

Summary
Nova hantavirus (NVAV) was first identified in a single European mole (Talpa europaea), captured in Hungary. Analysis of lung tissues from 94 moles captured in France revealed NVAV in 50%. Based on the genetic diversity of the cytochrome b mtDNA, moles collected in Poitiers and Bordeaux were more closely related to the Iberian mole (T. occidentalis), a species previously assumed to be restricted to the Iberian Peninsula. Several hypotheses are discussed to explain these observations: 1) presence of hitherto unnoticed T. occidentalis in southwestern France; 2) existence of an ancient mitochondrial introgression phenomenon between the two Talpa species, producing a particular phenotype in some hybrids; 3) existence of a hybrid zone between the two species; and 4) existence of a new Talpa species. NVAV was not detected in the southwestern moles, which begs the question of the potential presence of a particular Hantavirus sp. in this population and/or in the Iberian moles.
Keywords: Talpa europaea, Talpa occidentalis, Hantavirus, NVAV, phylogenetics, phylogeography
Introduction
Hantaviruses (family Bunyaviridae), long known to be harbored by rodents (rats, mice, voles, etc.) of the Muridae and Cricetidae families, have been detected in multiple species of shrews (Soricidae) and moles (Talpidae) (Yanagihara et al., 2014). More recently, highly divergent lineages of hantaviruses have also been discovered in insectivorous bats (Gu et al., 2014b; Guo et al., 2013; Yanagihara et al., 2014; Table 1). In-depth studies of non-rodent hosts, which are heavily represented among micro-mammals all around the world, may provide a better understanding about their evolutionary origins. Since some hantaviruses may infect and cause diseases in humans, such studies may also interest epidemiologists (Pontier & Fouchet, 2013; Reynes, 2013; Tordo et al., 2013).
Table 1. Checklist of the hantaviruses hosted by Soricomorpha and Chiroptera.
Since the report of Thottapalayam virus in 1971, more than 30 other hantavirus strains have been detected either from Soricomorpha (Soricidae, Talpidae) or Chiroptera, from: the Palearctic (19), Nearctic (5), Oriental (3) or Ethiopian (7) Biorealms, respectively. Accession numbers in GenBank are given when the short gene sequence (S) of the corresponding hantavirus strain is available.
| Virus name | Logo | Host species | Host family | Biorealm | Country | Collection year | Publication year | Genbank |
|---|---|---|---|---|---|---|---|---|
| Thottapalayam | TPMV | Suncus murinus | Soricidae | Oriental | India | 1964 | 1971 | AY526097 |
| Tanganya | TGNV | Crocidura theresae | Soricidae | Ethiopian | Guinea | 2004 | 2007 | EF050455 |
| Cao Bang | CBNV | Anourosorex squamipes | Soricidae | Oriental | Vietnam | 2006 | 2007 | EF543524 |
| Ash River | ARRV | Sorex cinereus | Soricidae | Nearctic | USA | 1994 | 2008 | EF650086 |
| Amga | MGAV | Sorex caecutiens | Soricidae | Palearctic | Russia | 2006 | 2008 | - |
| Jemez Spring | JMSV | Sorex monticolus | Soricidae | Nearctic | USA | 1996 | 2008 | FJ593499 |
| Kenkeme | KKMV | Sorex roboratus | Soricidae | Palearctic | Russia | 2006 | 2009 | GQ306148 |
| Kathmandu | TPMV | Suncus murinus | Soricidae | Oriental | Nepal | 1996 | 2010 | HQ831363 |
| Imjin | MJNV | Crocidura lasiura | Soricidae | Palearctic | South Korea | 2004 | 2010 | EF641804 |
| Qian Hu Shan | QHSV | Sorex cylindricauda | Soricidae | Palearctic | China | 2005 | 2010 | GU566023 |
| Asagny | AZGV | Crocidura obscurior | Soricidae | Ethiopian | Côte d'Ivoire | 2009 | 2011 | JF276226 |
| Longwan | TPMV | Suncus murinus | Soricidae | Palearctic | China | 2009 | 2011 | JF784177 |
| Uluguru | ULUV | Myosorex geata | Soricidae | Ethiopian | Tanzania | 1996 | 2012 | JX193695 |
| Kilimanjaro | KMJV | Myosorex zinki | Soricidae | Ethiopian | Tanzania | 2002 | 2012 | JX193698 |
| Seewis | SWSV | Sorex araneus | Soricidae | Palearctic | Switzerland | 2006 | 2012 | EF636024 |
| Jeju | JJUV | Crocidura shantungensis | Soricidae | Palearctic | South Korea | 2007 | 2012 | HQ834695 |
| Bowé | BOWV | Crocidura douceti | Soricidae | Ethiopian | Guinea | 2012 | 2013 | KC631782 |
| Lianghe | CBNV | Anourosorex squamipes | Soricidae | Palearctic | China | 2011 | 2013 | JX465395 |
| Yakeshi | YKSV | Sorex isodon | Soricidae | Palearctic | China | 2006 | 2013 | JX465423 |
| Asikkala | ASIV | Sorex minutus | Soricidae | Palearctic | Germany | 2010 | 2013 | KC880343 |
| Boginia | BOGV | Neomys fodiens | Soricidae | Palearctic | Poland | 2011 | 2013 | - |
| Asama | ASAV | Urotrichus talpoides | Talpidae | Palearctic | Japan | 2008 | 2008 | EU929070 |
| Nova | NVAV | Talpa europaea | Talpidae | Palearctic | Hungary | 1999 | 2009 | FJ539168 |
| Oxbow | OXBV | Neurotrichus gibbsii | Talpidae | Nearctic | USA | 2003 | 2010 | FJ539166 |
| Dahonggou Creek | DHCV | Scaptonyx fusicaudus | Talpidae | Palearctic | China | - | 2010 | HQ616595 |
| Rockport | RKPV | Scalopus aquaticus | Talpidae | Nearctic | USA | 1986 | 2011 | HM015223 |
| Magboi | MGBV | Nycteris hispida | Nycteridae | Ethiopian | Sierra Leone | 2010 | 2012 | - |
| Mouyassué | MOYV | Neoromicia nanus | Vespertilionidae | Ethiopian | Côte d'Ivoire | 2011 | 2012 | - |
| Xuan Son | XSV | Hipposideros pomona | Hipposideridae | Oriental | Vietnam | 2012 | 2013 | KF704712 |
| Longquan | LQUV | Rhinolophus spp | Rhinolophidae | Palearctic | China | 2011 | 2013 | JX465422 |
| Huangpi | HUPV | Pipistrellus abramus | Vespertilionidae | Palearctic | China | 2011 | 2013 | JX473273 |
Between October 2012 and March 2013, we sampled European moles, Talpa europaea, in various regions of France. The purpose of this investigation was: 1) to verify the presence of Nova hantavirus (NVAV), originally described in a single specimen of T. europaea from Hungary (Kang et al., 2009); 2) to assess the prevalence and genetic variability of NVAV; and 3) to compare NVAV genetic diversity with mole population genetics. The study revealed the presence of NVAV in France and the existence of a genetic variability of the virus linked to the localities where the moles were captured (Gu et al., 2014a). Analysis of the cytochrome b mtDNA produced an unexpected finding: that is, although only one mole species (T. europaea) was considered to be present in France, some moles collected in the southwestern part of the country were genetically more closely related to T. occidentalis, a different species previously believed to be endemic in the Iberian Peninsula. Details of these results are presented and discussed.
Material and Methods
Collecting moles
Approximately 350 moles were captured in different localities in France (Figure 1) from October 2012 to March 2013, using Putange traps and following a protocol approved by French professional mole catchers (Dormion, 2012).
Figure 1. Map of collection localities.
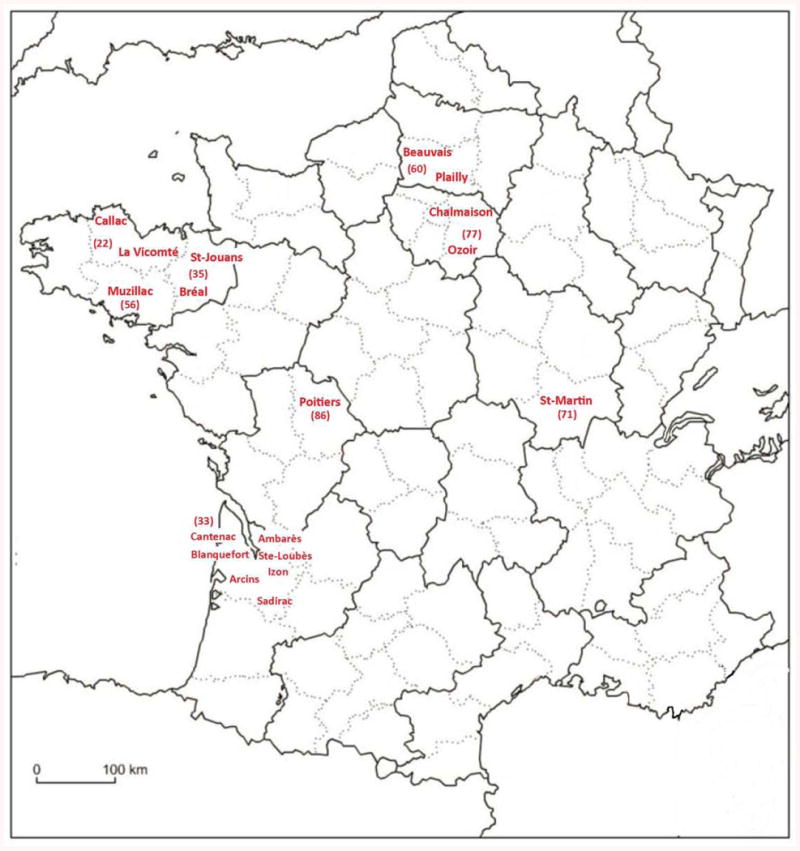
Map of France, showing localities where European moles were captured during October 2012 to March 2013. The results presented in this manuscript were obtained from specimens collected in the localities indicated in red (Blanquefort, St-Loubès, Sadirac, Ambarès, Arcins and Izon are small cities situated on either side of the Gironde estuary, in the vicinity of Bordeaux).
Research of hantavirus
A subset of 94 moles captured either in the Golf of Ozoir-la-Ferrière (48.7700°N, 2.6800°E), or in the Beauvais agreement park called “Le Plan d'eau du Canada” (49°452698 N, 2°058234 E), were studied. We also analyzed 24 mole specimens collected between Bordeaux and Poitiers and genetically different from T. europaea. After capture, moles were frozen at -20°C; for tissue dissection, moles were partially thawed and lung tissues were removed using ethanol-cleaned instruments and placed in RNAlater® RNA Stabilization Reagent (Qiagen Inc., Valencia, CA).
Hantavirus detection and sequencing
Total RNA was extracted from mole lung tissues, using the PureLink Micro-to-Midi total RNA purification kit (Invitrogen, San Diego, CA), and cDNA was prepared using the SuperScript III First-Strand Synthesis System (Invitrogen) and random hexamers. PCR was performed as described previously using oligonucleotide primers designed from NVAV and other soricomorph-borne hantaviruses (Song et al., 2007; Arai et al., 2008) and DNA sequencing was performed using an ABI Prism 377XL Genetic Analyzer (Applied Biosystems, Foster City, CA).
Mitochondrial DNA sequencing
DNA was extracted from ethanol-preserved tissues by the CTAB method (Winnepenninckx et al., 1983). The cytochrome b gene was amplified from 40 mole specimens using polymerase chain reaction (PCR) primers L14723 and H15915 (Ducroz et al., 2001; Nicolas et al., 2008).
Sequence alignment
Alignment of the coding region of the hantavirus S segment, or the cytochrome b of the moles was performed using CLUSTAL-X automatic procedure (Thompson et al., 1994) then improved manually using SEAL v2.0a11 (Rambaut, 1996) and validated using the amino acid translation.
Phylogenetic analyses
Evolutionary relationships among sequences were estimated by constructing phylogenetic trees using maximum parsimony (MP) and Bayesian Markov chain Monte Carlo phylogenetic analyses (MCMC). MP analysis was performed with PAUP 4b10 (Swofford, 2000) and Bayesian analysis with MrBayes 3.1.2 (Huelsenbeck and Ronquist, 2000). The MP analysis was performed with tree-bisection-reconnection (TBR) branch swapping option and 10 random addition replicates. We estimated the robustness of internal nodes by 1,000 bootstrapping replicates (each with a single replication of random addition of taxa). An equal weighting of character-state transformations was applied. In all MCMC analyses three heated chains and one single cold chain were employed, and runs were initiated with random trees. Two independent MCMC runs were conducted with six million generations per run; trees (and parameters) sampled every 100 generations. Stationarity was assessed by: examining the average standard deviation of split frequencies and, the Potential Scale Reduction Factor (Ronquist et al., 2005). For each run, the first 25% of sampled trees were discarded as burn-in. A consensus tree was computed using the “halfcompat” option, equivalent to the 50% majority rule and rooted using the “midpoint root” option. Proportion values of posterior probability of bipartition were used for evaluation of robustness of the nodes.
Results
Prevalence of NVAV in moles
Hantavirus RNA was detected in 47 moles (50%) by RT-PCR (Gu et al., 2014). Of 36 and 28 moles captured in Ozoir-la-Ferrière on October 18, 2012 and February 21, 2013, 15 and 14, respectively, were positive, while 18 of 30 moles captured in Beauvais on March 3, 2013 were positive (Gu et al., 2014a). NVAV was not detected in the 24 southwestern moles.
Phylogenetic analysis of hantavirus found in micro-mammals
Phylogenetic analysis, using Bayesian methods (Figure 2) showed that: 1) NVAV from France and prototype NVAV from Hungary grouped together in a particular lineage; 2) NVAV strains from France segregated along geographic-specific lineages; 3) NVAV clade was strongly associated with another clade, comprising hantaviruses detected in insectivorous bats; 4) other hantaviruses hosted by soricomorphs were distributed into two separate clades: i) the most divergent included seven strains from shrews captured in India, South Korea, China and Tanzania; ii) the second clade included strains from moles and shrews captured in North America, China, Japan and Europe, and associated with hantaviruses hosted by Murinae rodents; 5) two other clades grouping hantaviruses hosted by Sigmodontinae-Neotominae and Arvicolinae rodents, respectively; 6) a single mole-borne hantavirus, Rockport virus from the eastern mole in North America, was situated between the Sigmodontinae and Arvicolinae clades. All the basal nodes of the cladogram had the maximum posterior probability of bipartition value (= 1).
Figure 2. Phylogeny of hantaviruses hosted by Chiroptera, Soricomorpha or Rodents.
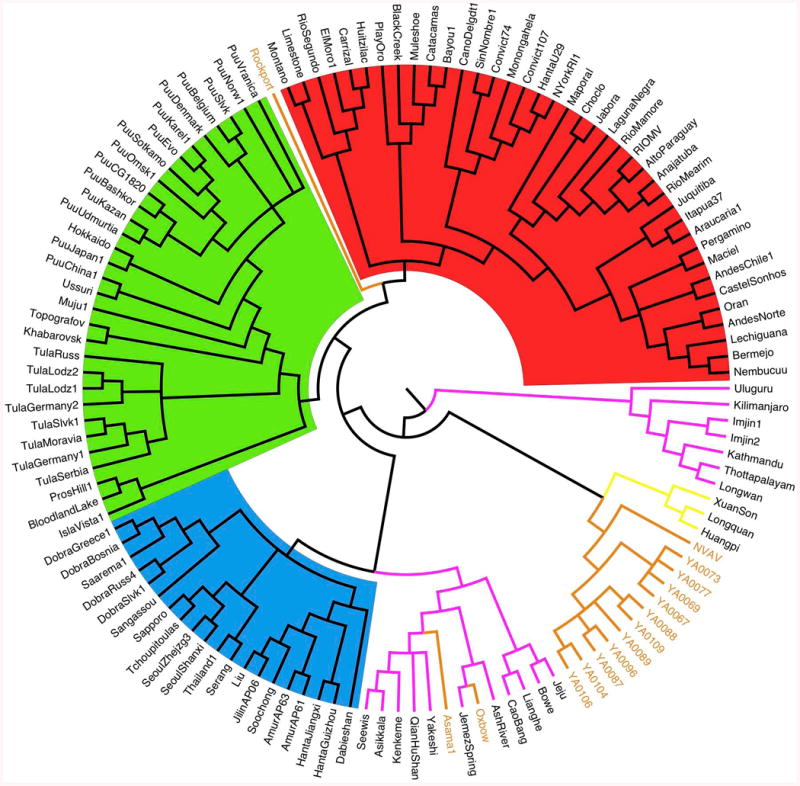
Cladogram resulting of Bayesian (MCMC) analysis (GTR+I+G model), based on the entire coding region of the S gene (nucleotides 43–1341). Hantaviruses hosted by different host groups are highlighted in: red (Sigmodontinae-Neotominae), green (Arvicolinae), blue (Murinae), pink (Soricidae), light brown (Talpidae) and yellow (Chiroptera). The analysis used 1) an extent data set of the S gene coding region available on GenBank, including most of the hantavirus strains detected either in rodents or non-rodent mammals (Soricomorpha and Chiroptera), and 2) 11 complete S-segment sequences from moles captured either in Ozoir (4) or Bauvais (7). GenBank accession numbers: FJ539168, NVAV from Hungary; KF010573–KF010576, NVAV strains from Ozoir-la-Ferrière; KF010565–KF010571, NVAV strains from Beauvais. For Chiroptera and Soricomorpha hosts, see Table 1; for rodent hosts, see Herbreteau et al., 2007, Table 16.1.
Genetic affinities within the Family Talpidae
Phylogenetic analysis, using Bayesian methods, of all available sequences from Talpidae species (Figure 3) showed that: 1) the genus Talpa may be considered a monophyletic group, in which each different species is recognized as a distinct clade; 2) T. europaea and T. occidentalis are sister taxa; 3) specimens captured in Brittany, Northern France and Saône-et-Loire clustered with strains identified as T. europaea and collected from different European countries (Sweden, Denmark, Switzerland, Hungary, Germany, Italy and Greece); 4) specimens captured in the vicinity of Bordeaux, or Poitiers, clustered with specimens captured either from Spain or Portugal and identified as T. occidentalis, the Iberian mole. All the nodes of the cladogram corresponding to a particular mole species had the maximum posterior probability of bipartition value (= 1).
Figure 3. Phylogenetic analysis of Talpa spp. and closely related taxa.

Bayesian (MCMC) analysis (GTR+I+G model) based on the entire coding region of the cytochrome b gene. The analysis was rooted using as outgroups several taxa included within the Talpidae: Urotrichus, Dymecodon, Mogera, Euroscaptor, Uropsilus, Desmana and Galemys. All the sequences available in GenBank for these taxa or for Talpa spp. were included. The tip labels indicate the GeneBank accession number and the name of the species (for the Talpa spp.), the name of the genus for the other taxa. Our own mole samples are labeled using the field collection number + the name of the locality of capture and highlighted in red.
Discussion
Mole infection
Prevalence of anti-viral antibodies and/or viral antigens in rodent species known to harbor disease-causing hantaviruses generally varies from less than 1% to more than 25%, depending on seasonal factors, reservoir population density, geographical area and ecological diversity (Lee et al., 1978; Dobly et al., 2012). Recently, a similarly high prevalence of NVAV infection, as evidenced by RT-PCR and confirmed by DNA sequencing, has been found in European moles captured in central Poland, suggesting that NVAV is widespread throughout the vast distribution of T. europaea (Gu et al., 2014a). The high prevalence of this hantavirus suggests that the mechanisms of transmission between individuals are very efficient.
NVAV was not detected in the southwestern moles, but few specimens (24) were available and the RNA quality was suboptimal. Thus, future studies are warranted to ascertain if NVAV or NVAV-related hantaviruses are harbored by moles in southwestern France. These investigations will be extended to mole specimens collected from different parts of the Iberian Peninsula. Until now no human infection due to NVAV has been recorded. However, moles have a high propensity to occupy arable fields, deciduous woodland and permanent pastures, and hantavirus are known to be able to survive for prolonged periods (12-15 days) in external environment (Kallio et al., 2006; Hardestam et al., 2007). Any unusual clinical syndromes, recorded among individuals reporting contact with European moles, should be thoroughly studied by physicians and public health workers.
Host and hantavirus coevolution
Hantaviruses were traditionally considered to have codiverged (co-speciatiated) with their rodent hosts (Herbreteau et al., 2006, 2007; Hentonnen et al., 2008; Guo et al., 2013; Yanagihara et al., 2014) and some evidence for such codivergence is apparent here. In particular, rodent-borne hantaviruses clustered according to whether their hosts were members of the Murinae, Arvicolinae or Sigmodontinae-Neotominae subfamilies. However, codivergence alone cannot explain the clustering of hantaviruses hosted by Soricomorpha and Chiroptera, which exist in several paraphyletic clades with no correspondence between the association of the viruses, their geographic origin and/or host taxonomy (Figure 2; Tordo et al., 2013).
Presence of genetically distinct moles in France
Identification of two genetically divergent mole lineages in France was unexpected. Until now, only T. europaea was considered to be present in France. In the Iberian Peninsula, the situation is quite different and two species have been recorded for a long time. T. europaea is present in Northeast Spain (Cataluña, Navarra and the Eastern part of Pais Vasco) while the closely related T. occidentalis is present in the Northwest and South of Spain and in Portugal (Palomo et al., 2007). To explain this presence of mitochondrial haplotypes related to T. occidentalis in the Southwest of France, the simplest hypothesis is to admit that T. occidentalis was able to cross the mountains, leaving Spain and partly colonizing a territory extending from the Pyrenees to the river Loire. However, this hypothesis doesn't fit well with the comparison of the morphology of the different populations. Table 2 shows that: i) if the measurements of the specimens clustering with the other European moles fit with the values attributed to T. europaea (Palomo et al., 2007; Aulagnier et al., 2008); conversely, ii) the comparison of the measurement of the southwestern mole specimens with the values attributed to T. occidentalis (Palomo et al., 2007; Aulagnier et al., 2008) are incompatible; iii) comparison between the two data sets identified in France shows that moles captured in the southwest reach the highest size values recorded for T. europaea and are significantly heavier. Thus, moles from southwestern France may be considered giant specimens while T. occidentalis from Iberia may be considered a dwarf species. These morphological results do not support the simplest hypothesis of the presence of T. occidentalis in France. To interpret our observations, three alternative hypotheses could be raised: 1) an ancient phenomenon of mitochondrial introgression occurred between the two species; 2) existence of a hybrid zone between the two species; and, 3) existence of a new undescribed Talpa species in southwestern France. To better understand the presence of mitochondrial haplotypes related to T. occidentalis in this part of France, it would be necessary to perform additional molecular analyses, especially of more specimens from various other localities in France and in the Iberian Peninsula and also to sequence nuclear markers. These new investigations will be accompanied by concurrent research on the presence of NVAV, or another related hantavirus species, in moles genetically identified as T. occidentalis.
Table 2. Comparison of the measurements of the moles captured in France, following their genetic affinities.
The measurements shown are those currently used when trapping mammals: Weight, Length of head and body, Length of tail, Length of foot, and Length of skull. A total of 24 moles (20 females and 4 males) were captured in the vicinity of Bordeaux and Poitiers, and 330 (173 females and 157 males) were captured in other localities in France. The measurements of T. europaea and T. occidentalis are after Palomo et al., 2007 and Aulagnier et al., 2008.
| Weight | H & Body | Tail | Foot | Head | Weight | H & Body | Tail | Foot | Head | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MAX | 118,0 | 166,0 | 40,1 | 30,8 | 49,0 | MAX | 147,0 | 165,0 | 35,0 | 26,0 | 49,0 |
| MIN | 48,0 | 122,8 | 14,0 | 16,6 | 31,6 | MIN | 69,0 | 141,0 | 19,0 | 19,0 | 39,0 |
| MOY | 78,0 | 145,5 | 26,8 | 20,8 | 43,6 | MOY | 92,5 | 153,2 | 27,5 | 21,3 | 45,8 |
| 330 moles from different sites in France | 24 moles from South-west of France | ||||||||||
| Talpa europaea | Talpa occidentalis | ||||||||||
| MAX | 130,0 | 165,0 | 51,0 | 25,0 | 38,0 | MAX | 70,0 | 135,0 | 35 | 18,0 | 32,0 |
| MIN | 36,0 | 100,0 | 20,0 | 16,0 | 30,0 | MIN | 30,0 | 90,0 | 16 | 14,0 | 28,0 |
| MOY | 83,0 | 132,5 | 35,5 | 20,5 | 34,0 | MOY | 50,0 | 112,5 | 25,5 | 16,0 | 30,0 |
Conclusions
Our work confirms the presence of NVAV in France, in association with its specific host, T. europaea. This result and the high prevalence observed in the analyzed specimens gives additional evidence for the probable distribution of NVAV throughout the geographic range of T. europaea. Although NVAV infection has not been recorded yet in humans, the presence and abundance of this hantavirus increases the likelihood of human exposure and should alert medical practitioners and public health workers to be vigilant for unusual clinical syndromes that may be caused by NVAV.
Hantaviruses are found in small mammals all around the World with the exception of the Australian and Antartica Biorealms. An extended phylogenetic analysis of the hantavirus strains hosted by rodents, soricomorphs or bats suggests coevolution between hantavirus and their hosts and implicates complex mechanisms including codivergence (cophylogeny), host switching and/or geographic isolation. The presence in France of two genetically different mole lineages begs the question of the taxonomic position of the southwestern moles and, also, of the potential circulation of a novel hantavirus species in the Iberian mole (T. occidentalis). Further investigations are warranted to clarify this possibility.
Acknowledgments
This work would have been impossible without the help of the professionals of the Taupgreen network (http://www.taupegreen.com/). We especially thank: J Dormion, JF Barbot, F Burgot, F Cairey, M Derosier, Y Dupont, JM Georgeon, M Giroud, JL Laclamette, EJ Laffont, G Moncel, B Pradier, L Quemener and R Fons for their help in mole capture. Molecular analyses were performed at the “Service de Systématique Moléculaire” (UMS 2700) of the MNHN. This work was also supported in part by grant number R01AI075057 from the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
(Communication présentée le 22 mai 2014)
References
- Arai S, Ohdachi SD, Asakawa M, Kang HJ, Mocz G, Arikawa J, Okabe N, Yanagihara R. Molecular phylogeny of a newfound hantavirus in the Japanese shrew mole (Urotrichus talpoides) Proc Natl Acad Sci USA. 2008;105(42):16296–16301. doi: 10.1073/pnas.0808942105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulagnier S, Haffner P, Mitchell-Jones AJ, Moutou F, Zima J. In: Guide des mammifères d'Europe, d'Afrique du Nord et du Moyen-Orient. Delachaux, Niestlé, editors. Paris: 2008. p. 272. [Google Scholar]
- Dormion J. Editions Eugen Ulmer; Paris: 2012. Le piégeage traditionnel des taupes; p. 67. Available from: http://www.taupegreen.com/methodes-eradication.htm. [Google Scholar]
- Ducroz JF, Volobouev V, Granjon L. An assessment of the systematics of Arvicanthine rodents using mitochondrial DNA sequences: evolutionary and biogeographical implications. J Mammal Evol. 2001;8:173–206. [Google Scholar]
- Dobly A, Yzoard C, Cochez C, Ducoffre G, Aerts M, Roels S, Heyman P. Spatiotemporal dynamics of Puumala hantavirus in suburban reservoir rodent populations. J Vector Ecol. 2012;37(2):276–283. doi: 10.1111/j.1948-7134.2012.00228.x. [DOI] [PubMed] [Google Scholar]
- Gu SH, Dormion J, Hugot JP, Yanagihara R. High prevalence of hantavirus infection in the European mole (Talpa europaea) in France. Epidemiol Infect. 2014a;142(6):1167–71. doi: 10.1017/S0950268813002197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu SH, Lim BK, Kadjo B, Arai S, Kim JA, Nicolas V, Lalis A, Denys C, Cook JA, Dominguez SR, Holmes KV, Urushadze L, Sidamonidze K, Putkaradze D, Kuzmin IV, Kosoy MY, Song JW, Yanagihara R. Molecular phylogeny of hantaviruses harbored by insectivorous bats in Côte d'Ivoire and Vietnam. Viruses. 2014b;6(5):1897–1910. doi: 10.3390/v6051897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo WP, Lin XD, Wang W, Tian JH, Cong ML, Zhang HL, Wang MR, Zhou RH, Wang JB, Li MH, Xu J, Holmes EC, Zhang YZ. Phylogeny and origins of hantaviruses harbored by bats, insectivores, and rodents. PLoS Pathog. 2013;9(2):e1003159. doi: 10.1371/journal.ppat.1003159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardestam J, Simon M, Hedlund KO, Vaheri A, Klingström J, Lundkvist Å. Ex vivo stability of the rodent-borne Hantaan virus in comparison to that of arthropod-borne members of the Bunyaviridae family. Applied and Environmental Microbiology. 2007;73(8):2547–2551. doi: 10.1128/AEM.02869-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henttonen H, Buchy P, Suputtamongkol Y, Jittapalapong S, Herbreteau V, Laakkonen J, Chaval Y, Galan M, Charbonnel N, Michaux J, Cosson JF, Morand S, Hugot JP. Recent discoveries of new hantaviruses widen their range and question their origins. Ann NY Acad Sc. 2008;1149:84–89. doi: 10.1196/annals.1428.064. [DOI] [PubMed] [Google Scholar]
- Herbreteau V, Gonzalez JP, Hugot JP. Implication of phylogenetic systematics of rodent-borne hantaviruses on their distribution. Ann NY Acad Sc. 2006;1081:39–56. doi: 10.1196/annals.1373.004. [DOI] [PubMed] [Google Scholar]
- Herbreteau V, Henttonen H, Yoshimatsu K, Gonzalez JP, Suputtamongkol Y, Hugot JP. Chapter 16: Hantavirus coevolution with their rodent hosts. In: Tibayrenc M, editor. Encyclopedia of Infectious Diseases Modern Methodologies. Wiley & Sons; 2007. pp. 243–264. [Google Scholar]
- Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. Available from: http://mrbayescsitfsuedu/: [DOI] [PubMed] [Google Scholar]
- Kallio ER, Klingström J, Gustafsson E, Manni T, Vaheri A, Henttonen H, Vapalahti O, Lundkvist Å. Prolonged survival of Puumala hantavirus outside the host: Evidence for indirect transmission via the environment. J Gen Virol. 2006;87(8):2127–2134. doi: 10.1099/vir.0.81643-0. [DOI] [PubMed] [Google Scholar]
- Kang HJ, Bennett SN, Sumibcay L, Arai S, Hope AG, Mocz G, Song JW, Cook JA, Yanagihara R. Evolutionary insights from a genetically divergent hantavirus harbored by the European common mole (Talpa europaea) PLoS One. 2009;4(7):e6149. doi: 10.1371/journal.pone.0006149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HW, Lee PW, Johnson KM. Isolation of the etiologic agent of Korean hemorrhagic fever. J Infect Dis. 1978;137(3):298–308. doi: 10.1093/infdis/137.3.298. [DOI] [PubMed] [Google Scholar]
- Nicolas V, Mboumba JF, Verheyen E, Denys C, Lecompte E, Olayemi A, Missoup AD, Katuala P, Colyn M. Phylogeographical structure and regional history of Lemniscomys striatus (Rodentia: Muridae) in tropical Africa. J Biogeogr. 2008;35:2074–2089. [Google Scholar]
- Palomo L, Gisbert J, Blanco JC. Atlas y Libro Rojo de los Mamíferos Terrestres de España. Dirección General para la Biodiversidad-SECEM-SECEMU; Madrid: 2007. p. 588. [Google Scholar]
- Pontier D, Fouchet D. Animal health perceived in the light of ecology and evolution. Bull Acad Vét France. 2013;166(4):354–363. [Google Scholar]
- Rambaut A. University of Oxford; Oxford, UK: 1996. Se-Al. Sequence Alignment Editor. Ver. 2.0a11. Available from : http://tree.bio.ed.ac.uk/software/seal/ [Google Scholar]
- Reynes JM. Hantavirus taxonomy and situation in France. Bull Acad Vét France. 2013;166(2):155–162. [Google Scholar]
- Ronquist F, Huelsenbeck JP, Van der Mark P. MrBayes 3.1 Manual. 2005 Available from http://mrbayes.csit.fsu.edu/mb3.1_manual.pdf.
- Song JW, Gu SH, Bennett SN, Arai S, Puorger M, Hilbe M, Yanagihara R. Seewis virus, a genetically distinct hantavirus in the Eurasian common shrew (Sorex araneus) Virol J. 2007;4:114. doi: 10.1186/1743-422X-4-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford D. PAUP: phylogenetic analysis using parsimony Version 4b10 ed. Washington DC: Smithsonian Institution; Sinauer Associates; Sunderland, MA, USA: 2001. [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL-W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position, specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tordo N, Castel G, Filippone C, Marianneau P. Recent data on hantaviruses and perspectives for research Bull Acad Vét France. 2013;166(4):364–371. [Google Scholar]
- Winnepenninckx B, Backeljau T, De Wachter R. Extraction of high molecular weight DNA from molluscs. Trends Genet. 1993;9:407. doi: 10.1016/0168-9525(93)90102-n. [DOI] [PubMed] [Google Scholar]
- Yanagihara R, Gu SH, Arai S, Kang HJ, Song JW. Hantaviruses: Rediscovery and new beginnings. Virus Res. 2014 Jan 8; doi: 10.1016/j.virusres.2013.12.038. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]