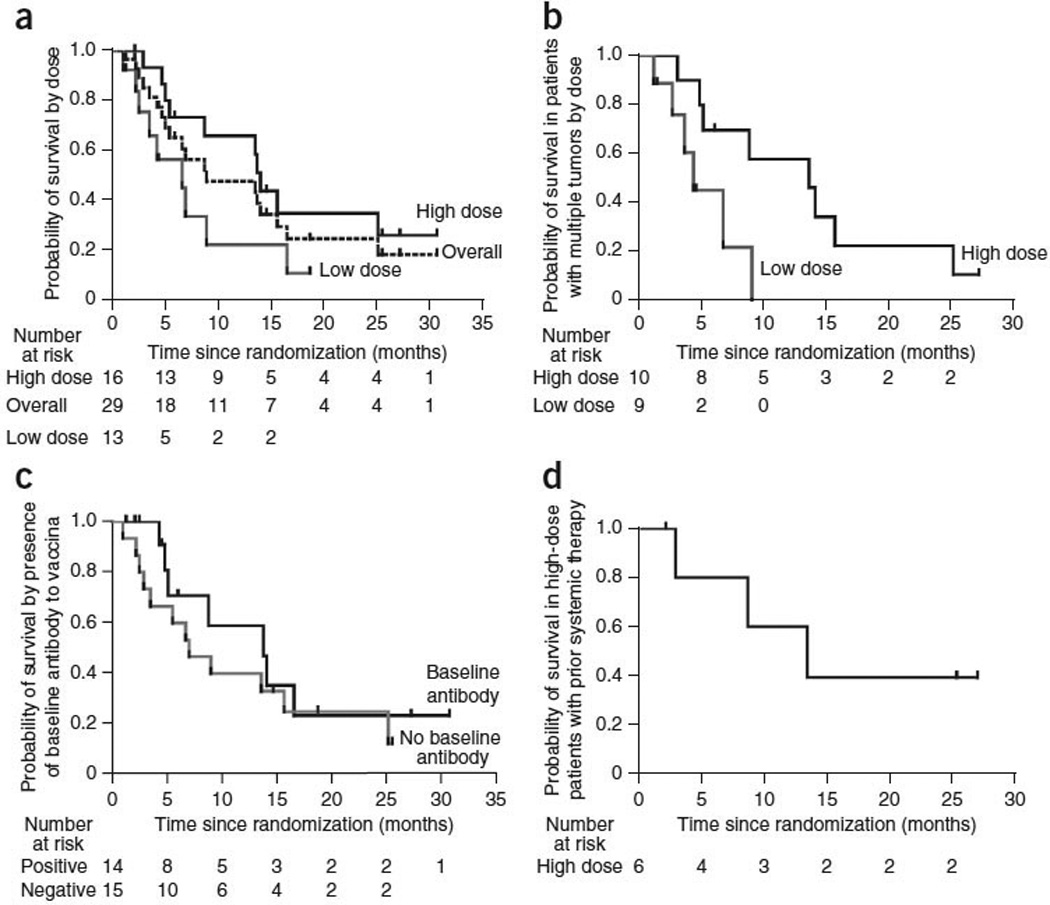
Kaplan-Meier analysis of overall survival. (a) Overall survival in the entire evaluable study population (dashed line) and by dose group (n = 29 total). (b) Overall survival in subjects with multiple tumors at baseline (ten high-dose and nine low-dose subjects). (c) Overall survival in the entire evaluable study population by the presence or absence of baseline neutralizing antibody status. (d) Overall survival in high-dose subjects having previously failed systemic therapy (n = 6).
Six high-dose subjects had previously failed systemic therapy at the time of study enrollment, as described above; four in this group had failed previous treatment with sorafenib. The median survival for this subject population was estimated to be ~2–4 months on the basis of data from randomized phase 3 trials with sorafenib19,20. High-dose subjects in this systemic therapy failure subgroup had a median survival of 13.6 months, and two such subjects were still alive after more than 2 years (Fig. 4d).
DISCUSSIOn
We report here for the first time, to our knowledge, several key findings for JX-594 and the field of oncolytic immunotherapies. First, we demonstrated that JX-594 dose was an important determinant of overall survival duration in subjects with advanced carcinoma; given that oncolytic viruses replicate in tumor cells, it has been predicted that maximizing the dose to the subject would be less important than it is with other therapies in cancer. Second, to our knowledge, this is the first randomized clinical trial showing that an oncolytic virus or gene therapy agent was associated with significantly improved overall survival duration. Of note, the low dose in this trial had clear anticancer efficacy, and therefore, showing a significant survival impact for the high-dose group compared to low-dose active controls was presumably a higher hurdle than a comparison to placebo might have been. Third, we demonstrated the active induction of functional antitumor immunity with an oncolytic virus in multiple subjects within a homogenous population. JX-594 treatment induced a polyclonal humoral immune response resulting in antibody-dependent CDC. In a melanoma clinical trial with herpes simplex virus (HSV) hGM-CSF (T-Vec; Amgen), phenotypic analysis of T cells derived from tumor samples suggested distinct differences from peripheral blood T cells. Compared to T cells derived from nontreated control patients, there was an increase in melanoma-associated antigen recognized by T cells (MART-1)-specific T cells in tumors undergoing regression after vaccination; functional antitumoral immunity, however, was not assessed1.
These trial results address a number of key questions, but others remain unanswered. Although data on diverse MOA were obtained, including JX-594 replication and immune stimulation, the relative importance of each MOA remains to be determined. The only variable that correlated with overall survival duration, besides dose group, was acute peak JX-594 concentration in the blood; of note, blood concentrations in the high-dose group were above the threshold concentration required for i.v. delivery in phase 1 (ref. 15), whereas low-dose concentrations were not. Hence, these data suggest that systemic tumor control and improved survival may be achieved with JX-594 through high-dose i.v. administration. As predicted on the basis of this hypothesis, the survival benefit of high-dose JX-594 was most pronounced in patients with the most extensive tumor burdens. Replication and hGM-CSF transgene expression were associated with three MOA: acute vascular disruption in tumors, tumor oncolysis and necrosis and antitumor immunity. The long-term survival of high-dose patients (~35% at 2 years) after dosing over only 4 weeks suggests a potential durable systemic benefit. Although not proven, this durable efficacy could reflect chronic viral oncolysis or immune-mediated effects, as demonstrated by tumor-lysing antibody generation and lymphocyte infiltration into tumors. Further study will be needed to assess the relative importance of each MOA in specific patient populations. Likewise, although helper T cell engagement was demonstrated by IgG class switching and the induction of T cells specific for the β-gal transgene, further data on tumor-specific T lymphocyte induction will need to be obtained in future trials. In addition, larger trials will allow the exploration of correlations between immune and virus-replication endpoints and patient survival. Choi response criteria include changes in tumor density and maximum diameter. The application of mRECIST and Choi criteria in HCC trials is increasing, and preliminary data suggest that these criteria may be better predictors of overall survival in HCC than the standard RECIST criteria21,22. Nevertheless, more data are needed to know how to interpret these mRECIST and Choi responses.
The survival analysis from this stratified and randomized clinical trial demonstrated a significant dose-dependent survival prolongation. However, these findings should be extended in larger randomized trials with a placebo control arm included. We selected the sample size of 30 patients to give sufficient power for toxicity analyses in both arms. Despite the relatively small sample size and an active control-group treatment, a statistically significant survival benefit was demonstrated because of the large effect size. Prognostic factors were well-balanced between the two arms, with the exception that more high-dose patients had the poor prognostic factor of having failed previous systemic therapy, including sorafenib treatment; the survival duration in this subgroup on high-dose therapy was similar to that of systemic treatment–naive patients. Therefore, the survival benefit of high-dose JX-594 cannot be explained by imbalances in known prognostic factors.
Large randomized trials adequately powered for overall survival effects are underway or have been planned with JX-594 in patients with advanced HCC. In a randomized trial involving patients having failed treatment with sorafenib, patients are being randomized to either JX-594 plus the best supportive (palliative) care or to the best supportive care alone (ClinicalTrials.gov, NCT01387555). Trials in other solid tumor populations are underway, including colorectal carcinomas with a mutant K-RAS genotype. Furthermore, targeted oncolytic vaccinia viruses such as JX-594 can be engineered to express diverse biologics with varied and potentially synergistic MOA; these include cytokines, tumor antigens, checkpoint inhibitors, prodrug-activating enzymes, single-chain antibodies and tumor antigens (reviewed in ref. 6). In particular, exploration of tumor antigen expression is warranted given our demonstration in this trial that a T cell response was induced to the JX-594 transgene β-gal. Given the large transgene-encoding capacity of vaccinia viruses, multiple tumor antigens, as well as complementary cytokines (for example, GM-CSF or interleukin-2 (IL-2)), could be expressed from the same virus backbone. In summary, the oncolytic and immunotherapeutic vaccinia virus JX-594 (Pexa-Vec) holds promise for the treatment of advanced solid tumors, and further clinical trials are warranted.
METHODS
Methods and any associated references are available in the online version of the paper.
Supplementary Material
ACKNoWLeDGMeNTS
J.-M. Limacher, M. Homerin, B.M. Bastien and M. Lusky (all from Transgene SA) gave insightful comments on the manuscript. T.-H.H. and M.K.K. were supported by a grant of the Korea Healthcare technology Research and Development Project, Ministry for Health, Welfare and Family Affairs, Republic of Korea (A091047). C.R. was supported by a pilot grant from the Dan Duncan Cancer Center. M.N. was supported by the Robert and Janice McNair Foundation and Baylor Research Advocates for Student Scientists Fund. J.C.B. is supported by the Ontario Institute for Cancer Research and the Terry Fox Foundation. Funding was provided by Jennerex, Transgene SA (Illkirch, France) and the Green Cross Corporation; grants to T.-H.H. from Korea Healthcare technology R&D Project, Ministry for Health, Welfare and Family Affairs, Republic of Korea; to J.C.B. from the Terry Fox Foundation and the Canadian Institute for Health Research (CIHR); and a pilot grant to C.R. from the Dan Duncan Cancer Center. This trial was registered with ClinicalTrials.gov, number NCT00554372.
Footnotes
AUThOR CONTRIBUTIONS
D.H.K. and R.P. designed the study. C.J.B., D.H.K., T.-H.H., A.M., R.P., T.H., K.D., J.C.B., R.L., L.L., B.-G.R., M.C., C.R. and J.B. analyzed the data and wrote the manuscript. Y.S.L., M.K.K., M.D. and M.N. performed bioanalytical analyses. J.H., T.R., L.R., S.R., M.B., H.Y.L., H.C.C. and C.W.K. enrolled and managed the patients. C.J.B. and D.H.K. had access to all the data in the trial. D.H.K. made the final decision to submit for publication.
CoMPeTING FINANCIAL INTeReSTS
The authors declare competing financial interests: details are available in the online version of the paper.
References
- 1.Kaufman HL, et al. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann. Surg. Oncol. 2010;17:718–730. doi: 10.1245/s10434-009-0809-6. [DOI] [PubMed] [Google Scholar]
- 2.Kirn D, Martuza RL, Zwiebel J. Replication-selective virotherapy for cancer: biological principles, risk management and future directions. Nat. Med. 2001;7:781–787. doi: 10.1038/89901. [DOI] [PubMed] [Google Scholar]
- 3.Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat. Clin. Pract. Oncol. 2007;4:101–117. doi: 10.1038/ncponc0736. [DOI] [PubMed] [Google Scholar]
- 4.Chiocca EA. Oncolytic viruses. Nat. Rev. Cancer. 2002;2:938–950. doi: 10.1038/nrc948. [DOI] [PubMed] [Google Scholar]
- 5.Heise C, Kirn DH. Replication-selective adenoviruses as oncolytic agents. J. Clin. Invest. 2000;105:847–851. doi: 10.1172/JCI9762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat. Rev. Cancer. 2009;9:64–71. doi: 10.1038/nrc2545. [DOI] [PubMed] [Google Scholar]
- 7.Kim JH, et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol. Ther. 2006;14:361–370. doi: 10.1016/j.ymthe.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 8.Parato KA, et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol. Ther. 2012;20:749–758. doi: 10.1038/mt.2011.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hwang TH, et al. A mechanistic proof-of-concept clinical trial with JX-594, a targeted multi-mechanistic oncolytic poxvirus, in patients with metastatic melanoma. Mol. Ther. 2011;19:1913–1922. doi: 10.1038/mt.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heo J, et al. Sequential therapy with JX-594, a targeted oncolytic poxvirus, followed by sorafenib in hepatocellular carcinoma: preclinical and clinical demonstration of combination efficacy. Mol. Ther. 2011;19:1170–1179. doi: 10.1038/mt.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thorne SH, et al. Rational strain selection and engineering creates a broad-spectrum, systemically effective oncolytic poxvirus, JX-963. J. Clin. Invest. 2007;117:3350–3358. doi: 10.1172/JCI32727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wein LM, Wu JT, Kirn DH. Validation and analysis of a mathematical model of a replication-competent oncolytic virus for cancer treatment: implications for virus design and delivery. Cancer Res. 2003;63:1317–1324. [PubMed] [Google Scholar]
- 13.Senzer NN, et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor–encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J. Clin. Oncol. 2009;27:5763–5771. doi: 10.1200/JCO.2009.24.3675. [DOI] [PubMed] [Google Scholar]
- 14.Park BH, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 2008;9:533–542. doi: 10.1016/S1470-2045(08)70107-4. [DOI] [PubMed] [Google Scholar]
- 15.Breitbach CJ, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477:99–102. doi: 10.1038/nature10358. [DOI] [PubMed] [Google Scholar]
- 16.Lencioni R, Llovet JM. Modified RECIST (mRECIST) assessment for hepatocellular carcinoma. Semin. Liver Dis. 2010;30:52–60. doi: 10.1055/s-0030-1247132. [DOI] [PubMed] [Google Scholar]
- 17.Liu TC, Hwang T, Park BH, Bell J, Kirn DH. The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol. Ther. 2008;16:1637–1642. doi: 10.1038/mt.2008.143. [DOI] [PubMed] [Google Scholar]
- 18.Choi H, et al. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J. Clin. Oncol. 2007;25:1753–1759. doi: 10.1200/JCO.2006.07.3049. [DOI] [PubMed] [Google Scholar]
- 19.Llovet JM, et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 20.Cheng AL, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]
- 21.Faivre S, et al. Changes in tumor density in patients with advanced hepatocellular carcinoma treated with sunitinib. Clin. Cancer Res. 2011;17:4504–4512. doi: 10.1158/1078-0432.CCR-10-1708. [DOI] [PubMed] [Google Scholar]
- 22.Edeline J, et al. Comparison of tumor response by Response Evaluation Criteria in Solid Tumors (RECIST) and modified RECIST in patients treated with sorafenib for hepatocellular carcinoma. Cancer. 2012;118:147–156. doi: 10.1002/cncr.26255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.