

Abstract
Notch signaling has emerged as a key player in skeletal muscle development and regeneration. Simply stated, Notch signaling inhibits differentiation. Accordingly, fine-tuning the pathway is essential for proper muscle homeostasis. This review will address various aspects of Notch signaling, including our current views of the core pathway, its effects in muscle, its interactions with other signaling pathways, and its relationship with ageing.
Keywords: Notch, Muscle
Notch: the core pathway
Notch signaling is a critical regulator of metazoan development. The name is derived from a mutant fly with notched wings, described in the early 1900s. In 1985, the Drosophila Notch gene was cloned and shown to encode a single-pass transmembrane protein. Two additional genes, Delta and Serrate, which interact with Notch genetically, were later found to encode proteins that interact with Notch physically. It was quickly appreciated that Notch functions as a cell-surface receptor and mediates cell–cell communication by engaging with ligands (Delta and/or Serrate) on neighboring cells to initiate an intracellular signaling cascade. The pathway is highly conserved; in mammals, there are four Notch receptors (Notch1–4) and five Notch ligands (Jagged 1 and 2 and Delta-like (Dll) 1, 3 and 4). The prototypical effect of Notch signaling in both invertebrate and vertebrate development centers on the control binary cell fate decisions, with Notch often acting to inhibit the differentiation of specific cell types [1].
Notch is activated through three sequential proteolytic cleavage events [2]. The first (S1) is constitutive and is mediated in the Golgi apparatus by a Furin-like protease. S1 cleavage occurs in what eventually becomes the Notch extracellular domain, and generates a meta-stable heterodimer at the plasma membrane. Ligand presented by a neighboring cell is thought either to physically disrupt the heterodimer or to alter its structure such that the second (S2) and third (S3) cleavage events can occur. S2 is mediated by a membrane-associated Adam family metalloprotease while S3 is carried out by the presenillin-containing γ-Secretase complex. The resulting product is the Notch intracellular domain (dubbed NICD or NIC), which is liberated from the plasma membrane and free to enter the nucleus where it activates transcription of target genes (Fig. 1). Mouse embryos homozygous for a Notch1 (N1) mutation that eliminates S3 cleavage are indistinguishable from those that lack a functional N1 gene, arguing that proteolysis is necessary for all Notch-mediated signaling events [3].
Fig. 1.

Schematic representation of Notch signaling in myoblasts and its interactions with other pathways. CSL interacts with transcriptional corepressors (CoR) in the absence of signaling and with coactivators, including Mastermind (MAML), when bound by NICD. This process is enhanced by the binding of Smad proteins (activated by TGFß), Hif1α (stabilized by hypoxia) and Foxo1 (inactivated by Insulin). Relevant gene targets include those encoding Hey1, MyoR and possibly many others. Protein products of the Notch target genes repress transcription of pro-myogenic genes, shown here to be activated by MyoD and Mef2C, through multiple mechanisms, including targeted repression by Hey1.
Genetic studies from Drosophila combined with assays using cultured cells have defined roughly the mechanism of NICD-mediated transcription [4]. Nuclear NICD associates with the DNA binding protein CSL (CBF1, Suppressor of Hairless, Lag-1; also known as RBP-J), which binds the promoters and/or enhancers of Notch-responsive genes. In the absence of signaling, CSL is thought to associate with corepressors, including N-CoR, SMRT, SHARP, and CtIP/CtBP [5-8]. While corepressor-bound CSL may actively repress transcription of Notch targets in the absence of NICD, direct in vivo evidence for this is limited [7]; moreover, CSL may not bind all genes constitutively [9]. Nevertheless, binding of NICD results in the displacement of the corepressors and recruitment of transcriptional coactivators such as Mastermind-like (MAML) and CBP/p300 to target genes [10].
While much is known about Notch activation, less is known about how activity is terminated to ensure proper temporal control of signaling. At one level, activation and termination are linked through the regulation of NICD protein stability. Coincident with activation, MAML recruits the kinase CDK8, which phosphorylates NICD and thus accelerates it degradation by the proteosome [11]. The half-life of NICD has been estimated to be roughly 3 h. Hence, without continued availability of receptor and/or ligand, Notch signaling will quickly diminish. However, a cell culture model showed that with ample ligand Notch signaling can persist for days, which argues against a general mechanism for feedback inhibition [12]. Notch is negatively regulated by the cytoplasmic protein Numb (and the related Numb-like), through a mechanism linked to the process of endocytosis [13,14]. Upon mitosis, Numb can be asymmetrically distributed into newly formed daughter cells, giving rise to asymmetric Notch activity and distinct fate choices for the two daughters [15]. In certain contexts, Notch can down-regulate the expression of Numb and Numb-like, rendering itself resistant to inhibition [16].
Notch and muscle
Notch signaling plays critical roles in the regulation of embryonic and post-natal skeletal myogenesis. It has been known for well over a decade that forced activation of Notch in cultured myoblasts inhibits their differentiation [17], and more recent work has shown this to be true in vivo as well [18-21]. In post-natal myogenesis, muscle injury results in the activation of Notch signaling, as evidenced by increased levels of cleaved Notch1, which may result from the induction of the Notch ligand Dll1 [18]. Experiments in muscle explants demonstrated further that the Notch inhibitor Numb is asymmetrically segregated in dividing myogenic progenitors. Numb-positive daughter cells were found to express lineage progression markers, such as Myf-5 and Desmin, but not the earlier pre-myoblast marker Pax3, while Numb-negative cells had the opposite expression profile. These data suggested that cessation of Notch activity (Numb positive) correlated with progression down the myogenic pathway, while sustained signaling (Numb negative) correlated with maintenance of the undifferentiated state. While this correlative evidence is compelling, a role for Numb in muscle regeneration has yet to be confirmed genetically. (The importance of this is underscored by the recent observation that forced expression of Numb in muscle progenitors neither decreases Notch signaling nor induces precocious differentiation in somites [22]). Additional studies in primary myoblast cultures showed that artificial activation of Notch via retrovirally expressed NICD resulted in enhanced proliferation, while siRNAs targeting Notch1 led to compromised proliferation. Together, these findings argue that activation of Notch signaling following muscle injury promotes the expansion of satellite cells or myogenic precursors and prevents differentiation, while termination of Notch activity allows for subsequent progression down the myogenic lineage (Fig. 2).
Fig. 2.
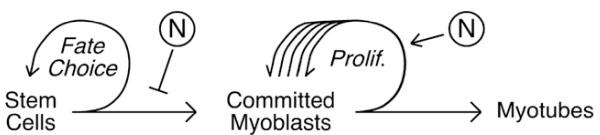
Points of control exerted by Notch signaling during muscle regeneration. Notch has been implicated in 1) the cell fate choice that governs the progression of muscle stem cells to committed progenitors and 2) the proliferation of progenitors prior to terminal differentiation into myotubes.
Studies employing Notch gain-of-function and loss-of-function in vivo strengthened these conclusions. Inhibiting Notch signaling with soluble Jagged was shown to impair regeneration, while enhancing Notch activation with a Notch1-specific antibody facilitated the repair process [19].
Notch signaling has also been implicated in events that distinguish true muscle stem cells (Pax7+/Myf5−) from committed progenitors (Pax7+/Myf5+). These two cell types were observed as newly generated pairs (sisters), with the stem cell more likely to be closely apposed to the muscle fiber and the committed progenitor positioned further away [23]. Interestingly, Notch3 was enriched in the stem cell while Dll1 was expressed primarily in the progenitor. Numb was expressed equally in both cells. These findings suggest that elevated levels of Notch signaling in muscle stem cells, and maintenance of the pool, could result from asymmetric expression of ligand in daughter cells (Fig. 2).
Genetic approaches have been used to demonstrate a role for Notch in embryonic muscle development. Prior studies using various vertebrate models showed that Notch affects somitogenesis, by either helping to specify somite borders or to impose anterior–posterior polarity [24]. Some of these conclusions were based on the analysis of mice harboring gene mutations in various components of the Notch pathway. However, due to the disruption of the somites and embryonic lethality, such mice could not be used to study events further downstream, specifically those involving the actual generation and maintenance of skeletal muscle. To circumvent embryonic lethality caused by a homozygous null mutation in Dll1, mice carrying one null allele and one hypomorphic allele were analyzed [20]. These mice survived until birth, but displayed striking defects in the skeletal muscle lineage. The central phenotype was a dramatic hypotrophy of muscle tissue, which appeared to reflect premature, accelerated muscle differentiation, accompanied by loss of the Pax3+/Pax7+ myogenic progenitor cell pool. A transient excess of myogenic cells was observed early in the embryo, but myogenesis terminated too soon to allow for the ultimate generation of normal musculature. These data argue that Notch signaling prevents precocious differentiation and thus allows the maintenance and expansion of the progenitor pool. Similar findings and conclusions were reported using conditional CSL knockout mice [21]. Genetic deletion of CSL in either somitic or migrating myogenic precursors resulted in premature loss of these progenitors due to precocious differentiation. An absence of satellite cells was also observed in these animals, consistent with previous proposals that the Pax3+/Pax7+ pool of progenitors represents the cellular origin of this stem cell population.
Mechanisms of inhibition
The molecular mechanisms by which Notch exerts its effects in both muscle and other tissues are not well understood. In the last fifteen years, the question of how Notch represses muscle differentiation has been tackled by a number of different studies but has remained controversial and resistant to solution. In 1994, Weintraub et al. observed that forced expression of NICD repressed myoblast differentiation and MyoD-mediated fibroblast conversion in culture [17]. This inhibitory effect was also observed upon ligand-mediated activation of the pathway, in which myoblasts were co-cultured with Jagged1-expressing cells [25]. The block to differentiation imposed by NICD could be overcome by expression of a MyoD-VP16 fusion protein but not a MyoD–E47 tethered dimer [17]. These initial results suggested that NICD might target the inherent transcriptional activity of MyoD without affecting its dimerization or DNA binding. Such conclusions, however, rested upon the use of an artificial MyoD fusion construct and did not directly assess MyoD occupancy at target gene promoters.
Subsequent findings led to an alternative model of Notch action in muscle, whereby Notch would signal through the transcriptional repressor protein Hes1. NICD was shown to directly activate the Hes1 promoter via CSL [4], and forced expression of Hes1 mimicked the inhibitory effects of NICD on fibroblast conversion [26]. It appeared that Hes1 functioned by disrupting the in vitro DNA binding of MyoD/E47 heterodimers, likely by binding to and sequestering E47. While initially appealing, this model relied on overexpressing NICD and using very high levels of transiently transfected Hes1. Indeed, later work demonstrated that Jagged1-mediated Notch signaling in C2C12 myoblasts repressed myogenesis without inducing Hes1 transcription and that stable myoblast lines expressing Hes1 exhibited normal differentiation [27], calling into question the significance of Hes1 induction. In vivo data also revealed that myogenesis was not perturbed in Hes1 knockout mice [28].
Support for a possible MyoD-independent effect of Notch signaling was also provided recently by studies of Hey1 [29]. Hey1 transcription was induced by Notch signaling in myoblasts and constitutive expression of Hey1 was sufficient to block myogenesis. Like Hes1, Hey1 is a bHLH protein and has been reported to form heterodimers with MyoD and/or E47. However, under conditions sufficient to block myogenesis, Hey1 did not disrupt endogenous MyoD:E47 heterodimers. Furthermore, Hey1 did not inhibit MyoD transcriptional activity per se, but rather exerted promoter-specific repression. Hey1 associated with and repressed activity of the promoters of both the Myogenin and Mef2C genes (precise binding sites were not identified) arguing that it acts to target key myogenic loci and not the MyoD protein.
Other studies have proposed that Notch inhibits the activity of additional components necessary for myogenesis. One showed that NICD can interact physically with and inhibit Mef2C in reporter assays [30]. Another focused on the signaling pathway involving the MAP Kinase p38. In those experiments NICD was shown to induce expression of MKP-1, a phosphatase that inactivates p38 [31]. The validity of both studies, however, is uncertain since both employed over-expressed NICD and neither confirmed the results using physiological levels of signaling. A third report showed that MAML can serve as a coactivator for Mef2C and postulated that NICD sequesters MAML away from Mef2C to shut down myogenesis [32]. While an intriguing model, it remains to be shown that MAML is limiting within the nucleus. MAML1 knockout mice exhibit muscle defects and die within a few days of birth, but heterozygous mice are phenotypically normal [33].
A CSL-independent pathway for Notch activity has also been proposed [27,34]. However, the genetic studies mentioned above argue against this. The phenotype of Dll1 null/hypomorph mice was similar to that of the conditional CSL knockout mice, indicating that CSL-dependent signaling accounts for the majority of Notch activity [20,21].
The lack of a clear mechanistic model for Notch’s effects on myogenesis may be due to the presence of multiple, redundant pathways. Evidence for this was derived from a preliminary analysis of ligand inducible genes in C2C12 myoblasts [35]. Over 100 genes were induced within 6 h of exposure of cells to ligand. Several of these had no effect on myogenesis when expressed constitutively, arguing that their induction is irrelevant in myoblasts. Two others, Hey1 and MyoR, blocked myogenesis when expressed constitutively. siRNA-mediated knock-down of Hey1, MyoR or both Hey1 and MyoR had no effect on Notch’s ability to repress myogenesis, arguing that more pathways exist. The identification of Notch-responsive genes in Drosophila, has provided additional evidence that the pathway is robust and indeed may generate different outcomes in different cell types [36].
Crosstalk with other pathways
The view that Notch signaling may act quite generally to maintain stem cells in their undifferentiated state has led to the proposal that it may also be linked to the mechanisms underlying other signaling pathways that inhibit differentiation. Indeed, this has now been demonstrated for BMP signaling and for hypoxia, both of which inhibit myogenesis (Fig. 1). BMPs are members of the TGFß superfamily of ligands. Binding of BMPs/TGFßs to their cognate receptors induces the phosphorylation of receptor Smads (Smad1, Smad2 and Smad3), which activate transcription of target genes as heterodimers with Smad4. Interestingly, the ability of BMP4 to inhibit myogenesis was found to be significantly reversed when Notch signaling was compromised using either a γ-Secretase inhibitor or a dominant-negative version of CSL [37]. BMP4 was shown to induce expression of Hes1 and Hey1 mRNAs in a Notch dependent manner and to synergize with Notch to activate transcription of the Hey1 promoter. This synergy also occurred with artificial promoters lacking potential SMAD binding sites. These data, along with the demonstration of a physical interaction between NICD and SMAD1, led to a model in which SMADs stimulate expression of Notch-responsive genes by augmenting NICD-mediated transcription. The background level of Notch signaling in myoblasts may not be sufficient to significantly inhibit myogenesis, but this low-level activity is key for the inhibitory activity of BMP4. While these pathways clearly interact in cell culture models of myogenesis, it remains to be shown that this interaction is important in vivo.
Hypoxia also inhibits myogenesis and, as was the case for BMP4, was also shown to require Notch signaling. Hypoxia, as compared to a normal level of oxygen, leads to the stabilization of Hif proteins (Hif1α, Hif2α and Hif3α), which activate transcription of target genes as heterodimers with ARNT (Hif1ß Hif2ß and Hif3ß). In myoblasts, hypoxia was shown to induce Hes1 mRNA and to stimulate transcription from both a Hes1 promoter and an artificial promoter containing multiple CSL binding sites [38]. Hif1α was shown further to interact with physically and stabilize NICD and to be recruited to the Hey2 promoter. (Other Notch-responsive promoters were not evaluated due to technological limitations.) Thus, like SMAD1, Hif1a is recruited by NICD to Notch-responsive promoters and this augments an otherwise inconsequential level of Notch signaling. Factor inhibiting HIF1 (FIH-1) can also inhibit the activity of Notch, suggesting an additional level of interaction between the two pathways [39]. The interplay between hypoxia and Notch signaling has yet to be evaluated genetically.
Notch has also been linked, albeit somewhat indirectly, to the Insulin signaling pathway. Insulin initiates a complex cascade of intracellular events, one of which is the phosphorylation and inactivation (through nuclear export) of members the Foxo family of transcription factors. Mutant Foxo proteins that cannot be phosphorylated are constitutively active (nuclear) and have been shown to inhibit myogenesis of cultured C2C12 myoblasts [40]. Interestingly, this inhibition was partially relieved when Notch signaling was inhibited. Constitutively active Foxo1 was shown to stimulate transcription of several Notch target genes, to interact physically with CSL, and to greatly stimulate the recruitment of NICD to its targets (Fig. 1). Given these observations, one would expect Insulin to inhibit Notch signaling by removing Foxo proteins from the nucleus. This prediction has yet to be tested directly.
Finally, Notch has also been linked to Wnt signaling, but in this case inversely. Recent work has suggested that canonical Wnt signaling functions in later phases of a normal regenerative response to promote myogenic differentiation [41]. In the canonical pathway Wnt inactivates GSK3ß, yet Notch signaling was found to sustain the kinase in a form associated with activation. While this suggests a nodal point at which Notch and Wnt may function antagonistically during myogenesis, what governs the necessary transition from Notch signaling to Wnt signaling is not known. By contrast, non-canonical Wnt signaling has been implicated in maintaining satellite cells in their undifferentiated state [42]. A possible relationship between non-canonical Wnt signaling and Notch signaling has not been addressed.
Notch and ageing
The ability to regenerate muscle declines with age. This was shown to be reflected in vitro by the inability of satellite cells (CD34+/M-cadherin+) to proliferate and generate myotubes, but not by an inherent inability to fuse or by a decrease in overall cell numbers [19]. Interestingly, induction of Dll1 in response to injury was diminished in old mice and this led to the hypothesis that impaired Notch signaling might underlie some of the age-related effects. Indeed, forced activation of Notch largely restored regeneration in old muscle. Subsequent experiments using parabiotic parings (in which the circulatory system is shared between mice) established that the blood of young mice could restore regeneration in old mice and that serum from young mice could restore both Dll1 expression and proliferation in old myoblasts in culture [43]. Hence, Notch signaling may play a major role in defining the differences between young and old satellite cells.
Old myoblasts produce elevated levels of TGF-ß1, which, like BMP4, inhibits differentiation [44]. Notch may restore proliferation to old myoblasts in part by negating some of the anti-proliferative effects of TGF-ß. Specifically, TGF-ß, acting through Smad3, was found to induce the expression of several CDK inhibitors, including p15, p16, p21 and p27 [45]. Increased Notch signaling diminished expression of these genes, whereas decreased Notch signaling further induced their expression. NICD interacts physically with Smad3 and this somehow reduces recruitment of Smad3 to a subset of these promoters, suggesting a molecular mechanism by which Notch antagonizes the growth inhibitory properties of TGF-ß.
These findings with Notch and TGF-ß raise a conundrum. On one hand, Notch negates certain effects of TGF-ß, while it supports the effects of BMP4. (The downstream pathways are essentially the same.) This illustrates the difficulty in sorting through the myriad effects of these pathways; for example, Notch may both antagonize the anti-proliferative effects of TGF-ß and support the ability of TGF-ß, through Smads, to induce classic Notch-responsive genes. Furthermore, the ability of Notch to inhibit myogenesis isn’t necessarily linked to its effects on proliferation. Notch signaling does not overcome G1 arrest in C2C12 myoblasts transduced by a p21-expressing Adenovirus, yet it still inhibits differentiation (Shara Kabak and Tom Kadesch, unpublished). Thus, it is likely that the effects of Notch on myoblasts are multiple and diverse.
Concluding remarks
While it is clear that Notch signaling functions at multiple stages of muscle development and regeneration, many questions remain. For example, how can Notch act to maintain a population of uncommitted satellite cells while promoting the expansion of more committed progenitors? What are the key target genes that distinguish these two activities? What are the molecular events underlying the crosstalk that occurs between Notch and other signaling pathways and how, ultimately, are these pathways regulated? Given the central role of Notch in muscle regeneration, answering these questions will be critical if we ever want to manipulate the process and to generate stem cells that can be used therapeutically.
REFERENCES
- [1].Artavanis-Tsakonas S, Matsuno K, Fortini ME. Notch signaling. Science. 1995;268:225–232. doi: 10.1126/science.7716513. [DOI] [PubMed] [Google Scholar]
- [2].Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Huppert SS, Le A, Schroeter EH, Mumm JS, Saxena MT, Milner LA, Kopan R. Embryonic lethality in mice homozygous for a processing-deficient allele of Notch1. Nature. 2000;405:966–970. doi: 10.1038/35016111. erratum appears in Nature 2000 Nov 30;408(6812):616. [DOI] [PubMed] [Google Scholar]
- [4].Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signalling downstream of activated mammalian Notch. Nature. 1995;377:355–358. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- [5].Kao HY, Ordentlich P, Koyano-Nakagawa N, Tang Z, Downes M, Kintner CR, Evans RM, Kadesch T. A histone deacetylase corepressor complex regulates the Notch signal transduction pathway. Genes Dev. 1998;12:2269–2277. doi: 10.1101/gad.12.15.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Oswald F, Kostezka U, Astrahantseff K, Bourteele S, Dillinger K, Zechner U, Ludwig L, Wilda M, Hameister H, Knochel W, Liptay S, Schmid RM. SHARP is a novel component of the Notch/RBP-Jkappa signalling pathway. EMBO J. 2002;21:5417–5426. doi: 10.1093/emboj/cdf549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Oswald F, Winkler M, Cao Y, Astrahantseff K, Bourteele S, Knochel W, Borggrefe T. RBP-Jkappa/SHARP recruits CtIP/CtBP corepressors to silence Notch target genes. Mol. Cell. Biol. 2005;25:10379–10390. doi: 10.1128/MCB.25.23.10379-10390.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Barolo S, Stone T, Bang AG, Posakony JW. Default repression and Notch signaling: Hairless acts as an adaptor to recruit the corepressors Groucho and dCtBP to Suppressor of Hairless. Genes Dev. 2002;16:1964–1976. doi: 10.1101/gad.987402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Krejci A, Bray S. Notch activation stimulates transient and selective binding of Su(H)/CSL to target enhancers. Genes Dev. 2007;21:1322–1327. doi: 10.1101/gad.424607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fryer CJ, Lamar E, Turbachova I, Kintner C, Jones KA. Mastermind mediates chromatin-specific transcription and turnover of the Notch enhancer complex. Genes Dev. 2002;16:1397–1411. doi: 10.1101/gad.991602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fryer CJ, White JB, Jones KA. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol. Cell. 2004;16:509–520. doi: 10.1016/j.molcel.2004.10.014. [DOI] [PubMed] [Google Scholar]
- [12].Ross DA, Kadesch T. Consequences of Notch-mediated induction of Jagged1. Exp. Cell Res. 2004;296:173–182. doi: 10.1016/j.yexcr.2004.02.003. [DOI] [PubMed] [Google Scholar]
- [13].Berdnik D, Torok T, Gonzalez-Gaitan M, Knoblich JA. The endocytic protein alpha-Adaptin is required for numb-mediated asymmetric cell division in Drosophila. Dev. Cell. 2002;3:221–231. doi: 10.1016/s1534-5807(02)00215-0. [DOI] [PubMed] [Google Scholar]
- [14].McGill MA, Dho SE, Weinmaster G, McGlade CJ. Numb regulates post-endocytic trafficking and degradation of Notch1. J. Biol. Chem. 2009;284:26427–26438. doi: 10.1074/jbc.M109.014845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gonczy P. Mechanisms of asymmetric cell division: flies and worms pave the way. Nat. Rev. Mol. Cell Biol. 2008;9:355–366. doi: 10.1038/nrm2388. [DOI] [PubMed] [Google Scholar]
- [16].Chapman G, Liu L, Sahlgren C, Dahlqvist C, Lendahl U. High levels of Notch signaling down-regulate Numb and Numblike. J. Cell Biol. 2006;175:535–540. doi: 10.1083/jcb.200602009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kopan R, Nye JS, Weintraub H. The intracellular domain of mouse Notch: a constitutively activated repressor of myogenesis directed at the basic helix–loop–helix region of MyoD. Development. 1994;120:2385–2396. doi: 10.1242/dev.120.9.2385. [DOI] [PubMed] [Google Scholar]
- [18].Conboy IM, Rando TA. The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev. Cell. 2002;3:397–409. doi: 10.1016/s1534-5807(02)00254-x. [DOI] [PubMed] [Google Scholar]
- [19].Conboy IM, Conboy MJ, Smythe GM, Rando TA. Notch-mediated restoration of regenerative potential to aged muscle. Science. 2003;302:1575–1577. doi: 10.1126/science.1087573. [DOI] [PubMed] [Google Scholar]
- [20].Schuster-Gossler K, Cordes R, Gossler A. Premature myogenic differentiation and depletion of progenitor cells cause severe muscle hypotrophy in Delta1 mutants. Proc Natl Acad Sci U S A. 2007;104:537–542. doi: 10.1073/pnas.0608281104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Vasyutina E, Lenhard DC, Wende H, Erdmann B, Epstein JA, Birchmeier C. RBP-J (Rbpsuh) is essential to maintain muscle progenitor cells and to generate satellite cells. Proc Natl Acad Sci U S A. 2007;104:4443–4448. doi: 10.1073/pnas.0610647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jory A, Le Roux I, Gayraud-Morel B, Rocheteau P, Cohen-Tannoudji M, Cumano A, Tajbakhsh S. Numb promotes an increase in skeletal muscle progenitor cells in the embryonic somite. Stem Cells. 2009;27:2769–2780. doi: 10.1002/stem.220. [DOI] [PubMed] [Google Scholar]
- [23].Kuang S, Kuroda K, Le Grand F, Rudnicki MA. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell. 2007;129:999–1010. doi: 10.1016/j.cell.2007.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lewis J, Hanisch A, Holder M. Notch signaling, the segmentation clock, and the patterning of vertebrate somites. J. Biol. 2009;8:44. doi: 10.1186/jbiol145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lindsell CE, Shawber CJ, Boulter J, Weinmaster G. Jagged: a mammalian ligand that activates Notch1. Cell. 1995;80:909–917. doi: 10.1016/0092-8674(95)90294-5. [DOI] [PubMed] [Google Scholar]
- [26].Sasai Y, Kageyama R, Tagaawa Y, Shigemoto R, Nakanishi S. Two mammalian helix–loop–helix factors structurally related to Drosophila hairy and Enhancer of split. Genes Dev. 1992;6:2620–2634. doi: 10.1101/gad.6.12b.2620. [DOI] [PubMed] [Google Scholar]
- [27].Shawber C, Nofziger D, Hsieh JJ, Lindsell C, Bogler O, Hayward D, Weinmaster G. Notch signaling inhibits muscle cell differentiation through a CBF1- independent pathway. Development. 1996;122:3765–3773. doi: 10.1242/dev.122.12.3765. [DOI] [PubMed] [Google Scholar]
- [28].Ishibashi M, Ang SL, Shiota K, Nakanishi S, Kageyama R, Guillemot F. Targeted disruption of mammalian hairy and Enhancer of split homolog-1 (HES-1) leads to up-regulation of neural helix–loop–helix factors, premature neurogenesis, and severe neural tube defects. Genes Dev. 1995;9:3136–3148. doi: 10.1101/gad.9.24.3136. [DOI] [PubMed] [Google Scholar]
- [29].Buas MF, Kabak S, Kadesch T. The Notch effector Hey1 associates with myogenic target genes to repress myogenesis. J. Biol. Chem. 2010;285:1249–1258. doi: 10.1074/jbc.M109.046441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rawls J, Molkentin JD, Black BL, Olson EN. Activated notch inhibits myogenic activity of the MADS-Box transcription factor myocyte enhancer factor 2C. Mol. Cell. Biol. 1999;19:2853–2862. doi: 10.1128/mcb.19.4.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kondoh K, Sunadome K, Nishida E. Notch signaling suppresses p38 MAPK activity via induction of MKP-1 in myogenesis. J. Biol. Chem. 2007;282:3058–3065. doi: 10.1074/jbc.M607630200. [DOI] [PubMed] [Google Scholar]
- [32].Shen H, McElhinny AS, Cao Y, Gao P, Liu J, Bronson R, Griffin JD, Wu L. The Notch coactivator, MAML1, functions as a novel coactivator for MEF2C-mediated transcription and is required for normal myogenesis. Genes Dev. 2006;20:675–688. doi: 10.1101/gad.1383706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wu L, Maillard I, Nakamura M, Pear WS, Griffin JD. The transcriptional coactivator Maml1 is required for Notch2-mediated marginal zone B-cell development. Blood. 2007;110:3618–3623. doi: 10.1182/blood-2007-06-097030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nofziger D, Miyamoto A, Lyons KM, Weinmaster G. Notch signaling imposes two distinct blocks in the differentiation of C2C12 myoblasts. Development. 1999;126:1689–1702. doi: 10.1242/dev.126.8.1689. [DOI] [PubMed] [Google Scholar]
- [35].Buas MF, Kabak S, Kadesch T. Inhibition of myogenesis by Notch: evidence for multiple pathways. J. Cell. Physiol. 2009;218:84–93. doi: 10.1002/jcp.21571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Krejci A, Bernard F, Housden BE, Collins S, Bray SJ. Direct response to Notch activation: signaling crosstalk and incoherent logic. Sci. Signal. 2009;2:ra1. doi: 10.1126/scisignal.2000140. [DOI] [PubMed] [Google Scholar]
- [37].Dahlqvist C, Blokzijl A, Chapman G, Falk A, Dannaeus K, Ibanez CF, Lendahl U. Functional Notch signaling is required for BMP4-induced inhibition of myogenic differentiation. Development. 2003;130:6089–6099. doi: 10.1242/dev.00834. [DOI] [PubMed] [Google Scholar]
- [38].Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, Ruas JL, Poellinger L, Lendahl U, Bondesson M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev. Cell. 2005;9:617–628. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- [39].Zheng X, Linke S, Dias JM, Zheng X, Gradin K, Wallis TP, Hamilton BR, Gustafsson M, Ruas JL, Wilkins S, Bilton RL, Brismar K, Whitelaw ML, Pereira T, Gorman JJ, Ericson J, Peet DJ, Lendahl U, Poellinger L. Interaction with factor inhibiting HIF-1 defines an additional mode of cross-coupling between the Notch and hypoxia signaling pathways. Proc Natl Acad Sci U S A. 2008;105:3368–3373. doi: 10.1073/pnas.0711591105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kitamura T, Kitamura YI, Funahashi Y, Shawber CJ, Castrillon DH, Kollipara R, DePinho RA, Kitajewski J, Accili D. A Foxo/Notch pathway controls myogenic differentiation and fiber type specification. J Clin Invest. 2007;117:2477–2485. doi: 10.1172/JCI32054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Brack AS, Conboy IM, Conboy MJ, Shen J, Rando TA. A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell. 2008;2:50–59. doi: 10.1016/j.stem.2007.10.006. [DOI] [PubMed] [Google Scholar]
- [42].Le Grand F, Jones AE, Seale V, Scime A, Rudnicki MA. Wnt7a activates the planar cell polarity pathway to drive the symmetric expansion of satellite stem cells. Cell Stem Cell. 2009;4:535–547. doi: 10.1016/j.stem.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760–764. doi: 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- [44].Liu D, Black BL, Derynck R. TGF-beta inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev. 2001;15:2950–2966. doi: 10.1101/gad.925901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Carlson ME, Hsu M, Conboy IM. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature. 2008;454:528–532. doi: 10.1038/nature07034. [DOI] [PMC free article] [PubMed] [Google Scholar]