

Abstract
While it is well documented iron oxide can reduce soot through burnout in the oxidative regions of flames, it may also impact molecular growth and particle inception. The role of Fe2O3 nanoparticles in mass growth of soot from 1-methylnapthalene (1-MN) was studied in a dual-zone, high-temperature flow reactor. An iron substituted, dendrimer template was oxidized in the first zone to generate ~5 nm Fe2O3 nanoparticles, which were seeded into the second zone of the flow reactor containing 1-MN at 1100°C and ϕ = 1.4–5.0. Enhanced molecular growth in the presence of Fe2O3 nanoparticles resulted in increased yields of polycyclic aromatic hydrocarbons (PAH) and soot compared to purely gas-phase reactions of 1-MN at identical fuel–air equivalence ratios. This also resulted in an increase in soot-number concentration and a slight shift to smaller particles with increasing addition (from no addition to 3 mM) of Fe2O3. Introduction of Fe2O3 nanoparticles resulted in the formation of stabilization of environmentally persistent free radicals (EPFRs), including benzyl, phenoxyl, or semiquinone-type radicals as well as carbon-centered radicals, such as cyclopentadienyl or a delocalized electron in a carbon matrix. At the high concentrations in the flow reactor, these resonance-stabilized free radicals can undergo surface-mediated, radical–radical, molecular growth reactions which may contribute to molecular growth and soot particle inception.
Keywords: Transition metal, Fine particles, PM2.5, Pollution
1. Introduction
Iron compounds, often used for soot reduction, have been extensively studied for their potential to oxidize soot, but have also been observed to enhance some aspects of soot formation [1–24]. For example, addition of iron pentacarbonyl ((Fe(CO)5) to a sooting diffusion flame resulted in an enhanced rate of soot oxidation in the soot burnout regime [6,15]. Doping ferrocene (Fe(C5H5)2) into a flame led to the formation of solid iron oxide particles and enhanced soot oxidation rates near the flame terminus [23]. Although, laminar premixed ethylene flames doped with ferrocene were found to exhibit an overall reduction in mass yield of soot, they also exhibited an increase in both soot particle size and number density [24]. Iron oxide particles have also been observed to shorten the time for soot to first appear in the flame [2,23]. Similarly, a threefold increase in soot formation has been reported when 200 ppm of iron was doped into the fuel of a laminar, sooting, premixed ethlylene/oxygen/nitrogen flame [5]. These studies demonstrate iron compounds are effective in reducing overall soot yields due to efficient burnout of soot under oxidizing conditions rather than by inhibition [1,5,6,13,15,24]. However, the data may also suggest iron compounds promote soot inception in some regions of the flames under both pyrolysis and oxidative pyrolysis conditions. The question is then: What is the reason for this dichotomous behavior?
Some combustion-generated, ultra-fine particles are formed via post-combustion condensation of relatively non-volatile species vaporized within the high-temperature flame zone [14,20,25,26]. These include suboxides of silica and alumina, SO2 converted to non-volatile sulfates, moderately volatile species ofcommon metals such as Ni, V, Fe, Zn, and Cu, and some high-molecular weight organics [27–29]. The presence of seed nuclei are generally accepted to be necessary (due to the Kelvin effect which reduces the vapor pressure of species on surfaces) for volatile species to condense into particles. Although sulfate anions are the most frequently identified seed nuclei, there is evidence the counter metal cations are equally capable of mediating condensation of volatile species into particles [7,27]. For example, metal cations associated with nanoparticles of metals and their oxides have shown enhanced surface reactivity and increased adsorption due to increased interfacial tension and surface free energy with decreasing particle size [30].
To determine the mechanism by which metal nanoparticles may enhance certain aspects of molecular growth and soot formation, we conducted a study to determine if Fe2O3 nanoparticles promote soot inception from a high sooting fuel, 1-MN, under oxidative pyrolysis conditions.
2. Experimental
A two-zone, fused silica, heterogeneous-flow reactor system was developed in our laboratory and utilized to generate soot from the oxidative pyrolysis of 95% 1-MN (Sigma–Aldrich M56808-100G). Zone 1 generated the metal oxide nanoparticles in situ from the oxidation of a polypropylenimine tetra-hexacontaamine dendrimer complex with iron(III) nitrate nonahydrate. A methanolic solution of the dendrimer–metal complex was delivered at 85 μL/h with a syringe pump. The gas-phase residence time in Zone 1 was maintained at 60 s. The nanoclusters produced in Zone 1 were continually introduced into Zone 2 of the reactor where they could impact the otherwise gas-phase reactions of 1-MN (cf. Fig. 1). The dendrimer-methanolic solution, containing no iron(III) nitrate nonahydrate, served as the baseline standard. McMillan Mass Flow Controllers and a DryCal DC-2 gilibrator (Bios International Corp.) were utilized to ensure stable flow of carrier gases through the two-zone flow reactor system. The gas-phase residence time in Zone 2 was 1.0 s.
Fig. 1.

Diagram of 2-zone reactor system. Zone 1 of the reactor is used to generate metal-oxide nanoparticles, which are seeded into Zone 2 in the presence of a 1-MN fuel.
All samples were collected on Advantec 46 mm, 0.8 μm pore size mixed cellulose ester filters from the end of the dual zone reactor with the aid of a filter holder and rotary pump. Immediately after collection, the samples were subjected to electron paramagnetic resonance (EPR) analyses for free radicals. These filters were then subjected to a washing in excess chloroform followed by sonnication to remove all soluble organic material. This extract was subjected to GC–MS analysis. The remaining soot was removed by dissolving the filter in acetone and separating the soluble filter from the soot through excess washing and centrifugation. This soot was then analyzed for free radicals with EPR spectroscopy.
EPR measurements were performed using a Bruker EMX- 20/2.7X-band EPR spectrometer. Typical parameters included a frequency of 9.655, an attenuation of 20.0 dB, receiver gain of 1.00 × 104, a modulation frequency of 100.00 kHz, and a modulation amplitude of 4.0 G. The g-values and spins/gram values were determined by comparison with 2, 2- diphenyl-1-picrylhydrazyl (DPPH).
The chloroform extracts were analyzed for polycyclic aromatic hydrocarbons (PAHs) using a Varian Chrompack CP-3800 gas chromatograph coupled to a Varian Saturn Series 2000 MS equipped with a capillary column (Rtx-5MS, 30 m × 0.25 μm × 0.25). All extracts were concentrated before injection and quantified with standards obtained from Sigma Aldrich.
A JEOL JEM-1011 High Resolution Transmission Electron Microscope equipped with a LaB6 source, with 200 kV accelerating voltage and magnification of 400–800X (HRTEM) was used to characterize the particle size and morphology of the particles. The samples were prepared by thermophoretic collection of the particles on a TEM Grid (Holey Carbon Coated Copper Grid) at the outlet of reactor 1 or reactor 2.
A Differential Mobility Analyzer (DMA TSI Model 3081 or Model 3085) coupled to an Ultra-fine Condensation Particle Counter (UCPC Model 3776) was utilized to determine the concentration of particles and particle diameter. An aerosol flow rate of 1.5 lpm and sheath flow rate of 15.0 lpm was maintained in the DMA. Carbon analyses were performed by MicroAnalysis Inc. with an EA 112 CHN elemental analyzer.
3. Results
In Zone 1, iron-oxide nanoparticles were generated in situ from the oxidation of the genera-tion-4 polypropylenimine tetra-hexacontaamine dendrimer complex tethered with iron(III) nitrate nonahydrate. Dendrimers serve as a structure-directing template to provide control of the size and stability of nanoparticles generated from the collapse of the core structure. The increasing size associated with increasing dendrimer generation as well as the relationship between metal ions and terminal dendrimer ends allowed for the specific tailoring of nanoparticle size [31–33].
The fully doped dendrimer, where theoretically each terminus end binds one iron ion, was oxidized at 700 °C. Upon oxidation the dendrimer collapsed, resulting in the formation of ~5 nm Fe2O3 nanoclusters as seen in micrographs presented in Fig. 2. Since these nanoparticles were captured thermophoretically on a TEM grid; a Differential Mobility Analyzer (DMA) was used for in situ, near real-time measurements of the size distribution of the Fe2O3 nanoparticles. DMA measurements indicated the presence of primarily ~2–7 nm Fe2O3 nanoparticles (cf. Fig. 2) which was roughly consistent with size distributions obtained through TEM micrographs.
Fig. 2.
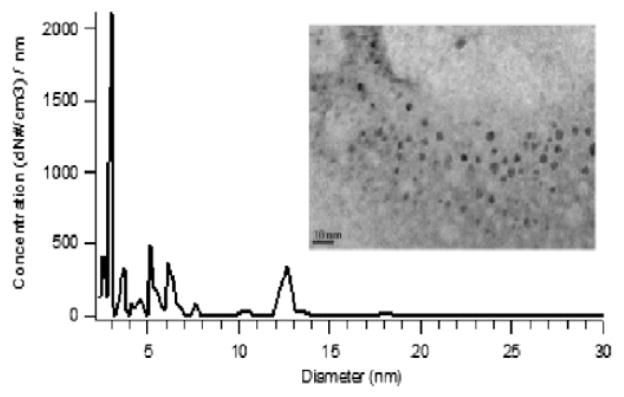
Size distribution of Fe2O3 nanoparticles generated from Zone 1 of the reactor.
Introduction of 1-MN into Zone 2 of the reactor in the presence of Fe2O3 nanoparticles resulted in the formation of ~20 nm diameter, spherical, coalesced amorphous aggregates, accumulated into chains of several hundred nanometers in length (cf. Fig. 3A). Higher magnification microscopy revealed the semi-crystalline, pseudo-onion skin structure, characteristic of individual soot nanoparticles. (cf. Fig. 3B) [35]. Microscopy of the thermophoretically condensed products, from the oxidative-pyrolysis of 1-MN, established the dual zone flow reactor was capable of generating soot in the presence of redox-active transition metal nanoparticles. Although TEM was utilized to elucidate the morphology of soot nanoparticles, elemental analysis of the insoluble fraction provided the soot’s chemical composition. Since soot is often characterized by its elemental composition, particles generated in the dual zone flow-reactor were subjected to elemental analysis to provide further evidence the particles generated in the dual zone reactor were comparable to soot generated from combustion sources. Soot generated in the dual zone reactor was determined to be composed of 81.8% carbon, 2.9% hydrogen, and 1.2% nitrogen. Both TEM micrographs and elemental analysis of the particulate generated in the dual zone reactor confirm the identity of the condensable material generated in the dual zone reactor from the oxidative-pyrolysis of 1-MN as soot.
Fig. 3.

(A) TEM micrographs of soot agglomerates formed at 1100°C and ϕ = 2.5. (B) Nanostructure of ultrafine soot.
The effect of Fe2O3 nanoparticle introduction on the size and morphology of soot condensed from the dual zone reactor at various fuel/air equivalence ratios were examined through TEM microscopy (cf. Fig. 4). Amorphous particles, suspected of containing soluble organic precursors were evident at ϕs as small as ϕ = 1.4. A decrease in the oxygen resulted in the transformation of the amorphous carbon precursors into concentrically wrapped nanoparticles agglomerated into larger chains. The introduction of Fe2O3 nanoparticles at ϕ = 2.5 resulted in formation of what appeared to be soot nanoparticles, whereas the condensable material generated with no Fe2O3 addition under the same conditions resulted in the formation of amorphous agglomerates with no soot nanoparticles. The presence of randomly oriented amorphous carbon structures at lower equivalence ratios, and the appearance of ~20 nm, spherical, semi-crystalline, aggregate chains of several hundred nanometers in length at higher equivalence ratios, indicates the introduction of Fe2O3 nanoparticles induced the formation of soot at lower equivalence ratios than purely gas-phase reactions. The addition of Fe2O3 nanoparticles also resulted in increased agglomeration and larger soot particles.
Fig. 4.

TEM micrographs of soot morphology formed from the pyrolysis of 1-MN at ϕ = 1.4–5.0 and 1100°C, with and without the addition of ~5 nm Fe2O3 nanoparticles.
These observations were supported by GC/MS analysis of the PAHs associated with the soluble organic fraction. At ϕ = 2.5, the introduction of Fe2O3 nanoparticles led to the formation of higher molecular weight PAHs. Both chrysene and coronene were observed in ppm quantities in soot generated with Fe2O3 nanoparticles but were absent in soot generated with no Fe2O3 addition. Concentrations of phenanthrene, fluoranthene, pyrene, 4H-cyclopentaphenanthrene, benzo[e]phenanthrene were observed to increase with Fe2O3 doping while concentrations of anthracene and benzo[a]pyrene decreased. These results suggested the introduction of Fe2O3 nanoparticles resulted in the formation of larger PAHs. As a result of PAHs being formed and destroyed with similar rates in the molecular growth process, a linear increase in all PAH concentrations was not observed as a function of Fe2O3 introduction.
Previous research has shown PAHs and other pollutants can be formed by reactions of environmentally persistent free radicals (EPFRs) formed on transition metal containing particles [44–47]. Furthermore, the EPFRs may be more toxic than their molecular precursors [42–47]. Based on the structural differences observed in the TEM micrographs at ϕ = 2.5, samples of soot were generated for EPR analysis as a function of increased Fe2O3 doping at 1100°C and ϕ = 2.5 (cf. Fig. 5). The large, broad signal (ΔHp–p = 9.5–10 G) was indicative of multiple radical species. The EPR radical signal was observed to strikingly increase with increasing addition of Fe2O3 (from no addition to 3 mM). Normalization of the radical intensity to the soot mass (spins/g of soot) confirmed the formation of more paramagnetic species as a result of the introduction of Fe2O3. Even after removing the soluble organic fraction, the large broad signal persisted. A similar correlation between free radical concentration and iron concentration in atmospheric aerosols has been previously observed [48].
Fig. 5.

EPR spectra of soot generated at 1100°C and ϕ = 2.5 as a function of addition of Fe2O3 nanoparticles. g-factor = 2.00298, ΔHp–p = 9.5–10 G, and ΔHtotal = 50 G.
The half-lives of the radicals were determined by allowing the soot to age under ambient conditions with periodic measurement of the EPR signal (cf. Fig. 6). The initial concentration of radicals was only 30% greater in the presence of iron; however, the half-lives were much longer. Two radical signals were observed for soot generated with Fe2O3 addition. One exhibited a halflive of 2.9 days and a g-value of 2.0035. The other decayed very slowly over 50 days and exhibited a g-value of 2.0025. The g-value of the shorter lived radical was consistent with that of a resonance-stabilized semiquinone-, phenoxyl, or possibly benzyl-type radical [43,49], while the g-value of the longer lived radical of 2.0025 was typical of a carbon-centered radical, such as cyclopentadienyl or a delocalized electron in a carbon matrix [50–53]. Soot radicals generated from the combustion of pure 1-MN exhibited a substantially shorter half-live, 1.8 days, than soot generated with the introduction of Fe2O3 nanoparticles. After 48 h, the concentration of radicals within soot generated with pure 1-MN were below detection limit. Based on these observations, radicals were stabilized by the introduction of Fe2O3 nanoparticles, which is consistent with previously reported studies of EPFR formation [43–45].
Fig. 6.

Measurement of half-lives of radicals associated with soot from the pyrolysis of 1-MN in the presence of Fe2O3 nanoparticles and pure 1-MN.
The size distribution and concentration of soot formed as a result of increasing addition of Fe2O3 were determined by DMA (cf. Fig. 7). Particles with a diameter of 40–320 nm were observed from the thermal degradation of 1-MN. An increase in number concentration was observed with increasing addition (from no addition to 3 mM) of Fe2O3, which correlated with the increase in soot yield due to Fe2O3 addition. As the iron concentration was increased, the size distribution shifted slightly to smaller soot particles.
Fig. 7.
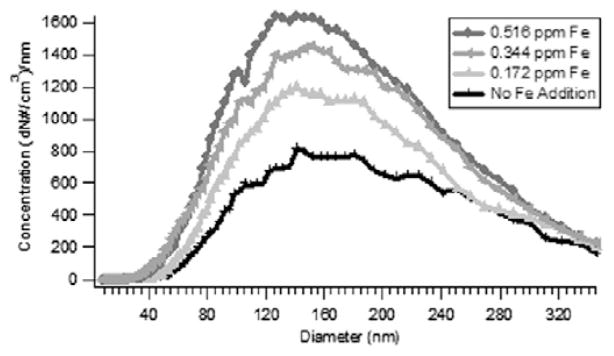
Increase in number concentration and decrease in the median diameter of soot particles as a function of doping of Fe2O3 nanoparticles at 1100°C and ϕ = 2.5.
4. Discussion
The soot generated from the introduction of 1- MN into Zone 2 of the reactor in the presence of Fe2O3 nanoparticles was consistent with soot generated from various combustion sources in both morphology and chemical composition [34,36– 40]. Elemental analysis of the insoluble fraction indicated that the soot from the dual zone reactor was similar to the elemental composition of soot generated in flame studies of various hydrocarbon fuels [22,41]. Soot generated from the combustion of 1-MN (C11H10) was most similar in chemical composition to soot from the combustion of diesel fuel, a blend of C10–C19 aliphatic and aromatic hydrocarbons. TEM images of the particulate morphology and elemental composition provided additional validation that the soot generated in the dual zone flow reactor was consistent with soot generated from combustion sources.
This new method of metal-oxide nanoparticle generation and pyrolysis of 1-MN in a dual zone reactor has provided a method for studying molecular growth and particle nucleation in the presence of redox-active, transition metal-oxide clusters. Our data suggests redox-active transition metal nanoparticles can mediate formation of surface- stabilized radicals and resultant surface-mediated, radical-radical and/or radical-molecule, molecular growth, which may contribute to PAH and soot formation.
Based on review of the literature and our own studies of CuO and Fe2O3 mediated formation of pollutants, Fe2O3 induces chemical reactions resulting in the formation of PAHs as well as polychlorinated dibenzo-p-dioxins and dibenzofurans (PCDD/F) from simple aromatic precursors at temperatures as low as 200°C [43–47]. Research has demonstrated the mechanism involves the formation of EPFRs. When the EPFRs are in low concentration in the post-combustion, cool-zone, at temperatures <300°C, they can survive and be emitted into the atmosphere. In high concentrations, in the post-flame, thermal zone, at temperatures of 200–600°C, the EPFRs react with other EPFRs or surface adsorbed molecular species to form PAH and PCDD/F. It was presumed in these previously reported studies that, due to the reversibility of the chemisorption reaction, the molecular precursors would preferentially desorb at temperatures >600°C, further reducing the reaction yields. However, chemically activated intermediates, such as those formed by chemisorption, can undergo a rapid secondary reaction, such as “polymerization,” resulting in deactivation and stabilization of the initially formed adduct (cf. Fig. 8). Thus, when molecular growth reactions are possible, raising the temperature of the system can overcome the tendency for the initially formed complex to desorb.
Fig. 8.

Chemically-activated surface reactions demonstrating how subsequent molecular growth steps can stabilize initially formed surface adducts and lead to high molecular weight products at elevated temperatures.
Radicals are formed by initial physisorption, followed by chemisorption to a metal site, and electron transfer from the organic sorbate to the metal to form an organic EPFR and a reduced metal. (cf. Fig. 9, Path A). The formation of stabilized free radicals has been demonstrated in previous studies for hydroquinones, catechols, chlorinated phenols, chlorinated benzenes and copper-oxide surfaces under post-flame, cool-zone conditions [54]. Similar studies have indicated that Fe2O3 nanoparticles undergo a reduction of the surface oxidation state when incorporated with a soot matrix [55]. A subsurface layer of Fe+2 resulted from the addition of acetylene to combustion- synthesized Fe2O3 nanoparticles [56–58]. Once the initial radical is formed, successive radical– molecule or radical–radical reactions may continue molecular growth to form PAHs and soot.
Fig. 9.

Pathways of radical formation from 1-methylnaphthalene by chemisorption and electron transfer to a metal oxide surface.
Toluene and naphthalene have been reported to interact with metal and metal oxide surfaces by the charge transfer from the π orbitals of the aromatic ring to the metal center, suggesting a parallel orientation of the reacting molecule with the surface [59–62]. (cf. Fig. 9, Path B) Hydroxylated metal oxide surfaces interact with the aromatic ring through OH-π electrons backbonding [63]. These interactions lead to the destabilization of the aromatic ring and substituent groups and result in surface assisted hydrogen abstraction [60,61]. These initially formed radicals may react directly or tautomerize to form more stable radicals [63], oxygenated species [64–66] PAHs [62], and soot. Decomposition may also occur via either pathway to form purely aromatic radicals and carbonyls (cf. Fig. 14, Path C).
Along with gas-phase reactions and PAH polymerization, the formation of surface associated radical species with redox-active transition-metal nanoparticles must be considered as a potential route capable of molecular growth of hydrocarbon precursors into soot. However, research has shown the dominant impact of redox-active iron addition to flames is soot burnout. Inhibition of flames by Fe(CO)5 has been extensively studied in premixed and diffusion flames [15–17]. The overall effectiveness of Fe(CO)5 in chemical inhibition was found to be dependent on doping concentration [16]. Other studies found the soot particle size and number density increased at shorter residence times and resulted in enhanced soot formation. A decrease in soot particle size and number density were observed in the soot burnout regime with an overall net effect of reduction of soot yield [1,2]. These observed differences in inhibition of Fe(CO)5 in premixed and diffusion flames was associated with absence of a soot burnout region or oxidation region in premixed flame configurations [6]. The overall effectiveness of inhibition was dependent upon iron concentration, form of iron present, particle size, residence time, flame temperature, and the drag and thermophoretic forces in the flame [17].
Regardless of the evaluation of iron-containing compounds as a soot inhibitor or soot promoter, the introduction of Fe(CO)5 to premixed flames resulted in the formation of Fe2O3 species dispersed throughout the soot particle matrix [1,2,4]. The nucleation of iron-based compounds before soot particle inception resulted in nanoparticle formation and increased surface area for soot formation and growth to occur [6,20,24]. The iron-oxide nanoparticles became incorporated into the soot matrix or agglomerated on the soot surface leading to the formation of active sites for both soot surface growth and soot oxidation to occur [6].
In regions of poor fuel–air mixing, where there are limited chances for oxidation, Fe2O3 can be initially reduced through reactions with substituted PAHs to form reduced iron and larger PAHs and soot. Iron-oxide nanoparticles, not encased by soot, may agglomerate onto the surface of soot and be re-oxidized back to Fe2O3. These highly oxidized species then may participate in the oxidative burnout of soot in more oxidizing regions of the system. This is supported by the presence of both metallic and oxidized iron species incorporated into the soot matrix [67].
5. Conclusions
Fe2O3 nanoparticles can mediate the formation of surface-stabilized radicals and, through subsequent surface-mediated, radical–radical and/or radical–molecule molecular growth, impact the mechanism of PAH formation, soot particle inception, and surface growth in hydrocarbon combustion systems. While Fe2O3 may not cause an overall net increase in soot formation, other less oxidizing transition metal nanoparticles may have a net promoting effect on soot formation.
Acknowledgments
This work was partially supported by the NIEHS Superfund Research Program under grant 5 P42 ES013648-03 and the Patrick F. Taylor Chair.
References
- 1.Bonczyk PA. Combust Sci Technol. 1988;59:143. [Google Scholar]
- 2.Bonczyk PA. Combust Flame. 1991;87:233. [Google Scholar]
- 3.Braun A, Huggins FE, Kelly KE, Mun BS, Ehrlich SN, Huffman GP. Carbon. 2006;44:2904. [Google Scholar]
- 4.Charalampopoulos TT, Hahn DW, Chang H. Appl Opt. 1992;31:6519. doi: 10.1364/AO.31.006519. [DOI] [PubMed] [Google Scholar]
- 5.Feitelberg AS, Longwell JP, Sarofim AF. Combust Flame. 1993;92:241. [Google Scholar]
- 6.Hahn DW, Kim KB, Masiello KA. Combust Flame. 2008;154:164. [Google Scholar]
- 7.Haynes BS, Neville M, Quann RJ, Sarofim AF. J Colloid Interf Sci. 1982;87:266. [Google Scholar]
- 8.Hirasawa T, Sung CJ, Yang ZW, Joshi A, Wang H. Combust Flame. 2004;139:288. [Google Scholar]
- 9.Kasper M, Sattler K, Siegmann K, Matter U, Siegmann HC. J Aerosol Sci. 1999;30:217. [Google Scholar]
- 10.Kasper M, Siegmann K. Combust Sci Technol. 1998;140:333. [Google Scholar]
- 11.Kayes D, Hochgreb S. Environ Sci Technol. 1999;33:3968. [Google Scholar]
- 12.Lee DG, Miller A, Park KH, Zachariah MR. Int J Automot Technol. 2006;7:667. [Google Scholar]
- 13.Li ZS, Tian K, Staude S, et al. Proc Combust Inst. 2009;32:445. [Google Scholar]
- 14.Linak WP, Srivastava RK, Wendt JOL. Combust Sci Technol. 1994;101:7. [Google Scholar]
- 15.Linteris GT, Babushok VI. Proc Combust Inst. 2009;32:2535. doi: 10.1016/j.proci.2018.06.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Linteris GT, Katta VR, Takahashi F. Combust Flame. 2004;138:78. doi: 10.1016/j.combustflame.2016.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linteris GT, Rumminger MD, Babushok VI. Prog Energy Combust. 2008;34:288. [Google Scholar]
- 18.Marsh ND, Preciado I, Eddings EG, Sarofim AF, Palotas AB, Robertson JD. Combust Sci Technol. 2007;179:987. [Google Scholar]
- 19.Miller A, Ahlstrand G, Kittelson D, Zachariah M. Combust Flame. 2007;149:129. [Google Scholar]
- 20.Miller AL, Stipe CB, Habjan MC, Ahlstrand GG. Environ Sci Technol. 2007;41:6828. doi: 10.1021/es070999r. [DOI] [PubMed] [Google Scholar]
- 21.Rumminger MD, Linteris GT. Combust Flame. 2002;128:145. [Google Scholar]
- 22.Stanmore BR, Brilhac JF, Gilot P. Carbon. 2001;39:2247. [Google Scholar]
- 23.Zhang J, Megaridis CM. Combust Flame. 1996;105:528. [Google Scholar]
- 24.Ritrievi KE, Longwell JP, Sarofim AF. Combust Flame. 1987;70:17. [Google Scholar]
- 25.Keskinen J, Ronkko T, Virtanen A, Kannosto J, Lappi M, Pirjola L. Environ Sci Technol. 2007;41:6384. doi: 10.1021/es0705339. [DOI] [PubMed] [Google Scholar]
- 26.Lighty JS, Veranth JM, Sarofim AF. J Air Waste Manage. 2000;50:1565. doi: 10.1080/10473289.2000.10464197. [DOI] [PubMed] [Google Scholar]
- 27.Allouis C, Beretta F, D’Alessio A. Chemosphere. 2003;51:1091. doi: 10.1016/S0045-6535(02)00714-2. [DOI] [PubMed] [Google Scholar]
- 28.Sarofim AF, Howard JB, Padia AS. Combust Sci Technol. 1977;16:187. [Google Scholar]
- 29.Zappoli S, Andracchio A, Fuzzi S, et al. Atmos Environ. 1999;33:2733. [Google Scholar]
- 30.Grassian VH. J Phys Chem C. 2008;112:18303. [Google Scholar]
- 31.Bosman AW, Janssen HM, Meijer EW. Chem Rev. 1999;99:1665. doi: 10.1021/cr970069y. [DOI] [PubMed] [Google Scholar]
- 32.Floriano PN, Noble CO, Schoonmaker JM, Poliakoff ED, McCarley RL. J Am Chem Soc. 2001;123:10545. doi: 10.1021/ja010549d. [DOI] [PubMed] [Google Scholar]
- 33.Lomnicki SM, Wu HY, Osborne SN, et al. Mater Sci Eng B-Adv. 2010;175:136. doi: 10.1016/j.mseb.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alfe M, Apicella B, Rouzaud JN, Tregrossi A, Ciajolo A. Combust Flame. 1959;2010:157. [Google Scholar]
- 35.Kis VK, Posfai M, Labar JL. Atmos Environ. 2006;40:5533. [Google Scholar]
- 36.Wal RLV, Tomasek AJ. Combust Flame. 2004;136:129. [Google Scholar]
- 37.Winans RE, Tomczyk NA, Hunt JE, et al. Energy Fuel. 2007;21:2584. [Google Scholar]
- 38.Palotas AB, Rainey LC, Feldermann CJ, Sarofim AF, VanderSande JB. Microsc Res Techniq. 1996;33:266. doi: 10.1002/(SICI)1097-0029(19960215)33:3<266::AID-JEMT4>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 39.Rouzaud JN, Clinard C. Fuel Process Technol. 2002;77:229. [Google Scholar]
- 40.Ciajolo A, Alfe M, Apicella B, Barbella R, Rouzaud JN, Tregrossi A. Proc Combust Inst. 2009;32:697. [Google Scholar]
- 41.Horn AB, Daly HM. Phys Chem Chem Phys. 2009;11:1069. doi: 10.1039/b815400g. [DOI] [PubMed] [Google Scholar]
- 42.Anton R. J Mater Res. 1837;2005:20. [Google Scholar]
- 43.Dellinger B, Lomnicki S, Truong H, Vejerano E. Environ Sci Technol. 2008;42:4982. doi: 10.1021/es071708h. [DOI] [PubMed] [Google Scholar]
- 44.Dellinger B, Masko Z, Khachatryan L, Lomnicki S. Free Radical Biol Med. 2009;47:S187. [Google Scholar]
- 45.Dellinger B, Nganai S, Lomnicki S. Environ Sci Technol. 2009;43:368. doi: 10.1021/es8022495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dellinger B, Nganai S, Lomnicki SM. Environ Sci Technol. 2011;45:1034. doi: 10.1021/es102948f. [DOI] [PubMed] [Google Scholar]
- 47.Dellinger B, Vejerano E, Lomnicki S. Environ Sci Technol. 2011;45:589. doi: 10.1021/es102841s. [DOI] [PubMed] [Google Scholar]
- 48.Aboukais A, Ledoux F, Zhilinskaya EA, Courcot D, Puskaric E. Atmos Environ. 2004;38:1201. [Google Scholar]
- 49.Neta P, Fessenden RW. J Phys Chem-Us. 1974;78:523. [Google Scholar]
- 50.Allard P, Barra AL, Andersson KK, Schmidt PP, Atta M, Graslund A. J Am Chem Soc. 1996;118:895. [Google Scholar]
- 51.Barclay LRC, Cromwell GR, Hilborn JW. Can J Chem. 1994;72:35. [Google Scholar]
- 52.Tian LW, Koshland CP, Yano JK, et al. Energy Fuel. 2009;23:2523. doi: 10.1021/ef8010096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dalal NS, Smirnov AI, Belford RL. Appl Spectrosc. 1997;51:1429. [Google Scholar]
- 54.Alderman SL, Dellinger B. J Phys Chem A. 2005;109:7725. doi: 10.1021/jp051071t. [DOI] [PubMed] [Google Scholar]
- 55.Alderman SL, Farquar GR, Poliakoff ED, Dellinger B. Environ Sci Technol. 2005;39:7396. doi: 10.1021/es050063n. [DOI] [PubMed] [Google Scholar]
- 56.Jasinski J, Pinkerton KE, Kennedy IM, Leppert VJ. Sensor Actuat B-Chem. 2005;109:19. [Google Scholar]
- 57.Jasinski J, Pinkerton KE, Kennedy IM, Leppert VJ. Microsc Microanal. 2006;12:424. doi: 10.1017/S1431927606060491. [DOI] [PubMed] [Google Scholar]
- 58.Leppert VJ, Pinkerton KE, Jasinski J. Mater Res Soc Symp Proc. 2004;839:17. [Google Scholar]
- 59.Haber J, Tokarz R, Witko M. Stud Surf Sci Catal. 1994;82:739. [Google Scholar]
- 60.Irigoyen B, Juan A, Larrondo S, Amadeo N. J Catal. 2001;201:169. [Google Scholar]
- 61.Irigoyen B, Juan A, Larrondo S, Amadeo N. Surf Sci. 2003;523:252. [Google Scholar]
- 62.Taylor SH, Ntainjua E. Top Catal. 2009;52:528. [Google Scholar]
- 63.Nagao M, Suda Y. Langmuir. 1989;5:42. [Google Scholar]
- 64.Boix AV, Serra RM, Miro EE. Micropor Mesopor Mater. 2010;127:182. [Google Scholar]
- 65.Cardona-Martinez N, Mendez-Roman R. Catal Today. 1998;40:353. [Google Scholar]
- 66.Freitag C, Besselmann S, Loffler E, Grunert W, Rosowski F, Muhler M. Catal Today. 2004;91–2:143. [Google Scholar]
- 67.Zachariah MR, Kim SH, Fletcher RA. Environ Sci Technol. 2005;39:4021. doi: 10.1021/es048828z. [DOI] [PubMed] [Google Scholar]