

Abstract
Identifying novel allosteric inhibitors of G protein-coupled receptor kinases (GRKs) would be of considerable use in limiting both the extent of desensitization of GPCRs as well as downstream positive regulation through GRKs. Several peptides have previously been identified as inhibitors of specific GRKs, but to date there have been few comparisons of the selectivities of these materials on the seven GRKs, modifications to allow cell penetration, or off-target activities. The goal of this study was to determine if a panel of peptides mimicking domains on either GPCRs or GRKs would exhibit selective inhibition of GRKs 2, 5, 6 and 7 phosphorylation of rhodopsin. Peptides included sequences from GRK5; helices 3, 9, and 10 (α3, α9, and α10) in the RH domain, and the N-terminal peptide (N-Ter), as well as the intracellular loop 1 (iL1) of the β2-adrenergic receptor (β2AR), and the Gα transducin C-tail (TCT). While some selectivity for individual GRKs was found, overall selectivity was limited and often not reflective of structural predictions. Off-target effects were probed by determining peptide inhibition of adenylyl cyclase (AC) and PKA, and while peptides had no effect on AC activity, N-Ter, iL1, and α10 were potent inhibitors of PKA. To probe inhibition of GRK activity in intact cells, we synthesized TAT-tagged peptides, and found that TAT-α9-R169A and TAT–TCT inhibited isoproterenol-stimulated GRK phosphorylation of the β2AR; however, the TAT peptides also inhibited isoproterenol and forskolin stimulation of AC activity. Our findings demonstrate potent peptide inhibition of GRK activities in vitro, highlight the differences in the environments of biochemical and cell-based assays, and illustrate the care that must be exercised in interpreting results of either assay alone.
Keywords: Peptide inhibitors, GRKs, GPCRs, Cell-penetrating peptide
Introduction
G protein-coupled receptor kinases (GRKs) are a family of seven protein kinases, two visual (GRKs 1 and 7) and five non-visual (GRKs 2–6), that phosphorylate GPCRs on serine and threonine residues and are involved in GPCR desensitization as well as other novel functions (Gurevich et al. 2012). At present we know very little about the mechanism of allosteric activation of GRKs by GPCRs. Importantly, X-ray crystallographic structures of GRKs 1, 2 and 6 (Boguth et al. 2010; Lodowski et al. 2003, 2006; Singh et al. 2008) are providing clues to this process. The structure of the closed, presumably active state of GRK6 shows that the N-terminus binds intramolecularly to a cleft formed between the small lobe and the active site tether loop of the kinase domain (Boguth et al. 2010). Extensive site-directed mutagenesis of the N-terminus and its sites of interaction with the kinase domain of GRKs 1, 2, 5 and 6 demonstrated that these interactive domains are crucial for rhodopsin- and β2AR-mediated activation of GRKs (Huang et al. 2009; Noble et al. 2003; Pao et al. 2009; Yu et al. 1999). These findings suggest that the N-terminus interacts with GPCRs in addition to its intramolecularly binding to the kinase domain. Comparison of the structure of the N-terminal open-faced domain of GRK6 to the structure of a peptide derived from the C-terminal tail of Gα transducin bound to opsin (Scheerer et al. 2008), led to the proposal that the N-termini of GRKs interact with GPCRs, and is the main contributor to allosteric stimulation of GRKs by GPCRs (Huang et al. 2011; Boguth et al. 2010). In addition to these studies, it was found that mutations of highly conserved sites in helices 3, 9, and 10 in the RH domain of GRK5/6, severely impaired both GPCR activation of these GRKs and basal activity (Baameur et al. 2010), demonstrating an important role of the RH domain in maintaining the active state of GRKs.
Because of the ubiquitous role of GRKs in desensitization of many GPCRs, and other possible roles, the availability of specific inhibitors of these enzymes could aid in elucidation of their cellular function and provide a tool for inhibiting desensitization. It is possible that the interaction of receptors with GRKs can be probed and blocked with peptide-based allosteric inhibitors that mimic domains of either of the partners involved in, or crucial for, GPCR activation of GRKs. For the GPCR/GRK interaction it was shown that peptides based on the N-termini of GRK2 and GRK5 inhibited only their cognate kinase phosphorylation of GPCRs in cell free systems (Noble et al. 2003; Pao et al. 2009). Extending prior findings regarding peptide mimetics of β2AR intracellular loops (Benovic et al. 1990; Onorato et al. 1991), it was shown that a peptide derived from intracellular loop 1 (iL1) of the β2AR, inhibited the phosphorylation of rhodopsin by GRKs 2, 3, and 5, and increased cAMP accumulation in A431 cells following permeabilization with digitonin (Winstel et al. 2005), indicating a partial inhibition of desensitization. It was also found that this peptide partially blocked isoproterenol (ISO)-stimulated desensitization of cAMP accumulation in HEK293 cells, measured by whole cell patch clamping, that allowed intracellular perfusion of the peptide (Xin et al. 2008). A peptide derived from helix 9 in the RH domain of GRK5, as well as an analog chemically locked into a helix, inhibited GRK5 and GRK6 phosphorylation of rhodopsin (Baameur et al. 2010). While these studies collectively suggest the potential utility of peptides for inhibition of GRK activities, they have yet to be thoroughly examined for specificity either for inhibition of other protein kinases, or for inhibition of AC activity, and none of the peptides are cell permeable which can potentially impact utility in probes of cellular GPCR/GRK signaling.
To further the development of potentially specific and permeable peptide inhibitors of GRK activity, we report here characterization of a panel of peptide inhibitors on in vitro GRK phosphorylation of light-activated rhodopsin and tubulin by four GRK isoforms, PKA phosphorylation of Kemptide, and AC activity in membranes. The sequences of these peptides are based on helices 3, 9, and 10 in the RH domain of GRK5, the N-terminus of GRK5/6, iL1 of the β2AR (Winstel et al. 2005), and the C-Tail of Gα transducin (TCT) (Aris et al. 2001; Hamm et al. 1988; Martin et al. 1996) as shown in Table 1. The locations of the helices of GRK6 (Boguth et al. 2010) and TCT (Scheerer et al. 2008) from which the inhibitors were derived are shown in Fig. 1. Additionally, to examine intracellular activity of the peptides, we appended the TAT cell-penetration sequence to select peptides (Richard et al. 2005; Sanders et al. 2011; Sarko et al. 2010), and characterized their effects on cAMP accumulation, and GRK and PKA phosphorylation of the β2-adrenergic receptor.
Table 1.
Sequences of GRK5, Gα TCT, and β2AR iL1 peptides
| Peptide | Sequence |
|---|---|
| α3 (61–71) | Ac-PIGRLLFRQFC-NH2 |
| R169A (164–178)a | Ac-SNleFFDAFLQWKWLER-NH2 |
| α10 (507–517)a | Ac-VSIPWQNENleIE-NH2 |
| N-Ter (1–16) | MELENIVANTVLLKAR-NH2 |
| TCT (340–350)b | Ac-ILENLKDCGLF-NH2 |
| iL1 (59–74E)c | AKFERLQTVTNYFITSE |
Residues modified from original sequence are in bold
Norleucine (Nle) was substituted for methionine 165 (R169A) and M515 (α10)
TCT: K341L (Scheerer et al. 2008)
iL1: truncated and modified β2AR iL1 (Winstel et al. 2005)
Fig. 1.
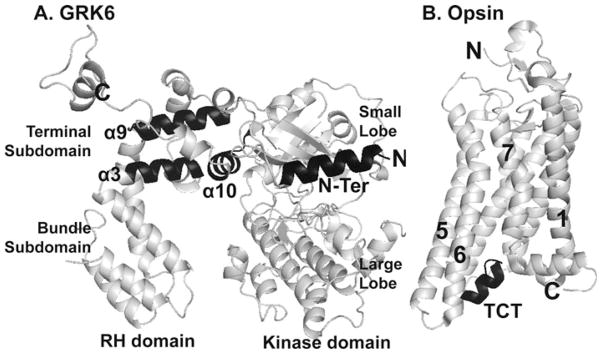
Location of GRK peptides and Gα transducin C-tail. a GRK6 structure [PDB code 3NYN] (Boguth et al. 2010) showing the location of RH domain peptides α3, α9 and α10, and the N-terminal peptide in black. b Opsin in its likely G-protein-interacting conformation [PDB code 3DQB] (Scheerer et al. 2008); the Gα TCT peptide is highlighted in black
Materials and Methods
Materials
Cell culture reagents are from Mediatech (Herdon, VA). Peptide N-glycosidase F was from New England Biolabs (Beverly, MA). Polyclonal primary antibodies to pS-(355,356) C-Tail of the β2AR, and to GRK2, GRK5, and GRK6 are from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal antibody 2G3 for pS-(262) of the β2AR was as described (Tran et al. 2004). N-terminal 6 His-tagged, recombinant, full-length human GRK6, and GRK7 were purchased from Millipore (Dundee, UK). Human His6-tagged GRK5 was a kind gift from John Tesmer (University of Michigan, Ann Arbor, MI). The HRP-conjugated secondary antibody was from BioRad. Enhanced chemiluminescence SuperSignal reagent was purchased from Thermo Scientific (Rockford, IL), and blue X-ray film was from Phenix (Candler, NC). SP-Sepharose Fast Flow was purchased from GE Healthcare (Piscataway, NJ). Sf-900 II SFM insect cell culture medium was purchased from Gibco (Grand Island, NY). Purified tubulin from bovine brain was purchased from Cytoskeleton (Denver, CO). [2,8-3H]adenine was purchased from Perkin Elmer. Protease inhibitor cocktail was from Gold Biotechnology (St. Louis, MO).
Cell Culture
HEK293 cells stably overexpressing FLAG-tagged wild type β2AR (WT-β2AR) (Tran et al. 2004) at 2–4 pmol/mg membrane were grown in 5 % CO2 at 37 °C in Dulbecco’s modified eagle’s medium containing 10 % fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 200 μg/ml G418. When seeding cells for experiments, dishes were coated with poly-L-lysine to aid attachment.
Purification of GRK2
Sf9 cells were infected with pFastBacGRK2-His6 (S670AFL) bacculovirus (gift from Dr. Tesmer, University of Michigan). GRK2 was purified as previously described (Benovic 1991; Singh et al. 2008; Sterne-Marr et al. 2009), with some modifications. Pellets from 1 l of cells were resuspended in 100 ml of lysis buffer (20 mM Hepes, pH 7.4, 250 mM NaCl, 0.02 % Triton X-100, 2 mM β-mercaptoethanol, and 1 tablet of EDTA-free protease inhibitor cocktail). Cells were lysed using a Parr bomb at 550 PSI for 30 min on ice (4 °C). Cell lysate was clarified by centrifugation in a SS34 rotor at 16,000 rpm for 20 min. High speed supernatant fraction was generated by centrifugation in a SW32 rotor at 30,000 rpm for 60 min, diluted and applied to an IMAC Ni-column washed and eluted with imidazole step gradient (5, 12, 50, 250 mM). One milliliter fractions were collected at a flow rate of 1 ml/min. Aliquots were electrophoresed to check for expression and purity of GRK2 by coomassie staining. Highly purified fractions were pooled, dialyzed in lysis buffer, and concentrated by Amicon pressure dialysis. Protein concentration was assessed by both protein assay and by reference to standard curves generated with purified GST-GRK2.
Peptide Synthesis
Peptides were synthesized as previously described (Baameur et al. 2010) on polydimethylacrylamide-based PL-DMA resin (Varian, Inc.) (Arshady et al. 1981) using automated methods and Fmoc chemistry on an AAPPTEC 348 multiple synthesizer with a 16-well reactor block. Peptides, with the exceptions of N-Ter and iL1, were acetylated at their N-termini by addition of acetic anhydride on the automated synthesizer. Peptides were cleaved from their resins with TFA:triethylsilane:H2O (95:2.5:2.5) for 2 h, the volumes were reduced, the products were precipitated in Et2O and collected by centrifugation and dried. Peptides were purified by reverse phase HPLC using gradients of acetonitrile in H2O (both solvents containing 0.1 % TFA). All peptides were >95 % pure and gave the correct mass by ESI-TOF mass spectrometry. Because of the large number of peptides used in this study we did not examine the helical nature of these peptides in solution, nor were scrambled peptides for each deemed necessary. For the α9 peptides we had previously demonstrated that chemically locking the peptide did not significantly alter its inhibition of GRK5, suggesting that this peptide assumes a helical structure in solution (Baameur et al. 2010). Also in our previous studies several minor modifications of α9 helix were inactive at 100 μM and we believe this provided sufficient control for a scrambled peptide.
Phosphorylation of Rhodopsin
Phosphorylation of rhodopsin was determined as previously described (Baameur et al. 2010) with minor modifications as indicated. Urea-stripped bovine rod outer segments (ROS) were a gift from Dr. Kevin Ridge (University of Texas Medical School, Houston, TX). To examine peptide inhibition of GRK phosphorylation of light-activated rhodopsin, peptides were dissolved in DMSO and the peptide concentrations calculated based on absorbance at 280 nm. Peptides were used at final concentrations of 1–100 μM with DMSO at 2–4 %. Purified GRKs were diluted to final concentrations of 10–100 nM. The reaction buffer contained 0.5 μM rhodopsin, 20 mM Tris–HCl, pH 7.5, 1 mM EDTA, 5 μM DTT, 10 mM MgCl2, and 100 μM [γ32P]ATP (500–1,000 dpm/pmol). Rhodopsin was activated by illumination (475 nm) for 30 s (Ridge et al. 2006) just prior to incubation. Reactions were stopped after 5 min by addition of 4X SDS-sample buffer, and samples electrophoresed on a 12 % SDS-PAGE. After transfer to nitrocellulose membranes, 32P-labeled proteins were visualized by autoradiograms. With these levels of GRKs and rhodopsin, phosphorylation was linear for 0–30 min. Control incubations lacking either GRK or activated rhodopsin revealed no activity over background. 32P-rhodopsin bands were quantified either by densitometry directly from autoradiograms, by using a Storm Molecular Dynamics Phosphorimager (GE Healthcare), or by direct counting of excised bands; comparable results were obtained from these measurements.
GRK Phosphorylation of Tubulin
Tubulin phosphorylation was measured by incubation in a buffer containing 20 mM Tris–HCl, pH 7.5, 1.0 mM EDTA, 10 mM MgCl2, 5 μM DTT, 100 μM [γ32P]ATP (~5,000 dpm/pmol), 500 nM tubulin and either 100 nM GRK2 or GRK5. Samples were incubated for 60 min at 30 °C and stopped by addition of 5× SDS-sample buffer and run on a 12 % SDS-PAGE. 32P-labeled tubulin was assessed by two methods; gels were either stained with coomassie and tubulin bands excised and counted in a scintillation counter, or transferred to nitrocellulose membranes and 32P-labeled tubulin quantified by densitometry from autoradiograms.
PKA Activity
PKA activity was measured by phosphorylation of the peptide substrate kemptide (LRRASLG). 50 μM of kemptide was incubated with 2.5 nM (5 ng) PKA catalytic subunit (mouse recombinant PKA kindly provided by Dr. Susan Taylor, UC San Diego, San Diego, CA) for 10 min at 30 °C with shaking in 10 mM Hepes, pH 7.4, 5 mM MgCl2, 200 μM 32P-ATP [~5,000 dpm/pmol] in the absence (2.0 % DMSO) or presence of the peptides at the given concentrations. Reactions were stopped by pipetting samples onto p81 phosphocellulose paper, and immediately plunged in water. This was followed by five rinses with water, two rinses with acetone and scintillation counting (Kunkel et al. 1989).
Adenylyl Cyclase Activity
Membranes from WT-β2AR cells containing 5 μg membrane protein were incubated for 10 min at 30 °C in buffer containing 40 mM Hepes, pH 7.7, 1 mM EDTA, 6 mM MgCl2, 100 μM ATP, 1 μM GTP, 0.1 mM 3-isobutyl-1-methylxanthine (IBMX), 8 mM creatine phosphate, 16 U/ml creatine phosphokinase, and 2 μCi of [α32P]ATP in the absence or presence of 30–100 μM peptides or TAT-peptides (2.0 % final DMSO). AC activity was measured after stimulation with either 1 μM ISO or 20 μM forskolin (FSK) (Tran et al. 2007a).
Intact-cell Phosphorylation of the β2AR
WT-β2AR cells growing in 12 well plates were treated with DMSO, 100 μM of TAT alone, or TAT-peptides for 20 min. Cells were then stimulated with 20 nM ISO dissolved in the carrier, 0.1 mM ascorbate/1 mM thiourea pH 7 (AT), or AT alone for 5 min at 37 °C, then solubilized as previously described (Tran et al. 2007b). Samples were resolved on 12 % SDS-PAGE, transferred to nitrocellulose membrane and immunoblotted with either anti-pS(355,356) (GRK site) or anti-pS-262 (PKA site) antibodies, then stripped and reprobed with anti-C-Tail antibody. Results were normalized to the β2AR levels (anti-C-Tail).
Intact-cell cAMP Measurement
Human airway smooth muscle cells (HASM), passage 6–10 (kindly provided by Dr. Robert Moore, Baylor College of Medicine, Houston, TX) were grown to confluence in F12-HAM medium in 12-well plates. The growth medium was removed and replaced with 0.3 ml of NaHCO3-free, 25 mM Hepes-buffered medium (pH 7.4) containing 12 μCi of [2,8-3H]adenine at 37 °C. After 2.5 h, IBMX was added to a final concentration of 1 mM for 20 min. The cells were then pretreated for 20 min with 1 % DMSO or peptides at a final concentration of 1 % DMSO, followed by 5–20 min treatments with 100 nM ISO. To stop the reaction, the medium was removed and [3H]-cAMP in cells determined as previously described (Clark et al. 1986).
Results
Effects of Substitutions and Truncations of α9 on GRK5 Phosphorylation of Rhodopsin
It was previously reported that a peptide mimicking helix 9 of the RH domain of GRK5/6 (α9) inhibited GRK5 phosphorylation of rhodopsin non-competitively. Incorporation of side-chain to side-chain bridges to induce helical constraints did not significantly impair inhibition (Baameur et al. 2010). The Evolutionary Trace program, which first identified helix 9 as a potential inhibitor, also identified specific residues that were highly conserved and were therefore hypothesized to be important for activity (Baameur et al. 2010). To explore the importance of these amino acids, we made alanine substitutions and assayed for effects on rhodopsin phosphorylation by GRK5 (Table 2). The R169A, Q172A, and L176A substitutions all showed potent inhibition as compared to the native sequence of α9 helix. Surprisingly the R169A analog exhibited increased potency compared to the native sequence (IC50 ~7 μM), suggesting that a hydrophobic surface on GRK5 does not accommodate the arginine well. Both F166A and W173A substitutions showed reduced inhibition (~42 and 26 % respectively at 100 μM), which suggests these conserved hydrophobic residues are important for binding. To assess whether shorter α9 peptides would affect potency, residues were deleted from the C-terminus. Deletion of either the C-terminal dipeptide (T2) or tripeptide (T3), resulted in increased potency (IC50s = 5.2 ± 2 and 6.1 ± 4 respectively) relative to α9 (IC50 of 15.9 ± 4). Deletion of the C-terminal W175 (T4) reduced inhibition. Deletion of SNle from the N-terminus (T5) further reduced the potency while not eliminating inhibition. These findings collectively indicate the importance of hydrophobic residues in the α9 peptides.
Table 2.
Inhibition of GRK5 phosphorylation of rhodopsin by truncation and alanine substitutions of α9 peptide

| |||
|---|---|---|---|
| Peptide | Sequencea | % Inhibition (100 μM) | IC50 (μM) |
| α9 (164–178) | Ac-SNleFFDRFLQWKWLER-NH2 | 89 | 15.9 ± 4.3 |
| F166A | Ac-SNleAFDRFLQWKWLER-NH2 | 42 | ND |
| R169A | Ac-SNleFFDAFLQWKWLER-NH2 | 98 | 7.2 ± 4.8 |
| Q172A | Ac-SNleFFDRFLAWKWLER-NH2 | 95 | 15.1 ± 7.3 |
| W173A | Ac-SNleFFDRFLQAKWLER-NH2 | 26 | ND |
| L176A | Ac-SNleFFDRFLQWKWAER-NH2 | 74 | ND |
| T1 | Ac-FFDRFLQWKWLER-NH2 | 95 | 23.9 ± 16.0 |
| T2 | Ac-SNleFFDRFLQWKWL-NH2 | 98 | 5.2 ± 2.0 |
| T3 | Ac-SNleFFDRFLQWKW-NH2 | 97 | 6.1 ± 4.4 |
| T4 | Ac-SNleFFDRFLQWK-NH2 | 72 | ND |
| T5 | Ac-FFDAFLQWK-NH2 | 22 | ND |
The α9 helix is shown in the inset; conserved residues are highlighted in black
ND not determined
Alanine substitutions of α9 are in bold. Nle was substituted for M165 to avoid sulfur oxidation
Peptide Inhibition of GRK Phosphorylation of Rhodopsin
In addition to peptides derived from helix 9, we evaluated the inhibitory properties of peptides based on helices 3 and 10 in the RH domain of GRK5/6 (α3 and α10), the N-terminus of GRK5 (Noble et al. 2003; Pao et al. 2009) (N-Ter), and the iL1 of the β2AR, peptide 59–74E (Winstel et al. 2005) (iL1). The binding of the C-Tail of Gαt peptide K341L (TCT) to rhodopsin has been characterized and the structure of its complex with opsin determined; therefore, we included this peptide to monitor the effect of direct peptide binding to rhodopsin (Aris et al. 2001; Scheerer et al. 2008). To obtain a comparison of the inhibitory actions of this panel of peptides on a set of GRK isoforms, we determined their inhibition of the phosphorylation of rhodopsin by GRKs 2, 5, 6, and 7. The amino acid sequences of the peptides and results are given in Fig. 2 and Table 2. Peptide α3 inhibited GRKs 2, 5 and 7, but showed little activity against GRK6. Both R169A and α10 inhibited GRKs 5, 6 and 7 with similar potencies, but showed less inhibition of GRK2. N-Ter was a reasonably potent inhibitor of GRKs 5 and 7, but a less potent inhibitor of GRKs 2 and 6. Surprisingly, the TCT, that was expected to inhibit all GRKs similarly due to its binding to rhodopsin, showed inhibition of GRKs 5, 6, and 7, but was relatively inactive on GRK2. Peptide iL1 showed potent inhibition of all GRK phosphorylations of rhodopsin, consistent with previous findings for the inhibition of GRK activity both in vitro and in cells (Winstel et al. 2005; Xin et al. 2008).
Fig. 2.

Inhibition of GRKs 2, 5, 6 and 7 by the panel of peptides. a GRK2, b GRK5, c GRK6, and d GRK7. Phosphorylation of light-activated rhodopsin (0.5–4 μM) was determined by incubation with purified GRKs in the absence (Ctrl) or presence of peptide inhibitors α3, R169A (α9), α10, N-Ter, TCT and iL1 (10, 30 and 100 μM) as described in “Materials and Methods” section. Data shown are the mean ± SEM for four or more experiments performed in duplicate and normalized to the % of control. Comparison to Ctrl was performed by One-Way ANOVA; ***P < 0.001, **P < 0.01, and *P < 0.05
Peptide Inhibition of GRK Phosphorylation of Tubulin
As mentioned, GPCRs activate serine and threonine kinase activity of GRKs. To examine whether the inhibitors act on “basal” kinase activity, we probed the effect of peptides on tubulin phosphorylation by GRKs 2 and 5 in the absence of GPCRs (Fig. 3). The data show that at a concentration of 100 μM, both R169A and α3 inhibited these GRKs 60–80 %, with slightly better IC50s for GRK5 (~30 μM), consistent with their binding to GRKs. Peptide α10 significantly inhibited GRK5 (~45 % at 100 μM), but its inhibition of GRK2 was not significant. N-Ter, TCT, and iL1 had no significant effect on basal activity, suggesting their effects on rhodopsin phosphorylation were not caused by peptide binding to the GRKs, although the possibility remains that peptides bind to GRK sites with no effect on basal activity.
Fig. 3.

Peptide inhibition of GRK2 (a), and GRK5 (b) phosphorylation of tubulin. Phosphorylation of tubulin (0.5 μM) was determined by incubation with purified GRK2 and GRK5 in the absence (Ctrl) or presence of peptide inhibitors at the indicated concentrations as previously described (Baameur et al. 2010). Data shown are the mean ± SEM for four or more experiments performed in duplicate and normalized to the % of control. Comparison to Ctrl was performed by One-Way ANOVA; ***P < 0.001, **P < 0.01, and *P < 0.05
Lack of Peptide Inhibition of Adenylyl Cyclase
As a control for potential off-target effects on GPCR signal transduction pathway components, we determined the effects of the peptides on ISO and FSK stimulation of AC. Sucrose gradient purified membranes from WT-β2AR cells were treated with 100 μM peptides, stimulated with ISO and FSK, and AC activities determined (Fig. 4). None of the six peptides showed any significant inhibition of AC, establishing that they had no effect on either receptor or Gs activation of AC.
Fig. 4.
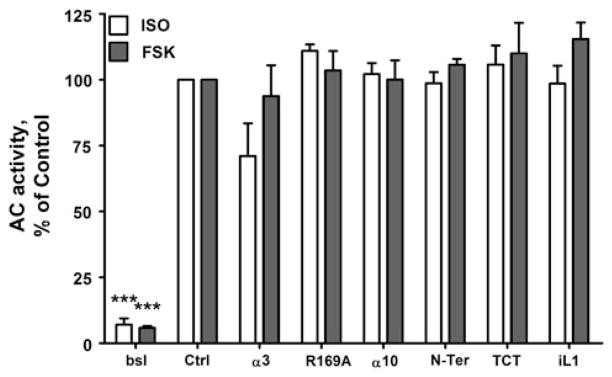
Lack of peptide inhibition of AC activity. Membranes from WTβ2AR cells were incubated for 10 min at 30 °C in the absence (Ctrl) or presence of either 1 μM ISO or 20 μM FSK. Basal AC activity (bsl) is as indicated. Peptide concentrations were 100 μM. AC activity was determined as previously described (Tran et al. 2007a). Data shown are the mean ± SEM for three or more experiments performed in triplicate and normalized to the % of control (***P < 0.001 by One-Way ANOVA)
Peptide Inhibition of PKA Phosphorylation
Since the GRKs are members of the AGC family of kinases, as a test of specificity we probed the panel of peptides for their effects on PKA catalytic activity by measuring phosphorylation of the substrate Kemptide. R169A, α3, and TCT inhibited PKA, but weakly with IC50s of ~60–80 μM (Fig. 5), revealing selectivity for GRKs. Unexpectedly, α10, N-Ter, and iL1 showed potent inhibition of PKA phosphorylation of kemptide with IC50 values of 5, 3.2, and 5.3 μM respectively. These findings reveal a lack of specificity for these three peptides, as their potencies for PKA inhibition are either equal to or greater than the most potent effects of these peptides on GRKs.
Fig. 5.

Peptide inhibition of PKA. Phosphorylation of the PKA substrate Kemptide peptide was determined by incubation with 2.5 nM recombinant mouse PKA catalytic subunit in the absence (Ctrl) or presence of peptide inhibitors (concentrations ranging from 1 to 100 μM) as described in “Materials and Methods” section. Data shown are the mean ± SEM for four or more experiments performed in duplicate and normalized to the % of control
Inhibition of Isoproterenol-stimulated GRK and PKA Site Phosphorylation of β2ARs in Intact Cells
An important question is whether the inhibitory peptides developed from in vitro assays of rhodopsin phosphorylation could be shown to inhibit GRK kinase activity in intact cells. To facilitate peptide delivery into the cells we appended the TAT cell penetration sequence and a three glycine linker (GRKKRRQRRRPPQGGG) to the N-termini of R169A, TCT, and N-Ter (Fawell et al. 1994). To monitor peptide delivery into the cells, we also synthesized peptides that were tagged at the N-termini with FITC; e.g., FITC–TAT–TCT (FITC-βAla-GRKKRRQRRRPPQGGG-ILENLKDCGLF-NH2). Fluorescence microscopy revealed that uptake of the FITC-TAT-peptides was complete by 10 min (data not shown).
The effects of the TAT-tagged peptides on the phosphorylation of β2AR were assessed in intact WT-β2AR cells. GRK and PKA phosphorylations were probed with anti-GRK site pS-(355,356) and anti-PKA site pS-262 antibodies. Following 20 min incubation with either TAT or TAT-peptides, cells were treated with 20 nM ISO for 10 min. We found that both TAT-R169A and TAT–TCT at 100 μM significantly inhibited ISO-induced GRK phosphorylation of pS-(355,356) of the β2AR by 30 and 60 % respectively (Fig. 6a), whereas neither TAT nor the TAT-N-Ter peptide had any effect. TAT–TCT, but not TAT-R169A, displayed significant inhibition at 30 μM (data not shown). As a control, we found that all TAT-peptides inhibited GRKs 5 and 7 phosphorylation of rhodopsin to the same extent as their non-tagged homologs, demonstrating that TAT tagging did not interfere with this activity (data not shown). Phosphorylation of S262, the PKA site in the 3rd loop of the β2AR, was not significantly affected (Fig. 6b) by any of the TAT-peptides. Thus in intact cells, the peptides are selective for GRKs over PKA.
Fig. 6.

TAT-tagged peptides inhibition of ISO stimulation of GRK phosphorylation of the β2AR. WT-β2AR cells were treated in the absence (Ctrl) or presence of 100 μM TAT or TAT-tagged peptides for 20 min prior to stimulation either with or without 20 nM ISO for 5 min. GRK site phosphorylation was determined using the anti-pS( 355,356) antibody (a), and PKA site phosphorylation using anti-pS262 (b) as previously described (Tran et al. 2004). Data shown are the mean ± SEM for four or more experiments performed in duplicate and normalized to the % of control (***P < 0.001, **P < 0.01, and *P < 0.05 by One-Way ANOVA)
TAT-peptide Inhibition of cAMP Accumulation
To assess the specificity of the cell-penetrating peptides, we determined whether TAT-R169A and TAT–TCT directly interfere with receptor activation of G-protein by measuring cAMP accumulation after ISO stimulation of primary HASM. In these cells ISO stimulation produces much greater signal to noise relative to the WT-β2AR cells that have a very high basal activity due to the overexpression of the receptor. Surprisingly, both TAT-R169A and TAT–TCT, but neither TAT nor TAT-N-Ter, significantly inhibited cAMP formation after 20 min stimulation (Fig. 7), showing a pattern similar to the inhibition of ISO-stimulated GRK-site phosphorylation of β2AR in WT-β2AR cells. Similar results were obtained following either 5, 10, or 20 min ISO stimulation (data not shown). These findings demonstrate that TAT-R169A and TAT–TCT interfered with receptor activation of Gs/AC. We also examined the effects of TAT and the TAT-tagged peptides on AC in sucrose gradient-purified membrane preparations from WT-β2AR cells. As with the intact HASM cells, TAT-R169A and TAT–TCT at 100 μM inhibited ISO- and FSK-stimulated AC (70–85 %) while TAT and TAT-N-Ter were without effect (data not shown). Since none of the non-TAT modified peptides inhibited AC our data suggested that the TAT modification was causing uncoupling of AC confounding results with the intact cell data.
Fig. 7.

Peptide inhibition of cAMP accumulation in HASM cells. HASM cells were first incubated with either TAT alone or TAT-tagged peptides (100 μM) for 20 min, then treated with AT (bsl) or 100 nM ISO for 20 min. cAMP accumulation was measured in the presence of 200 μM IBMX as described in “Materials and Methods” section. Data shown are the mean ± SEM for three or more experiments performed in duplicate and normalized to the % of TAT (***P < 0.001 by One-Way ANOVA)
TAT-peptide Inhibition of PKA Phosphorylation
Given that the N-Ter was found to inhibit PKA potently, the three TAT-peptides were also examined for inhibition of Kemptide phosphorylation by PKA. The TAT-N-Ter and TAT–TCT, inhibited PKA with IC50s of ~20 and 30 μM respectively, whereas the TAT alone as a control showed an IC50 of ~100 μM (Fig. 8). Most strikingly, the TAT-R169A peptide inhibited with an IC50 of ~200 nM even though the non-TAT tagged R169A peptide showed very weak inhibition of PKA (IC50 of ~70 μM) (Fig. 5). We also found that the potency of TAT-R169A inhibition of PKA was further increased at lower ATP concentrations (20 μM, data not shown) suggesting possible competition with ATP binding. As shown above, the ability of TAT-R169A to inhibit isolated PKA in biochemical assays did not translate to intact cells.
Fig. 8.
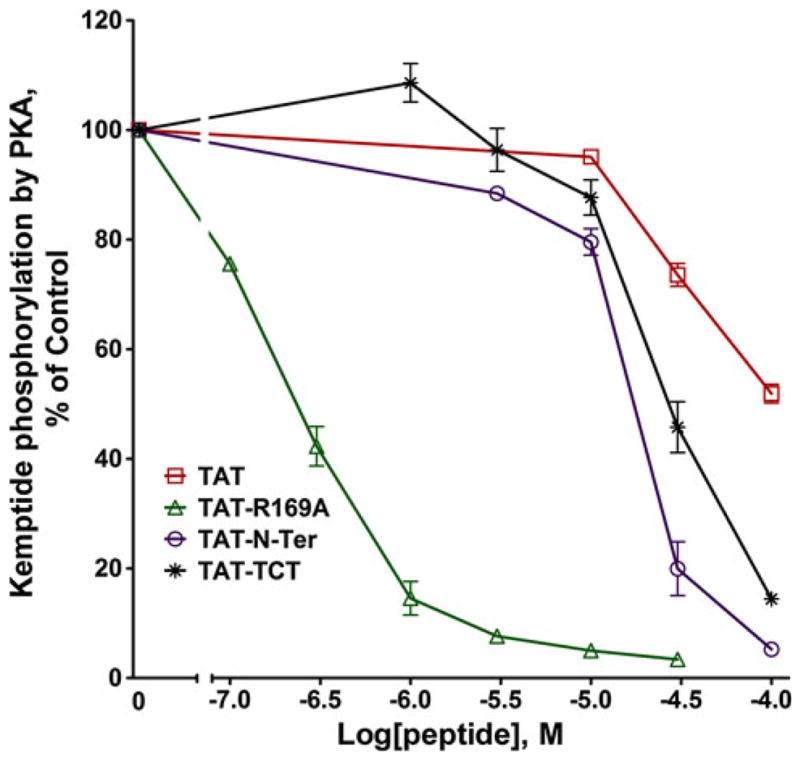
TAT-peptide inhibition of PKA. Phosphorylation of the PKA substrate Kemptide was determined as described in the legend for Fig. 5. Data shown are the mean ± SEM for three or more experiments performed in duplicate and normalized to the % of control
Discussion
At present little is understood about the complex activation of GRKs by ligand-activated GPCRs, including the nature of the allosteric activation, as well as the overall dynamics of GRK structural changes. The interaction involves elements of GRKs that differentially localize them to the plasma membrane, compose the receptor/GRK interaction site, and position the GPCR domain to be multiply phosphorylated by the GPCR/GRK–ATP complex. As discussed above, numerous studies of evolutionarily conserved sites on GRKs revealed several highly conserved domains unique to GRKs both in the kinase and RH domains. Mutagenesis of these domains profoundly inhibited GRK activities, opening up the possibility that allosterically acting peptides could be specific GRK inhibitors, and thus avoid blocking activity by targeting the ATP/receptor C-tail substrate binding pocket.
In a prior study the α9 helix of GRK5 was found to be a potent inhibitor of GRK5, and conformationally “locked” peptides were equivalent in activity indicating that both locked and unlocked peptides shared equivalent helicity. These findings suggested that several of the highly conserved residues on one face of the proposed helix were important for activity as previously reported (Baameur et al. 2010). In the present study, a partial alanine scan of conserved residues revealed that substitutions of three conserved hydrophobic residues (F166, W173, and L176) reduced inhibitory activity of GRK5. Only the R169A peptide showed an increased potency relative to the native α9 peptide, and therefore it was used in subsequent studies of GRK phosphorylation. Of further interest, truncation of the C-terminal three residues (LER) also resulted in increased potency. While this suggests that a smaller peptide could be further developed it was not pursued in this study.
In the second phase of this study, we performed a comprehensive screen of the panel of six peptides for inhibition of GRKs from the three subfamilies, GRKs 2, 5, 6 and 7. In assessing the overall pattern of GRK inhibition by the peptides mimicking GRK5 helices (3, 9, and 10) and the N-terminus (Fig. 2), several notable features emerge concerning specificity. First, all the peptides showed strong inhibition of GRK5 phosphorylation of rhodopsin, not surprisingly given that they were GRK5-based. However, they unexpectedly showed equivalent inhibition of GRK7 even though all the peptides differed in several key residues between GRKs 5 and 7. Several of the peptides showed little inhibition of GRK 2 (α10, TCT and N-Ter), and of GRK6 (α3 and N-Ter). R169A and α10 showed potent inhibition of GRKs 5, 6 and 7, but were less active against GRK2. Interestingly in our previous study of the α9 peptide we found no inhibition of GRK6, suggesting that the R169A modification reduced specificity for this GRK. The N-Ter peptide of GRK5 inhibited both GRKs 5 and GRK7 (IC50s ~5–10 μM), but less potently GRKs 2 and 6. This result is consistent with a prior study showing that, at similar concentrations of the peptides, the GRK5N-Ter was effective against GRK5, but not GRK2 (Noble et al. 2003), as well as the converse (Pao et al. 2009). However, since the N-Ter peptides in GRK5 and 6 are identical it was expected that their inhibitions would be similar. The α3 peptide was strong against GRKs 2, 5, and 7, but was weak against GRK6 even though GRKs 5 and 6 peptide sequences are identical except for a Q to E substitution.
Considering the non-GRK5 peptides TCT and iL1, we expected that TCT, which was shown to bind to rhodopsin and opsin (Aris et al. 2001; Hamm et al. 1988; Scheerer et al. 2008), would inhibit all GRKs. However, it showed a potency range of GRK5 (IC50 ~10 μM) ≥ GRK6 = GRK7>GRK2. The IC50 for GRK5 was about 10 fold higher than that reported by Aris et al. (2001); however, binding of TCT in these prior studies was assayed by stabilization of metarhodopsin II at 5.4 °C making direct comparisons difficult. The iL1 peptide showed similar inhibition of rhodopsin phosphorylation by all GRKs. This result extends prior findings that it blocked in vitro activities of GRKs 2, 3 and 5 and cellular desensitization of the β2AR in several cell systems (Winstel et al. 2005; Xin et al. 2008). These anomalies between the actions of N-Ter and α3 on GRKs 5 and 6, and TCT on all GRKs, could reflect differential specificity of GRK binding to either Rho*, to ROS membrane (Homan et al. 2013), or by differing GRK specificities for rhodopsin phosphorylation sites. Other possibilities are interference with substrate binding, and non-specific binding to sites unrelated to the locale of the peptides in their respective GRKs.
With regard to the tubulin assay of basal GRKs 2 and 5 activity, both α3 and R169A (and to a lesser extent α10 peptide) inhibited these GRKs with potencies (IC50s ~30 μM), similar to the pattern of their inhibitions of rhodopsin phosphorylation. These results suggest that both α3 and R169A inhibit rhodopsin phosphorylation pre-dominantly through binding to these GRKs. In contrast neither the N-Ter, TCT, or iL1 peptides showed significant inhibition of tubulin, suggesting they bind to rhodopsin rather than GRKs 2 or 5, although we cannot rule out binding to an allosteric GRK site that inhibits rhodopsin phosphorylation but not basal activity. The proposed mechanism of action of the N-Ter is that it first binds to the receptor and subsequently docks intramolecularly onto the GRK (Boguth et al. 2010; Huang et al. 2011). It was recently shown that GRK1 engineered such that the N-Ter was cross-linked to the kinase domain through a disulfide bond, activates GRK1 phosphorylation of rhodopsin C-terminal peptide (peptide C) (Huang et al. 2011). Our result is more consistent with peptide binding to the receptor, since it did not inhibit basal GRK activities. To further support the proposed mechanism, the N-Ter should show competition with rhodopsin. While we have not examined this aspect, previous work with the GRK2N-Ter showed no competition (Pao et al. 2009). Thus, uncertainty remains in defining the complex actions of the N-Ter in GPCR stimulation of GRKs.
Determining off-target activities of the panel of peptides was an important aspect of the present study. To approach this we first examined effects of the peptides on in vitro AC activity. Given that GPCRs activate G proteins, this was a notable deficiency in prior studies. In the absence of crystal structures of a GPCR/GRK complex it is simply not known to what extent there is overlap of key residues for the GPCR binding partners Gs and GRKs. To address the issue of possible peptide blockade of β2AR stimulation of Gs, we examined the inhibition of ISO- and FSK-stimulated AC by the six peptides and found that none significantly affected this activity, demonstrating that this aspect of specificity of the peptides was strong.
To further probe specificity it was important to characterize inhibition of a closely related class AGC protein kinase, and for that PKA catalytic activity was chosen. We found that in biochemical assays iL1, α10 and the N-Ter peptides all showed remarkable potencies for inhibition of PKA catalytic subunit activity (IC50s ~3–5 μM). Our result with iL1 is not consistent with Winstel et al. who reported only a 20 % inhibition of PKA by 100 μM iL1 (Winstel et al. 2005). Other peptides were not examined in this study. At present we do not understand the discrepancy with their iL1 results, although our assays were performed with recombinant PKA, whereas Winstel et al. used PKA purified from bovine heart. While R169A, α3, and TCT were much less potent inhibitors of PKA, our findings demonstrate a lack of peptide specificity for iL1, α10 and the N-Ter inhibition of GRKs. This demonstration is of considerable importance since, at least for the β2AR, PKA plays an important role in desensitization as it both phosphorylates and partially desensitizes the β2AR and activates PDE (Tran et al. 2004; Xin et al. 2008). In our previous study of iL1 inhibition of ISO-induced desensitization we assumed that it was specific for GRK, since its effects were additive with PDE inhibitors on blocking desensitization (Xin et al. 2008). This study was based on whole cell patching of HEK293 cells transfected with the cyclic nucleotide-gated channel allowing injection of iL1. Desensitization was determined following treatment with high concentrations of ISO, conditions in which the GRK/arrestin/internalization pathway of desensitization pre-dominates, nulling out a significant effect on receptor-level desensitization by PKA. However, iL1 clearly could have been limiting PKA activation of PDE and hydrolysis of cAMP since it was introduced into cells through the patch pipette, confounding interpretation of desensitization as assessed by cAMP turnover.
Having characterized peptide inhibition of GRKs in biochemical assays, we evaluated three of our peptides for their activities in intact cells by adding the TAT sequence to the N-termini of R169A, N-Ter, and the TCT to shuttle the peptides to the cell interior as was previously demonstrated in unrelated studies (Richard et al. 2005; Sanders et al. 2011; Sarko et al. 2010). We found that both TAT-R169A and TAT–TCT inhibited GRK phosphorylation of β2AR in WT-β2AR cells, whereas they failed to inhibit PKA phosphorylation of the β2AR. TAT and TAT-N-Ter did not inhibit either GRK or PKA phosphorylation of β2AR. Assaying for off-target effects we found TAT-R169A and TAT–TCT inhibited both intact-cell cAMP accumulation and cell-free AC activities in response to either ISO or FSK at the same concentrations as for inhibition of phosphorylation. These findings suggest either these peptides block receptor activation of both GRK phosphorylation and G-protein activation, or they possibly uncouple all receptor activation by nonspecific effects on the membrane. The latter is consistent with the equivalent effects on ISO and FSK stimulations. However, since inhibition of β2AR stimulation of GRKs should not be affected by inhibition of AC, as its actions are G protein-independent (Green and Clark 1981), it is possible the TAT peptides block GRK phosphorylation, but that it occurs in a background of membrane uncoupling of AC. The lack of effect on PKA phosphorylation of the β2AR in these cells is likely the result of the large amplification of PKA phosphorylation (Tran et al. 2004), and the incomplete inhibition of AC by the TAT peptides. Given that the non-TAT-tagged peptides showed no inhibition of AC, our conclusion is that TAT conjugation alters the peptides such that they act non-specifically by uncoupling AC. Additionally, the TAT-peptides also showed inhibition of in vitro PKA activity, in particular TAT-R169A (Fig. 8) the potency of which was dramatically increased relative to R169A action on PKA (Fig. 5). We also found that TAT-R169A (1.0 μM) inhibition of PKA was reduced with increasing concentrations of ATP suggesting possible interference with substrate binding. We are not aware of any other groups that have explored effects of their TAT-peptides on membrane-bound enzymes or PKA, and our study suggests these effects should be routinely investigated.
In summary, our findings demonstrate that while some of the panel of peptides show notably potent inhibition of rhodopsin phosphorylation there was a considerable level of non-specificity. Inhibition of PKA activity by TCT, N-Ter, iL1 and TAT-R169A, as well as inhibition of AC by TAT-tagged peptides further demonstrates non-specificity. These findings highlight the differences in the environments of biochemical and cell-based assays and illustrate the care that must be exercised in interpreting results of either assay alone. Clearly real challenges remain towards the development of cell-permeable peptides that would be useful as specific inhibitors of select GRKs.
Acknowledgments
This work was supported by the National Institute of Health National Institute of General Medical Sciences [Grant ARRA GM031208] to R.B.C. and J.S.M.; and the R. A. Welch Foundation Chemistry and Biology Collaborative Grant from the John S. Dunn Gulf Coast Consortium for Chemical Genomics. This work was supported by the National Institute for General Medicine RO1-GM031208. We acknowledge the Cancer Center Support Grant P30-CA16672 at MDACC for support of the Translational Chemistry Core Facility which provided mass spectrometry analysis. We thank Dr. John Tesmer (U. of Michigan) for providing us plasmids and GRK5 protein. We are grateful to Dr. Choel Kim and Jeong Joo Kim (Baylor College of Medicine) for their help with GRK purification. We thank Kevin Ridge posthumously for providing us ROSs. We also thank Drs Carmen Dessauer and Jeff Frost for aiding our efforts in so many ways.
Footnotes
Conflict of interest Faiza Baameur, Richard A. Hammitt, Jacqueline Friedman, John S. McMurray, and Richard B. Clark declare that they have no conflicts of interest.
Contributor Information
Faiza Baameur, Integrative Biology and Pharmacology, University of Texas Medical School-Houston, Houston, TX 77030, USA.
Richard A. Hammitt, M. D. Anderson Cancer Center, University of Texas, Houston, TX 77030, USA
Jacqueline Friedman, Integrative Biology and Pharmacology, University of Texas Medical School-Houston, Houston, TX 77030, USA.
John S. McMurray, Email: jsmcmur@mdanderson.org, M. D. Anderson Cancer Center, University of Texas, Houston, TX 77030, USA
Richard B. Clark, Integrative Biology and Pharmacology, University of Texas Medical School-Houston, Houston, TX 77030, USA
References
- Aris L, Gilchrist A, Rens-Domiano S, Meyer C, Schatz PJ, Dratz EA, et al. Structural requirements for the stabilization of metarhodopsin II by the C terminus of the alpha subunit of transducin. J Biol Chem. 2001;276(4):2333–2339. doi: 10.1074/jbc.M002533200. [DOI] [PubMed] [Google Scholar]
- Arshady R, Atherton E, Clive DL, Sheppard RC. Peptide synthesis. Part 1. Preparation and use of polar supports based on poly(dimethylacrylamide) J Chem Soc Perkin Trans. 1981;1:529–537. [Google Scholar]
- Baameur F, Morgan DH, Yao H, Tran TM, Hammitt RA, Sabui S, et al. Role for the regulator of G-protein signaling homology domain of G protein-coupled receptor kinases 5 and 6 in beta 2-adrenergic receptor and rhodopsin phosphorylation. Mol Pharmacol. 2010;77(3):405–415. doi: 10.1124/mol.109.058115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benovic JL. Purification and characterization of beta-adrenergic receptor kinase. Methods Enzymol. 1991;200:351–362. doi: 10.1016/0076-6879(91)00152-m. [DOI] [PubMed] [Google Scholar]
- Benovic JL, Onorato J, Lohse MJ, Dohlman HG, Staniszewski C, Caron MG, et al. Synthetic peptides of the hamster beta 2-adrenoceptor as substrates and inhibitors of the beta-adrenoceptor kinase. Br J Clin Pharmacol. 1990;30(Suppl 1):3S–12S. doi: 10.1111/j.1365-2125.1990.tb05462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boguth CA, Singh P, Huang CC, Tesmer JJ. Molecular basis for activation of G protein-coupled receptor kinases. EMBO J. 2010;29(19):3249–3259. doi: 10.1038/emboj.2010.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RB, Goka TJ, Proll MA, Friedman J. Homologous desensitization of beta-adrenergic receptors in lymphoma cells is not altered by the inactivation of Ni (Gi), the inhibitory guanine nucleotide regulatory protein. Biochem J. 1986;235(2):399–405. doi: 10.1042/bj2350399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, et al. Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci USA. 1994;91(2):664–668. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DA, Clark RB. Adenylate cyclase coupling proteins are not essential for agonist-specific desensitization of lymphoma cells. J Biol Chem. 1981;256(5):2105–2108. [PubMed] [Google Scholar]
- Gurevich EV, Tesmer JJ, Mushegian A, Gurevich VV. G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol Ther. 2012;133(1):40–69. doi: 10.1016/j.pharmthera.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm HE, Deretic D, Arendt A, Hargrave PA, Koenig B, Hofmann KP. Site of G protein binding to rhodopsin mapped with synthetic peptides from the alpha subunit. Science. 1988;241(4867):832–835. doi: 10.1126/science.3136547. [DOI] [PubMed] [Google Scholar]
- Homan KT, Glukhova A, Tesmer JJ. Regulation of G protein-coupled receptor kinases by phospholipids. Curr Med Chem. 2013;20(1):39–46. [PubMed] [Google Scholar]
- Huang CC, Yoshino-Koh K, Tesmer JJ. A surface of the kinase domain critical for the allosteric activation of G protein-coupled receptor kinases. J Biol Chem. 2009;284(25):17206–17215. doi: 10.1074/jbc.M809544200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Orban T, Jastrzebska B, Palczewski K, Tesmer JJ. Activation of G protein-coupled receptor kinase 1 involves interactions between its N-terminal region and its kinase domain. Biochemistry. 2011;50(11):1940–1949. doi: 10.1021/bi101606e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel MW, Friedman J, Shenolikar S, Clark RB. Cell-free heterologous desensitization of adenylyl cyclase in S49 lymphoma cell membranes mediated by cAMP-dependent protein kinase. FASEB J. 1989;3(9):2067–2074. doi: 10.1096/fasebj.3.9.2545497. [DOI] [PubMed] [Google Scholar]
- Lodowski DT, Barnhill JF, Pitcher JA, Capel WD, Lefkowitz RJ, Tesmer JJ. Purification, crystallization and preliminary X-ray diffraction studies of a complex between G protein-coupled receptor kinase 2 and Gbeta1gamma2. Acta Crystallogr D. 2003;59(Pt 5):936–939. doi: 10.1107/s0907444903002622. [DOI] [PubMed] [Google Scholar]
- Lodowski DT, Tesmer VM, Benovic JL, Tesmer JJ. The structure of G protein-coupled receptor kinase (GRK)-6 defines a second lineage of GRKs. J Biol Chem. 2006;281(24):16785–16793. doi: 10.1074/jbc.M601327200. [DOI] [PubMed] [Google Scholar]
- Martin EL, Rens-Domiano S, Schatz PJ, Hamm HE. Potent peptide analogues of a G protein receptor-binding region obtained with a combinatorial library. J Biol Chem. 1996;271(1):361–366. doi: 10.1074/jbc.271.1.361. [DOI] [PubMed] [Google Scholar]
- Noble B, Kallal LA, Pausch MH, Benovic JL. Development of a yeast bioassay to characterize G protein-coupled receptor kinases. Identification of an NH2-terminal region essential for receptor phosphorylation. J Biol Chem. 2003;278(48):47466–47476. doi: 10.1074/jbc.M308257200. [DOI] [PubMed] [Google Scholar]
- Onorato JJ, Palczewski K, Regan JW, Caron MG, Lefkowitz RJ, Benovic JL. Role of acidic amino acids in peptide substrates of the beta-adrenergic receptor kinase and rhodopsin kinase. Biochemistry. 1991;30(21):5118–5125. doi: 10.1021/bi00235a002. [DOI] [PubMed] [Google Scholar]
- Pao CS, Barker BL, Benovic JL. Role of the amino terminus of G protein-coupled receptor kinase 2 in receptor phosphorylation. Biochemistry. 2009;48(30):7325–7333. doi: 10.1021/bi900408g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard JP, Melikov K, Brooks H, Prevot P, Lebleu B, Chernomordik LV. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J Biol Chem. 2005;280(15):15300–15306. doi: 10.1074/jbc.M401604200. [DOI] [PubMed] [Google Scholar]
- Ridge KD, Marino JP, Ngo T, Ramon E, Brabazon DM, Abdulaev NG. NMR analysis of rhodopsin–transducin interactions. Vis Res. 2006;46(27):4482–4492. doi: 10.1016/j.visres.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Sanders WS, Johnston CI, Bridges SM, Burgess SC, Willeford KO. Prediction of cell penetrating peptides by support vector machines. PLoS Comput Biol. 2011;7(7):e1002101. doi: 10.1371/journal.pcbi.1002101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarko D, Beijer B, Garcia Boy R, Nothelfer EM, Leotta K, Eisenhut M, et al. The pharmacokinetics of cell-penetrating peptides. Mol Pharm. 2010;7(6):2224–2231. doi: 10.1021/mp100223d. [DOI] [PubMed] [Google Scholar]
- Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455(7212):497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- Singh P, Wang B, Maeda T, Palczewski K, Tesmer JJ. Structures of rhodopsin kinase in different ligand states reveal key elements involved in G protein-coupled receptor kinase activation. J Biol Chem. 2008;283(20):14053–14062. doi: 10.1074/jbc.M708974200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterne-Marr R, Leahey PA, Bresee JE, Dickson HM, Ho W, Ragusa MJ, et al. GRK2 activation by receptors: role of the kinase large lobe and carboxyl-terminal tail. Biochemistry. 2009;48(20):4285–4293. doi: 10.1021/bi900151g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TM, Friedman J, Qunaibi E, Baameur F, Moore RH, Clark RB. Characterization of agonist stimulation of cAMP-dependent protein kinase and G protein-coupled receptor kinase phosphorylation of the beta2-adrenergic receptor using phosphoserine-specific antibodies. Mol Pharmacol. 2004;65(1):196–206. doi: 10.1124/mol.65.1.196. [DOI] [PubMed] [Google Scholar]
- Tran TM, Friedman J, Baameur F, Knoll BJ, Moore RH, Clark RB. Characterization of beta2-adrenergic receptor dephosphorylation: comparison with the rate of resensitization. Mol Pharmacol. 2007a;71(1):47–60. doi: 10.1124/mol.106.028456. [DOI] [PubMed] [Google Scholar]
- Tran TM, Jorgensen R, Clark RB. Phosphorylation of the beta2-adrenergic receptor in plasma membranes by intrinsic GRK5. Biochemistry. 2007b;46(50):14438–14449. doi: 10.1021/bi700922h. [DOI] [PubMed] [Google Scholar]
- Winstel R, Ihlenfeldt HG, Jung G, Krasel C, Lohse MJ. Peptide inhibitors of G protein-coupled receptor kinases. Biochem Pharmacol. 2005;70(7):1001–1008. doi: 10.1016/j.bcp.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Xin W, Tran TM, Richter W, Clark RB, Rich TC. Roles of GRK and PDE4 activities in the regulation of beta2 adrenergic signaling. J Gen Physiol. 2008;131(4):349–364. doi: 10.1085/jgp.200709881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu QM, Cheng ZJ, Gan XQ, Bao GB, Li L, Pei G. The amino terminus with a conserved glutamic acid of G protein-coupled receptor kinases is indispensable for their ability to phosphorylate photoactivated rhodopsin. J Neurochem. 1999;73(3):1222–1227. doi: 10.1046/j.1471-4159.1999.0731222.x. [DOI] [PubMed] [Google Scholar]