

Abstract
Background and Objective
The aims of the present study were to investigate the thermal-dose dependent effect of heat stress on hepatocyte and HCC cell death mechanisms using clinically relevant experimental heat stress conditions in vitro and to investigate apoptotic cell death induced by laser thermal ablation in vivo.
Study Design/Materials and Methods
Institutional Animal Care and Use Committee approved all studies. Hepatocyte and HCC cell lines were heat stressed from 37°C to 60°C for 2 or 10 minutes and assessed for viability, cytotoxicity and caspase-3/7 activity at 6 and/or 24 hours post-treatment (N=3). Viability experiments were repeated with the RIPK1 inhibitor Necrostatin-1 to block necroptosis (N=3). Rats with orthotopic HCC tumors stably expressing luciferase (N1S1luc2) were randomized to US-guided laser ablation (3W-45s for an intentional partial ablation; N=6) or sham (N=6) and followed by post-ablation caspase-3/7 bioluminescence imaging at 6 and 24 hours and cleaved caspase-3 immunostaining. p<0.05 was considered statistically significant.
Results
Heat-stress induced apoptosis and necrosis in hepatocytes and HCC cells in a thermal dose and cell-type dependent manner. Inhibition of RIPIK1-mediated necroptosis induced a significant, differential increase in HCC cell viability under physiologic and hyperthermic heat stress (p<0.001). Intentional partial laser thermal ablation induced a significant increase in caspase-3/7 activity in the laser versus sham ablation groups at both 6 hours (10.1-fold, p<0.01) and 24 hours (16.7 fold, p<0.02). Immunohistochemistry confirmed increased cleaved caspase-3 staining at the tumor ablation margin 24 hours post-ablation.
Conclusions
Both regulated and non-regulated cell death mechanisms mediate heat stress-induced HCC cell killing and vary between hepatocytes and HCC subtypes. Apoptosis is a significant mechanism of cell death at the HCC tumor ablation margin.
Keywords: Apoptosis, Necroptosis, Necrosis
INTRODUCTION
Imaged-guided thermal ablative therapies including laser ablation have emerged as safe and effective, minimally invasive treatment options in the multidisciplinary care of non-transplantable patients with hepatocellular carcinoma (HCC) (1–4). Analysis of several clinical trials has demonstrated excellent local control rates and improved overall survival for HCC patients treated with thermal ablation for tumors smaller than 2–3 centimeters (5–7). However, as tumor size increases, thermal ablation of HCC is plagued by high local recurrence and tumor progression rates, particularly for tumors beyond 2–3 centimeters in size, and overall survival remains poor for these patients (5,8–10). The ablation margin is a frequent site of local tumor progression due to sublethal thermal injury, thereby resulting in incomplete treatment and viable residual disease (10–13).
The primary mechanism of action of thermal ablative therapies is cytotoxic heat stress with the goal of achieving focally high temperatures (>60°C) in a relatively short period of time (≤10 minutes) to induce irreversible cell injury throughout the tumor volume (14). However, the threshold temperature and/or exposure time at which heat stress induces cell death varies by tissue and cell type, thus cell-type specific mechanisms may confer sensitivity or resistance to heat stress-induced cell death (15). Previous in vitro studies have demonstrated that heat stress induces both rapid and slow forms of cell death, suggesting that heat stress may induce cell death via multiple mechanisms with varying kinetics (16). Non-programmed coagulation necrosis has been considered the primary mechanism of HCC cell death induced by thermal ablative therapies (17,18). However, the role of programmed or genetically controlled cell death mechanisms such as apoptosis and necroptosis in the regulation of heat stress-induced HCC cell killing is less well understood (19).
Given the variation in temperatures achieved at the ablation margin, uniform coagulation necrosis throughout the entire tumor volume is unlikely and some regions may receive a lower thermal dose (15,20). Consequently, injured neoplastic cells may or may not progress to irreversible cell injury depending on the regulation of cell death. Dysregulation or loss of endogenous cell death mediators may limit the efficacy of thermal ablative therapies for HCC (21). As such, there remains a need to further delineate the basic mechanisms of heat stress-induced HCC cell death, particularly at the ablation margin, in order to develop therapeutic strategies for enhancing thermal ablation-induced HCC cell killing.
The aims of the present study were to investigate the thermal-dose dependent effect of heat stress on hepatocyte and HCC cell death mechanisms using clinically relevant experimental heat stress conditions in vitro and to investigate apoptotic cell death induced by laser thermal ablation throughout the ablation zone in vivo.
MATERIALS AND METHODS
Cell Lines
The Clone9 rat hepatocyte and N1S1 rat HCC (ATCC, Manassas, VA) and AS30D rat HCC (DSMZ, Braunschweig, Germany) cell lines were cultured according to supplier recommendations.
In Vitro Heat Stress Protocol
The indicated cell lines were suspended in complete media in microcentrifuge tubes and heat stressed at the indicated temperatures (°C) in an isothermic water bath (22). Treatment temperature was monitored using an Omega HH41 digital thermometer (Omega Engineering, Stamford, CT) and maintained to within ±0.05°C.
Cell Death Assay
Clone9, N1S1 and AS30D cells were heat stressed at the indicated temperatures for exposure times of 2 or 10 minutes and plated in 96-well tissue culture plates (N=3). At 6 and/or 24 hours post heat stress, the ApoTox-Glo™ Triplex assay (Promega) was performed per manufacturer instruction. Briefly, the assay combines two fluorogenic substrates—one live cell protease and one dead cell protease— to measure cell viability and cytotoxicity and one luminogenic caspase-3/7 substrate to measure apoptosis within a single assay well (23,24). Fluorescence (GF-AFC, bis-AAF-R110) and luminescence were measured on a DTX 880 microplate reader (Beckman Coulter). Florescence and luminescence data were normalized to the non-heat stressed (37°C) control to determine relative cell viability, cytotoxicity and apoptosis and plotted versus temperature.
Necroptosis Inhibition
N1S1 and AS30D cells were pre-treated with the RIPK1 inhibitor necrostatin-1 (10μM; Sigma Aldrich) or vehicle control (0.1% DMSO) for one hour followed by heat stress at temperatures ranging from 37°C–50°C for 10 minutes (N=3) (25). Cells were analyzed for viability using WST-1 assay (Roche) per manufacturer’s instruction at 24 hours post-heat stress. Absorbance data were normalized to the 37°C vehicle control to determine relative cell viability.
In Vivo Ablation Study
Luciferase Plasmid Reporter Transfection
N1S1 cells were stably transfected with a pGL4.51[luc2/CMV/Neo] luciferase reporter vector (Promega) using X-tremeGENE HP DNA Transfection reagent (Roche) followed by positive selection in G418 (Life Technologies) and establishment of clonal populations using limiting dilution method as previously described (26).
Animal Model
Studies were approved by the Institutional Animal Care and Use Committee. N1S1 orthotopic HCC model was developed as previously described using N1S1luc2 cells stably expressing firefly luciferase (N=12) (26,27).
Pre-Ablation Imaging
N1S1luc2 tumor-bearing rats were anesthetized and imaged using non-contrast enhanced 3T magnetic resonance imaging (MRI; GE Healthcare) to confirm tumor size and location as previously described (27). To assess baseline tumor function, two-dimensional bioluminescence imaging (BLI) and three-dimensional diffuse luminescence tomography (DLIT) were performed beginning 10 minutes after a subcutaneous injection of sterile D-luciferin (150 mg/kg; Gold BioTechnology) using an IVIS200 (Caliper, a PerkinElmer Company) optical imaging system as previously described (26).
Ultrasound (US)-guided Laser Ablation
Rats were randomized to thermal ablation (N=6) or sham ablation (N=6). All ablation experiments were performed using an FDA-approved 980-nm laser generator (Visualase, Houston, TX) (26,27). Under ultrasound-guidance (logiq E9 Ultrasound, GE Healthcare), a bare 400μm core optical laser fiber with a 1.0 cm diffusing tip was inserted at the tumor margin. For the ablation group, tumors were ablated at a power setting of 3 watts for 45 seconds under continuous US-monitoring in order to generate an intentional partial ablation. The laser was not activated for sham-ablated animals.
Post-Ablation Imaging
Rats underwent repeat 2D BLI and 3D DLIT imaging at 6 and 24 hours post-ablation following an intraperitoneal injection of VivoGlo™ Caspase-3/7 Substrate (100 mg/kg; Promega). Z-DEVD–Aminoluciferin is a prosubstrate containing the DEVD tetrapeptide sequence recognized by caspase-3 and -7. The DEVD peptide is cleaved in the presence of activated caspase-3 or -7 thereby liberating the aminoluciferin to react with luciferase and generate light. Thus, the light output is a sensitive and specific measure of real-time intratumoral caspase-3/7 activity (28,29).
Immunohistochemistry
Following the last imaging session, rats were euthanized using CO2 inhalation. Liver/tumor tissue was removed and all specimens were placed in 10% neutral buffered formalin, embedded in paraffin and sectioned with a microtome for immunohistochemical analysis. Paraffin-embedded sections were stained with cleavage specific caspase-3 antibody (#9661; Cell Signaling Technology) per manufacturer instruction using methods previously described (30). All sections were reviewed by an experienced pathologist (>20 years) in a blinded and random fashion to assess for tumor/liver immunostaining as previously described (30). Digital images were captured using a Leica DMLB microscope (Leica Microsystems) equipped with a MicroPublisher 3.3 RTV camera (Q-Imaging, Surrey, BC), and MetaVue Imaging System (V.6.3r2; Universal Imaging Corp, Downington, PA).
Image Analysis
MRI data sets were analyzed as previously described (26,27). Tumor dimensions were measured from the FSE T2 images in the axial, coronal and sagittal planes. Tumor volumes (mm3) were calculated as previously described (26,27). Two-dimensional BLI and 3D DLIT image data sets were analyzed using Living Imaging Software 4.2 (Caliper, a PerkinElmer Company) as previously described (26). Regions of interest were automatically segmented onto the 2D planar BLI image with a 25% maximum threshold and the mean radiance (photons/s/cm2/sr) calculated for each animal as a measure of tumor viability (D-luciferin) or caspase-3/7 activity (Z-DEVD–Aminoluciferin).
Statistical Analysis
Statistical analyses were performed by using Prism 5.0 (GraphPad Software, Inc., La Jolla, CA). Differences between treatment groups were compared with an unpaired t test (or Exact Mann-Whitney test) or one-way analysis of variance (ANOVA) followed by post-hoc pairwise comparison using an unpaired t test. p<0.05 was considered statistically significant.
RESULTS
Heat stress induces differential effects on apoptotic and necrotic cell death in a thermal dose and cell-type dependent manner
To determine the effect of thermal dose (temperature, exposure time) on the kinetics and mechanism(s) of hepatocyte and HCC cell death, heat stress of hepatocyte (Clone9) and HCC (N1S1 and AS30D) cells at temperatures from 37°C to 60°C for 2 or 10 minutes followed by assessment for viability, cytotoxicity and caspase-3/7 at 6 and/or 24 hours post heat stress was performed. The Clone9 (Figure 1A, 1D), N1S1 (Figure 1B, 1E) and AS30D (Figure 1C, 1F) dose-response curves demonstrate that heat stress induces differential effects on cell death mechanisms in a thermal-dose and cell-line dependent manner. As evidenced by increased caspase-3/7 activity with decreasing viability and increasing cytotoxicity relative to the non heat-stressed control, there was early induction of apoptosis at lower temperatures (45°C to 50°C) for a given exposure time that was greater at 6 hours than 24 hours and different in magnitude across all three cell lines (Figure 1, 2). In general, as temperature increased for a given exposure time, there was a transition from apoptosis to apoptosis±necrosis to active or late necrosis alone (≥47.5°C to 52.5°C) but the absolute temperature transitions occurred in a cell-line dependent manner (Figure 1, 2).
Figure 1.

Effect of heat stress temperature and exposure time on induction of apoptosis and necrosis 24 hours post heat stress in hepatocyte and HCC cells in vitro. (A, D) Clone9 hepatocyte, (B, E) N1S1 HCC and (C, F) AS30D HCC cells heat-stressed at temperatures from 37°C–60°C for (A, B, C) 2 minutes or (D, E, F) 10 minutes were assessed for viability (black line), cytotoxicity (red line) and caspase-3/7 activity (blue line) at 24 hours post-heat stress using the ApoTox-Glo™ Triplex assay (Promega). Data were normalized to 37°C control and presented as mean±SEM of 3 independent experiments. NE= no effect. EA= early apoptosis. A= apoptosis. N= necrosis. LN= late necrosis. D= dead
Figure 2.

Effect of heat stress temperature and exposure time on induction of caspase-3/7 activity 6 and 24 hours post heat stress in hepatocyte and HCC cells in vitro. (A, D) Clone9 hepatocyte, (B, E) N1S1 HCC and (C, F) AS30D HCC cells heat-stressed at temperatures from 37°C–60°C for 2 (blue line) or 10 (red line) minutes were assessed for caspase-3/7 activity at (A, B, C) 6 hours and (D, E, F) 24 hours post-heat stress using the CaspaseGlo 3/7 assay (Promega). Data were normalized to 37°C control and presented as mean±SEM of 3 independent experiments.
Previous studies have suggested that N1S1 HCC cells are more resistant to heat stress than AS30D HCC cells (22). Further analysis of the early temporal evolution of heat stress induced cell death in N1S1 HCC cells demonstrated active necrosis at 6 hours post heat stress at temperatures ≥55°C and ≥50°C for 2 and 10 minute exposure times, respectively, with progressive necrosis at temperatures ≥52.5°C and ≥47.5°C for 2 and 10 minute exposure times by 24 hours post heat stress (Figure 3). In addition, there was evidence of early induction of apoptosis at 6 hours post heat stress at temperatures in the range of 45°C to 50°C for 2 and 10 minute exposure times but limited progression to apoptosis at lower temperatures by 24 hours (Figure 3).
Figure 3.

Effect of heat stress temperature and exposure time on induction of apoptosis and necrosis 6 and 24 hours post-heat stress in N1S1 HCC cells. N1S1 cells heat-stressed at temperatures from 37°C–60°C for (A, B) 2 or (C, D) 10 minutes were assessed for viability (black line), cytotoxicity (red line) and caspase-3/7 activity (blue line) at (A, C) 6 hours and (B, D) 24 hours post-heat stress using the ApoTox-Glo™ Triplex assay (Promega). Data were normalized to 37°C control and presented as mean±SEM of 3 independent experiments. NE= no effect. EA= early apoptosis. A= apoptosis. N= necrosis. LN= late necrosis. D= dead.
Inhibition of necroptotic cell death induces differential effects on HCC cell viability under physiologic and hyperthermic heat stress
Preliminary experiments suggested that heat stress-induced cell death transitions from apoptosis to necrosis in the range from 45°C–50°C and that the kinetics of necrosis are slower at 50°C than at higher temperatures (Figure 1–3). To determine if necroptosis mediated by receptor-interacting protein kinase 1 (RIPK1) may in part regulate heat stress-induced HCC cell death at intermediate thermal doses, pre-treatment of N1S1 and AS30D cells with Necrostatin-1, an ATP-competitive, allosteric inhibitor of RIPK1 followed by heat stress at temperatures from 37°C–50°C for 10 minutes and assessment of viability at 24 hours was performed (Figure 4) (25,31,32). Inhibition of RIPK1 resulted in a significant increase in cell viability relative to vehicle treated cells under both physiologic (37°C) and hyperthermic (45°C–50°C) temperatures in the N1S1 cell line (p<0.001; Figure 4A) but only under hyperthermic temperatures in the AS30D cell line (p<0.001; Figure 4B).
Figure 4.
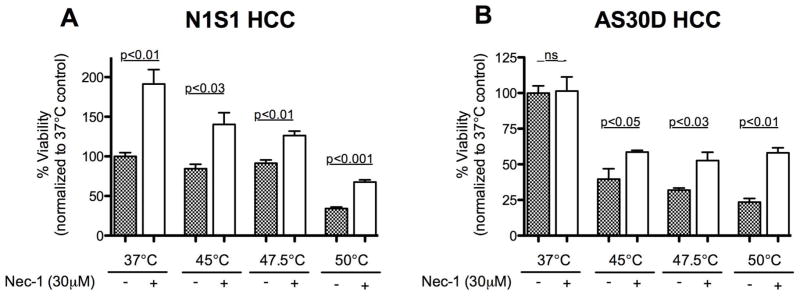
Effect of inhibition of necroptosis mediator RIPK1 on N1S1 and AS30D HCC cell viability following heat stress. (A) N1S1 and (B) AS30D HCC cells pre-treated with the RIPK1 inhibitor necrostatin-1 (30μM) or vehicle control (0.1% DMSO) for one hour followed by heat stress at the indicated temperatures ranging from 37°C–50°C for 10 minutes were assessed with WST-1 viability assay at 24 hours post-heat stress. Data were normalized to 37°C control and analyzed using one-way analysis of variance (ANOVA) followed by unpaired t-test. Data are presented as mean±SEM (N=3).
Intentional partial tumor ablation induces caspase-3/7 activity at the tumor ablation margin in vivo
Previous studies have demonstrated that lower thermal doses are achieved at the thermal ablation margin in vivo and the in vitro data herein suggested that apoptosis is induced at lower thermal doses (Figure 1–3) (15,20). To determine if laser thermal ablation induces apoptosis at the tumor ablation margin in vivo, an intentional partial laser ablation or sham ablation was performed in rats bearing orthotopic N1S1luc2 HCC tumors with pre- and post-ablation bioluminescence imaging.
At baseline, there was no difference in tumor volume (mm3), 2D radiance (photons/sec/cm2/sr) or 3D flux (photons/sec) between the laser ablation and the sham ablation groups (p>0.05, all; Figure 5). However, intentional partial thermal ablation induced a significant increase in in vivo caspase-3/7 activity in the laser ablation v. the sham ablation group that was elevated at both 6 hours (10.1-fold; 1.37×106± 9.0×105 v. 1.35×105± 8.7×104 p/s/cm2/sr, respectively; p<0.01) and 24 hours (16.7 fold, 6.8×105± 5.0×105 v. 4.1×104± 2.6×104 p/s/cm2/sr, respectively; p<0.02) (Figure 6). The average radiance was higher at 6 hours than 24 hours in the laser ablation group although the data did not reach statistical significance (p=0.1). Representative 2D images from baseline tumor function and post ablation caspase-3/7-activity imaging at 6 and 24 hours are shown in Figure 7. Post-ablation cross-sectional 3D DLIT caspase-3/7 imaging was feasible in laser ablation group but not the sham ablation group due to lack of induction of bioluminescent signal (Figure 8). Immunohistochemical (IHC) staining at 24 hours post-ablation demonstrated minimal cleaved caspase-3 staining within the tumor or at the margin between tumor and liver in the sham ablation group (Figure 9A, 9B). On the other hand, IHC staining demonstrated increased cleaved caspase-3 staining at the tumor ablation margin that decreased radially from the ablation margin in the laser ablation group (Figure 9C, 9D).
Figure 5.

Baseline N1S1 tumor volume and bioluminescence in sham versus laser ablation groups. N1S1luc2 tumor-bearing rats randomized to sham ablation (N=6) or laser ablation (N=6) underwent pre-ablation non-contrast enhanced 3 Tesla magnetic resonance imaging (MRI; GE Healthcare) to confirm tumor size and location as well as two-dimensional bioluminescence imaging (BLI) and three-dimensional diffuse luminescence tomography (DLIT) using an IVIS200 optical imaging system (Caliper, a PerkinElmer Company) to assess tumor function. Representative pre-ablation axial fast spin echo (FSE) T2-weighted MR images of N1S1 tumors in (A) sham and (B) laser ablation groups demonstrate hyperintense T2-weighted N1S1 tumors (denoted by white arrowhead). (C) Tumor volumes were calculated from MR images, (D) average radiance (photons/sec/cm2/sr) from 2D BLI images and (E) total flux (photons/sec) from 3D DLIT images and compared between sham and laser ablation groups using an unpaired t-test (or Exact Mann Whitney test). There were no significant differences in any of the baseline parameters between groups (p>0.05). Data are presented as mean±SEM.
Figure 6.
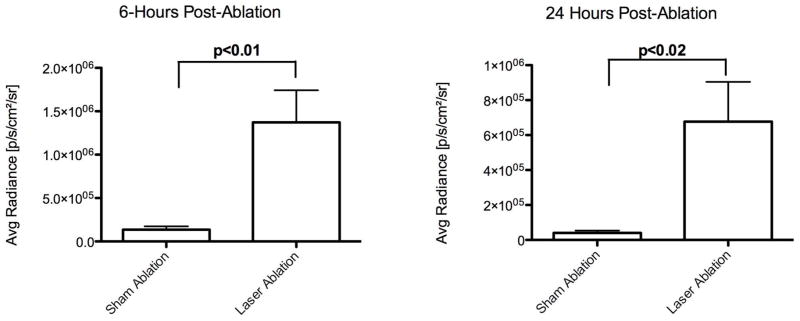
Quantitation of real-time N1S1 tumor caspase-3/7 activity 6- and 24-hours post-ablation in sham versus laser ablation groups. N1S1luc2 tumor-bearing rats underwent two-dimensional caspase-3/7 bioluminescence imaging (BLI) (A) 6 hours and (B) 24 hours post-sham or laser ablation to assess apoptosis. Average radiance (photons/sec/cm2/sr) was measured from 2D BLI images and compared between sham and laser ablation groups using an unpaired t-test (or Exact Mann Whitney test). Data are presented as mean±SEM.
Figure 7.

Representative two-dimensional (2D) bioluminescent images of baseline tumor function and 6- and 24-hour post-ablation caspase-3/7 activity in sham and laser ablation groups. 2D BLI images from (top row) laser and (bottom row) sham ablation groups demonstrate similar baseline tumor function (A, D) but increased caspase-3/7-activity at 6 (B, E) and 24 hours (C, F) post ablation in the laser ablation group (B, C) but not the sham ablation group (E, F). All images on same bioluminescent scale.
Figure 8.

Representative (A) coronal, (B) sagittal, (C) planar and (D) axial images of real-time intratumoral caspase-3/7 activity from three-dimensional (3D) diffuse luminescence tomography (DLIT) 6-hours post laser ablation.
Figure 9.

Representative cleaved caspase-3 immunostaining of N1S1 tumors from sham and laser ablation groups. A, low power (40x) and B, high power (100x) photomicrographs from the sham ablation group demonstrate very few cells staining positive (brown) for cleaved caspase-3 in the tumor (*), background liver or at the liver-tumor margin (denoted by black arrowheads). C, low power (40x) and D, high power (100x) photomicrographs from the laser ablation group demonstrate focal areas of markedly increased cleaved caspase-3 immunostaining at the tumor ablation margin (denoted by black arrowheads) with decreased immunostaining further from the ablation margin toward the non-ablated tumor.
DISCUSSION
Anti-cancer therapies including thermal ablation induce cellular stress within neoplastic cells resulting in reversible cell injury with recovery or irreversible cell injury with cell death. Apoptosis and necrosis have been the most widely recognized modes of cell death in mammalian cells, including liver cell types (33–35). However, further research into molecular basis of cell death has revealed that there are numerous cell death mechanisms with distinct morphologic, biochemical and molecular features and regulated by divergent and interrelated signaling pathways under both physiologic and stress states (34,36). Recent studies have focused on the interplay between endogenous necrosis, necroptosis and apoptosis mediating tumor response to anti-cancer therapies but their roles in the regulation of heat stress-induced HCC cell death remain unclear (37).
The present in vitro studies provide evidence that clinically relevant heat stress conditions induce both apoptotic and necrotic cell death in both hepatocytes and HCC cells in a thermal dose and cell-type dependent manner. Above a certain thermal dose threshold, these data suggest that necrosis is the principal mechanism of heat stress-induced cell death for both hepatocytes and HCC cells and that the higher the temperature for a given exposure time, the more rapid the necrosis, findings consistent with previous thermal ablation studies (17). Although loss of cell membrane integrity may be the final common endpoint for necrosis, the increasing rate of necrosis induction seen with increasing thermal dose suggested that heat stress-induced cell death may transition from non-programmed, non-regulated necrosis at higher thermal doses to programmed, regulated necrosis or necroptosis at lower thermal doses.
Interestingly, inhibition of RIPK1, the principal kinase mediating induction of necroptosis, increased HCC cell viability under both physiologic (37°C) and hyperthermic (45–50°C) temperatures in the N1S1 cell line but only under hyperthermic conditions in the AS30D HCC cell line. These data suggest that necroptosis may in part regulate heat stress-induced HCC cell death as well as basal HCC cell growth in a cell-line specific manner. Molecular profiling studies have determined that HCC is heterogeneous malignancy comprised of different prognostic subtypes resulting from dysregulation of multiple signaling pathways (38,39). Previous studies have shown that the N1S1 HCC cell line is a more rapidly proliferating, metastatic HCC model whereas the AS30D cell line is a slower growing, non-metastatic HCC model (27,30). Taken together, these data suggest that necroptosis may differentially regulate cell fate in different HCC subtypes, thereby underscoring the importance of utilizing biologically diverse HCC model systems for investigating the regulation of endogenous and heat stress-induced HCC cell death to improve the translational relevance of the findings.
Additionally, these data provide evidence that below a thermal dose threshold the mechanism of heat stress-induced cell death transitions from necrosis to apoptosis, findings consistent with previous studies in other non-HCC model systems (40,41). The in vivo data demonstrate that an intentional partial laser ablation induces a significant, time-dependent increase in caspase-3/7 activity, greater at 6 hours than 24 hours post-ablation. These findings are consistent with the in vitro data as well as previous studies demonstrating that apoptosis is induced in the early post-ablation period in both normal liver and breast cancer models (42–44). Of note, the immunohistochemical staining of the post-ablation zone confirms that the increased cleaved caspase-3 activity is preferentially localized to the tumor ablation margin, similar to findings by Yang and Solazzo in a breast cancer model (43,44). As such, these data suggest that apoptosis may be a significant mechanism of cell death at the tumor ablation margin.
However, there are several considerations. First, the thresholds for transition from necrosis to apoptosis are not uniform across hepatocyte and HCC cell lines. Second, the magnitude of apoptosis induction appears to be greater in the normal hepatocytes relative to the HCC cells. Third, above a critical thermal dose threshold, caspase-3/7 activity is inhibited, suggesting loss of function of the apoptotic machinery. Lastly, the data do not provide evidence for progressive caspase-3/7 activity at lower temperatures in HCC cells over time. These findings raise the question, do anti-apoptotic mechanisms inhibit apoptosis induced by heat stress over time? One of the hallmarks of neoplastic cells is evasion of apoptotic cell death (45). In HCC, the anti-apoptotic proteins Bcl-XL, Mcl-1, c-IAP1, XIAP and survivin are frequently over-expressed, thereby promoting an anti-apoptotic phenotype and resulting in loss of endogenous apoptosis (46). Given the frequent dysregulation of apoptotic cell death pathways and upregulation of anti-apoptotic mechanisms in HCC, reliance on apoptosis to kill HCC, particularly at the ablation margin, may result in incomplete HCC cell killing and subsequent tumor progression (47,48). As such, development of adjuvant therapeutic strategies inhibiting anti-apoptotic mechanisms or targeting alternative cell death pathways may restore or enhance endogenous cell death mechanisms induced by thermal ablation in HCC (21,37,43,44).
There are limitations to these studies. The in vitro experiments utilized high throughput screening assays to identify experimental heat stress conditions that induce necrosis and apoptosis. Moreover, the use of RIPK1 inhibitor necrostatin to interrogate heat stress necroptosis provides preliminary evidence of a role for necroptosis in the regulation of heat stress-induced HCC cell death. However, these findings raise several questions for further investigation: what signaling mechanisms upstream of RIPK1 are induced by heat stress? Do anti-necroptotic mechanisms exist to prevent heat stress-induced cell death? Does loss of one programmed cell death pathway such as necroptosis shift cells toward other mechanisms such as apoptosis? Does necroptosis occur at the tumor ablation margin in vivo? Additional methods for assessing the morphologic, biochemical and molecular features of heat stress-induced necrosis, necroptosis and apoptosis are needed to more specifically interrogate their regulation in different HCC cell lines (49). Moreover, given the known crosstalk between apoptotic and necroptotic cell death pathways, their interrelated regulation of heat stress-induced HCC cell death at lower thermal doses warrants further investigation (31). Additionally, the role of autophagy as both a pro-death and a pro-survival mechanism in the regulation of heat stress-induced HCC cell death warrants investigation in this model system (37). Lastly, the in vivo ablation experiments were limited to the N1S1 model (27). Given the differences in heat stress-induced cell death between the N1S1 and AS30D cell lines in vitro, these studies warrant further investigation in the AS30D model as well (30). Despite these limitations, the experiments herein provide a framework for further studies examining the molecular mechanisms of heat stress-induced HCC cell death in biologically diverse HCC models.
These experiments suggest that both regulated and non-regulated cell death mechanisms mediate heat stress-induced HCC cell killing and that the regulation of heat stress-induced cell death may vary between hepatocytes and different HCC subtypes. Overall these findings raise the question, does HCC response to thermal ablation vary between types of tumors due to differences in the regulation of heat stress induced cell death pathways? Further studies examining the molecular basis of heat stress-induced HCC cell death may aid in the development of individualized therapeutic strategies to enhance thermal ablation-induced HCC cell killing at lower temperatures, particularly at the ablation margin.
Acknowledgments
Funding Information
This publication was supported by CTSA Grant Number TL1 TR000137 from the National Center for Advancing Translational Science (NCATS). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. Additional research support provided in part by the American Society for Lasers in Medicine and Surgery (ASLMS) and the Society of Interventional Radiology (SIR) Foundation. Infrastructure support provided by NIH construction grant NIH C06 RR018898.
Footnotes
The results of this work were partially presented at the “33rd ASLMS Annual Conference” held in Boston, MA, April 2013.
Contributor Information
Scott M. Thompson, Medical Scientist Training Program, Mayo Clinic, 200 First Street SW Rochester, MN, 55905
Matthew R. Callstrom, Department of Radiology, Mayo Clinic, 200 First Street SW Rochester, MN, 55905
Kim A. Butters, Department of Radiology, Mayo Clinic, 200 First Street SW Rochester, MN, 55905
Bruce Knudsen, Department of Laboratory Medicine and Pathology, Mayo Clinic, 200 First Street SW Rochester, MN, 55905
Joseph P. Grande, Department of Laboratory Medicine and Pathology, Mayo Clinic, 200 First Street SW Rochester, MN, 55905
Lewis R. Roberts, Division of Gastroenterology and Hepatology, Mayo Clinic, 200 First Street SW Rochester, MN, 55905
David A. Woodrum, Department of Radiology, Mayo Clinic, 200 First Street SW Rochester, MN, 55905
References
- 1.Lencioni R, Crocetti L. Local-regional treatment of hepatocellular carcinoma. Radiology. 2012;262(1):43–58. doi: 10.1148/radiol.11110144. [DOI] [PubMed] [Google Scholar]
- 2.Francica G, Petrolati A, Di Stasio E, Pacella S, Stasi R, Pacella CM. Effectiveness, safety, and local progression after percutaneous laser ablation for hepatocellular carcinoma nodules up to 4 cm are not affected by tumor location. AJR Am J Roentgenol. 2012;199(6):1393–1401. doi: 10.2214/AJR.11.7850. [DOI] [PubMed] [Google Scholar]
- 3.Arienti V, Pretolani S, Pacella CM, Magnolfi F, Caspani B, Francica G, Megna AS, Regine R, Sponza M, Antico E, Di Lascio FM. Complications of laser ablation for hepatocellular carcinoma: a multicenter study. Radiology. 2008;246(3):947–955. doi: 10.1148/radiol.2463070390. [DOI] [PubMed] [Google Scholar]
- 4.Pacella CM, Francica G, Di Lascio FM, Arienti V, Antico E, Caspani B, Magnolfi F, Megna AS, Pretolani S, Regine R, Sponza M, Stasi R. Long-term outcome of cirrhotic patients with early hepatocellular carcinoma treated with ultrasound-guided percutaneous laser ablation: a retrospective analysis. J Clin Oncol. 2009;27(16):2615–2621. doi: 10.1200/JCO.2008.19.0082. [DOI] [PubMed] [Google Scholar]
- 5.Tiong L, Maddern GJ. Systematic review and meta-analysis of survival and disease recurrence after radiofrequency ablation for hepatocellular carcinoma. Br J Surg. 2011;98(9):1210–1224. doi: 10.1002/bjs.7669. [DOI] [PubMed] [Google Scholar]
- 6.Wang JH, Wang CC, Hung CH, Chen CL, Lu SN. Survival comparison between surgical resection and radiofrequency ablation for patients in BCLC very early/early stage hepatocellular carcinoma. J Hepatol. 2012;56(2):412–418. doi: 10.1016/j.jhep.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 7.Cho YK, Kim JK, Kim MY, Rhim H, Han JK. Systematic review of randomized trials for hepatocellular carcinoma treated with percutaneous ablation therapies. Hepatology. 2009;49(2):453–459. doi: 10.1002/hep.22648. [DOI] [PubMed] [Google Scholar]
- 8.Yin XY, Xie XY, Lu MD, Xu HX, Xu ZF, Kuang M, Liu GJ, Liang JY, Lau WY. Percutaneous thermal ablation of medium and large hepatocellular carcinoma: long-term outcome and prognostic factors. Cancer. 2009;115(9):1914–1923. doi: 10.1002/cncr.24196. [DOI] [PubMed] [Google Scholar]
- 9.Ng KK, Poon RT, Lo CM, Yuen J, Tso WK, Fan ST. Analysis of recurrence pattern and its influence on survival outcome after radiofrequency ablation of hepatocellular carcinoma. J Gastrointest Surg. 2008;12(1):183–191. doi: 10.1007/s11605-007-0276-y. [DOI] [PubMed] [Google Scholar]
- 10.Liu Y, Zheng Y, Li S, Li B, Zhang Y, Yuan Y. Percutaneous microwave ablation of larger hepatocellular carcinoma. Clin Radiol. 2013;68(1):21–26. doi: 10.1016/j.crad.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 11.Harrison LE, Koneru B, Baramipour P, Fisher A, Barone A, Wilson D, Dela Torre A, Cho KC, Contractor D, Korogodsky M. Locoregional recurrences are frequent after radiofrequency ablation for hepatocellular carcinoma. J Am Coll Surg. 2003;197(5):759–764. doi: 10.1016/S1072-7515(03)00750-6. [DOI] [PubMed] [Google Scholar]
- 12.Kei SK, Rhim H, Choi D, Lee WJ, Lim HK, Kim YS. Local tumor progression after radiofrequency ablation of liver tumors: analysis of morphologic pattern and site of recurrence. AJR Am J Roentgenol. 2008;190(6):1544–1551. doi: 10.2214/AJR.07.2798. [DOI] [PubMed] [Google Scholar]
- 13.Liu CH, Arellano RS, Uppot RN, Samir AE, Gervais DA, Mueller PR. Radiofrequency ablation of hepatic tumours: effect of post-ablation margin on local tumour progression. Eur Radiol. 2010;20(4):877–885. doi: 10.1007/s00330-009-1610-4. [DOI] [PubMed] [Google Scholar]
- 14.Stauffer PR, Goldberg SN. Introduction: thermal ablation therapy. Int J Hyperthermia. 2004;20(7):671–677. doi: 10.1080/02656730400007220. [DOI] [PubMed] [Google Scholar]
- 15.Mertyna P, Hines-Peralta A, Liu ZJ, Halpern E, Goldberg W, Goldberg SN. Radiofrequency ablation: variability in heat sensitivity in tumors and tissues. J Vasc Interv Radiol. 2007;18(5):647–654. doi: 10.1016/j.jvir.2007.02.033. [DOI] [PubMed] [Google Scholar]
- 16.Vidair CA, Dewey WC. Two distinct modes of hyperthermic cell death. Radiat Res. 1988;116(1):157–171. [PubMed] [Google Scholar]
- 17.McGahan JP, Browning PD, Brock JM, Tesluk H. Hepatic ablation using radiofrequency electrocautery. Invest Radiol. 1990;25(3):267–270. doi: 10.1097/00004424-199003000-00011. [DOI] [PubMed] [Google Scholar]
- 18.Goldberg SN, Gazelle GS, Halpern EF, Rittman WJ, Mueller PR, Rosenthal DI. Radiofrequency tissue ablation: importance of local temperature along the electrode tip exposure in determining lesion shape and size. Acad Radiol. 1996;3(3):212–218. doi: 10.1016/s1076-6332(96)80443-0. [DOI] [PubMed] [Google Scholar]
- 19.Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, Bao JK. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012;45(6):487–498. doi: 10.1111/j.1365-2184.2012.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mertyna P, Dewhirst MW, Halpern E, Goldberg W, Goldberg SN. Radiofrequency ablation: the effect of distance and baseline temperature on thermal dose required for coagulation. Int J Hyperthermia. 2008;24(7):550–559. doi: 10.1080/02656730802035662. [DOI] [PubMed] [Google Scholar]
- 21.Reed JC. Drug insight: cancer therapy strategies based on restoration of endogenous cell death mechanisms. Nat Clin Pract Oncol. 2006;3(7):388–398. doi: 10.1038/ncponc0538. [DOI] [PubMed] [Google Scholar]
- 22.Thompson SM, Callstrom MR, Butters KA, Sutor SL, Knudsen B, Grande JP, Roberts LR, Woodrum DA. Role for putative hepatocellular carcinoma stem cell subpopulations in biological response to incomplete thermal ablation: in vitro and in vivo pilot study. Cardiovasc Intervent Radiol. 2013 doi: 10.1007/s00270-013-0828-3. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niles AL, Moravec RA, Eric Hesselberth P, Scurria MA, Daily WJ, Riss TL. A homogeneous assay to measure live and dead cells in the same sample by detecting different protease markers. Anal Biochem. 2007;366(2):197–206. doi: 10.1016/j.ab.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 24.Niles AL, Moravec RA, Riss TL. In vitro viability and cytotoxicity testing and same-well multi-parametric combinations for high throughput screening. Curr Chem Genomics. 2009;3:33–41. doi: 10.2174/1875397300903010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4(5):313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thompson SM, Callstrom MR, Knudsen BE, Anderson JL, Sutor SL, Butters KA, Kuo C, Grande JP, Roberts LR, Woodrum DA. Molecular Bioluminescence Imaging as a Noninvasive Tool for Monitoring Tumor Growth and Therapeutic Response to MRI-Guided Laser Ablation in a Rat Model of Hepatocellular Carcinoma. Invest Radiol. 2013;48(6):413–421. doi: 10.1097/RLI.0b013e31827a4a3f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thompson SM, Callstrom MR, Knudsen B, Anderson JL, Carter RE, Grande JP, Roberts LR, Woodrum DA. Development and Preliminary Testing of a Translational Model of Hepatocellular Carcinoma for MR Imaging and Interventional Oncologic Investigations. J Vasc Interv Radiol. 2012;23(3):385–395. doi: 10.1016/j.jvir.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hickson J, Ackler S, Klaubert D, Bouska J, Ellis P, Foster K, Oleksijew A, Rodriguez L, Schlessinger S, Wang B, Frost D. Noninvasive molecular imaging of apoptosis in vivo using a modified firefly luciferase substrate, Z-DEVD-aminoluciferin. Cell Death Differ. 2010;17(6):1003–1010. doi: 10.1038/cdd.2009.205. [DOI] [PubMed] [Google Scholar]
- 29.Scabini M, Stellari F, Cappella P, Rizzitano S, Texido G, Pesenti E. In vivo imaging of early stage apoptosis by measuring real-time caspase-3/7 activation. Apoptosis. 2011;16(2):198–207. doi: 10.1007/s10495-010-0553-1. [DOI] [PubMed] [Google Scholar]
- 30.Thompson SM, Callstrom MR, Knudsen B, Anderson JL, Butters KA, Grande JP, Roberts LR, Woodrum DA. AS30D Model of Hepatocellular Carcinoma: Tumorigenicity and Preliminary Characterization by Imaging, Histopathology, and Immunohistochemistry. Cardiovasc Intervent Radiol. 2013;36(1):198–203. doi: 10.1007/s00270-012-0466-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11(10):700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 32.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 33.Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, Kroemer G. Cell death modalities: classification and pathophysiological implications. Cell death and differentiation. 2007;14(7):1237–1243. doi: 10.1038/sj.cdd.4402148. [DOI] [PubMed] [Google Scholar]
- 34.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA, Malorni W, Nunez G, Peter ME, Tschopp J, Yuan J, Piacentini M, Zhivotovsky B, Melino G. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16(1):3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malhi H, Gores GJ, Lemasters JJ. Apoptosis and necrosis in the liver: a tale of two deaths? Hepatology. 2006;43(2 Suppl 1):S31–44. doi: 10.1002/hep.21062. [DOI] [PubMed] [Google Scholar]
- 36.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W, Mehlen P, Nunez G, Peter ME, Piacentini M, Rubinsztein DC, Shi Y, Simon HU, Vandenabeele P, White E, Yuan J, Zhivotovsky B, Melino G, Kroemer G. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19(1):107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Long JS, Ryan KM. New frontiers in promoting tumour cell death: targeting apoptosis, necroptosis and autophagy. Oncogene. 2012;31(49):5045–5060. doi: 10.1038/onc.2012.7. [DOI] [PubMed] [Google Scholar]
- 38.Hoshida Y, Toffanin S, Lachenmayer A, Villanueva A, Minguez B, Llovet JM. Molecular classification and novel targets in hepatocellular carcinoma: recent advancements. Semin Liver Dis. 2010;30(1):35–51. doi: 10.1055/s-0030-1247131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JS, Heo J, Libbrecht L, Chu IS, Kaposi-Novak P, Calvisi DF, Mikaelyan A, Roberts LR, Demetris AJ, Sun Z, Nevens F, Roskams T, Thorgeirsson SS. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med. 2006;12(4):410–416. doi: 10.1038/nm1377. [DOI] [PubMed] [Google Scholar]
- 40.Harmon BV, Corder AM, Collins RJ, Gobe GC, Allen J, Allan DJ, Kerr JF. Cell death induced in a murine mastocytoma by 42–47 degrees C heating in vitro: evidence that the form of death changes from apoptosis to necrosis above a critical heat load. Int J Radiat Biol. 1990;58(5):845–858. doi: 10.1080/09553009014552221. [DOI] [PubMed] [Google Scholar]
- 41.VanderWaal R, Malyapa RS, Higashikubo R, Roti Roti JL. A comparison of the modes and kinetics of heat-induced cell killing in HeLa and L5178Y cells. Radiat Res. 1997;148(5):455–462. [PubMed] [Google Scholar]
- 42.Vanagas T, Gulbinas A, Sadauskiene I, Dambrauskas Z, Pundzius J, Barauskas G. Apoptosis is activated in an early period after radiofrequency ablation of liver tissue. Hepatogastroenterology. 2009;56(93):1095–1099. [PubMed] [Google Scholar]
- 43.Yang W, Ahmed M, Elian M, Hady el SA, Levchenko TS, Sawant RR, Signoretti S, Collins M, Torchilin VP, Goldberg SN. Do liposomal apoptotic enhancers increase tumor coagulation and end-point survival in percutaneous radiofrequency ablation of tumors in a rat tumor model? Radiology. 2010;257(3):685–696. doi: 10.1148/radiol.10100500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Solazzo SA, Ahmed M, Schor-Bardach R, Yang W, Girnun GD, Rahmanuddin S, Levchenko T, Signoretti S, Spitz DR, Torchilin V, Goldberg SN. Liposomal doxorubicin increases radiofrequency ablation-induced tumor destruction by increasing cellular oxidative and nitrative stress and accelerating apoptotic pathways. Radiology. 2010;255(1):62–74. doi: 10.1148/radiol.09091196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 46.Fabregat I. Dysregulation of apoptosis in hepatocellular carcinoma cells. World J Gastroenterol. 2009;15(5):513–520. doi: 10.3748/wjg.15.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fabregat I, Roncero C, Fernandez M. Survival and apoptosis: a dysregulated balance in liver cancer. Liver Int. 2007;27(2):155–162. doi: 10.1111/j.1478-3231.2006.01409.x. [DOI] [PubMed] [Google Scholar]
- 48.Schattenberg JM, Schuchmann M, Galle PR. Cell death and hepatocarcinogenesis: Dysregulation of apoptosis signaling pathways. J Gastroenterol Hepatol. 2011;26 (Suppl 1):213–219. doi: 10.1111/j.1440-1746.2010.06582.x. [DOI] [PubMed] [Google Scholar]
- 49.Kepp O, Galluzzi L, Lipinski M, Yuan J, Kroemer G. Cell death assays for drug discovery. Nat Rev Drug Discov. 2011;10(3):221–237. doi: 10.1038/nrd3373. [DOI] [PubMed] [Google Scholar]