

Abstract
Heterologous expression is an efficient alternative to conventional extraction to produce a specific Buthus martensii Karsch (BmK) peptide. In this work, BmK1 was successfully expressed in Escherichia coli after genetic codon optimization, but BmK1 content was <6% of total cellular protein. To improve BmK1 expression, a trc promoter library with a wide relative strength was constructed, and three promoters, PpJF136 (0.55), PpJF325 (1.29), and PpJF288 (2.31), were selected to control BmK1 expression. A higher BmK1 expression (>13.9% of total protein) was obtained using a stronger promoter, PpJF325. Furthermore, a maximum BmK1 content (>21.7% of total protein) was obtained by combining promoter PpJF325 and three copies of the BmK1 gene. The yield of the purified BmK1 achieved 196.74 mg L−1 in E. coli BL21(DE3) pJF431, which was improved 2.09-fold compared with the control. This was the highest reported production of scorpion peptides in E. coli.
Keywords: BmK1, heterologous expression, promoter engineering, copy number, scorpion venom
1. Introduction
Channelopathies are diseases caused by functional disturbances of ion channels subunits or the proteins that regulate them. Each channelopathy can play an important role in a number of different diseases, for example, the sodium channelopathies, including paramyotonia congenita, adynamia episodica hereditaria, and myotonia fluctuans. Multiple drugs targeting at the specific ion channels have been designed to treat these diseases 1. Chinese scorpion Buthus martensii Karsch (BmK) venom is a source of neurotoxins that bind to various ion channels with high affinity and specificity. These neurotoxins are widely used to modulate signal transduction and channel gating 2. For example, BmK1, a key chlorotoxin-like peptide and an alpha-like toxin, is a novel blocker of the sodium ion channel 3. As potential therapeutic agents, BmK peptides have received increasing attention in recent years. In China, scorpion toxin as a traditional Chinese medicine could be traced to almost 2,000 years ago. Although BmK is a scorpion species that has widespread distribution from the north of the Yangzi River in China to Mongolia and the Korean peninsula, traditional extraction from BmK is still not a simple and economic method to massively produce a specific BmK peptide for treating a specific disease. Thus, heterologous expression is a potential alternative for the production of various BmK peptides 4–6.
The common host for heterologous expression of BmK toxin is Escherichia coli 4, and only a few studies have used yeast and plant as expression hosts 7,8. However, all studies have been carried out on heterologous expression of BmK peptide toxins at a low level (<10 mg L−1) in microbial hosts 2,5,6, and there also has been no systematic investigation into improving heterologous expression of BmK peptides. To improve heterologous small peptide expression, gene dosage strategy is one of the major methods, and multicopy strains always significantly improve the expression level of recombinant proteins. Kim et al. 9 deployed this strategy to achieve the maximum expression level (∼60 mg L−1) at the four copies of lactoferricin gene. However, in some cases, it could reduce protein expression with an increase in gene dosage, for example, cationic antimicrobial peptide lactoferricin 9, suggesting that an optimal copy number should be considered because of other potential bottlenecks of protein expression such as metabolic burden, product toxicity, and transcriptional stability 10, as well as protein, translation, and degradation 11.
Promoter engineering has been widely used in the metabolic engineering area in the past decade 12,13. For example, a characterized library of promoter was used to assess the effect of phosphoenolpyruvate carboxylase levels on cell growth and deoxy-xylulose-P synthase levels for lycopene production in E. coli 13. The multifaceted characterization of promoter strength enables the identification of optimal expression levels of targeted genes to achieve the best desired cellular performance. Promoter engineering enables precise control of gene expression in a broad range of activities and identifies an optimal gene expression level for the desired product. Promoter engineering is thus a valuable toolbox for fine-tuning gene expression at the transcriptional level, especially in the case of the peptide expression process to alleviate the severe metabolic burden on the hosts.
In this work, by means of BmK1 as a model, we combined promoter engineering and gene dosage strategy to systematically optimize the expression level of BmK1 in E. coli, and eventually, the actual yield of purified BmK1 was 196.74 mg L−1. To our knowledge, this is the first report on improving expression of BmK peptides in E. coli through promoter engineering and gene dosage strategy.
2. Materials and Methods
2.1. Strains, expression vectors, and other materials
All primers and plasmids used in this study are listed in Table1. E. coli DH10B (Promega, Beijing, China) was used for plasmid construction, promoter library construction, and characterization. E. coli BL21(DE3) was used for peptide expression. Plasmid pTrcHis2B (Invitrogen, Shanghai, People's Republic of China) harboring trc promoter and its mutated promoter library was constructed and preserved in our laboratory. PrimeStar DNA polymerase, restriction endonucleases, T4 DNA ligase, and protein ladder were purchased from Takara Biotechnology (Dalian, People's Republic of China). Plasmid DNA isolation and DNA gel purification were performed using AxyPrep Plasmid Miniprep and AxyPrep DNA gel extraction kits (Axygen Biosciences, Union City, USA). DNA primers, bovine serum albumin (BSA), and all other regents were provided by Sangon Biotech (Shanghai, People's Republic of China). The working concentration of ampicillin was 100 mg L−1.
Table 1.
Plasmids and primers used in this study
| Name | Information |
|---|---|
| pTrcHis2B | Invitrogen plasmid with wild-type trc promoter |
| pJF07 | pTrcHis2B-derived plasmid with a gfp gene inserted into BamHI and EcoRI |
| pJF136 | pTrcHis2B-derived plasmid with a mutated trc promoter (PpJF136, 0.55) and a gfp gene inserted into BamHI and EcoRI |
| pJF288 | pTrcHis2B-derived plasmid with a mutated trc promoter (PpJF288, 2.31) and a gfp gene inserted into BamHI and EcoRI |
| pJF325 | pTrcHis2B-derived plasmid with a mutated trc promoter (PpJF325, 1.29) and a gfp gene inserted into BamHI and EcoRI |
| pJF391 | pTrcHis2B plasmid with a BmK1 gene inserted into NcoI and HindIII |
| pJF392 | Removed gfp gene from pJF136 and inserted BmK1 gene into NcoI and HindIII |
| pJF393 | Removed gfp gene from pJF325 and inserted BmK1 gene into NcoI and HindIII |
| pJF396 | Removed gfp gene from pJF288 and inserted BmK1 gene into NcoI and HindIII |
| pJF430 | Two copy expression cassette, inserted additional copy of BmK1 gene into pJF393 |
| pJF431 | Three copy expression cassette, inserted additional copy of BmK1 gene into pJF430 |
| pJF432 | Four copy expression cassette, inserted additional copy of BmK1 gene into pJF431 |
| BmK1-F | 5′CTAGTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATATACATATGCACCATCATCATCATCA3′ |
| BmK1-R | 5′CCCAAGCTTCTGCAGACTAGTTTAGTCAGAACGACCCGCCGG3′ |
| pJF07-F | 5′CGGGATCCAATGCGTAAAGGAGAAGAAC3′ |
| pJF07-R | 5′ATAAGAATGCGGCCGCATGATGTCGGCGCAAAAAACATTATC3′ |
| trc-F | 5′ATAAGAATGCGGCCGCAACGGTTCTGGCAAATATTCTGAAAT3′ |
| trc-R | 5′TCTCCTTTACGCATTGGATCCATGG3′ |
Restriction sites are underlined and sequence between restriction sites in italics is the RBS region.
2.2. General DNA manipulation of plasmids and construction of promoter library
General DNA manipulations of plasmids and promoter library construction, competent cell preparation, and transformation were performed according to the standard protocols 14. The codon-optimized gene of BmK1 was synthesized by Generay Biotech (Shanghai, People's Republic of China) and ligated into the NcoI and HindIII sites of pTrcHis2B to create pJF391 (Table1). Error-prone PCR amplification of trc promoter and RBS region in pTrcHis2B was performed using a JBS Error-Prone Kit PP101 (Jena Science, Jena, Germany) with trc-F and trc-R primer pair. The backbone of reporter vector pJF07 was PCR amplified using PrimeStar DNA polymerase with pJF07-F and pJF07-R primer pair. Both PCR products were gel purified, ligated together, and transformed into DH10B-competent cells. The transformants were spread onto a LB agar plate and cultivated overnight at 37 °C for 16 H for further screening.
To fine-tune gene expression of BmK1 peptide, a DNA fragment of the BmK1 gene from pJF391 was inserted into the NcoI and HindIII sites of pJF136, pJF325, and pJF288 containing various mutated trc promoters to construct pJF392, pJF393, and pJF396, respectively. To construct the multicopy expression cassettes, the gene BmK1 plus RBS region was PCR amplified from pJF391 using BmK1-F and BmK1-R primer pair. The purified PCR product was digested by XbaI and HindIII and inserted into the SpeI and HindIII sites of pJF393 to construct pJF430. Plasmids pJF431 and pJF432 were created by additional one and two copies of BmK1 gene insertion into pJF430. The identity of each construct was confirmed by DNA sequencing.
2.3. trc promoter library construction and characterization
For primary screening, transformants were picked into a 48-deep-well plate and cultivated at 37 °C and 250 rpm for 8 H with each well harboring 0.5 mL LB medium (0.1 mM IPTG). The OD600 value and green fluorescent signal (excitation/emission wavelength, 485/535 nm) of 100 µL culture were quantified by a Thermo Scientific microplate spectrophotometer (Shanghai, People's Republic of China ). For the convenience of comparison, we used the relative strength 15 to represent the strength of a mutated sequence, which was defined and calculated as
 |
where S is the relative strength of the promoter, F is the intensity of the fluorescent signal, pTrcHis2B is the control, and pJF07 represents the wild-type trc promoter.
Fifteen clones with a wide and scattered strength span were selected and precisely quantified by flow cytometry (FACSCalibur flow cytometer; Bection Dickinson, Shanghai, People's Republic of China) using a clone containing pTrcHis2B as control. For flow cytometry assay, 1% of overnight culture was inoculated into 1 mL LB medium containing 0.1 mM IPTG and 100 µg mL−1 ampicillin in 15 × 150 mm tube, and cultivated at 37 °C for 3 H. For promoter leaky expression quantification, no IPTG was added during the cultivation. The culture was diluted to a concentration of 106–107 cells mL−1 with ice-cold phosphate-buffered saline buffer. Each clone was sampled with 20,000 events, and the geometric mean (Gmean) of fluorescent signal was calculated by statistics. The relative strength 15 of promoter was calculated by the following formula:
2.4. Peptide BmK1 expression
All plasmids containing the BmK1 gene were transformed into E. coli BL21 (DE3) cells for peptide expression. A single clone was preinoculated into 2 mL LB medium containing 100 µg mL−1 ampicillin and grown at 37 °C overnight at 220 rpm. One-hundred microliters of preculture was inoculated into 10 mL of fresh LB medium containing 100 µg mL−1 ampicillin and the desired concentration of IPTG for induction. The culture was incubated at 37 °C and 220 rpm, and sampled at 0.5, 1, 1.5, 2.0, 3, 4, 5, and 6 H after induction, respectively. The large-scale expression process for BmK1 purification was carried out in a 2 L shake flask containing 1 L of LB medium and induced with 0.1 mM IPTG. To avoid the peptide degradation, all samples were centrifuged and cell pellets were stored at −20 °C in a freezer immediately.
2.5. Cell growth assay, BmK1 purification, and quantification
Cell growth was determined by measuring optical density at 600 nm (OD600). Tricine–SDS-PAGE 16 was used for BmK1 separation because of its low molecular weight of 9.94 kDa. Cell pellets were resuspended and adjusted to the same concentration with distilled water. The initial voltage of the electrophoresis was 30 V. When the sample completely entered the stacking gel, the voltage was changed to 90 V until the bromophenol blue band reached to the bottom of the gel. The gel was stained with coomassie dye and decolorized using a destained solution containing 5% acetic acid and 10% ethanol. The destained gel was imaged by the Tanon 2500R gel-imaging system (Tanon Science & Technology, Shanghai, People's Republic of China). To rapidly evaluate the BmK1 expression level in different constructs, the relative content of BmK1 (the relative ratio of BmK1 to total cellular protein) was quantified using the integral analysis method with the GIS 1-D software (Tanon), which was used for the quantification of recombinant BmK peptides by Fu et al. 17.
To purify BmK1 peptide, 1 L of culture was centrifuged and cell pellets were resuspended in 50 mL NTA-0 buffer (20 mM Tris–HCl, pH 7.9, 0.5 M NaCl). The cells were lysed by a high-pressure homogenizer (ATS Engineering, Shanghai, People's Republic of China), and the supernatant was separated by centrifugation (12,000g, 20 Min) at 4 °C. Two milliliters of Ni2+-NTA agarose resin (Invitrogen) was added into the supernatant, and the BmK1 peptide was absorbed at 4 °C for 30 Min with slightly shaking. Then, Ni2+-NTA agarose resin was loaded onto the column (16 × 200 mm) and the column was sequentially washed with five column volumes of NTA-0 buffer, NTA-20 buffer (NTA-0 buffer with 20 mM imidazole), and NTA-60 buffer (NTA-0 buffer with 60 mM imidazole). BmK1 peptide was selectively eluted with five column volumes of NTA-250 buffer (NTA-0 buffer with 250 mM imidazole). The Bradford method was used to directly quantify the purified BmK1 peptide using BSA as the standard 6.
3. Results
3.1. BmK1 expression in E. coli
A DNA sequence of BmK1 deposited in EMBL database (accession number: AAD39510) was optimized for E. coli expression by a Web-based program optimizer 18. Rare codons of the BmK1 gene were optimized, including nine codons for glycine and six codons for arginine (Fig.1A). Additionally, to simplify the purification process, the optimized BmK1 gene was fused upstream with a 6× His tag. To easily remove the His tag after purification, an enterokinase recognition site DDDDK was inserted between a 6× His tag and BmK1 peptide. The entire open reading frame of the recombinant BmK1 gene is shown in Fig.1B.
Figure 1.
Codon optimization and expression cassette construction. (A) DNA sequence alignment between original and optimized BmK1 gene. (B) The entire open reading frame of the recombinant BmK1 gene.
To achieve efficient expression of BmK1, a commercially available strong promoter trc was initially used to control the expression of a single-copy BmK1 gene. After 6 H of induction with 0.1 mM IPTG, BmK1 was successfully expressed, accounting for 4%–6% of total protein, with varied culture temperatures of 22, 30, and 37 °C, respectively (Fig.2A). It is indicated that the temperature does not significantly affect BmK1 expression, but the biomass of cultivation at 37 °C was much greater than those at the lower temperatures. We further investigated the effect of IPTG concentration at 37 °C. As for BmK1 expression, induction with 0.5 or 0.1 mM IPTG was obviously better than higher IPTG concentration (Fig.2B). Considering that cell growth of 0.1 mM IPTG was slightly better than 0.5 mM IPTG, the following study for BmK1 expression was carried out at 37 °C with 0.1 mM IPTG. The content of BmK1 reached 6.12% after 6 H induction using 0.1 mM IPTG at 37 °C; and cell growth slightly lagged behind the control (BL21[DE3] pTrcHis2B).
Figure 2.
Expression of BmK1 using commercially available trc promoter. (A) Effect of temperature on BmK1 expression. Lane C, control strain E. coli BL21 (DE3) pTrcHis2B at 37 °C; lanes 1–3, BmK1 expression at 22 , 30 , and 37 °C, respectively. (B) Effect of IPTG addition on BmK1 expression. Lane C, control strain; lanes 1–4, expression at 0.1, 0.5, 1.0, and 2.0 mM of IPTG, respectively.
3.2. trc promoter library construction and characterization
To achieve an improved expression of BmK1 through promoter engineering, a promoter library of trc (an IPTG inducible strong promoter) was constructed and precisely characterized by detection of the relative strength. Considering that mRNA transcription and protein translation are two major genetic processes for gene expression, a fragment of 224 bp DNA sequence containing −10 and −35 core region of the trc promoter and RBS region (from 187 to 410 bp on pTrcHis2B) was subjected to random mutagenesis by error-prone PCR. Fifteen mutated promoters isolated from >1,000 mutants by primary screening were further precisely strength quantified at the induced and uninduced stages, ranging from 0.03 to 2.31 of relative strength after IPTG induction (Fig.3). Promoters PpJF136 (0.55), PpJF325 (1.29), and PpJF288 (2.31) with a low level of leaky expression were selected to fine-tune BmK1 expression in the following study. The sequences of three selected promoters are shown in Text S1 in the Supporting Information.
Figure 3.
Precise characterization of trc promoter library.
3.2. Effect of promoter strength on BmK1 expression
There was a low level of BmK1 expression when a single copy of the BmK1 gene was under the control of a wild-type trc promoter. We thus made an attempt to enhance BmK1 expression through promoter engineering. A single copy of the BmK1 gene was constructed under the control of four different promoters with a relative strength of 0.55 (PpJF136), 1.0 (Ptrc), 1.29 (PpJF325), and 2.31 (PpJF288), respectively, to evaluate the effect of promoter strength on the BmK1 expression (Fig.4A). An interesting phenomenon was found that cell growth was deteriorated accompanied with upregulation of promoter strength (Fig.4B). The growth of E. coli BL21 (DE3) pJF396 was almost completely stopped after 1 H of a prompt initiation of BmK1 expression. As shown in Fig.4C, BmK1 expression was initiated immediately after IPTG addition. When only induced for 0.5 H, BmK1 content can reach 7.37% and 9.11% in E. coli BL21 (DE3) pJF393 (PpJF325, strong promoter) and pJF396 (PpJF288, strong promoter), which were higher than those of pJF391 (Ptrc, control) and pJF392 (PpJF136, weak promoter). As for pJF396 with promoter strength at 2.31, BmK1 content was significantly lower than that of pJF393; it slightly decreased at 1 H after induction, which might be attributed to the inhibition of cell growth caused by a higher BmK1 expression/BmK1 toxicity. During the whole process, all cultures reached their maximum BmK1 production less than 4 H after induction. A strong promoter can rapidly start BmK1 expression with a higher rate and relatively decrease peptide degradation, resulting in a higher BmK1 accumulation in the cell. The maximum BmK1 content of 13.93% was obtained at 2 H after induction using pJF393, which was improved 2.27-fold compared with that of the control (6.12% of BmK1 content in pJF391).
Figure 4.
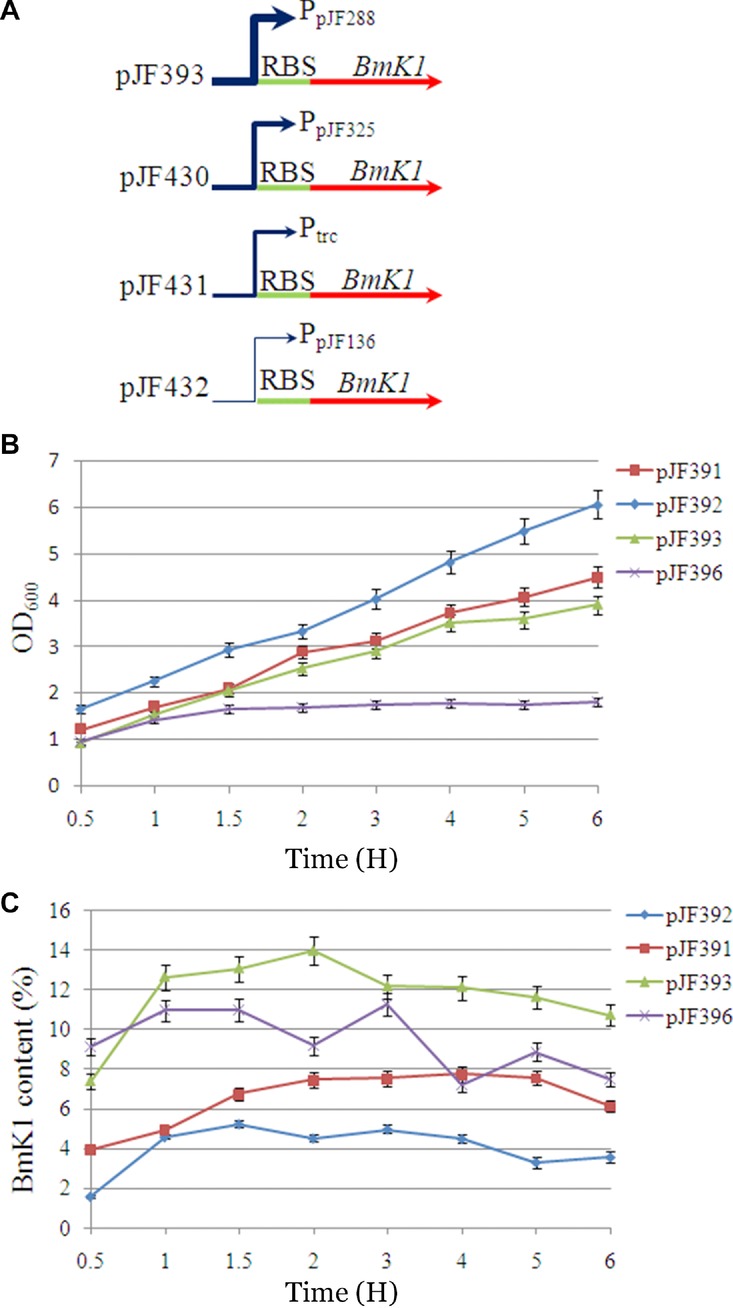
Effect of promoter strength on BmK1 expression. (A) Expression cassette construction using four different promoters. (B) Effect of promoter strength on cell growth. (C) Effect of promoter strength on BmK1 yield.
3.3. Combined promoter strength and gene dosage to further enhance BmK1 expression
When the best promoter strength was confirmed for BmK1 expression, gene dosage strategy was used to further increase the expression of BmK1. One, two, three, and four copies of BmK1 gene expression cassettes were assembled under the control of the best promoter (Fig.5A). The expression level of BmK1 with two, three, and four copies controlled by the promoter PpJF325 significantly increased along with a slight growth inhibition during the whole process (Figs.5B and 5C). The maximum BmK1 content of pJF430, pJF431, and pJF432 reached 20.6%, 21.79%, and 20.73%, respectively. The maximum BmK1 content using the optimal promoter strength and gene copy number (pJF431) was improved 3.56-fold compared with that of the control (pJF391) using the trc promoter and a single copy of gene, suggesting that it is a simple and efficient method to express BmK1 through promoter engineering and gene dosage strategy.
Figure 5.
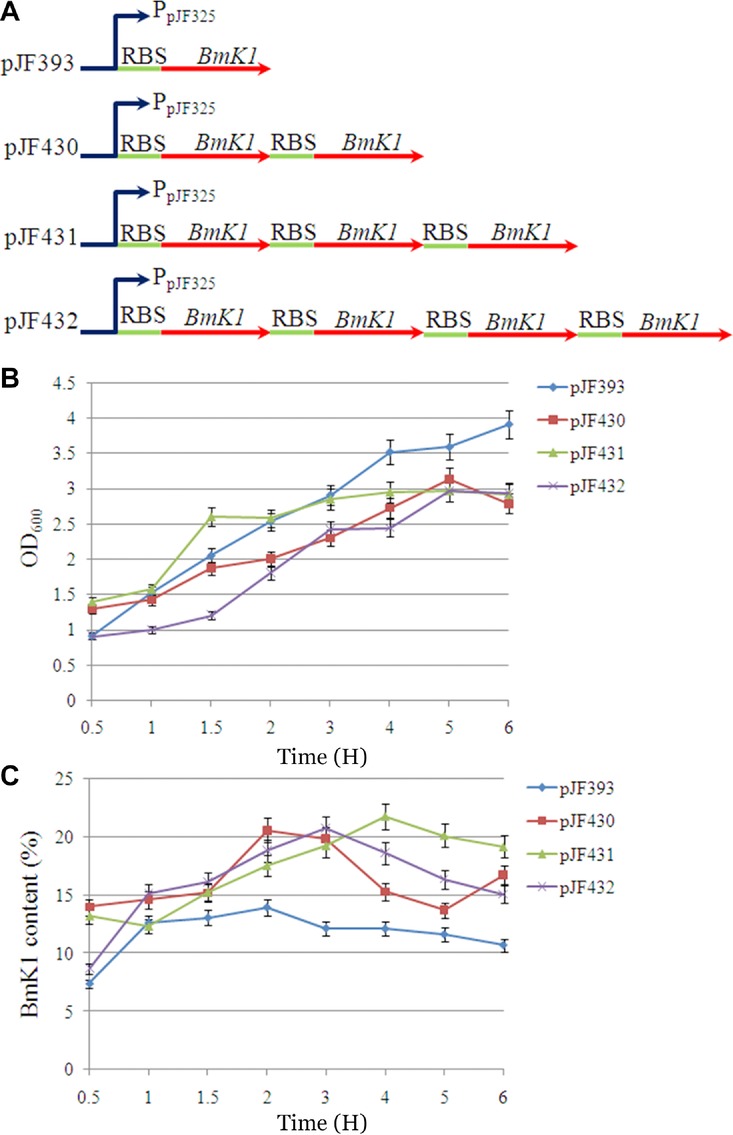
Effect of gene dosage on BmK1 expression. (A) Multicopy expression cassette construction. (B) Effect of gene dosage on cell growth. (C) Effect of gene dosage on BmK1 yield.
To eventually determine the actual yield of BmK1, the BmK1 peptide for pJF391, pJF393, and pJF431 was purified and quantified by Bradford method 6. The purification result is shown in Fig.6A with little impurities. The maximum yield of BmK1 was 196.74 mg L−1 in E. coli BL21 (DE3) pJF431 (Fig.6B), which was 2.09-fold improved as compared with the control E. coli BL21 (DE3) pJF391 (94.13 mg L−1). This was the highest reported production of scorpion peptides in a microbial cell factory.
Figure 6.
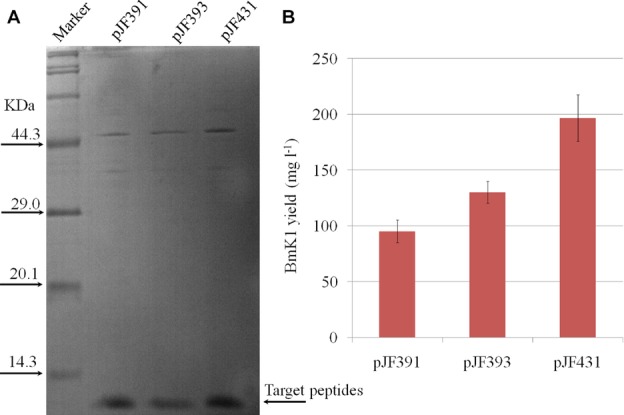
Quantification of soluble BmK1 peptide in culture. (A) SDS-PAGE analysis of the purified BmK1 peptide. (B) The yield of the purified BmK1 peptide.
4. Discussion
Compared with direct isolation from natural resources and chemical synthesis, heterologous expression is an efficient and economical alternative for the large-scale production of small peptides 10. Although heterologous expression systems such as yeast, insect cells, and mammalian cells have been well developed in recent years, a prokaryotic host E. coli remains the first choice for protein production, especially for small peptides 19,20. For prokaryotic expression of eukaryotic proteins, codon usage bias is one of the most important factors for the efficient expression 21. The elimination of rare codons and putative stable secondary structures of the target mRNA can significantly increase peptide expression. In this work, 15 rare codons for arginine and glycine were optimized (Fig.1A), and the optimal BmK1 gene was successfully expressed in E. coli BL21 (DE3) pJF391. The peptide content reached 4%–6% of total protein. The purified BmK1 from E. coli BL21 (DE3) pJF391 was examined for the electrophysiological effect by the voltage clamp method 22. The result indicated that BmK1 can bind to the sodium channel and block its inactivation (data not shown), suggesting that the recombinant BmK1 has similar bioactivity to the native BmK1.
With the rapid advance in synthetic biology, promoter engineering becomes a powerful tool to regulate the expression of key genes in a complicated metabolic network 13. Strong expression of a target gene always causes metabolic burden or accumulates toxic intermediates, and thus leads to cell growth inhibition. Therefore, choosing a suitable promoter is also an important alternative to efficiently express a target protein. Although many commercially available promoters (e.g., lac, tac, trc, and T7) were well developed and used for small peptide expression 23, many problems should still be considered, such as host inhibition caused by accumulation of toxic peptides and peptide properties affected by the expression rate including structure conformations, solubility, and stability. All these problems determine the ultimate yield of small peptides. Therefore, we constructed and characterized a trc promoter library to improve BmK1 expression based on the construct of pJF351 (Fig.3). As expected, when using a moderate strong and more tightly controlled promoter PpJF325, BmK1 peptide achieved the best expression, and peptide content was improved more than twofold compared with that of the trc promoter. But using a strongest promoter PpJF288, cell growth was inhibited and BmK1 yield was lower than that of PpJF325 (Fig.4), suggesting that fine-tuning expression of small peptide BmK1 is an effective approach that can be applied for other toxic peptide expression.
Currently, the production of most small peptides was still <100 mg L−1 10. The yield of heterologous expression of BmK peptide toxins in microbial hosts was also very low 5,6,14. For example, Shao et al. 6 have obtained 4.2 mg L−1 recombinant BmK AS peptide in E. coli. More importantly, all these studies have no systematic investigation. We thus have attempted to use the combined codon optimization strategy, promoter engineering, and multicopy strategy to further improve the production of the BmK peptide in E. coli. Ultimately, BmK1 content of pJF431 containing a stronger mutated promoter PpJF325 and three copy of the BmK1 gene achieved the maximum yield (21.79% of total protein, 196.74 mg L−1) after 4 H of induction. The yield of BmK1 was significantly improved, which was improved 2.09-fold as compared with its original expression (94.13 mg L−1 in pJF391). This is the first report on combining promoter engineering and multicopy strategy to improve the production of BmK toxins. BmK1 yield was also the highest yield reported so far.
5. Conclusion
In addition to the increase in gene dosage by construction of multicopy expression cassettes, the regulation of promoter strength is also an important option to increase heterologous expression of BmK peptide toxins. This work demonstrated that co-optimization of the promoter strength and gene dosage can effectively increase the yield of BmK peptide toxin. This strategy can also be used for other toxic peptide expression and further industrial production.
6. Acknowledgements
This work was supported by the National Basic Research Program of China (973 Program, grant no. 2012CB721104), the National High Technology Research and Development Program (“863”Program: 2012AA02A701), the National Natural Science Foundation of China (grants no. 31170101 and 31100073), and the major Projects of Knowledge Innovation Program of Chinese Academy of Sciences (grant no. KSCX2-EW-J-12).
Glossary
- BmK
Chinese scorpion Buthus martensii Karsch
- E. coli
Escherichia coli
- LB
Luria broth
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- Gmean
geometric mean
- BSA
bovine serum albumin
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
Supporting Information
Supporting Information is available in the online issue at wileyonlinelibrary.com.
7. References
- 1.Fan S, Sun Z, Jiang D, Dai C, Ma Y, Zhao Z, Liu H, Wu Y, Cao Z. Li W. Cancer Lett. 2010;291:158–166. doi: 10.1016/j.canlet.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 2.Fu YJ, An N, Chan KG, Wu YB, Zheng SH. Liang AH. Biotechnol. Lett. 2011;33:1309–1317. doi: 10.1007/s10529-011-0587-7. [DOI] [PubMed] [Google Scholar]
- 3.Chen J, Tan ZY, Zhao R, Feng XH, Shi J. Ji YH. Neurosci. Lett. 2005;390:66–71. doi: 10.1016/j.neulet.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Wang L, Cui Y, Song YB, Liu YF, Zhang R, Wu CF. Zhang JH. Biomed. Chromatogr. 2011;25:801–807. doi: 10.1002/bmc.1519. [DOI] [PubMed] [Google Scholar]
- 5.Hao CJ, Xu CG, Wang W, Chai BF. Liang AH. Biotechnol. Lett. 2005;27:1929–1934. doi: 10.1007/s10529-005-3905-0. [DOI] [PubMed] [Google Scholar]
- 6.Shao JH, Wang YQ, Wu XY, Jiang R, Zhang R, Wu CF. Zhang JH. Biotechnol. Lett. 2008;30:23–29. doi: 10.1007/s10529-007-9499-y. [DOI] [PubMed] [Google Scholar]
- 7.Pang SZ, Oberhaus SM, Rasmussen JL, Knipple DC, Bloomquist JR, Dean DH, Bowman KD. Sanford JC. Gene. 1992;116:165–172. doi: 10.1016/0378-1119(92)90512-n. [DOI] [PubMed] [Google Scholar]
- 8.Wang Z, Wang W, Shao Z, Gao B, Li J, Ma J, Li J, Che H. Zhang W. Mol. Cell. Biochem. 2009;330:97–104. doi: 10.1007/s11010-009-0104-7. [DOI] [PubMed] [Google Scholar]
- 9.Kim HK, Chun DS, Kim JS, Yun CH, Lee JH, Hong SK. Kang DK. Appl. Microbiol. Biotechnol. 2006;72:330–338. doi: 10.1007/s00253-005-0266-5. [DOI] [PubMed] [Google Scholar]
- 10.Li Y. Protein Expr. Purif. 2011;80:260–267. doi: 10.1016/j.pep.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 11.Hohenblum H, Gasser B, Maurer M, Borth N. Mattanovich D. Biotechnol. Bioeng. 2004;85:367–375. doi: 10.1002/bit.10904. [DOI] [PubMed] [Google Scholar]
- 12.Blazeck J. Alper HS. Biotechnol. J. 2013;8:46–58. doi: 10.1002/biot.201200120. [DOI] [PubMed] [Google Scholar]
- 13.Alper H, Fischer C, Nevoigt E. Stephanopoulos G. Proc. Natl. Acad. Sci. USA. 2005;102:12678–12683. doi: 10.1073/pnas.0504604102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sambrook J. Russell DW. Molecular Cloning: A Laboratory Manual. New York, NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 15.Kelly J, Rubin A, Davis J, Ajo-Franklin C, Cumbers J, Czar M, Mora K, Glieberman A, Monie D. Endy D. J. Biol. Eng. 2009;3:4. doi: 10.1186/1754-1611-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu YJ, Yin LT, Wang W, Chai BF. Liang AH. Biotechnol. Lett. 2005;27:1597–1603. doi: 10.1007/s10529-005-2514-2. [DOI] [PubMed] [Google Scholar]
- 17.Schagger H. Nat. Protoc. 2006;1:16–22. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
- 18.Puigbo P, Guzman E, Romeu A. Garcia-Vallve S. Nucleic. Acids Res. 2007;35:W126–W131. doi: 10.1093/nar/gkm219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yin J, Li G, Ren X. Herrler G. J. Biotechnol. 2007;127:335–347. doi: 10.1016/j.jbiotec.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 20.Piers KL, Brown MH. Hancock RE. Gene. 1993;134:7–13. doi: 10.1016/0378-1119(93)90168-3. [DOI] [PubMed] [Google Scholar]
- 21.Gustafsson C, Govindarajan S. Minshull J. Trends Biotechnol. 2004;22(7):346–353. doi: 10.1016/j.tibtech.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 22.Ji YH, Mansuelle P, Terakawa S, Kopeyan C, Yanaihara N, Hsu K. Rochat H. Toxicon. 1996;34:987–1001. doi: 10.1016/0041-0101(96)00065-7. [DOI] [PubMed] [Google Scholar]
- 23.Tegel H, Ottosson J. Hober S. FEBS J. 2011;278:729–739. doi: 10.1111/j.1742-4658.2010.07991.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.