

Abstract
High levels of fetal hemoglobin (Hb F) protect from many of the complications of sickle cell disease and lead to improved survival. Butyrate and other short chain fatty acids were previously shown to increase Hb F production in erythroid cells in vitro and in animal models in vivo. However, butyrates are also known to inhibit the proliferation of many cell types, including erythroid cells. Experience with the use of butyrate in animal models and in early clinical trials demonstrated that the Hb F response may be lost after prolonged administration of high doses of butyrate. We hypothesized that this loss of response may be a result of the antiproliferative effects of butyrate. We designed a regimen consisting of intermittent or pulse therapy in which butyrate was administered for 4 days followed by 10 to 24 days with no drug exposure. This pulse regimen induced fetal globin gene expression in 9 of 11 patients. The mean Hb F in this group increased from 7.2% to 21.0% (P < .002) after intermittent butyrate therapy for a mean duration of 29.9 weeks. This was associated with a parallel increase in the number of F cells and F reticulocytes. The total hemoglobin levels also increased from a mean of 7.8 g/dL to a mean of 8.8 g/dL (P < .006). The increased levels of Hb F were sustained in all responders, including 1 patient who has been on pulse butyrate therapy for more than 28 months. This regimen, which resulted in a marked and sustained increase in Hb F levels in more than two thirds of the adult sickle cell patients enrolled in this study, was well tolerated without adverse side effects. These encouraging results require confirmation along with an appropriate evaluation of clinical outcomes in a larger number of patients with sickle cell disease.
Sickle cell disease was first described as a distinct clinical entity in the early part of this century. Identification of its etiology as a single amino acid substitution of the β-globin chain of the hemoglobin molecule marked the beginning of the era of molecular medicine.1,2 Numerous investigations followed to elucidate the physico-chemical properties of sickle hemoglobin, and attempts were made to develop agents that would alter the mutant molecule to decrease red blood cell sickling in vivo.2–4 However, these approaches did not lead to the development of effective therapies that impacted on the course of the disease. It was also recognized early on that the disease does not become clinically manifest until fetal hemoglobin (Hb F) production is developmentally suppressed during infancy.1,2,5,6 When Hb F was added to sickle hemoglobin in vitro at concentrations exceeding 20%, polymerization of sickle hemoglobin was inhibited.2,5,7 Such high concentrations of Hb F were also noted to ameliorate the clinical complications of sickle cell disease in vivo.2,4–13 The large study of the natural history of this disease known as the Cooperative Study of Sickle Cell Disease (CSSCD) demonstrated that smaller increments in Hb F have some ameliorating effects on the clinical manifestations of the disease and that levels greater than 9% could prevent its early mortality.13 These observations were largely responsible for the shift of therapeutic emphasis to strategies to increase the level of Hb F in vivo in patients with sickle cell disease.
Initial efforts to stimulate Hb F production in sickle cell disease used S-phase–specific chemotherapeutic agents such as 5-azacytidine and hydroxyurea.2,14–25 The Multicenter Study of Hydroxyurea (MSH), a recently completed randomized, placebo-controlled, multicenter clinical trial demonstrated a significant reduction in the incidence of vaso-occlusive crises and acute chest syndrome in patients who received hydroxyurea.22 The Hb F levels increased in approximately half the adult subjects who received the drug from a mean of 5.1% to a mean of 8.6%.23 Although mean Hb F levels of 15% to 18% have been achieved in children with sickle cell disease, such levels were achieved only in about 25% of adult subjects receiving the same therapy in the MSH.23–27 Because of the modest increases in Hb F levels that were seen in adult patients treated with hydroxyurea and because of lingering concerns about potentially serious side effects of chronic administration of chemotherapeutic agents, development of safe and effective inducers of fetal globin gene expression has been encouraged.26
Reports from several laboratories had demonstrated that butyrate and other short chain fatty acids can selectively stimulate embryonic or fetal globin gene expression in a variety of experimental systems.28–36 Butyrate was shown to increase the expression of the embryonic ρ-globin gene in adult chickens. However, this activation of the embryonic ρ-globin gene required pretreatment with 5-azacytidine.37 In utero infusions of butyrate were later shown to delay the developmental switch from γ- to β-globin gene expression in sheep fetuses.29 Other studies demonstrated that butyrate can induce γ-globin gene expression in adult baboons and increase the expression of the human γ-globin gene in transgenic mice.31,32 Transfection experiments also showed that butyrate and other short chain fatty acids selectively stimulate the activity of the human γ-globin gene promoter in reporter assays.33,35 These laboratory observations provided a rational basis for the clinical studies of butyrate that followed.
A short-term safety trial of arginine butyrate in 6 patients with β-globin disorders demonstrated induction of γ-globin mRNA, γ-globin chain synthesis, and F reticulocytes in all 6 patients after 2 to 3 weeks of therapy.38 A hematologic response consisting of more than 4 g/dL increase in total hemoglobin was seen in 1 patient homozygous for Hb Lepore who received extended, lower-dose therapy for 7 weeks.38 In a second 10-week study by Sher et al,39 arginine butyrate was administered to 5 patients with sickle cell disease and 5 patients with β-thalassemia at high doses (2,000 mg/kg/d) by continuous infusion (24 h/d, 6 d/wk). Increases in Hb F levels were observed in three of the 5 patients with sickle cell disease, with the mean Hb F increasing from 5.1% at baseline to a peak of 9.6%.39 In other studies, sodium 4-phenylbutyrate was also shown to increase Hb F in one third of sickle cell patients and total hemoglobin in 4 of 8 nontransfused patients with β-thalas-semia.40,41
In addition to the activation of fetal globin gene expression, butyrates are also known to induce growth arrest in the G1 phase of the cell cycle.42–46 Two studies in baboons demonstrated that continuous administration of high doses of butyrate (1,000 mg/kg) or prolonged administration of one of its metabolites, acetate, can result in loss of initial Hb F responses.47,48 In addition, our own experience with butyrate described in this report and the published experience of Sher et al39 demonstrated that the initial Hb F response to butyrate may be lost after extended periods of continuous therapy in patients with sickle cell disease. We hypothesized that the decrease in the high levels of Hb F after butyrate therapy may be a result of cumulative antiproliferative effects. Thus, we designed and evaluated an alternate regimen of therapy consisting of pulsed administration of butyrate to avoid inhibition of growth of erythroid cells from prolonged exposure to butyrate. More than two thirds of the adult patients with sickle cell disease that were enrolled on this regimen demonstrated a marked and sustained activation of Hb F production.
MATERIALS AND METHODS
Patients and treatment
Fifteen patients with sickle cell disease were enrolled in our butyrate studies, 6 on a pilot weekly regimen, and 11 on the pulse regimen. Two of the patients were initially enrolled on the weekly regimen and later re-enrolled on the pulse regimen. There were 6 male and 9 female patients with an age range of 17 to 55 years. These studies were approved by the Food and Drug Administration and by the Institutional Review Boards of the Mount Sinai School of Medicine and the Boston University School of Medicine (Boston, MA). Informed consent was provided according to the Declaration of Helsinki. Only patients with moderate to severe sickle cell disease, defined by three or more hospitalizations per year for sickling complications, were eligible for enrollment in these studies. None of the patients was on a chronic transfusion regimen during the 3 months preceding enrollment. The diagnosis of homozygous sickle cell disease was confirmed in all patients by hemoglobin electrophoresis on cellulose acetate. In the 1 patient with sickle β°-thalassemia, the diagnosis was confirmed by molecular analysis.
Arginine butyrate was prepared as a sterile, nonpyrogenic 5% or 10% solution. The drug was first infused through a central venous line over 8 to 12 hours at rates of 22 to 45 mg/kg/h, usually at night. If no side effects occurred, the infusion rate was increased gradually to 65 to 75 mg/kg/h. Patients who were enrolled on the weekly regimen were infused with arginine butyrate 5 nights per week in escalating doses that ranged from 166 to 666 mg of butyric acid/kg/d. The mean duration of therapy in patients enrolled in the weekly regimen was 15 weeks (range, 3 to 72 weeks). The patients who were enrolled on the pulse regimen received arginine butyrate infusions for 6 to 12 h/d to deliver between 250 and 500 mg/kg/d, with a usual maintenance dose of 500 mg/d. The drug was administered for 4 days, followed by 10 to 24 days with no drug exposure before the next cycle of treatment. The mean duration of therapy in the 11 patients enrolled on the pulse regimen was 30 weeks (range, 7 to 112 weeks). All patients enrolled on either regimens received ferrous sulfate (300 mg/d) on the days of butyrate infusions and 1 mg/d of folic acid for the duration of the study. Chemistry panels, complete blood counts, and reticulocyte counts were monitored at least once weekly during the week of butyrate therapy. Serum ferritin levels were determined at least once monthly during enrollment in this study.
Analysis of F reticulocytes and Hb F levels
The proportions of reticulocytes and mature erythrocytes that contained Hb F (F reticulocytes and F cells) were determined as previously described.19,38 Hb F levels in peripheral blood samples were quantified by electrophoresis on cellulose acetate, followed by scanning densitometry, because our experience and that of others10 showed that this method significantly underestimates high Hb F levels in the range achieved in this study. However, when the Hb F levels were less than 5%, the alkali denaturation method was used to confirm the densitometric data.
RESULTS
The first 6 patients that were enrolled in this study received butyrate infusions on a weekly basis. The Hb F levels increased in 3 of the 6 patients (Fig 1). The Hb F response of a very informative patient from this group who was enrolled in the weekly regimen over a period of 16 months is presented in detail in Fig 2. Within the first 4 months of butyrate therapy, the Hb F increased from 3.5% to 23%. The total Hb level increased in parallel with the Hb F level during therapy from 7.5 to 10 g/dL. The patient elected to withdraw from the study at that time and her Hb F level began to decrease. She requested re-enrollment in the study 2 months later and her Hb F level increased again while on butyrate to levels above 20%. However, while on weekly therapy (except for a 2-week interruption during the holidays), her Hb F levels gradually decreased to levels that were only slightly higher than her baseline.
Fig 1.

A bar graph that shows the initial Hb F levels (▨) and the increment in Hb F during therapy (■) in the 6 patients enrolled on the weekly butyrate regimen.
Fig 2.
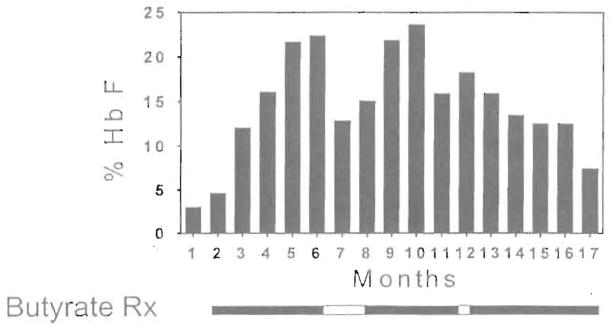
Bar graph illustrating the Hb F response in a patient receiving weekly butyrate therapy. Each vertical bar represents the average of approximately eight determinations of hemoglobin F per month. The periods of butyrate therapy are depicted as solid horizontal bars below the vertical bar graph and the periods of interruption of therapy are depicted as open horizontal bars.
The loss of the Hb F response during weekly therapy in this patient and the reported loss of responses in sickle cell patients studied by Sher et al39 were similar to the loss of Hb F response seen in baboons after high-dose and prolonged administration of butyrate.47,48 This suggested the possibility that cumulative toxicity resulting from continuous exposure to butyrate may be responsible for this loss of response. A pulse regimen that was designed to allow recovery from the antiproliferative effects of butyrate was investigated in 11 patients. The characteristics of these patients and their responses are summarized in Table 1. The Hb F levels of the 11 patients enrolled on this regimen increased from a mean of 7.2% at baseline to a mean of 21.0% during therapy (P = .002, paired t-test; Fig 3). The details of the Hb F response to butyrate in 3 of the 9 responsive patients are shown in Fig 4. Interestingly, the 2 patients who did not respond to butyrate had baseline Hb F levels less than 2%, whereas all 9 responders had baseline Hb F levels of 2% or more. The same was true of the patients who were enrolled on the weekly therapy as shown in Fig 1. The F reticulocytes levels increased from a mean of 14.0% before therapy to a mean of 26.2% during therapy (P = .003, paired t-test). The F cells also increased from a mean of 27.5% before therapy to a mean of 42.5% during therapy (P = .0004, paired t-test). There was a very significant correlation between the baseline Hb F levels and the peak Hb F levels during therapy (r = .87 with P = .002). An equally strong correlation was found between baseline and peak F cells (r = .93 with P < .0001) and F reticulocyte. The Hb level increased in approximately half of the patients from a mean of 7.8 g/dL before therapy to a mean of 8.8 g/dL during therapy (P = .006, paired t-test). However, there was no correlation between the baseline and the peak Hb levels achieved during therapy (r = .49 with P = .12). The absence of an increase of the total Hb levels in some patients may be related to partial inhibition of the proliferation of their erythroid progenitors in vivo. The initial HbF responses and the total hemoglobin responses were maintained after prolonged administration in all patients who received pulse butyrate. In 1 patient, the Hb F levels have been maintained in the 20% range for more than 28 months. Interestingly, of the 5 patients who failed to respond to the weekly or the intermittent regimen, 3 received hydroxyurea either before or after the completion of the butyrate study. All 3 had excellent Hb F responses to hydroxyurea, as shown in Fig 5.
Table 1.
Hematologic Responses to Pulse Butyrate Therapy
| Patient No. | Age (yr)/Sex | Time (wk) Treated | % F Reticulocytes
|
% F Cells
|
% Hb F
|
Total Hb (g/dL)
|
||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pre | Therapy | Pre | Therapy | Pre | Therapy | Pre | Therapy | |||
| 1 | 43/F | 13 | 18 | 29 | 32 | 57 | 3 | 25 | 7.0 | 8.7 |
| 2 | 55/F | 112 | 18 | 36 | 42 | 61 | 6.5 | 29 | 7.5 | 7.5 |
| 3 | 30/F | 20 | 19 | 46 | 53 | 73 | 8 | 33 | 7.7 | 9.8 |
| 4 | 35/F | 7 | 5 | 9 | 6 | 11 | 0.5 | 1.5 | 8.7 | 9.1 |
| 5 | 18/F | 9 | 1 | 7 | 10 | 15 | 1.2 | 1.8 | 8.2 | 8.2 |
| 6 | 30/M | 20 | 13 | 29 | 23 | 59 | 7.0 | 22 | 7.0 | 9.5 |
| 7 | 31/F | 52 | 26 | 35 | 58 | 70 | 22 | 35 | 6.8 | 7.8 |
| 8 | 24/F | 36 | 18 | 29 | 25 | 34 | 6.1 | 17 | 7.6 | 8.9 |
| 9 | 31/M | 14 | 10 | 17 | 10 | 18 | 2 | 17 | 7.7 | 9.5 |
| 10 | 28/F | 28 | 5 | 9 | 7 | 19 | 2 | 12.5 | 7.5 | 7.5 |
| 11 | 25/M | 18 | 21 | 42 | 37 | 50 | 20 | 37 | 10.1 | 10.2 |
| Mean | 31.8 | 29.9 | 14.0 | 26.2 | 27.5 | 42.5 | 7.1 | 21.0 | 7.8 | 8.8 |
| P value, paired t-test | .003 | .0004 | .002 | .006 | ||||||
Fig 3.
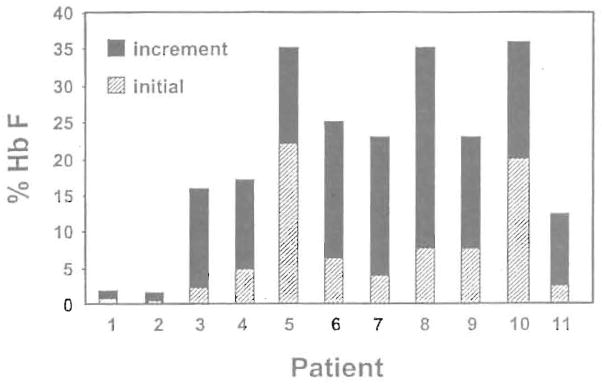
A bar graph that shows the initial Hb F levels (▨) and the increment in Hb F during therapy (■) in the 11 patients enrolled on the pulse butyrate regimen.
Fig 4.

Detailed representation of the Hb F responses to pulse butyrate therapy in 3 adult patients with sickle cell disease. Each bar represents the average of two fetal hemoglobin levels obtained during the week of therapy.
Fig 5.

A bar graph that shows the initial Hb F levels (▨) and the increment in Hb F during therapy (■) in the 3 butyrate nonresponsive patients who were treated with hydroxyurea either before or after the end of their butyrate therapy.
No symptoms attributed to arginine butyrate were observed at infusion rates of 65 to 75 mg/kg/h at the 500 mg/kg/d dose administered in this study. Antiemetics were usually not required, and some patients reported an increase, rather than a decrease, in their appetite while receiving the drug. No laboratory abnormalities were noted after frequent monitoring of liver function tests, serum creatinine levels, electrolyte levels, and coagulation assays. Blood urea nitrogen (BUN) levels were frequently elevated during the infusions, presumably due to the metabolic conversion of arginine to urea. These high BUN levels always decreased within a few hours of completing the infusions. No significant changes were seen in the white blood cell (WBC) counts, platelet counts, mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), and mean corpuscular hemoglobin concentration (MCHC) during the course of butyrate therapy. No adverse side effects were observed in greater than 230 patient-weeks of observation. Interestingly, despite iron supplementation, the serum ferritin levels did not increase in any patient, whereas a decrease was seen in some patients.
DISCUSSION
A number of clinical and experimental observations support the hypothesis that high levels of Hb F can reduce sickling and ameliorate the clinical severity of sickle cell disease.2,4–13 The observations that patients from the eastern provinces of Saudi Arabia and southern India with levels of Hb F greater than 20% generally have a very mild clinical course provided a compelling justification for therapeutic attempts to increase Hb F in adult life.8–11 This was further supported by extensive data collected from the multicenter CSSCD that showed a clinical benefit associated with any increase in Hb F with no threshold level that had to be exceeded before these benefits are realized.13 Nonetheless, the complete amelioration of sickling complications may require levels of Hb F in excess of 20%.12 The MSH represents a landmark clinical trial that demonstrated, for the first time, the clinical efficacy of an agent that stimulates Hb F production in patients with sickle cell disease.22 Yet, the mean increase in Hb F in the adult subject was only 4%.23 Even more surprisingly, there was no direct correlation between the Hb F responses and the clinical benefits in the patients who received hydroxyurea.49 These findings suggest that hydroxyurea may have additional beneficial effects that are not related to Hb F induction.23,49–51
Study of newborn infants born to diabetic mothers demonstrated that high plasma levels of α-amino-n-butyric acid during gestation can be associated with a delay in the switch from fetal to adult hemoglobin production.52 The Hb F inducing activity of butyrate was first demonstrated in a short-term, 2-week dose-evaluation trial of arginine butyrate in patients with hemoglobin disorders.38 However, a subsequent 10-week trial in which patients received high doses of butyrate for 24 h/d, 6 d/wk (with no iron supplementation) showed a significant but nonsustained increase in fetal hemoglobin in 3 of 5 patients with sickle cell disease.39 We also observed a decrease in Hb F levels after several months of weekly butyrate administration (at a much lower dose) in the patient whose course is shown in Fig 2. This decrease in Hb F after prolonged exposure to butyrate, considered in the context of the well-known growth-inhibitory activity of butyrate, suggested that an intermittent dosing schedule may prevent toxicity and allow more proliferation of the erythroid cells in which Hb F is induced. Thus, a pulse-dosing regimen in which butyrate was administered for 4 days followed by a variable period of no drug exposure was designed and tested. In addition, because some prior studies had suggested that pharmacologic induction of Hb F may be enhanced by iron supplements,20,21 we elected to administer ferrous sulfate to all patients on the days of butyrate infusion. Even though the majority of patients responded to this regimen, we cannot be certain that iron was necessary for this response, because this study was not designed to answer this question.
It is of interest to note that all 5 patients who did not respond to weekly or pulse butyrate in our studies had low baseline Hb F levels (<2%), whereas all 11 responders had higher baseline Hb F levels (≥2%). This striking correlation between baseline Hb F and response to butyrate suggests that induction of γ-globin expression by butyrate may require the fetal globin genes to be in a partially active or accessible state at the time of exposure to butyrate. This is consistent with experimental findings in chicken in which the induction of embryonic globin genes required prior exposure to 5-azacytidine.37 Butyrate and related compounds are believed to stimulate Hb F production at least in part by inhibition of histone deacetylase.53 Histone deacetylases have recently been shown to be recruited to specific DNA sequences by binding to transcription complexes through their interaction with transcriptional coactivators and corepressors.54,55 Thus, it is tempting to speculate that the butyrate effect requires transcriptionally active fetal globin genes whose regulatory elements are occupied by transcription factors. These transcription factors may be responsible for recruiting histone deacetylase and modulating the chromatin that surrounds the fetal globin genes. Such a mechanism may also account for the specificity of the enhancing effect of butyrate on fetal globin gene expression.
If these correlations between baseline Hb F and response are confirmed, the baseline Hb F levels may serve as a convenient predictor of response to butyrate therapy. The mean level of Hb F in patients enrolled in the CSSCD was 6.4%.13 In the MSH study in which enrollment was limited to patients with moderate to severe disease, the mean Hb F level at baseline was 5.1%, whereas 75% of patients had a baseline Hb F level greater than 2.5%.23 These Hb F levels are similar to the Hb F levels of patients enrolled in our study (mean, 7.1%). Therefore, if the correlation between initial Hb F and response to butyrate is confirmed, the two thirds of patients with sickle cell disease who have baseline Hb F levels greater than 2% may be responsive to treatment with arginine butyrate. However, these predictions require the assessment of butyrate-responsiveness in a much larger number of patients.
It should be noted that the aim of our study was to identify an effective dose-regimen of arginine butyrate rather than to formally compare two dose regimens. The two groups of patients that were enrolled on the weekly or the pulse regimens differed significantly in multiple respects. Thus, we cannot conclude that the pulse butyrate regimen is more effective in inducing Hb F than the weekly regimen. Interestingly, the 2 patients who were treated with both regimens had identical Hb F responses to both. However, a loss of Hb F response has not been seen in any patient enrolled on the pulse regimen. In addition, the pulse regimen is more acceptable to patients and requires significantly less resources than the weekly regimen. Thus, it would be difficult to justify the weekly administration of butyrate to sickle cell patients in future clinical studies.
The studies described in this report do not allow us to make statements about the relative efficacy of butyrate and hydroxyurea in inducing Hb F levels in patients with sickle cell disease. Nonetheless, a few important differences are apparent when the findings of our study are compared with those of the MSH and other hydroxyurea studies. First, the Hb F responses in patients treated with hydroxyurea usually require several months of treatment and in some patients may not peak before 2 years.27 In contrast, some increase in Hb F is usually seen within days of exposure to butyrate in the majority of responsive patients, and the peak is achieved within a few (3 to 4) months. This rapid response may help avoid long courses of unnecessary treatment in butyrate nonresponsive patients if these observations are confirmed in larger studies. Second, in the MSH, there was no correlation between the initial Hb F level and the response to therapy.23 However, in a recent study of hydroxyurea in children with sickle cell disease, a strong correlation was noted between the initial Hb F and the magnitude of the Hb F response. Similarly, the response to butyrate correlates with the initial Hb F and may require some baseline activity of the fetal globin genes.27 Third, all three butyrate nonresponsive patients who received hydroxyurea had excellent Hb F responses. This demonstrates that failure to respond to one Hb F inducing agent does not necessarily predict failure to respond to another and confirms that the two agents increase Hb F by different mechanisms. This also raises the possibility that combination or sequential administration of hydroxyurea followed by butyrate may be effective in patients who do not achieve a sufficient Hb F response to either agent alone.
We noted a marked decrease in the number of days of hospitalization for sickle cell disease-related complications during butyrate therapy in the 5 responsive patients who received treatment for 6 months or longer. In contrast, no decrease in hospital days was noted in the 2 patients whose Hb F levels did not increase during therapy. Two responsive patients who could not be transfused due to extensive alloantibodies underwent successful total hip replacements while on butyrate without prior transfusions. However, it should be noted that the study described in this report was not a randomized, placebo-controlled clinical trial designed to assess clinical end-points. Thus, despite the favorable observations noted here, definitive conclusions about clinical efficacy should be withheld until an appropriately designed larger clinical trial is conducted.
The pulse butyrate regimen in which therapy is limited to 4 days every 4 weeks (that can be administered in an outpatient setting) is acceptable to many patients with severe sickle cell disease who would otherwise require frequent hospitalizations for complications of sickle cell disease. Nonetheless, this regimen still requires prolonged intravenous administration of arginine butyrate through a central venous line with all the associated risks, costs, and inconvenience. Although home therapy has been used for more than 3 years in selected patients with thalassemia, this option is not suitable for all patients. Orally bioavailable compounds such as the recently identified fatty acid derivatives have the potential to make a significant impact on sickle cell disease.56,57
In summary, the studies described in this report have defined a therapeutic approach that results in sustained induction of Hb F to levels that have been demonstrated to result in significant amelioration of the clinical complications of sickle cell disease. The marked responses that were seen in the patients enrolled in these studies demonstrate that developmentally suppressed fetal globin genes can be reactivated to a very significant level by butyrate exposure in adult patients with sickle cell disease. These encouraging results should be confirmed in a larger study that will also include a prospective evaluation of the effects of pulse butyrate therapy on clinical outcome.
Acknowledgments
Supported by Grant No. FDR-001084 from the Food and Drug Administration, by gifts from Hannah and Lawrence Langsam, by the W.H. Roberts Foundation, and by Grants No. HL-54184, HL-37118, HL-15157, RR-00523, and RR-0007 to the General Clinical Research Center of Mount Sinai School of Medicine from the National Institutes of Health.
This manuscript is dedicated in memoriam to Wayne Turner. The authors appreciate the expert technical assistance of Abbie Mays and Shirley Purvis.
References
- 1.Conley CL. Sickle cell anemia—The first molecular disease. In: Wintrobe MM, editor. Blood, Pure and Eloquent: A Story of Discovery, of People, and of Ideas. New York, NY: McGraw-Hill; 1980. p. 319. [Google Scholar]
- 2.Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337:762. doi: 10.1056/NEJM199709113371107. [DOI] [PubMed] [Google Scholar]
- 3.Ingram VM. A specific chemical difference between the globins of normal human and sickle cell anemia haemoglobin. Nature. 1956;178:792. doi: 10.1038/178792a0. [DOI] [PubMed] [Google Scholar]
- 4.Brittenham GM, Schechter AN, Noguchi CT. Hemoglobin S polymerization. Primary determinant of the hemolytic and clinical severity of the sickling syndromes. Blood. 1985;65:183. [PubMed] [Google Scholar]
- 5.Wood WG, Bunch C, Kelly S, Gunn Y, Breckon G. Control of haemoglobin switching by a developmental clock. Nature. 1985;313:320. doi: 10.1038/313320a0. [DOI] [PubMed] [Google Scholar]
- 6.Stamatoyannopoulos JA, Nienhuis AW. Therapeutic approaches to hemoglobin switching in treatment of the hemoglobinopathies. Annu Rev Med. 1992;43:487. doi: 10.1146/annurev.me.43.020192.002433. [DOI] [PubMed] [Google Scholar]
- 7.Nagel RL, Bookchin RM. Structural bases of the inhibitory effects of Hb F and A2 on the polymerization of Hb S. Proc Natl Acad Sci USA. 1979;76:670. doi: 10.1073/pnas.76.2.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perrine RP. Benign sickle cell anemia. Lancet. 1972;2:1163. doi: 10.1016/s0140-6736(72)92592-5. [DOI] [PubMed] [Google Scholar]
- 9.Brittenham G, Lozoff B, Harris JW, Sharma VS, Narasimhan S. Sickle cell anemia and trait in a population in southern India. Am J Hematol. 1977;2:25. doi: 10.1002/ajh.2830020104. [DOI] [PubMed] [Google Scholar]
- 10.Wood WG, Pembrey ME, Serjeant GR, Perrine RP, Weatherall DJ. Hb F synthesis in sickle cell anaemia: A comparison of Saudi Arab cases with those of African origin. Br J Haematol. 1980;45:431. doi: 10.1111/j.1365-2141.1980.tb07163.x. [DOI] [PubMed] [Google Scholar]
- 11.Perrine RP. Natural history of sickle cell anemia in Saudi Arabs. Ann Intern Med. 1978;88:1. doi: 10.7326/0003-4819-88-1-1. [DOI] [PubMed] [Google Scholar]
- 12.Noguchi CT, Rodgers GP, Serjeant G, Schechter AN. Levels of fetal hemoglobin necessary for treatment of sickle cell disease. N Engl J Med. 1988;318:96. doi: 10.1056/NEJM198801143180207. [DOI] [PubMed] [Google Scholar]
- 13.Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in sickle cell disease: Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 14.DeSimone J, Heller P, Hall L, Zwiers D. 5-Azacytidine stimulates fetal hemoglobin synthesis in anemic baboons. Proc Natl Acad Sci USA. 1982;79:4428. doi: 10.1073/pnas.79.14.4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ley TJ, DeSimone J, Anagnou NP. 5-Azacytidine selectively increases γ-globin synthesis in a patient with β+ thalassemia. N Engl J Med. 1982;307:1469. doi: 10.1056/NEJM198212093072401. [DOI] [PubMed] [Google Scholar]
- 16.Ley TJ, DeSimone J, Noguchi CT. 5-Azacytidine increases γ-globin synthesis and reduces the proportion of dense cells in patients with sickle cell anemia. Blood. 1983;62:370. [PubMed] [Google Scholar]
- 17.McDonagh KT, Dover GJ, Donahue RE, Nathan DG. Hydroxyurea-induced Hb F production in anemic primates: Augmentation by erythropoietin, growth factors, and sodium butyrate. Exp Hematol. 1992;20:1156. [PubMed] [Google Scholar]
- 18.Dover GJ, Humphries RK, Moore JG. Hydroxyurea induction of hemoglobin F production in sickle cell disease: Relationship between cytotoxicity and F-cell production. Blood. 1986;67:735. [PubMed] [Google Scholar]
- 19.Charache S, Dover GJ, Moyer MA, Moore JW. Hydroxyurea-induced augmentation of fetal hemoglobin production in patients with sickle cell anemia. Blood. 1987;69:109. [PubMed] [Google Scholar]
- 20.Rodgers GP, Dover GJ, Uyesaka N, Noguchi CT, Schechter AN, Nienhuis AW. Augmentation by erythropoietin of the fetal-hemoglobin response to hydroxyurea in sickle cell disease. N Engl J Med. 1993;328:73. doi: 10.1056/NEJM199301143280201. [DOI] [PubMed] [Google Scholar]
- 21.Nagel RL, Vichinsky E, Shah M. F-reticulocyte response in sickle cell anemia treated with recombinant human erythropoietin: A double blind study. Blood. 1993;81:9. [PubMed] [Google Scholar]
- 22.Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, McMahon RP, Bonds DR. Multicenter study of hydroxyurea in sickle cell anemia: Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. N Engl J Med. 1995;332:1317. doi: 10.1056/NEJM199505183322001. [DOI] [PubMed] [Google Scholar]
- 23.Steinberg MH, Lu ZH, Barton FB, Terrin ML, Charache S, Dover GJ Multicenter Study of Hydroxyurea. Fetal hemoglobin in sickle cell anemia: Determinants of response to hydroxyurea. Blood. 1997;89:1078. [PubMed] [Google Scholar]
- 24.Ohene-Frempong HK, Bulgarelli W, Schursky HM. Hydroxyurea increases Hb F production in children with sickle cell disease. Blood. 1993;82:3258. [Google Scholar]
- 25.Ferster A, Vermylen C, Cornu G. Hydroxyurea for treatment of severe sickle cell anemia: A pediatric trial. Blood. 1996;88:1960. [PubMed] [Google Scholar]
- 26.Schechter AN, Rodgers G. Hydroxyurea in sickle cell disease. N Engl J Med. 1996;334:333. doi: 10.1056/NEJM199602013340516. [DOI] [PubMed] [Google Scholar]
- 27.Maier-Redelsperger M, deMontalembert M, Flahault A, Neonato MG, Ducrocq R, Masson M-P, Girot R, Elion J. Fetal hemoglobin and F-cell responses to long-term hydroxyurea treatment in young sickle cell patients. Blood. 1998;91:4472. [PubMed] [Google Scholar]
- 28.Partington GA, Yarwood NJ, Rutherford TR. Human globin gene transcription in injected Xenopus oocytes: Enhancement by sodium butyrate. EMBO J. 1984;3:2787. doi: 10.1002/j.1460-2075.1984.tb02210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perrine SP, Rudolph A, Faller DV. Butyrate infusions in the ovine fetus delay the biologic clock for globin gene switching. Proc Natl Acad Sci USA. 1988;85:8540. doi: 10.1073/pnas.85.22.8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perrine SP, Miller BA, Faller DV, Cohen RA, Vichinsky EP, Hurst D, Lubin BH, Papayannopoulou T. Sodium butyrate enhances fetal globin gene expression in erythroid progenitors of patients with Hb SS and β thalassemia. Blood. 1989;74:454. [PubMed] [Google Scholar]
- 31.Constantoulakis P, Knitter G, Stamatoyannopoulous G. On the induction of fetal hemoglobin by butyrates: in vivo and in vitro studies with sodium butyrate and comparison of combination treatments with 5-azaC and araC. Blood. 1989;74:1963. [PubMed] [Google Scholar]
- 32.Blau A, Constantoulakis P, Shaw CM, Stamatoyannopoulos G. Fetal hemoglobin induction with butyric acid: Efficacy and toxicity. Blood. 1993;81:529. [PubMed] [Google Scholar]
- 33.Safaya S, Ibrahim A, Reider RF. Augmentation of γ globin gene promoter activity by carboxylic acids and components of the human β-globin locus control region. Blood. 1994;84:3929. [PubMed] [Google Scholar]
- 34.Liakopoulou E, Blau C, Li Q. Stimulation of fetal hemoglobin production by short chain fatty acids. Blood. 1995;86:3227. [PubMed] [Google Scholar]
- 35.Perrine SP, Dover GH, Daftari P, Walsh CT, Jin YX, Mays A, Faller DV. Isobutyramide, an orally bioavailable butyrate analogue, stimulates fetal globin gene expression in vitro and in vivo. Br J Haematol. 1994;88:555. doi: 10.1111/j.1365-2141.1994.tb05073.x. [DOI] [PubMed] [Google Scholar]
- 36.Burns LJ, Glauber JG, Ginder GD. Butyrate induces selective transcriptional activation of a hypomethylated embryonic globin gene in adult erythroid cells. Blood. 1988;72:1536. [PubMed] [Google Scholar]
- 37.Ginder GD, Whitters MJ, Pohlman JK. Activation of a chicken embryonic globin gene in adult erythroid cells by 5-azacytidine and sodium butyrate. Proc Natl Acad Sci USA. 1984;81:3954. doi: 10.1073/pnas.81.13.3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perrine SP, Ginder GD, Faller DV, Dover GH, Ikuta T, Witkowska HE, Cai SP, Vichinsky EP, Olivieri NF. A short-term trial of butyrate to stimulate fetal-globin-gene expression in the β-globin disorders. N Engl J Med. 1993;328:81. doi: 10.1056/NEJM199301143280202. [DOI] [PubMed] [Google Scholar]
- 39.Sher GD, Entsuah B, Ginder G, Dover G, Berkovitch M, Lewis N, Perrine S, Olivieri NF. Intravenous arginine butyrate increases γ-globin mRNA, F-reticulocytes and hemoglobin F in patients with sickle cell disease. 19th Annual Meeting of the National Sickle Cell Disease Program; 1994. p. 177. [Google Scholar]
- 40.Dover GJ, Brusilow S, Charache S. Induction of fetal hemoglobin production in subjects with sickle cell anemia by oral sodium phenylbutyrate. Blood. 1994;84:339. [PubMed] [Google Scholar]
- 41.Collins AF, Pearson HA, Giardina P, McDonagh KT, Brusilow SW, Dover GJ. Oral sodium phenylbutyrate therapy in homozygous β thalassemia: A clinical trial. Blood. 1995;85:43. [PubMed] [Google Scholar]
- 42.Wintersberger E, Mudrak I, Wintersberger U. Butyrate inhibits mouse fibroblasts at a control point in the G1 phase. J Cell Biochem. 1983;21:239. doi: 10.1002/jcb.240210306. [DOI] [PubMed] [Google Scholar]
- 43.Toscani A, Soprano DR, Soprano KJ. Molecular analysis of sodium butyrate induced growth arrest. Oncogene Res. 1988;3:223. [PubMed] [Google Scholar]
- 44.Charollais RH, Buguet C, Mester J. Butyrate blocks the accumulation of CDC2 mRNA in late G1 phase but inhibits both the early and late G1 progression in chemically transformed mouse fibroblasts BP-A31. J Cell Physiol. 1990;145:46. doi: 10.1002/jcp.1041450108. [DOI] [PubMed] [Google Scholar]
- 45.Fibach E, Prosmanne J, Rodgers GP, Samid D. Enhanced fetal hemoglobin production by phenylacetate and 4 phenylbutyrate in erythroid precursors derived from normal donors and patients with sickle cell anemia and β-thalassemia. Blood. 1993;82:2203. [PubMed] [Google Scholar]
- 46.Vaziri C, Stice LL, Faller DV. Butyrate-induced G1 arrest results from p21-independent disruption of retinoblastoma-mediated signals. Cell Growth Differ. 1998;9:465. [PubMed] [Google Scholar]
- 47.Faller DV, Perrine SP. Butyrate in the treatment of sickle cell disease and β thalassemia. Curr Opin Hematol. 1995;2:109. doi: 10.1097/00062752-199502020-00002. [DOI] [PubMed] [Google Scholar]
- 48.Stamatoyannopoulos G, Blau CA, Nakamoto B, Josephson B, Li Q, Liakopoulou E, Pace B, Papayannopoulou T, Brusilow SW, Dover G. Fetal hemoglobin induction by acetate, a product of butyrate catabolism. Blood. 1994;84:3198. [PubMed] [Google Scholar]
- 49.Charache S, Barton FB, Moore RD. Hydroxyurea and sickle cell anemia: Clinical utility of a myelosuppressive “switching” agent: The Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Medicine. 1996;75:300. doi: 10.1097/00005792-199611000-00002. [DOI] [PubMed] [Google Scholar]
- 50.Orringer EP, Blythe DS, Johnson AE, Phillips GJ, Dover GJ, Parker JC. Effects of hydroxyurea on hemoglobin F and water content in the red blood cells of dogs and of patients with sickle cell anemia. Blood. 1991;78:212. [PubMed] [Google Scholar]
- 51.Ballas SK, Dover GJ, Charache S. Effect of hydroxyurea on the rheological properties of sickle erythrocytes in vivo. Am J Hematol. 1989;32:104. doi: 10.1002/ajh.2830320206. [DOI] [PubMed] [Google Scholar]
- 52.Perrine SP, Greene MF, Faller DV. Delay in the fetal globin switch in infants of diabetic mothers. N Engl J Med. 1985;312:334. doi: 10.1056/NEJM198502073120602. [DOI] [PubMed] [Google Scholar]
- 53.McCaffrey PG, Newsome DA, Fibach E, Yoshida M, Su MS. Induction of γ-globin by histone deacetylase inhibitors. Blood. 1997;90:2075. [PubMed] [Google Scholar]
- 54.Heinzel T, Lavinsky RM, Mullen TM, Soderstrom M, Laherty CD, Torchia J, Yang WM, Brard G, Ngo SD, Davie JR, Seto E, Eisenman RN, Rose DW, Glass CK, Rosenfeld MG. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature. 1997;387:43. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- 55.Laherty CD, Yang WM, Sun JM, Davie JR, Seto E, Eisenman RN. Histone deacetylases associated with the mSin3 corepressor mediate mad transciptional repression. Cell. 1997;89:349. doi: 10.1016/s0092-8674(00)80215-9. [DOI] [PubMed] [Google Scholar]
- 56.Torkelson S, White GL, Phipps K. Erythroid progenitor proliferation is stimulated by phenoxyacetic and phenylalkyl acids. Blood Cells Mol Dis. 1996;20:150. doi: 10.1006/bcmd.1996.0022. [DOI] [PubMed] [Google Scholar]
- 57.Boosalis MS, Ikuta T, Pace BS. Abrogation of IL-3 requirements and stimulation of hematopoietic cell proliferation in vitro and in vivo by carboxylic acids. Blood Cells Mol Dis. 1997;23:434. doi: 10.1006/bcmd.1997.0162. [DOI] [PubMed] [Google Scholar]