

Abstract
Purpose
Ovarian cancer is the deadliest of the gynecological malignancies. Carcinogenic progression is accompanied by up-regulation of ubiquitin-dependent protein degradation machinery as a mechanism to compensate with elevated endogenous proteotoxic stress. Recent studies support the notion that deubiquitinating enzymes (DUBs) are essential factors in proteolytic degradation and that their aberrant activity is linked to cancer progression and chemoresistance. Thus, DUBs are an attractive therapeutic target for ovarian cancer.
Experimental Design
The potency and selectivity of RA-9 inhibitor for proteasome-associated DUBs was determined in ovarian cancer cell lines and primary cells. The anticancer activity of RA-9 and its mechanism of action was evaluated in multiple cancer cell lines in vitro and in vivo in immunodeficient mice bearing an intra-peritoneal ES-2 xenograft model of human ovarian cancer.
Results
Here we report the characterization of RA-9 as a small-molecule inhibitor of proteasome-associated DUBs. Treatment with RA-9 selectively induces onset of apoptosis, in ovarian cancer cell lines and primary cultures derived from donors. Loss of cell viability following RA-9 exposure is associated with an Unfolded Protein Response (UPR) as mechanism to compensate for unsustainable levels of proteotoxic stress. In vivo treatment with RA-9 retards tumor growth, increases overall survival and was well tolerated by the host.
Conclusions
Our preclinical studies support further evaluation of RA-9 as an ovarian cancer therapeutic.
Keywords: Ovarian cancer, ER stress, Proteasome Inhibitors, Deubiquitinating Enzymes, Ubiquitin Proteasome System (UPS), UPS-stress
Introduction
The ubiquitin-proteasome-system (UPS) is responsible for >80% of the intracellular protein degradation in eukaryotes and consists of three components: the proteasomes, the ubiquitin conjugating system and the deubiquitinating enzymes (DUBs) (1). While the ubiquitin-conjugating system is responsible for initiating the cascade leading to protein degradation, DUBs deconjugate ubiquitin from targeted proteins. This step is essential for protein degradation as de-ubiquitination is required for regulating proteasome-mediated protein degradation. The human genome encodes for ~100 DUBs that can be grouped based on their catalytic domain into two families; cysteine proteases and metalloproteases. Within the cysteine-based DUB family, Ubiquitin-Specific Proteases (USPs) and Ubiquitin C-terminal Hydrolases are the most represented members constituting >90% of the mammalian cell DUBs pool (2, 3). Importantly, members of both USP and UCH families are differentially expressed and activated in a number of cancer types and their aberrant activity linked to cancer progression, and the onset of chemoresistance. Thus, DUBs have been suggested as a potential therapeutic targets for cancer treatment (4-9).
Ovarian cancer is a heterogeneous disease with respect to histopathology and molecular biology (10), yet it is accompanied by progressive cellular adaptation to cope with increasing levels of metabolic stress inherent to the cancer phenotype (i.e rapid proliferation, elevated oxidative and proteotoxic stress) (11-14). Previously, we showed that ovarian cancer cells are under higher levels of ubiquitin-proteasome stress as compared to normal cells, rendering them selectively sensitive to inhibition of ubiquitin-dependent protein degradation (11, 12). Recent work from our laboratory suggests that the presence of an α-β carbonyl system constitutes a key molecular determinant for a new series of small-molecule inhibitors of cysteine-based DUBs, thus paving the way to the further development of inhibitors of this class of proteases for cancer treatment (15).
Here, we report the identification and characterization of RA-9 as a cell permeable, potent and specific inhibitor of the subset of deubiquitinating enzymes associated with the 19S regulatory particle (RP) of the proteasome. We show that RA-9 causes onset of apoptosis in ovarian cancer, both cell lines and primary cultures derived directly from clinical specimens. Furthermore, the loss of ovarian cancer cell viability following RA-9 treatment is associated with an unresolved Unfolded-Protein-Response (UPR) as a mechanism to compensate with increasing levels of proteotoxic stress. In addition, RA-9 inhibits human ovarian cancer xenograft growth in mice, prolonging their survival, and drug treatment is well tolerated. In light of its new mechanism, favorable toxicity profile and activity against a xenograft model, further evaluation of RA-9 alone, or in combination with conventional or other novel therapies, as a candidate treatment for ovarian cancer is warranted.
Materials and Methods
Reagents and plasmids
The deubiquitinating enzyme inhibitor RA-9 was synthesized and purified as we have previously described (15). The proteasome inhibitor Bortezomib was purchased from Cayman Chemical. Cisplatin (cis-Diamineplatinum(II) dichloride) was purchased from Sigma-Aldrich. The 2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxanilide inner salt (WST-1) was purchased from Roche Diagnostics GmbH. The CellTiter96® AQueous One Solution Cell Proliferation Kit was purchased from Promega. The 19S RP (Cat. # E-366), and the Ub-AMC substrate (Cat. # U-550) were purchased from Boston Biochem. The positive control deubiquitinating enzyme inhibitor b-AP15 was purchased from Bio Vision. The lentivirus vector pEF-GFP-SIN, the VSVG envelope and the delta-8.9 plasmids were kindly provided by Dr. Koho Iizuka (University of Minnesota, MN).
Cell lines and Transfection
Human ovarian cancer cell lines SKOV-3, TOV-21G, OVCAR-3 and ES-2 were obtained from American Type Cell Culture and cultured in DMEM supplemented with 10% fetal bovine serum, 100 IU/mL penicillin, and 100 μg/mL streptomycin at 5% CO2. The ovarian cancer cell line HEY was a generous gift from Dr. Sundaram Ramakrishnan (University of Minnesota, MN) and was cultured as described above. The MM cell lines RPMI8226 and ANBL6 and their Bortezomib resistant clones RPMI8226-V10R and ANBL6-V10R were provided by Dr. Robert Z. Orlowski (M.D. Anderson Cancer Center, TX) and were cultured as previously described (16). For in vivo experiments, subconfluent cultures of ES-2 ovarian cancer cells were infected with lentiviral particles expressing the GFP reporter as we have previously described (17, 18).
Ub-AMC protease assay on 19S RP
Residual 19S RP was measured on purified 19S RP as previously described (18). Briefly, 19S RP (5 nmol/L) was incubated in DUB buffer (20 mM HEPES 0.5 mM EDTA, 5mM DTT, and 0.1mg/ml BSA, pH 7.8) with the indicated concentration of drugs in a 100-μL-reaction volume for 60 minutes at room temperature, and the reaction was initiated by the addition of 500 nmol/L of the fluorogenic substrate Ub-AMC. Release of the AMC fluorophore was recorded using a plate-reading luminometer equipped with 380 nm excitation and 440 nm emission filters (Molecular Devices). All experiments were performed in triplicate.
Ub-AMC protease assay on whole cell lysate
To measure the inhibition of deubiquitinating enzyme activity on whole cell lysate, exponentially growing ES-2 cells were incubated with the indicated drug concentrations for 18 hours. Cells were lysed in DUB lysis buffer (25 mM HEPES, 5 mM, EDTA, 0.1% CHAPS, 5 mM ATP), the nuclei were removed by centrifugation and 100-μL of supernatant was incubated with equal volume of Ub-AMC (500 nmol/L) at room temperature for 30 minutes. Release of the AMC fluorophore was recorded using a plate-reading luminometer equipped with 380 nm excitation and 440 nm emission filters (Molecular Devices). All experiments were performed in triplicate.
Tissue collection
Clinical specimens from patients undergoing surgery for ovarian cancer or oophorectomy for benign conditions were obtained with informed consent by the University of Minnesota Tissue Procurement Facility (TPF) after Institutional Review Board Committee (IRB) approval. Ovarian Surface Epithelial (OSE) cells and primary ovarian cancer cells were isolated from ovarian specimens excised from patients undergoing oophorectomy for benign conditions and cultured as we have previously described (17, 19, 20).
Cell viability assay
Cell viability was determined by WST-1 or CellTiter96® AQueous One Solution Cell Proliferation assays as previously described (15-17). Briefly, cells were seeded at the concentration of 1,000 or 10,000 per well in 100 μL medium in 96-well plate and treated with the indicated concentrations of drugs. At the indicated time points, cells were incubated according to the manufacturer's protocol with the WST-1 or CellTiter96® labeling mixture. Formazan dye was quantified using a spectrophotometric plate (ELISA reader 190; Molecular Devices). All experiments were performed in triplicate.
Antibodies and Western Blot Analysis
Total cellular protein (10-20 μg) from each sample was separated by SDS-PAGE, transferred to PVDF membranes and subjected to Western blot analysis. Antibodies for Western blot analysis were obtained by the following commercial sources: anti-ubiquitin (Santa Cruz Biotechnology and Millipore), anti-PCNA (Abcam), anti-PARP (BD Pharmingen), anti-GRP78, anti-GCN2, anti-phospho-eIF2oc, anti-IRE1-α, anti-Ero1L-α, anti-caspase-3 (Cell Signaling), anti-β-actin (Sigma). Peroxidase-linked anti-mouse Immunoglobulin G and peroxidase-linked anti-rabbit Immunoglobulin G were from Amersham.
Flow cytometry
Cell cycle status was analyzed with a FACSCalibur flow cytometer (Becton Dickinson) by measuring fluorescence from cells stained with propidium iodide (PI; Sigma) following drug treatment. For active caspase-3 experiments, cells were treated for the indicated amount of time, harvested, and immediately stained with the FITC-conjugated anti-active caspase-3 antibody according to the protocol provided by the manufacturer.
Animals
Six-week-old female immunodeficient (NCr nu/nu) mice were obtained from National Cancer Institute-Frederick (Frederick, MD) and maintained in a pathogen-free animal facility at least 1 week before use. All animal studies were done in accordance with institutional guidelines following approval by the IACUC.
Xenograft murine model
Mice were inoculated i.p. with 100,000 ES-2 cells (in 100 μl DMEM) stably expressing GFP. When tumor was detectable (approx. 6 days post inoculation), mice were randomly assigned into two groups receiving RA-9 or 0.9% saline. Treatment with RA-9 was given i.p. on a one-day on, two-days off schedule. The control group received the vehicle alone at the same schedule. Working concentrations of RA-9 (10 mg/ml) were reached by dissolution in Cremophor EL and polyethylene glycol 400 (Sigma). Prior to each injection RA-9 was further diluted in 0.9% saline (working concentration 1mg/ml). To monitor for tumor growth, RA-9 treated and control mice were imaged with an IVIS SpectrumCT Pre-clinical in vivo imaging system (PerkinElmer) every other day. Animals were sacrificed when abdomens became distended to twice normal size.
Total blood cell count
Analysis of total blood cell count from saline and RA-9 treated mice was performed at the Veterinary Clinical Pathology Laboratory at the University of Minnesota and reviewed by a Board-Certified Clinical Pathologist.
Immunohistochemistry
Sections of (3-4 μm) of paraffin-embedded tissues were used for immunohistochemistry as we have previously described (11). Briefly, following deparaffinization and rehydratation, sections were incubated with the mouse monoclonal antibody to PCNA diluted 1:10.000 for 1 hour at room temperature. The avidin-biotin-peroxidase complex method from DAKO (Glostrup, Denmark) was used to visualize antibody binding, and the tissues were counterstained with hematoxylin. For immunohistochemical analyses of mouse tissues, tumors and organs from mice were excised, fixed in 10% neutral buffered formalin and embedded in paraffin. H&E staining was performed according to standard histologic procedure.
Terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling assay
Sections (3-4 μm) of paraffin-embedded tissues were processed for terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling (TUNEL) using an established method to assay for cell death-associated DNA double-strand breaks as we previously described (11).
Statistical analysis
Unless otherwise indicated, results are reported as mean ± standard deviation, and statistical significance was assessed by two-tailed Student's t tests using Prism (V.4 Graphpad, San Diego, CA) with the level of significance set at p<0.05. Survival was summarized using Kaplan Meier methods and compared using log-rank tests. The combination index (CI) of RA-9 and cisplatin was calculated using the method of Chou and Talaly (15). CI<1 indicates synergism, CI = 1 indicates additivity, and CI>1 indicates antagonism.
Results
RA-9 blocks proteasome-associated DUB activity in ovarian cancer cells
Within the UPS system, DUB expression and activity has been linked to cancer initiation, progression and onset of chemoresistance (7, 8, 21-24). Through multiple iterations of a new class of small-molecule UPS inhibitors, we have recently identified RA-9 (Figure 1A) as a promising inhibitor that blocks ubiquitin-dependent protein degradation without impacting 20S proteasome proteolytic activity (15). To test whether RA-9 is a DUB inhibitor, we initially evaluated its impact on protein ubiquitination in ES-2 ovarian cancer cells exposed to increasing doses of RA-9 over a period of 24 hours. As shown in Supplementary Figure 1A (left panel), RA-9 treatment resulted in a dose-dependent accumulation of poly-ubiquitinated proteins in ES-2 cells starting at 5 μM RA-9 treatment. Quantification of the changes in high-molecular weight ubiquitin species in treated cells versus control is given in Supplementary 1A (right panel).
Figure 1. RA-9 is an inhibitor of 19S-associated DUBs.
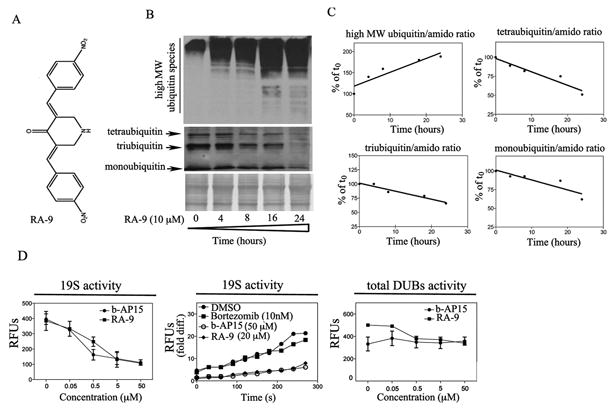
A, chemical structure of RA-9 (3E, 5E)-3,5-bis(nitrobenzylidene)piperidin-4-one. B, time-dependent accumulation of high-molecular weight ubiquitin species and concomitant reduction in mono, tri and tetraubiquitin species in ES-2 ovarian cancer cell line exposed to 10μM RA-9 for 4h, 8h, 16h and 24 h. Amido black staining was used as loading control. C, quantification of the ubiquitin/total protein (amido) ratio expressed as % of t0. D. Left panel, dose-dependent residual DUB activity in 19S RP exposed to the indicated doses of RA-9 expressed as Relative Fluorescence Units (RFUs). b-AP15 was used as positive control. Middle panel, time-dependent residual deubiquitinating enzyme activity in 19S proteasome particles exposed to the indicated dose of RA-9 expressed as fold Relative Fluorescence Units (RFUs) difference over time. b-AP15 was used as positive control. Bortezomib was used as negative control. Right panel, dose-dependent residual DUB activity in ES-2 cell lysate (total DUBs activity) exposed to the indicated doses of RA-9 expressed as Relative Fluorescence Units (RFUs). b-AP15 was used as positive control.
Next, we monitored the rate of poly-ubiquitinated protein accumulation in ES-2 ovarian cancer cells exposed to 10 μM RA-9 over a period of 24 hours. As shown in Figure 1B, immunoblot analysis revealed a rapid, time-dependent increase of high-molecular weight poly-ubiquitinated species accompanied by a decrease in monotri and tetra-ubiquitin forms. Quantification of the changes in high- and low-molecular weight ubiquitin species in treated cells versus control is given in Figure 1. Similar results were obtained with SKOV-3 and TOV-12G ovarian cancer cell lines (Supplementary Figure 1B).
The human genome encodes for at least ninety-eight DUBs, of which only Ubiquitin-Specific-Protease 14 (USP14) and Ubiquitin Carboxy-terminal Hydrolase 37 (UCH37) directly associate with the 19S RP (25). The structural similarities between RA-9 and previously described DUBs inhibitors (9, 26, 27), together with its lack of inhibition of the 20S catalytic activities (15), suggest that RA-9 may selectively inhibit 19S RP associated DUBs rather than acting broadly on the cellular pool of DUBs as a whole. To test this hypothesis, first we monitored the residual DUB activity in purified 19S RP exposed to increasing concentration of RA-9 (0-50 μM). As shown in Figure 1D, RA-9 treatment resulted in a dose- (left panel) and time- (middle panel) dependent reduction in 19S RP-associated DUB activity following RA-9 exposure. Second, to determine if RA-9 broadly inhibits cellular DUB activity as a whole, we next measured the total deubiquitinating enzyme activity in cell lysate from ES-2 ovarian cancer cells exposed to escalating doses of RA-9. As shown in Figure 1D (right panel), RA-9 treatment does not affect total DUB activity in ES-2 cell lysate. The 19S RP-associated DUB inhibitor b-AP15 and the 20S proteasome inhibitor Bortezomib were used as positive and negative control respectively. Taken together these observations suggest that RA-9 is a selective inhibitor of 19S RP-associated DUB activity.
RA-9 inhibits growth of ovarian cancer cell lines and primary cultures
We and others, have previously shown that cancer cells exhibit higher endogenous levels of proteotoxic stress inherent to the cancer phenotype (11, 12), as compared to normal cells. This renders ovarian cancer cells more dependent upon ubiquitin-mediated protein degradation than immortalized ovarian surface epithelial cells (IOSEs) (11, 12, 28). Therefore, we examined whether inhibition of 19S RP-associated DUBs via RA-9 treatment would selectively cause loss of viability of a panel of ovarian cancer cells as compared to normal ovarian epithelial cells. Specifically, we compared the effect of RA-9 on the viability of the cisplatin-sensitive TOV-21G and ES-2 cell lines, the cisplatin-resistant OVCAR-3 and HEY cell lines, primary ovarian cancer cell cultures (derived directly from clinical isolates of ovarian cancers) versus primary ovarian surface epithelial cell cultures (OSEs) isolated from ovaries of women undergoing oophorectomies for benign reasons. Our results indicate that exposure to increasing concentrations of RA-9 over a period of 48 hours compromised the viability of ovarian cancer cells in a dose-dependent fashion (Figure 2B-D) and to greater extent as compared to OSEs (Figure 2A). Importantly, while TOV-21G and ES-2 cell lines are traditionally considered cisplatin-sensitive, several studied have recently reported a certain degree platinum-resistance (29, 30). Thus, we measured the IC50 levels for cispatin treatment of ES-2 and TOV-21G cells. As shown in Supplementary Figure 2A, the IC50 levels for cispatin were 11.8 and 13.9 μM for ES-2 and TOV-21G ovarian cancer cell lines respectively. Furthermore, combination of cisplatin and RA-9 resulted in synergistic effect in terms of loss of cell viability in cisplatin resistant ovarian cancer cells (Supplementary Figure 2B).
Figure 2. Effect of RA-9 treatment upon OSEs, ovarian cancer and MM cells viability.
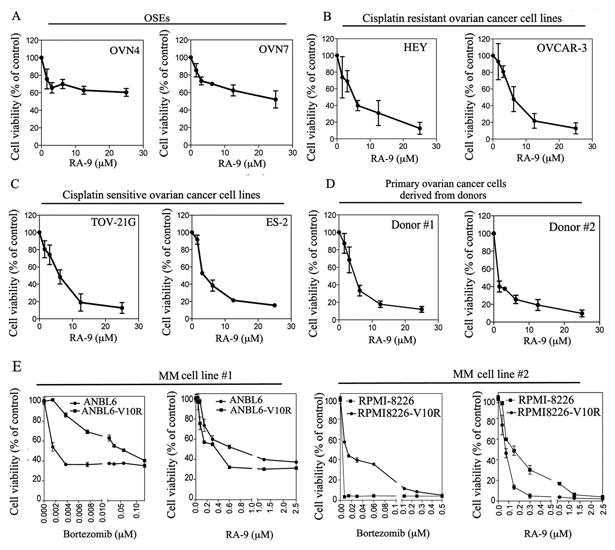
A, cell viability of OSEs derived from clinical specimens benign oophorectomies exposed at the indicated concentration of RA-9 for 48 hours. B, cell viability of the cisplatin-sensitive ovarian cancer cell lines TOV-21G and ES-2 exposed at the indicated concentration of RA-9 for 48 hours. C, cell viability of the cisplatin-resistant ovarian cancer cell lines HEY and OVCAR-3 exposed at the indicated concentration of RA-9 for 48 hours. D, cell viability of primary ovarian cancer cells derived from donors exposed at the indicated concentration of RA-9 for 48 h. E, cell viability of the MM cell lines ANBL6 and its Bortezomib-resistant parental cell line ANBL6-V10R (left panels) and RPMI-8226 and its Bortezomib-resistant parental cell line RPMI-8226-V10R (right panels) to Bortezomib or RA-9. Cell viability was measured by WST-1 or CellTiter96® assays following 48 hours drugs exposure and it is expressed as % of control.
Because RA-9 inhibits ubiquitin-mediated protein degradation independently from inhibition of the catalytic activities of the proteasomes (15) (Figure 1D), we reasoned that cancer cells that developed resistance to proteasome inhibitors would still remain sensitive to RA-9 treatment. To test this hypothesis, the multiple myeloma (MM) cell lines RPMI8226 and ANBL6 and their bortezomib resistant derivatives RPMI8226-V10R and ANBL6-V10R (16, 31) were exposed to either Bortezomib or RA-9 and the residual cell viability measured after 48 hours of treatment. Surprisingly, as shown in Figure 2E, RA-9 treatment was more effective against Bortezomib-resistant MM cell lines than their Bortezomib-sensitive parental cell lines.
Taken together, this suggests that the selectivity profile of RA-9 for ovarian cancer cells is dependent upon their levels of proteotoxic stress and independent from their histological subtype. This also suggests that RA-9 treatment is effective against cancer cells that developed resistance to the licensed proteasome inhibitor bortezomib. Lastly, the synergistic activity of RA-9 combined with cisplatin on the cell viability of ovarian cancer cells suggests that this combinatorial approach may be effective for ovarian cancer treatment, and that RA-9 might still be used to treat cisplatin-resistant disease.
RA-9 causes cell cycle arrest and caspase-mediated apoptosis in ovarian cancer cells
Ubiquitin-dependent protein degradation regulates the steady levels of key cell cycle regulatory proteins whose dysregulation is expected to affect cell cycle progression and viability (32). To test whether the reduced cell viability of ovarian cancer cell lines following RA-9 treatment is associated with cell cycle dysregulation, ES-2 cells were incubated with increasing concentrations of RA-9 and analyzed by flow cytometry after staining with PI. As shown in Figure 3A, 18 h exposure to RA-9 resulted in a dose-dependent increase in the fraction of ES-2 cells in the G2-M cell cycle phase in ES-2 treated cells. To investigate the fate of the cells accumulated in G2-M following RA-9 treatment, we analyzed them by flow cytometry after staining with the phycoerythrin (PE)-conjugated antibody specific for the active form of caspase-3, a marker of apoptosis. As shown in Figure 3B, treatment for 18 h with RA-9 produced a dose-dependent increase in the levels of active caspase-3. Further, we measured the levels of cleaved Poly (ADP-ribose) polymerase (PARP), a substrate of caspase-3. ES-2 cells exposed to 5 μM RA-9 for 24 h were analyzed by Western blot using an antibody recognizing both full-length and cleaved PARP. As shown in Figure 3C, RA-9 treatment resulted with time-dependent accumulation of the cleaved formed of PARP noticeable as early as 8 h post RA-9 treatment. Quantification of the ratio between full length and cleaved PARP in ES-2 cells treated for up to 24 h is given in Figure 3D. Taken together this data suggests that the loss of cell viability by ovarian cancer cell lines following RA-9 treatment is associated with caspase-3 mediated apoptotic cell death.
Figure 3. RA-9 induces G2-M cell cycle arrest and caspase-mediated apoptosis in ovarian cancer cells.
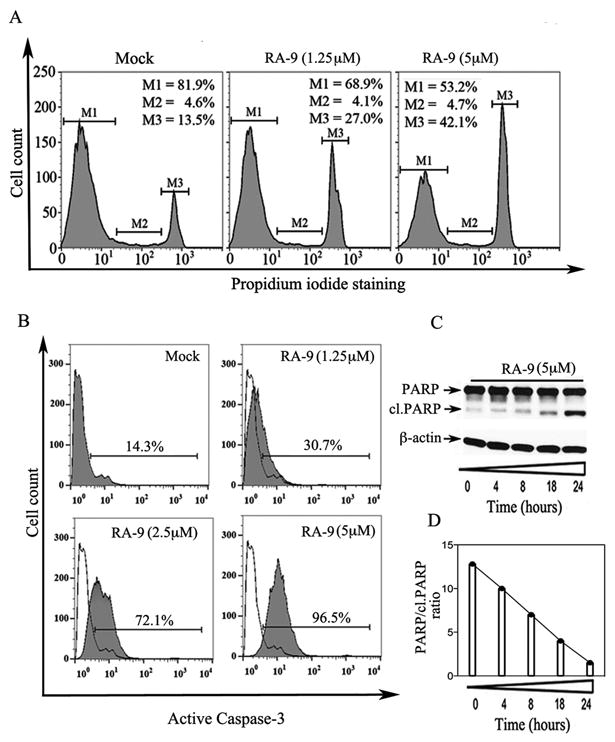
A, cell cycle distribution of ES-2 ovarian cancer cells either mock treated or treated with the indicated concentration of RA-9 for 18 h, was evaluated by PI staining and flow cytometric analysis. Percentage of cells in G1 phase (M1), S phase (M2) and G2-M phase (M3). B, ES-2 ovarian cancer cells either mock treated or treated with the indicated concentrations of RA-9 for 18 hours. Harvested cells were stained with a FITC-conjugated anti-active caspase-3 monoclonal antibody and analyzed by flow cytometry. C, cell lysate from ES-2 ovarian cancer cells exposed to 5 μM RA-9 over 24 hours was Western blotted with an antibody recognizing both the full-length and cleaved forms of PARP. Equivalent protein loading was verified by using an antibody directed against β-actin. D, quantification of the PARP/cl.PARP ratio in mock versus ES-2 treated cells.
RA-9 induces unfolded protein responses (UPR) in ovarian cancer cells
Activation of unfolded protein responses (UPR), including endoplasmic reticulum (ER) stress responses, is a cellular mechanism to compensate for increasing levels of proteotoxic stress (33, 34) typical of the malignant phenotype and upon proteasome inhibition. We have previously shown that sensitivity to inhibition of ubiquitin-mediated protein degradation in ovarian cancer cells is dependent upon the levels of ubiquitin-proteasome stress.
Because RA-9 treatment resulted in the rapid accumulation of poly-ubiquitinated proteins, we investigated its effect on ER-stress responses in a panel of ovarian cancer cell lines. ES-2, SKOV-3 and TOV-21G cells were exposed to 5μM RA-9 for 24 h and subjected to Western blot analysis for the levels of key markers of early and late ER-stress responses. As shown in Figure 4 (left panels), RA-9 exposure caused a time-dependent increase in the steady levels of the early ER-stress markers GRP-78 and PERK, as well as the late ER-stress markers IRE1-α and Ero1L-α. This increase was detectable as early as 4-hours following drug exposure, and is consistent with an attempt by the ovarian cancer cells to compensate for RA-9 induced proteotoxic stress by both inhibiting transcription and increasing protein degradation. Quantification of the changes in the steady state levels of these ER-stress markers over time is given in Figure 4 (right panels). Furthermore, as shown in Figure 4B, RA-9 treatment resulted with time-dependent increase in p-eIF2-α levels in ES-2 and TOV-21G ovarian cancer cells thus strengthening the evidence of a general halting in the cellular translational machinery following drug treatment. On the contrary, we did not observe increase in GCN2 expression levels following RA-9 treatment (Supplementary Figure 4). Consistent with our previous reports of greater dependency of ovarian cancer cells upon proteolytic degradation machinery (11, 15, 16), exposure of OSEs to RA-9 treatment resulted with only moderate increase in poly-ubiquitinated protein levels (Supplementary Figure 3A) and no sign of UPR stress as measured by Ero1L-α expression levels (Supplementary Figure 3B).
Figure 4. RA-9 induces ER-stress responses in ovarian cancer cells.
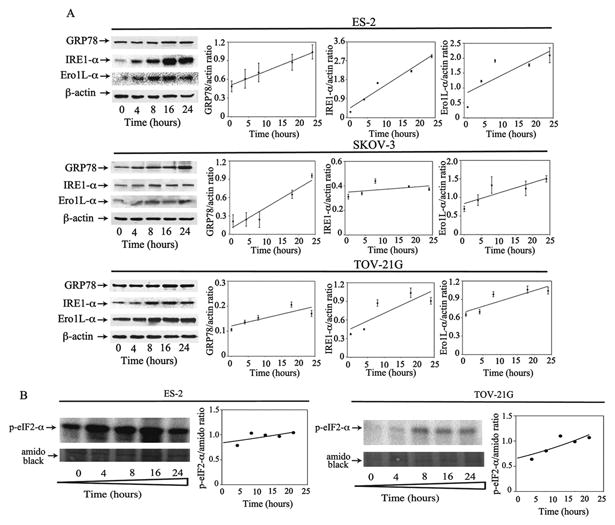
A, Left panels, ES-2 (top), SKOV-3 (middle) and TOV-21G (bottom) ovarian cancer cells were exposed to 5 μM of RA-9 over a period of 24 h following Western blot analysis with specific antibodies against the ER stress-associated proteins GRP-78, IRE-1α, and Ero1L-α. β-actin was used as loading control. Right panels, quantification of the ER stress-associated proteins/β-actin ratios for each cell line. B, ES-2 (left panels) and TOV-21G (right panels) ovarian cancer cells were exposed to 5 μM of RA-9 over a period of 24 h following Western blot analysis with specific antibody against p-eIF2-α. αmido black staining was used as loading control and for quantification of the p-eIF2-α/total protein ratios.
RA-9 inhibits human ovarian cancer cell growth in vivo and prolongs survival in a mouse model for ovarian cancer
Having shown that RA-9 treatment induces onset of apoptosis in ovarian cancer cells in vitro, we next investigated the efficacy of RA-9 in inhibiting ES-2 tumor growth in a mouse xenograft model of ovarian cancer. Specifically, NCr nu/nu mice were inoculated i.p. with 100,000 ES-2 ovarian cancer cells stably expressing the GFP reporter with 93% of the animals developing a detectable tumor within 6 days from inoculation. Mice were then randomly assigned to RA-9 treatment group (5mg/kg n=12) or 0.9% normal saline-treated controls (n=12) and treated by i.p. injections on a one-day on, two-day off schedule. The average tumor burden, as measured by fluorescence intensity, at the beginning of the treatment (day 0) was not significantly different in the RA-9 and control cohorts. However, a significant reduction in tumor burden was observed in mice treated with RA-9 versus the control group by day 5 of treatment (Supplementary Figure 5). The difference became more significant by day 12 (P < 0.002) as measured by fluorescence intensity (Figure 5A and B). A survival curve for mice in each group is shown in Fig. 5C, with a log-rank test revealing a significant prolongation in overall survival by RA-9 treatment (P < 0.0005). Notably, by day 17 of treatment, all of the control mice were sacrificed due to their tumor burden, whereas only 15% of mice in the RA-9 treatment arm had to be euthanized. Consistent with the reduction in tumor growth and increased overall survival, RA-9 treated mice also showed a reduction in tumor mass (Figure 5D, left panel) and in ascitic fluid (Figure 5D, right panel) as compared to the control cohort. Next we tested whether the effect of RA-9 on the increase in ER-stress and poly-ubiquitinated protein responses in vitro could be recapitulated in cancer cells derived from saline and RA-9 treated mice. Specifically, tumors from either vehicle or RA-9 treated mice were collected 8 hours after the last treatment and cell lysate subjected to Western blot analysis for levels of the ER-stress response protein GRP-78 and ubiquitin. As shown in Figure 5E, Western blot analysis revealed an increase in the levels of GRP-78 and high molecular weight poly-ubiquitinated proteins suggesting that RA-9 mediated anti-cancer activity in vivo occurs via inhibition of ubiquitin-proteasomal pathway and is associated to ER-stress responses activation.
Figure 5. RA-9 inhibits tumor growth in vivo.
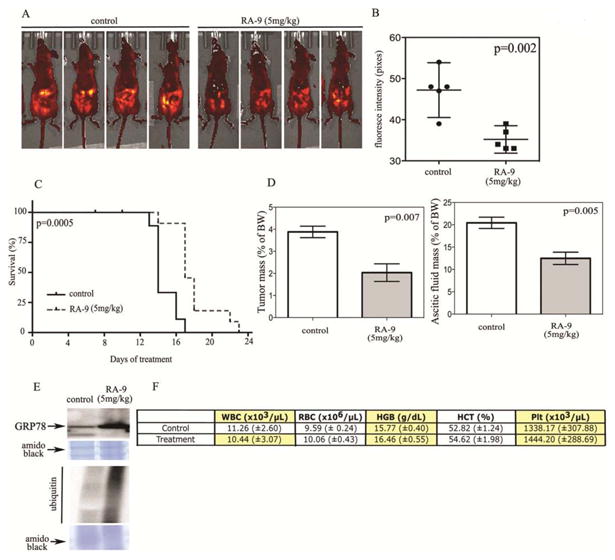
A, athymic nude (NCr nu/nu) mice were inoculated with 100,000 GFP expressing ES-2 ovarian cancer cell lines intraperitoneally. On detection of quantifiable tumors (day 0), mice were treated with i.p injection of 5mg/kg RA-9 (n=10) or saline (control n=9) on a one-day on, two-days off schedule. Representative images of control or RA-9 treated mice at day 12 of treatment. B, measurement of tumor growth in controls or RA-9 treated mice as assessed by fluorescence intensity quantification. C, effect of RA-9 treatment on survival of mice bearing and intra-peritoneal ES-2 xenograft that expresses GFP. Survival was evaluated from the first day of treatment until mice in control and RA-9 treated groups had to be sacrificed based upon a doubling of their girth. The statistical significance of the difference in overall survival between control and RA-9 treated group was determined by Wilcoxon signed-rank test. D, left tumor mass expressed as percentage of total body weight (BW) in saline versus RA-9 treated cohorts. Right, mass of ascitic fluid expressed as percentage of total body weight (BW) in saline versus RA-9 treated cohorts. E, increase in GRP-78 and ubiquitin protein levels in tumor derived from either control or treated mice as measured by Western blot analysis, and amido black staining was used as loading control. F, total blood cell count in RA-9 treated and control cohorts. White blood cells (WBC), red blood cells (RBC), hemoglobin (HGB), hematocrit (HCT), platelet (Plt), ± Standard Error.
RA-9 caused no significant toxicity on the host
RA-9 was well tolerated in mice at the dose of 5mg/kg with the difference in total body weight in treated and control cohorts never exceeding 3% (Supplementary Figure 5). Additionally, no statistically significant difference was found in terms of complete blood count (CBC) and white blood cell (WBC) differential in treated and control cohorts (Figure 5F). Importantly, no evidence of toxicity was found in treated and control cohorts as assessed by H&E staining on histological tissue sections of liver, spleen, heart and kidneys (Figure 6A).
Figure 6. Lack of RA-9 associated toxicity on the host.
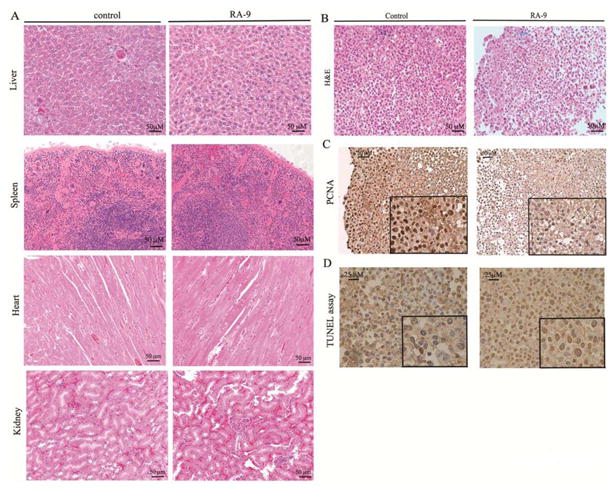
A, H&E staining of liver, spleen, heart and kidney from control and RA-9 treated mice (objective, 20X). B, H&E staining of control and RA-9 treated tumors (objective, 20X). C, PCNA staining in control and RA-9 treated tumors (objective, 20X). D, TUNEL assay in histologic specimens from control (left) and RA-9 treated (right) mice (objective 40X).
Next, tumor sections harvested from control and RA-9 treated mice were subjected to H&E or PCNA staining as well as TUNEL assay to evaluate for onset of apoptosis and cell proliferation status. As shown in Figure 6B, H&E staining reveled a pattern of tumor regression and the presence of apoptotic cells defined by cytoplasmic shrinkage and nuclear chromatin condensation in RA-9 treated versus control cohorts (Figure 6B). This was accompanied with a marked reduction in cellular proliferation and increased DNA fragmentation in tumor sections from RA-9 treated versus control as assessed by PCNA staining (Figure 6C) and TUNEL assay (Figure 6D) respectively.
Discussion
Targeting of metabolic pathways to selectively kill cancer cells has promise as an anti-cancer strategy. The use of the clinically available proteasome inhibitors Bortezomib and Carfilzomib resulted with improved clinical outcome in MM and mantle cell lymphoma patients. However, the use of proteasome inhibitors has also been linked to onset of side effects and resistance (13, 14, 35, 36) and Bortezomib has had limited activity against solid cancers, including ovarian cancer (37). This has prompt new efforts in the direction of developing second- and third-generation proteasome inhibitors currently in clinical trials or in preclinical characterization phase (38). This has also led efforts to generate molecules targeting ubiquitin-proteasome degradation pathways independently from inhibition of catalytic activities of the proteasome.
In this context, we have recently provided evidence that targeting of ubiquitin-dependent protein degradation upstream of the proteasome may represent a novel therapeutic approach for cancer treatment. This includes the DUB inhibitor b-AP15 and RA-190, a small-molecule inhibitor of ubiquitin receptor RPN13 (4, 5, 7, 9, 16, 21, 24, 39). To date, the feasibility of targeting DUBs for ovarian cancer treatment and the effect of DUBs inhibition on ovarian cancer cells in vitro and in vivo is unclear.
During a recent drug development effort screening for small-molecule inhibitors of ubiquitin-mediated protein degradation, we identified two distinct classes of compounds: i) compounds with selective inhibitory capacity toward the catalytic activities of the 20S proteasome (40) and ii) compounds that inhibit ubiquitin-mediated protein degradation independently of the 20S catalytic activity, suggesting a possible role as DUB inhibitors (15). In this study we describe and characterize RA-9 as a small-molecule inhibitor of 19S RP-associated DUBs, its mechanism of action on ovarian cancer cells in vitro and in vivo and its therapeutic potential for ovarian cancer treatment.
The presence of α-β carbonyl system has been suggested as the molecular determinant for inhibiting DUBs activity via interaction with the cysteine-based catalytic domain found in their active sites (27, 41-43). In this study we contribute to this view, by showing that RA-9 is capable of preventing ubiquitin-mediated protein degradation and free-ubiquitin recycling upstream of the 20S proteolytic activity. Furthermore, while RA-9 treatment resulted with inhibition of DUB activity in affinity-purified 19S RP, it fails to exert the same effect on total DUB activity measured in whole cell lysates. This indicates that RA-9 is not a general DUB inhibitor, but rather an inhibitor of 19S RP-associated DUBs, although we cannot rule out activity against a small fraction of other cellular DUBs at this time and further studies of its mechanism are needed.
We and others have previously shown that independently from the genetic mutations leading to cancer, malignant transformation is accompanied with progressive up-regulation of the metabolic cancer machinery to sustain the increased proliferation rate that is typical of cancer cells (11, 12, 14). This phenomenon renders ovarian cancer cells selectively sensitive to inhibition of ubiquitin-dependent protein degradation. In this study we show that, inhibition of 19S RP-associated DUBs via RA-9 treatment, selectively hinders the cell viability of our panel of primary ovarian cancer cultures and ovarian cancer cell lines, including those resistant to conventional chemotherapy. Indeed, RA-9 effectively inhibits the growth of these chemoresistant ovarian cancer cell lines at pharmacologically achievable doses. Reduction in cell viability following RA-9 exposure occurred in ovarian cancer cell lines containing either wild-type p53 or mutant p53, indicating that ovarian cancer cell sensitivity to RA-9 is independent of p53 mutation status (11). Importantly, RA-9 treatment caused marked reduction in cell viability of MM cells and their Bortezomib-resistant parental lines. This strongly suggesting that RA-9 could be used in the treatment of multiple cancers, including multiple myeloma. This may also include forms of cancer resistant to clinically available proteasome inhibitors based upon our in vitro studies of bortezomib-resistant multiple myeloma lines. Interestingly, the bortezomib-resistant multiple myeloma lines were both slightly more sensitive to RA-9 than their parental lines.
A major determinant of cell fate is regulation of cell cycle. Previous investigations have shown that DUB substrates include proteins involved in the regulation of the cell cycle progression (44, 45). In this study we show that treatment of ovarian cancer cells with RA-9 caused dose-dependent accumulation in G2-M phase within 8 h of drug exposure. Importantly our studies also show that failure to proceed through cell cycle leads to loss of cell viability associated with caspase-3 activation and apoptosis.
Activation of UPR is a cellular response to accumulation of unfolded or misfolded proteins in the lumen of the ER (28, 34, 46, 47). The UPR attempts to restore protein homeostasis by both halting translation and increasing protein degradation (48, 49). Here we show that greater levels of proteotoxic stress caused by RA-9 increase in the steady state levels of both early and late ER-stress response markers, including GRP-78, IRE1-α, Ero1L-α p-eIF2-α. This suggests that ovarian cancer cells attempt to escape cell death by slowing down protein translation and activating protein degradation in response to RA-9.
In addition to our in vitro studies, we also examined anti-ovarian cancer activity of RA-9 in vivo. Our results show that treatment of ovarian cancer xenograft with RA-9 significantly retarded tumor growth and increased overall survival. Consistent with the effect on UPR induction in vitro, we also observed increase in the levels of ER-stress associated and poly-ubiquitinated proteins in ES-2 tumor cells of treated mice. As seen for in vitro studies, this was accompanied by marked reduction in cellular proliferation and onset of apoptosis in tumor sections from RA-9 treated versus control. Importantly, our data show that RA-9 treatment was not associated with significant toxicity, difference in body weight, blood profile or histology of major organs was observed between RA-9 treated and control mice.
Collectively, our findings suggest that RA-9 has significant in vitro and in vivo anti-cancer activity at doses well tolerated in a mouse xenograft model, and warrants further exploration as a therapeutic agent for ovarian cancer.
Supplementary Material
Translational Relevance.
Ovarian cancer is the fifth leading cause of cancer deaths among American women. Ovarian cancer is associated with up-regulation of protein degradation machinery presumably as a mechanism to sustain the metabolic demands inherent to the malignant phenotype of ovarian cancer cells. In proteasome-mediated proteolysis, deubiquitinating enzymes (DUBs) are an essential component of the ubiquitin-dependent protein degradation. Owing to their recently described role during cancer progression and chemoresistance, deubiquitinating enzymes are an attractive new target for ovarian cancer treatment. Here we report the characterization of RA-9 as a small-molecule inhibitor of proteasome-associated DUB activity. We show that treatment with RA-9 selectively kills ovarian cancer cells including primary cells derived from patient tumors. Mechanistically, RA-9 exposure induces onset of apoptosis via an unresolved Unfolded-Protein-Response (UPR) including activation of ER-stress responses. Our preclinical studies using immunodeficient mice bearing an intra-peritoneal xenograft model of human ovarian cancer shows that treatment with RA-9 retards tumor growth and increases overall survival with no apparent toxicity on the host. Taken together our preclinical studies support further evaluation of this small-molecule inhibitor of proteasome-associated DUBs alone, or in combination with conventional or other novel therapies, for ovarian cancer treatment.
Acknowledgments
We would like to thank Drs. Linda Carson, Levi Downs, Rahel Gebhre, Melissa Geller, Peter Argenta, and Amy Jonson, Division of Gynecological Oncology, University of Minnesota for assisting with the selection of the tumor samples. We would like to thank the Tissue Procurement Facility of the University of Minnesota for their help with procurement of patient tissue samples.
Grant Support: This work was supported by the Department of Defense Ovarian Cancer Research Program (OCRP) OC093424 to MB, by the Randy Shaver Cancer Research and Community Fund to MB and by the Minnesota Ovarian Cancer Alliance (MOCA) to MB. RBSR and RA were supported by National Institutes of Health ATIP Program grant, P50 CA098252 and the HERA foundation. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Disclosure of Potential Conflict of Interests: The authors declare no conflict of interests.
Author's contributions: Conception and design: M. Bazzaro, RBS. Roden and R. Anchoori.
Development of methodology: M. Bazzaro, M.K. Lee, R.Z. Orlowski, K. Coughlin and R. Anchoori.
Acquisition of data: M. Bazzaro, K. Coughlin, R Anchoori, Y. Iizuka, L. MacNeill and J. Meintz.
Analysis and interpretation of data: M. Bazzaro, K. Coughlin, R Anchoori, Rachel Isaksson Vogel, Y. Iizuka, and L. MacNeill.
Writing, review and/or revision of the manuscript: M. Bazzaro, RBS Roden, K. Coughlin, R Anchoori, Y. Iizuka, and L. MacNeill.
Study Supervision: M. Bazzaro.
References
- 1.Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nature reviews Molecular cell biology. 2009;10:550–63. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 2.Love KR, Catic A, Schlieker C, Ploegh HL. Mechanisms, biology and inhibitors of deubiquitinating enzymes. Nat Chem Biol. 2007;3:697–705. doi: 10.1038/nchembio.2007.43. [DOI] [PubMed] [Google Scholar]
- 3.Burrows JF, Johnston JA. Regulation of cellular responses by deubiquitinating enzymes: an update. Front Biosci (Landmark Ed) 2012;17:1184–200. doi: 10.2741/3980. [DOI] [PubMed] [Google Scholar]
- 4.Ye Y, Scheel H, Hofmann K, Komander D. Dissection of USP catalytic domains reveals five common insertion points. Molecular bioSystems. 2009;5:1797–808. doi: 10.1039/b907669g. [DOI] [PubMed] [Google Scholar]
- 5.Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, Donato NJ. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. 2010;70:9265–76. doi: 10.1158/0008-5472.CAN-10-1530. [DOI] [PubMed] [Google Scholar]
- 6.Sun H, Kapuria V, Peterson LF, Fang D, Bornmann WG, Bartholomeusz G, et al. Bcr-Abl ubiquitination and Usp9x inhibition block kinase signaling and promote CML cell apoptosis. Blood. 2011;117:3151–62. doi: 10.1182/blood-2010-03-276477. [DOI] [PubMed] [Google Scholar]
- 7.Luise C, Capra M, Donzelli M, Mazzarol G, Jodice MG, Nuciforo P, et al. An atlas of altered expression of deubiquitinating enzymes in human cancer. PLoS One. 2011;6:e15891. doi: 10.1371/journal.pone.0015891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010;463:103–7. doi: 10.1038/nature08646. [DOI] [PubMed] [Google Scholar]
- 9.D'Arcy P, Brnjic S, Olofsson MH, Fryknas M, Lindsten K, De Cesare M, et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med. 2011;17:1636–40. doi: 10.1038/nm.2536. [DOI] [PubMed] [Google Scholar]
- 10.Kurman RJ, Shih Ie M. Pathogenesis of ovarian cancer: lessons from morphology and molecular biology and their clinical implications. Int J Gynecol Pathol. 2008;27:151–60. doi: 10.1097/PGP.0b013e318161e4f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bazzaro M, Lee MK, Zoso A, Stirling WL, Santillan A, Shih Ie M, et al. Ubiquitin-proteasome system stress sensitizes ovarian cancer to proteasome inhibitor-induced apoptosis. Cancer research. 2006;66:3754–63. doi: 10.1158/0008-5472.CAN-05-2321. [DOI] [PubMed] [Google Scholar]
- 12.Bazzaro M, Lin Z, Santillan A, Lee MK, Wang MC, Chan KC, et al. Ubiquitin proteasome system stress underlies synergistic killing of ovarian cancer cells by bortezomib and a novel HDAC6 inhibitor. Clin Cancer Res. 2008;14:7340–7. doi: 10.1158/1078-0432.CCR-08-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231–4. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Anchoori RK, Khan SR, Sueblinvong T, Felthauser A, Iizuka Y, Gavioli R, et al. Stressing the ubiquitin-proteasome system without 20S proteolytic inhibition selectively kills cervical cancer cells. PloS one. 2011;6:e23888. doi: 10.1371/journal.pone.0023888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anchoori RK, Karanam B, Peng S, Wang JW, Jiang R, Tanno T, et al. A bis-Benzylidine Piperidone Targeting Proteasome Ubiquitin Receptor RPN13/ADRM1 as a Therapy for Cancer. Cancer cell. 2013;24:791–805. doi: 10.1016/j.ccr.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sueblinvong T, Ghebre R, Iizuka Y, Pambuccian SE, Isaksson Vogel R, Skubitz AP, et al. Establishment, characterization and downstream application of primary ovarian cancer cells derived from solid tumors. PloS one. 2012;7:e50519. doi: 10.1371/journal.pone.0050519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iizuka YM, Somia NV, Iizuka K. Identification of NK cell receptor ligands using a signaling reporter system. Methods Mol Biol. 2010;612:285–97. doi: 10.1007/978-1-60761-362-6_19. [DOI] [PubMed] [Google Scholar]
- 19.Shepherd TG, Theriault BL, Campbell EJ, Nachtigal MW. Primary culture of ovarian surface epithelial cells and ascites-derived ovarian cancer cells from patients. Nat Protoc. 2006;1:2643–9. doi: 10.1038/nprot.2006.328. [DOI] [PubMed] [Google Scholar]
- 20.Pribyl L, Coughlin KA, Sueblinvong T, Shields K, Iizuka Y, Downs LS, et al. Method for Obtaining Primary Ovarian Cancer Cells From Solid Specimens. J Vis Exp. 2014;84 doi: 10.3791/51581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rolen U, Kobzeva V, Gasparjan N, Ovaa H, Winberg G, Kisseljov F, et al. Activity profiling of deubiquitinating enzymes in cervical carcinoma biopsies and cell lines. Mol Carcinog. 2006;45:260–9. doi: 10.1002/mc.20177. [DOI] [PubMed] [Google Scholar]
- 22.Fraile JM, Quesada V, Rodriguez D, Freije JM, Lopez-Otin C. Deubiquitinases in cancer: new functions and therapeutic options. Oncogene. 2012;31:2373–88. doi: 10.1038/onc.2011.443. [DOI] [PubMed] [Google Scholar]
- 23.Goncharov T, Niessen K, de Almagro MC, Izrael-Tomasevic A, Fedorova AV, Varfolomeev E, et al. OTUB1 modulates c-IAP1 stability to regulate signalling pathways. EMBO J. 2013 doi: 10.1038/emboj.2013.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McFarlane C, Kelvin AA, de la Vega M, Govender U, Scott CJ, Burrows JF, et al. The deubiquitinating enzyme USP17 is highly expressed in tumor biopsies, is cell cycle regulated, and is required for G1-S progression. Cancer Res. 2010;70:3329–39. doi: 10.1158/0008-5472.CAN-09-4152. [DOI] [PubMed] [Google Scholar]
- 25.Hu M, Li P, Song L, Jeffrey PD, Chenova TA, Wilkinson KD, et al. Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J. 2005;24:3747–56. doi: 10.1038/sj.emboj.7600832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D'Arcy P, Linder S. Proteasome deubiquitinases as novel targets for cancer therapy. Int J Biochem Cell Biol. 2012;44:1729–38. doi: 10.1016/j.biocel.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 27.Mullally JE, Fitzpatrick FA. Pharmacophore model for novel inhibitors of ubiquitin isopeptidases that induce p53-independent cell death. Mol Pharmacol. 2002;62:351–8. doi: 10.1124/mol.62.2.351. [DOI] [PubMed] [Google Scholar]
- 28.Nawrocki ST, Carew JS, Dunner K, Jr, Boise LH, Chiao PJ, Huang P, et al. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005;65:11510–9. doi: 10.1158/0008-5472.CAN-05-2394. [DOI] [PubMed] [Google Scholar]
- 29.Steg AD, Katre AA, Goodman B, Han HD, Nick AM, Stone RL, et al. Targeting the notch ligand JAGGED1 in both tumor cells and stroma in ovarian cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:5674–85. doi: 10.1158/1078-0432.CCR-11-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu P, Khurana A, Rattan R, He X, Kalloger S, Dowdy S, et al. Regulation of HSulf-1 expression by variant hepatic nuclear factor 1 in ovarian cancer. Cancer research. 2009;69:4843–50. doi: 10.1158/0008-5472.CAN-08-3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuhn DJ, Berkova Z, Jones RJ, Woessner R, Bjorklund CC, Ma W, et al. Targeting the insulin-like growth factor-1 receptor to overcome bortezomib resistance in preclinical models of multiple myeloma. Blood. 2012;120:3260–70. doi: 10.1182/blood-2011-10-386789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim JH, Park KC, Chung SS, Bang O, Chung CH. Deubiquitinating enzymes as cellular regulators. J Biochem. 2003;134:9–18. doi: 10.1093/jb/mvg107. [DOI] [PubMed] [Google Scholar]
- 33.Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Dunner K, Jr, Huang P, et al. Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res. 2005;65:11658–66. doi: 10.1158/0008-5472.CAN-05-2370. [DOI] [PubMed] [Google Scholar]
- 34.Rao R, Balusu R, Fiskus W, Mudunuru U, Venkannagari S, Chauhan L, et al. Combination of pan-histone deacetylase inhibitor and autophagy inhibitor exerts superior efficacy against triple-negative human breast cancer cells. Mol Cancer Ther. 2012;11:973–83. doi: 10.1158/1535-7163.MCT-11-0979. [DOI] [PubMed] [Google Scholar]
- 35.Chauhan D, Singh AV, Aujay M, Kirk CJ, Bandi M, Ciccarelli B, et al. A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood. 2010;116:4906–15. doi: 10.1182/blood-2010-04-276626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chauhan D, Tian Z, Zhou B, Kuhn D, Orlowski R, Raje N, et al. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin Cancer Res. 2011;17:5311–21. doi: 10.1158/1078-0432.CCR-11-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parma G, Mancari R, Del Conte G, Scambia G, Gadducci A, Hess D, et al. An open-label phase 2 study of twice-weekly bortezomib and intermittent pegylated liposomal doxorubicin in patients with ovarian cancer failing platinum-containing regimens. International journal of gynecological cancer: official journal of the International Gynecological Cancer Society. 2012;22:792–800. doi: 10.1097/IGC.0b013e318251051a. [DOI] [PubMed] [Google Scholar]
- 38.Lawasut P, Chauhan D, Laubach J, Hayes C, Fabre C, Maglio M, et al. New proteasome inhibitors in myeloma. Current hematologic malignancy reports. 2012;7:258–66. doi: 10.1007/s11899-012-0141-2. [DOI] [PubMed] [Google Scholar]
- 39.Theard D, Labarrade F, Partisani M, Milanini J, Sakagami H, Fon EA, et al. USP9x-mediated deubiquitination of EFA6 regulates de novo tight junction assembly. EMBO J. 2010;29:1499–509. doi: 10.1038/emboj.2010.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bazzaro M, Anchoori RK, Mudiam MK, Issaenko O, Kumar S, Karanam B, et al. alpha, beta-Unsaturated carbonyl system of chalcone-based derivatives is responsible for broad inhibition of proteasomal activity and preferential killing of human papilloma virus (HPV) positive cervical cancer cells. Journal of medicinal chemistry. 2011;54:449–56. doi: 10.1021/jm100589p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verbitski SM, Mullally JE, Fitzpatrick FA, Ireland CM. Punaglandins, chlorinated prostaglandins, function as potent Michael receptors to inhibit ubiquitin isopeptidase activity. J Med Chem. 2004;47:2062–70. doi: 10.1021/jm030448l. [DOI] [PubMed] [Google Scholar]
- 42.Rodriguez AM, Enriz RD, Santagata LN, Jauregui EA, Pestchanker MJ, Giordano OS. Structure-cytoprotective activity relationship of simple molecules containing an alpha, beta-unsaturated carbonyl system. J Med Chem. 1997;40:1827–34. doi: 10.1021/jm960280m. [DOI] [PubMed] [Google Scholar]
- 43.Kapuria V, Levitzki A, Bornmann WG, Maxwell D, Priebe W, Sorenson RJ, et al. A novel small molecule deubiquitinase inhibitor blocks Jak2 signaling through Jak2 ubiquitination. Cell Signal. 2011;23:2076–85. doi: 10.1016/j.cellsig.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 44.Wilkinson KD. DUBs at a glance. J Cell Sci. 2009;122:2325–9. doi: 10.1242/jcs.041046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.D'Andrea A, Pellman D. Deubiquitinating enzymes: a new class of biological regulators. Crit Rev Biochem Mol Biol. 1998;33:337–52. doi: 10.1080/10409239891204251. [DOI] [PubMed] [Google Scholar]
- 46.Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280:26729–34. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 47.Zang Y, Thomas SM, Chan ET, Kirk CJ, Freilino ML, Delancey HM, et al. The next generation proteasome inhibitors carfilzomib and oprozomib activate prosurvival autophagy via induction of the unfolded protein response and ATF4. Autophagy. 2012;8 doi: 10.4161/auto.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197:857–67. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.