

Abstract
Y459H and V492E mutations of cytochrome P450 reductase (CYPOR) cause Antley-Bixler Syndrome due to diminished binding of the FAD cofactor. To address whether these mutations impaired the interaction with drug metabolizing CYPs, a bacterial model of human liver expression of CYP1A2 and CYPOR was implemented. Four models were generated: PORnull, PORwt, PORYH, and PORVE, for which equivalent CYP1A2 and CYPOR levels were confirmed, except for PORnull, not containing any CYPOR. The mutant CYPORs were unable to catalyze cytochrome c and MTT reduction, and were unable to support EROD and MROD activities. Activity was restored by the addition of FAD, with V492E having a higher apparent FAD affinity than Y459H. The CYP1A2-activated procarcinogens, 2-aminoanthracene, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, and 2-amino-3-methylimidazo(4,5-f)quinoline, were significantly less mutagenic in PORYH and PORVE models than in PORwt, indicating that CYP1A2, and likely other drug-metabolizing CYPs, are impaired by ABS-related POR mutations as observed in the steroidogenic CYPs.
Keywords: NADPH-cytochrome P450 oxidoreductase, Antley-Bixler Syndrome, POR, Polymorphism, Cytochrome P450, CYP1A2, P450 1A2, Protein-protein interaction, Drug-metabolizing enzymes, Adverse drug reactions
Introduction
Antley-Bixler syndrome (ABS, OMIM#201750) is a genetic disorder characterized by severe midface hypoplasia, humeroradial synostosis, bowing and fracture of femora, and other severe developmental malformations [1]. The disease was initially attributed to fibroblast growth factor receptor 2 (FGFR2) mutations [2]. Miller’s group discovered a correlation between allelic variants of the POR gene (encoding NADPH-cytochrome P450 oxidoreductase or CYPOR, E.C. 1.6.2.4) and disordered steroidogenesis observed in patients with and without other clinical features of ABS [3]. Currently, two types of ABS patients are distinguished, namely Type I ABS patients with FGFR2 mutations, those with the most severe skeletal abnormalities but normal genitalia, and Type II ABS patients with POR mutations, who exhibit less severe skeletal abnormalities, but ambiguous genitalia [4].
The CYPOR protein is the obligate electron donor to 50 microsomal cytochrome P450s (CYPs) that catalyze steroidogenic and xenobiotic reactions [5]. Currently > 25 point mutations of the POR gene have been described in individuals with ABS and/or disordered steroidogenesis [3, 4, 6, 7]. These POR mutations are believed to be causal for ABS by limiting the function of CYPs involved in endogenous substrate metabolism (CYP families 4 to 51), in particular, in steroidogenesis. Miller’s group hypothesized that POR mutations were more common than the relatively low incidence of ABS suggested, and that milder mutations could result in disordered steroidogenesis or increased sensitivity to drugs and environmental toxins [3]. This more frequent incidence of POR polymorphisms seems to be confirmed by a very recent study of Hart et al [8]. In this cohort study several new POR polymorphisms were identified, several leading to amino acid changes, among which the L577P mutation was shown to alter the activity of several drug-metabolizing CYPs. For ABS-affected individuals, this altered drug-metabolizing capacity might be of particular relevance due to the more severe, functionally disruptive POR mutations affecting potential drug therapies for these patients. Recently, reduced metabolism of the pesticide paraquat and the anticancer drug mitomycin C by several ABS-related POR mutants was described [9, 10]. CYPOR bioactivates the latter two compounds directly through a one-electron reductive mechanism. The suggested milder mutations implicate a POR-polymorphism, which might be responsible for variations in effectiveness of anticancer drug therapies, as has been implicated for mitoxantrone, another CYPOR-dependent anti-leukemia prodrug [11]. So far, no data have been reported regarding the effect of ABS-CYPOR mutations on xenobiotic-metabolizing CYPs (families CYP1 to CYP3), which are involved in the majority of biotransformations of drugs and environmental contaminants in humans.
In a former study, we reported on the molecular characterization of two prominent ABS-related POR mutations, encoding the CYPOR alleles Y459H and V492E [12, 13]. The deleterious nature of these mutations on the function of CYPOR was demonstrated to be due to diminished binding of the prosthetic group FAD, essential for the electron-transfer function of CYPOR.
In the present study, we describe the extension of the characterization of these two ABS-related CYPORs, using full-length holoenzymes in combination with human CYP1A2, co-expressed in a bacterial cell-model, BTC [14]. Previously, this simple cell-model demonstrated the co-expression of CYPOR with human CYPs (including CYP1A2), via a bi-plasmid system [15]. The two human proteins are correctly expressed, membrane-anchored and are fully active, reflecting in vivo activities and representing the stoichiometry of the membrane-bound enzyme complex in the endoplasmic reticulum [16]. This is of importance, as exaggerated CYPOR:CYP ratios (≥ 2) lead to irrelevant enzyme kinetics and, thus, cell models containing such relative expression levels are not representative for studying the effects of ABS-CYPOR mutants on CYP activity. Such non-physiological conditions (CYPOR:CYP = 2:1) were used in reconstitution experiments, combining CYP17A1 with different ABS-POR mutants, using N-27 truncated CYPOR proteins [4].
The objective of this present study is to further explore our former findings for Y459H and V492E, using full-length holoenzymes coupled with a physiologically relevant redox-partner, namely CYP1A2, interacting in a relevant stoichiometry and in an appropriate membrane milieu. Although these two ABS-CYPOR variant enzymes seem to hamper the function of the steroidogenic CYP17A1 (in reconstitution experiments, vida supra) substantially, particular ABS-related CYPOR variants have been demonstrated to influence the function of CYPs differentially. The activity of CYP17A1 was significantly reduced when combined with the ABS-POR allele A287P, whilst this mutant protein did not affect the activity of CYP21A2 [17] or CYP19A1 [18]. The effects of the two prominent ABS-POR mutant-alleles, Y459H and V492E, on a different, drug-metabolizing CYP, such as CYP1A2, are presented here as a first approach to evaluating the effects of ABS-related POR gene variants on CYP-mediated xenobiotic metabolism.
Material and Methods
Abbreviations
2AA: 2-aminoanthracene; ABS: Antley-Bixler Syndrome; CYP: cytochrome P450; CYPOR: NADPH-cytochrome P450 oxidoreductase; EROD: ethoxyresorufin-o-dealkylation; FAD: flavin adenine dinucleotide; FGFR2: fibroblast growth factor receptor 2; FMN: flavin mononucleotide; IQ: 2-amino-3-methylimidazo(4,5-f)quinoline; MROD: methoxyresorufin-o-dealkylation; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NNK: 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone;
POR expression plasmids and BTC1A2 cell-models
(See Supplemental Material for detailed methodology.) The expression of the different PORs with CYP1A2 was based on the bi-plasmid coexpression system previously described [15, 19] (Supplemental Fig. 1). Plasmid pLCMhOR [15] contains the POR cDNA, which was modified to contain wild-type POR, the Y459H or the V492E allele, or no cDNA [plasmid pLCM [20]]. The 4 different plasmids were transfected into strain BTC [14], already containing pCWhCYP1A2, the expression plasmid for human CYP1A2 [20, 21], thus creating the humanized bacterial cell-models BTC1A2_PORWT, BTC1A2_PORY459H, BTC1A2_PORV492E and BTC1A2_PORNull, respectively.
CYP1A2- and POR-Detection
(See Supplemental Material for detailed methodology.) Culturing of the different BTC1A2 cell-models for CYP1A2 and POR expression, isolation of membranes and determination of CYP expression was performed as described before [19]. It is important to note that cell lysis for membrane preparation was performed using several short rounds (60 s) of low intensity sonication interspersed with 30 s ice bath submersion between sonication rounds, such that the temperature of the preparation never exceeded 8°C. This proved to be absolutely necessary for successful membrane preparations and “rescue” of the membrane-bound enzyme activities upon addition of FAD.
Membrane proteins (10 μg) of the four BTC1A2 cell-models were separated by SDS-PAGE gel electrophoresis (10% polyacrylamide gel) and either stained with Coomassie blue or electro-transferred to nitrocellulose and further processed for immuno-detection of CYPOR, using a polyclonal antibody purified by Protein-A Sepharose from rabbit serum raised against recombinant rat CYPOR.
Enzyme activities
(See Supplemental Material for detailed methodology.) NADPH-cytochrome c reductase activity of the membranes was performed as described before [15, 19]. When indicated, FAD was added to a final concentration of 10 μM, before starting the reaction with NADPH. Activities represent the mean of at least triplicate determinations.
NADPH-mediated MTT reduction: NADPH-dependent 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction activity of the membranes was determined in micro-plate format and based on the method described by Yim et al. [22]. Seven different FAD concentrations were used (up to 50 μM) and the kinetic parameters kcat and Km were determined by plotting the rates according to the Michaelis-Menten equation. The activity for each FAD concentration was determined at least in triplicate.
Methoxy- and ethoxy-resorufin O-dealkylation: the CYP1A2-mediated dealkylation activities of methoxy- and ethoxyresorfin of membrane preparations were determined in micro-plate format and based on the method described by Burke et al. [23]. Seven different substrate concentrations were used (up to 5 μM for ethoxy- and 2 μM of methoxyresorufin), maintaining the solvent DMSO concentration at 0.2% (v/v) throughout the experiment, preventing interference with CYP1A2 activity [24]. When indicated, FAD was added to a final concentration of 10 μM, and the kinetic parameters kcat and Km were determined by Michaelis-Menten plotting of velocity data. The activity for each substrate concentration was determined at least in triplicate.
Mutagenicity Assays
(See Supplemental Material for detailed methodology.) The whole cell mutagenicity assays were performed as previously described [25]. The CYP1A2-mediated bioactivation is expressed in mutagenic activities [L-Arginine prototrophic (revertant) colonies/nmol compound or in revertant colonies/μmol compound] with the BTC1A2 cell-models. The mutagenicity activity was determined from the linear portion of the dose-response curve, with the determination of mutagenicity of each dose level performed in triplicate.
Statistical analysis
Statical analysis was performed by applying Student t test, using GraphPad software.
Results
Expression of human CYP1A2 with the different POR alleles in the BTC1A2_POR cell-models
Initially, the BTC1A2_POR cell-models were characterized with respect to CYPOR and CYP1A2 expression. Membranes were derived from the four BTC1A2_POR strains and expression levels were evaluated (Table 1). The human CYP was expressed at levels of the same order of magnitude in all POR_strains, ranging from 25 to 48 pmol per mg protein, which represents contents normally obtained for CYP1A2 in this cell model. The expression of POR was verified by SDS PAGE analysis (Supplemental Fig. 2), which showed a band of around the expected size (77 kD) for CYPOR present in all BTC1A2-derived membranes, except for the PORNull strain. CYPOR expression was further analyzed by immuno-detection (Fig. 1). Several immuno-reactive proteins were detected, but signals can easily be distinguished between non-specific (E. coli, Null-lane) and specific signals (control lane). The wild type, Y459H, and V492E CYPOR-membranes showed, in addition to the expected full-length protein, a smaller protein that we have previously observed when expressing CYPOR in E. coli [12]. Although the two mutant proteins showed only background cytochrome c reductase activity (Table 1), Western-blot analysis indicated equal expression levels of the Y459H and the V492E CYPOR variant proteins, when compared with wild-type CYPOR (Fig. 1). This indicated that the mutations were not detrimental for protein expression and stability in the BTC1A2 cell-model. These results indicate that the two mutant alleles were co-expressed with CYP1A2 in stoichiometries which approximate the one of the wild-type CYPOR (ratio: 1 to ~5 CYPOR: CYP1A2).
Table 1.
Cytochrome c reduction, CYP and CYPOR content of BTC1A2_POR membranes
| Bacterial cell-model | CYP content* (pmol/mg) | cytochrome c reduction* (nmol reduced/[min.mg]) | CYPOR content* (pmol/mg) |
|---|---|---|---|
| BTC1A2_PORnull | 26 ± 1 | 3.1 ± 0.1 | -(a) |
| BTC1A2_PORWT | 34 ± 1 | 21.1 ± 0.6 | 6.6 ± 0.2 |
| BTC1A2_PORY459H | 48 ± 2 | 4.1 ± 0.6 | ~6.6(b) |
| BTC1A2_PORV492E | 25 ± 1 | 3.8 ± 0.7 | ~6.6(b) |
n ≥ 3
not detectable
values estimated through intensity comparison of CYPOR specific bands of Western blot, relative to BTC1A2_PORWT
Fig. 1.
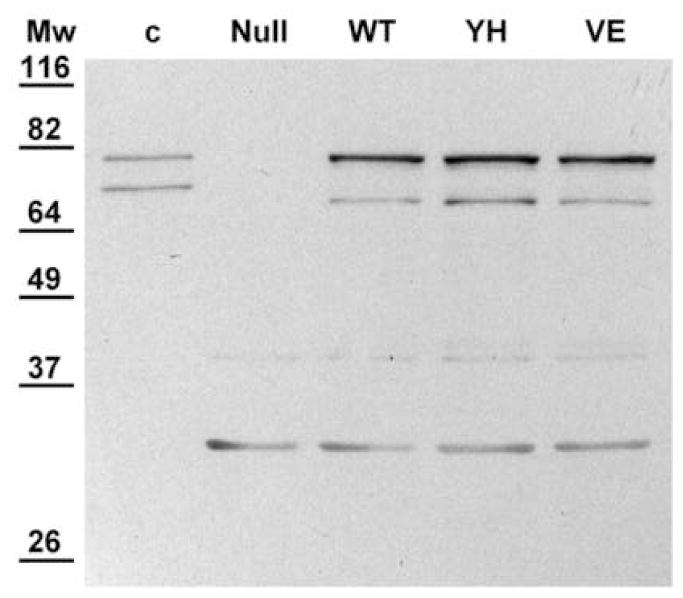
Immuno-detection of POR in membranes of the four BTC1A2_POR cell-models. Each lane contained 10 μg of membrane proteins except for the control lane (c), which contained 10 ng of purified fusion-protein ompA::POR (79 kD). Mw.: Molecular weight marker (Pre-stained Protein ladder, Invitrogen): values in kD
POR activity of the Y459H and V492E alleles
In our former study with purified proteins, it was demonstrated that the loss of function of the Y459H- and V492E-CYPOR variants was caused by a diminished binding affinity of these two proteins for the FAD prosthetic group [12]. In that study, this loss was overcome, to a large extent, by addition of excess FAD to the proteins. The present study was performed to verify that this recovery could also be obtained when these mutant proteins were inserted into the membrane milieu in combination with physiological redox partners, such as CYP1A2. When FAD was added to the membranes (10 μM), ~50% of the cytochrome c reductase activity could be recovered for the Y459H variant, whilst the recovery was 100% for the V492E CYPOR protein (Fig. 2).
Fig. 2.
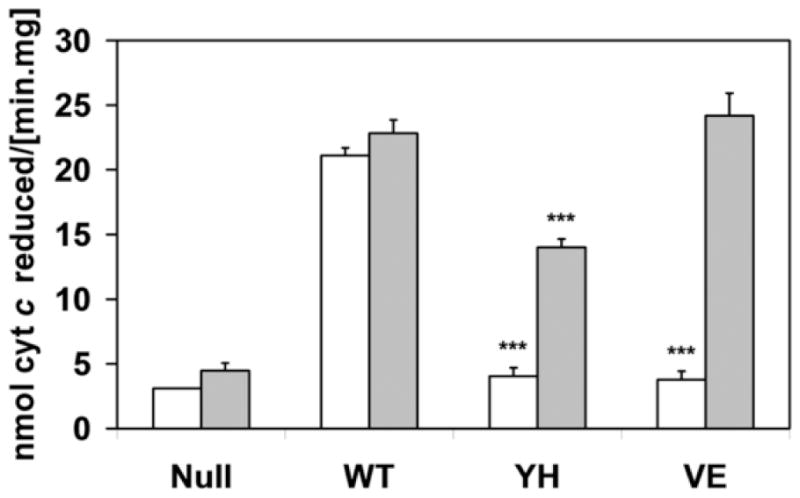
Histograms of cytochrome c reduction activity (in nmol cytochrome c reduced per min.mg membrane protein) of BTC1A2_POR membranes in the absence (white bars) or presence (gray bars) of 10 μM FAD. (Error bars represents the standard deviation to the mean of at least triplicate determination; statistical probability (p) is within the data-group, without or with FAD, relative to PORWT; ***: p<0.001;)
To investigate flavin-mediated redox functions of the two mutant CYPOR proteins further, we performed an extended evaluation of the effect of FAD, using a different non-physiologic CYPOR substrate, namely 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) [22] (see Supplemental Fig. 3). The derived kinetic parameters of this analysis further demonstrated full recovery of the V492E mutant protein by addition of FAD, with a maximal reaction velocity similar to that of the wild-type protein, while the Y459H variant could only be rescued by FAD to ~70% of the reaction velocity of PORwt (Fig. 3). The apparent affinities for FAD of the Y459H and V492E mutants, as derived from the saturating plots of activation versus [FAD], were 0.44 and 0.21 μM, respectively. The PORnull membranes did not show any detectable MTT reduction activity, either in the presence or absence of FAD.
Fig. 3.
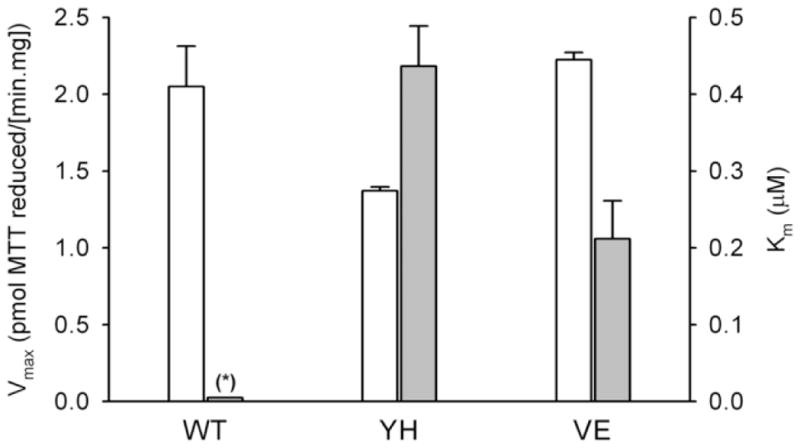
Histogram of Vmax (white bars) and apparent Km (grey bars) derived from the plots of MTT-reduction rate versus FAD concentration by BTC1A2_PORWT (WT), BTC1A2_PORY459H (YH) and BTC1A2_PORV492E (VE) membranes. (Error bars represents the standard deviation; see Supplemental Fig. 3 for Michaelis Menten plots; (*): no Km value could be derived from the plot for BTC1A2_PORWT membranes)
Effect of the Y459H and V492E POR mutations on CYP1A2 activity: membranes
The effect of CYPOR mutations on the interaction with its natural redox partner, CYP1A2, was then tested using BTC membrane preparations. For this purpose, we measured ethoxyresorufin-o-dealkylation (EROD) and methoxyresorufin-o-dealkylation (MROD) reactions, which are typically mediated by the CYP1A family [23]. The BTC1A2_PORWT membranes demonstrated typical Michaelis-Menten traces (see Supplemental Fig. 4) with kinetic parameters Km and kcat (Fig. 4, and Supplemental Table 1), as described previously [19]. This is in contrast to the CYP1A2 membranes containing either PORYH or PORVE, which showed very low, or no activity. When the same CYP kinetic studies were performed in the presence of 10 μM FAD, the CYP1A2-mediated EROD and MROD reactions were partially or fully recovered.
Fig. 4.
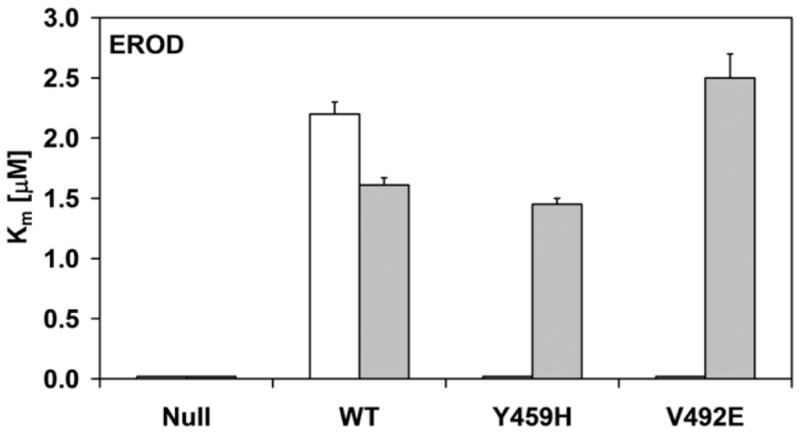
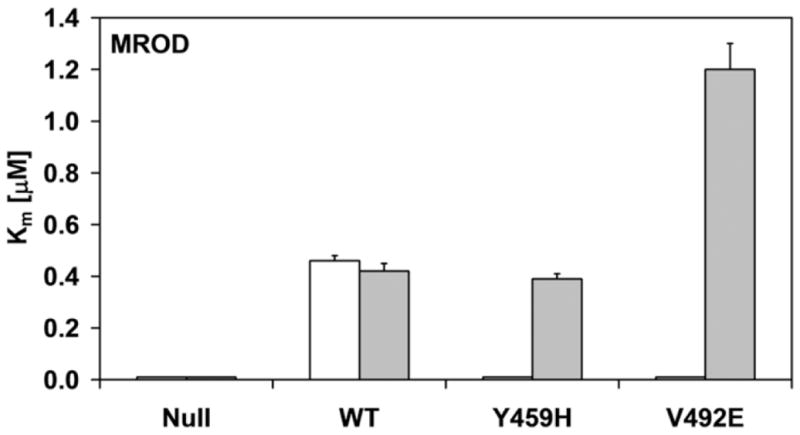
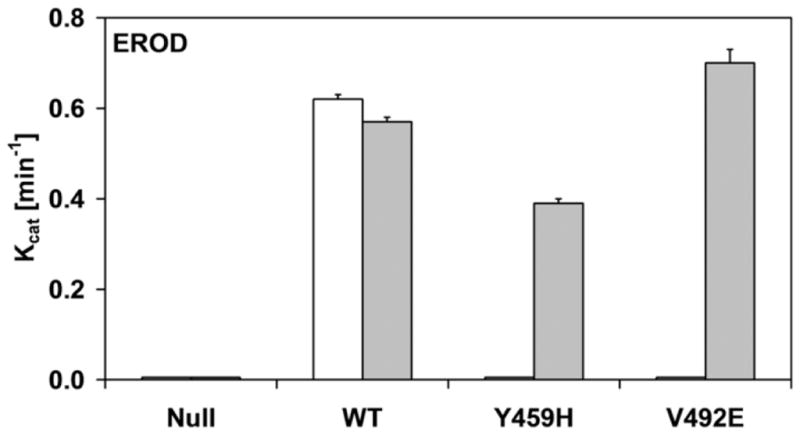
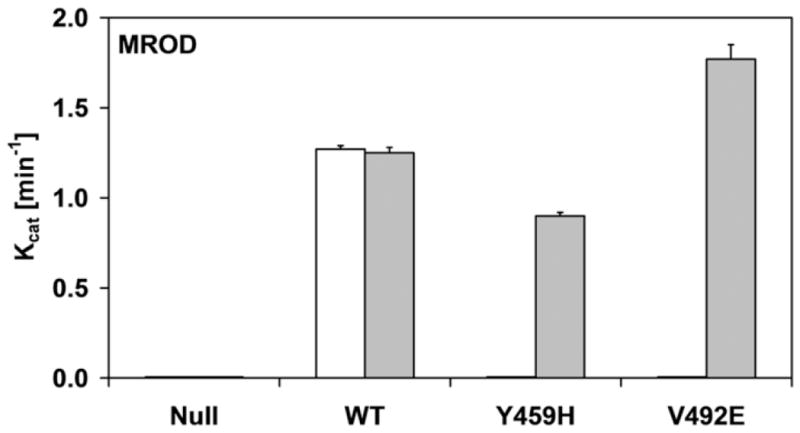
Histograms of the kinetic parameters of ethoxyresorufin-O-deethylation (a and c) and methoxyresorufin-O-demethylation (b and d) by BTC1A2_POR membranes, in absence or presence of 10 μM FAD. The affinity constant (Km) of CYP1A2 for ethoxyresorufin and methoxyresorufin are presented in a and b, respectively. The turnover number (kcat) of CYP1A2 for ethoxyresorufin and methoxyresorufin are presented in c and d, respectively (All Km and kcat values represented standard deviations (error-bars)< 10%, see Supplemental Table 1)
As was observed for the cytochrome c and MTT reduction assays, the addition of FAD totally recovered the activities in the BTC1A2_PORV492E membranes, for both EROD and MROD, but only partially recovered these activities in the BTC1A2_PORY459H membranes. This difference in FAD recovery between the two mutant proteins was more pronounced for MROD activity compared to EROD activity. By comparison, the addition of FAD had no significant effect on the two CYP1A2-mediated activities supported by PORwt (Fig. 4 and Supplemental material). Neither EROD nor MROD activity could be detected for BTC1A2_PORNull membranes, either in the absence or presence of FAD.
Effect of the Y459H and V492E POR mutations on CYP1A2 activity: whole cells assays
In order to evaluate the effect of the CYPOR mutants on xenobiotic metabolism, we made use of a whole cell mutagenicity assay utilizing the BTC1A2_POR bacteria [14]. We tested 3 pro-carcinogens that are bioactivated by CYP1A2, namely, 2-aminoanthracene (2AA), 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), and 2-amino-3-methylimidazo(4,5-f)quinoline (IQ) (Fig 5). The compound 2AA was highly mutagenic with the BTC1A2_PORWT cells, but BTC1A2_PORYH and BTC1A2_PORVE cells demonstrated low levels of mutagenicity for this compound, only slightly greater than that found for the BTC1A2_PORNull strain. More striking were the results obtained with NNK, which only the PORWT strain was able to bioactivate. On the other hand, the food carcinogen IQ was efficiently bioactivated by BTC1A2_PORWT cells whilst BTC1A2_PORYH- and BTC1A2_PORVE cells demonstrated only ~ 50% of the mutagenicity level of the PORWT for this compound.
Fig. 5.
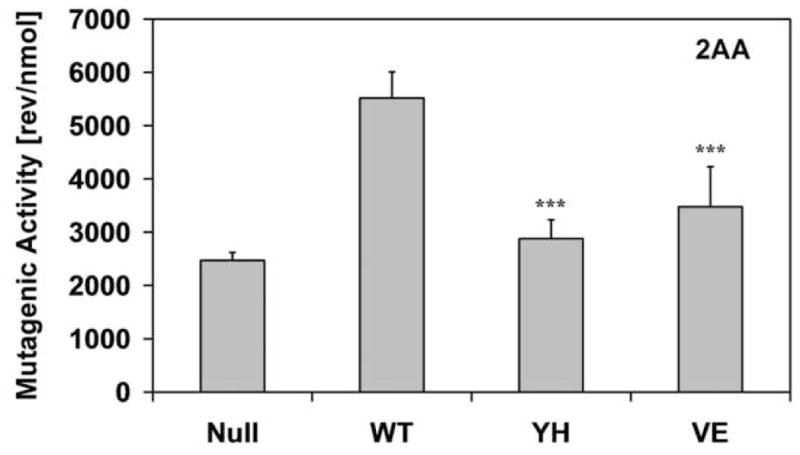
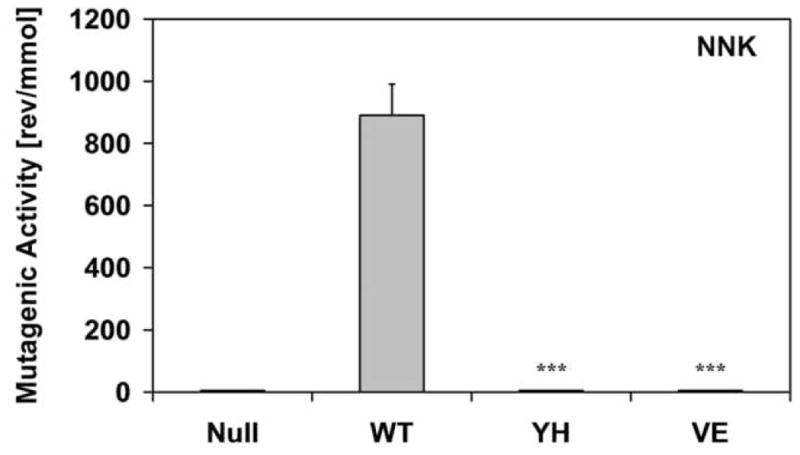
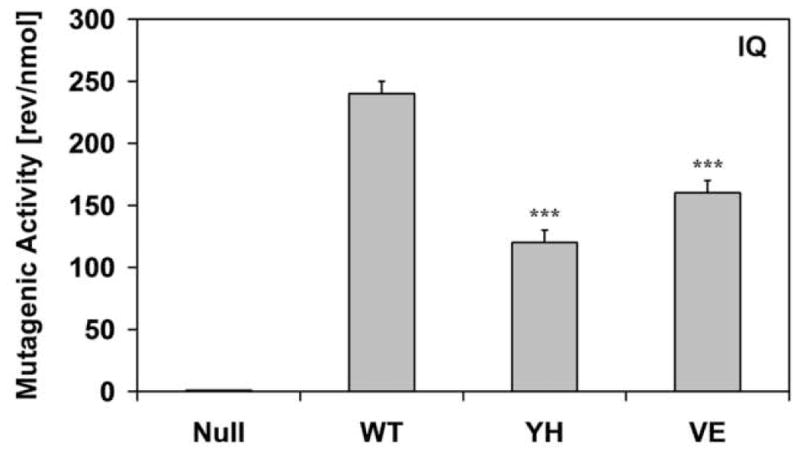
Histograms of the bioactivation capacity of the different BTC1A2_POR cells (in number of L-Arginine prototrophic (revertant) colonies per quantity of compound) of the pro-carcinogens 2-aminoanthracene (2AA), 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), and 2-amino-3-methylimidazo(4,5-f)quinoline (IQ). (Error-bars represent the standard deviation of the slope of the linear regression analysis; statistical probability is relative to PORWT; ***: p< 0.001).
Discussion
The ABS-related Y459H and the V492E CYPOR mutations were previously demonstrated, by use of reconstitution experiments applying non-physiological CYPOR: P450 ratios (>1), to be incapable of supporting the function of human CYP17A1 [4]. One may argue that coupling of these mutant proteins with any other CYP would lead to similar findings. However, ABS-related CYPOR mutations could differentially influence the function of CYPs, as suggested by Flück et al. [3]. Recently it was shown that the CYPOR mutation, A287P, substantially decreased the activity of CYP17A1, while CYP19A1 and CYP21A2 were hardly affected [17, 18]. Interestingly, residue A287 is located within one of the two stretches of the CYPOR protein considered to form the FAD binding-pocket, of which Y459 and V492 are part [26].
Membrane anchoring and a negatively-charged surface patch of CYPOR are considered to be major determinants of the proper alignment and protein-protein interaction of CYPOR and CYP (reviewed by Hlavica et al.) [27]. Although mitochondrial CYPs seem to have a signature of key basic amino acids on the proximal side for their interaction with the iron-sulfur protein, adrenodoxin, such signature sequences seem not to exist for microsomal CYPs in their interaction with CYPOR [28]. This implies diversity in (charged) amino acids on their proximal side and suggests the possibility of affinity differences of microsomal CYPs for CYPOR. With CYPOR in excess relative to CYP (ratio >1), such differences would have minor consequences, but the physiologically encountered stoichiometry of CYPOR and CYP (ratio 1: 5–10) implies competition among CYPs for CYPOR. The diversity of amino acids of microsomal CYPs at their CYPOR binding face, therefore, becomes a major determinant of CYP catalaytic activity. One might also conclude that mutations altering the CYPOR binding surface for CYP would have significant effects on specific CYP-mediated catalysis. On the other hand, it is also possible that CYPOR mutations distant from the CYP binding site could influence CYP activity indirectly as the binding of CYP induces conformational changes in CYPOR through a relay network, which affects its electron donor function [27]. As such, ABS-related CYPOR mutations, that are not directly involved in cofactor- (NADPH, FMN, FAD) or CYP-binding can have profound effects on CYP activity. The interaction between CYP and CYPOR is also affected by competitive binding with other ER membrane proteins such as cytochrome b5 [29] and possibly the progesterone receptor C-terminal membrane component 1 (PGRMC1) [30]
Although reconstitution experiments are valid in a first approach, a more informative evaluation of the effect of the ABS-related mutations of CYPOR on CYP activity would be obtained with CYPOR and CYP protein, membrane-anchored in a functionally relevant membrane environment, with a stoichiometry that reflects the physiologically relevant competition for reducing equivalents from CYPOR. Such an approach, as applied in this study, reflects the actual in vivo conditions in the coupling of CYPOR with CYP, minimizing the possibility of overlooking subtle conformational differences with highly significant functional effects of ABS-related CYPOR variant proteins, when coupled with particular CYPs.
Due to the high degree of amino acid sequence homology between rat and human CYPORs (~95%), human POR mutations have been interpreted in previous papers, using the crystal structure of the soluble di-flavin catalytic domain of rat CYPOR [26]. The active site of the enzyme is formed at the interface of the FMN-, FAD-, and NADPH-binding domains (Fig. 6). The protein structure of CYPOR influences the thermodynamics of flavin redox reactions by modulating midpoint potentials through hydrogen bonding and π-π interactions. Based on our findings, residues Y459 and V492 appear to play these roles in human CYPOR [12]. FAD recovery data, both in our former study and in the present one, indicate a more severe disturbance of FAD-binding by the Y459H mutation than by V492E. Addition of excess FAD led to total recovery of the V492E variant, while providing only partial rescue of the Y459H. In concordance with the protein model (Fig. 6), in which the tyrosine residue at position 459 is responsible for the correct positioning of the isoalloxazine ring of FAD by π-π stacking and a hydrogen bond, this residue provides optimal alignment with both NADPH and FMN for electron transfer to occur. Valine at position 492 makes hydrogen bonds to the pyrophosphate oxygen nearest to the adenosine portion of the FAD, obviously important for FAD-binding, but likely playing a lesser role in affecting electron transfer once FAD is bound. Early work on CYPOR FAD-dependence [31–33] demonstrated that FAD was very tightly bound to the wild type enzyme, with an estimated Kd <10 nM. Here, we estimated the affinity of the V492E and Y459H variants for FAD using the non-physiological substrate MTT, with apparent KdFAD values of 0.21 and 0.44 μM, respectively. These are three orders of magnitude higher than the FAD dissociation parameter suggested for the wild type CYPOR, and corroborate the more severe disturbance of structure and function by the Y459H mutation.
Fig. 6.
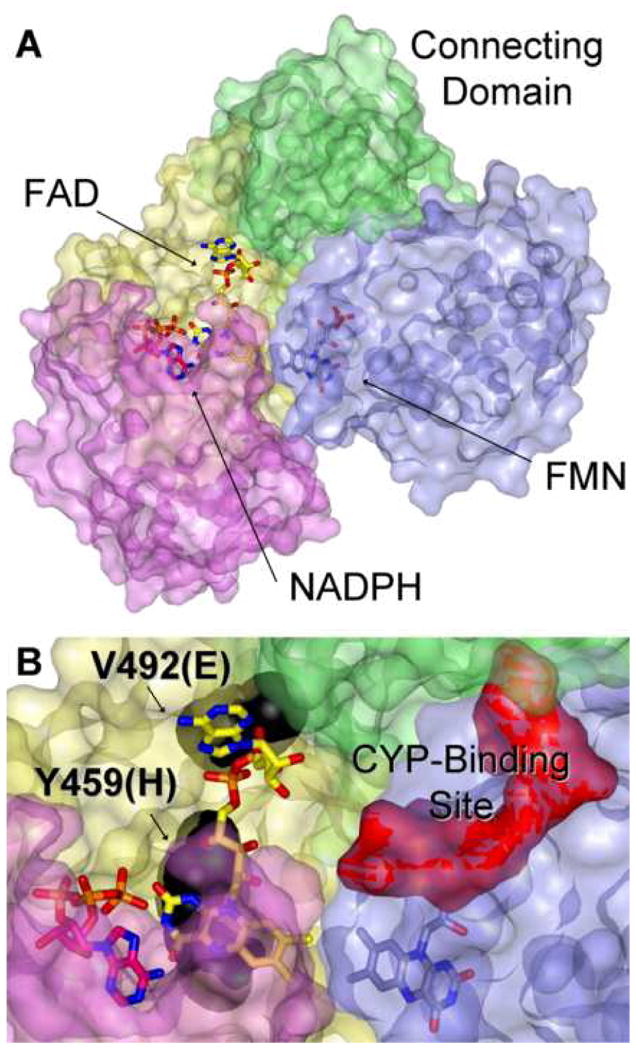
A – The crystal structure of rat POR [1AMO, [26]] demonstrating the overall domain arrangement of the enzyme with bound 2′ADP (an NADP+ analog). The solvent-exposed surfaces of the FMN-binding domain (light blue), the connecting domain (green), the FAD-binding domain (yellow), and the NADPH-binding domain (pink) are shown. FMN, FAD, and 2′ADP are drawn in stick configuration with carbons in light blue, yellow, and pink, respectively; oxygens are red and nitrogens are blue. B – A closer perspective of the active site showing residues Y456, the rat homolog of human Y459, and V489 (human V492) in black spacefill. The surface of the putative CYP-binding site, formed by two clusters of acidic residues within the FMN-binding domain, is rendered in red.
Full recovery with FAD of both the EROD- and MROD-activities mediated by CYP1A2 when supported by CYPORVE, relative to the only partial recovery of the CYPORYH, totally agrees with the cytochrome c and MTT reduction data. On the other hand, the protein-protein interaction between the mutant CYPORs and CYP1A2 was not affected differentially from the reductase-mediated activities (cytochrome c and MTT). Moreover, FAD is not hindered in its access to its binding-site when CYPOR is membrane-bound and in close vicinity with its natural redox partner CYP1A2. This free access of FAD to CYPOR suggests a solution-exposed surface for the FAD-binding domain, even when the enzyme is in complex with CYPs. This indicates either a non-overlap of the FAD- and CYP-binding domains of CYPOR, or sufficient time between CYP release and binding events for FAD to bind, or both.
The two mutants severely hindered the CYP1A2-mediated bioactivation of the three pro-carcinogens 2AA, NNK and IQ, confirming our EROD and MROD data. It is interesting to note how the bioactivation is differentially affected among the three compounds. The compound 2AA could be bioactivated by the BTC1A2_PORnull bacteria in contrast to NNK and IQ, which did not show any mutagenicity with these PORnull cells. 2AA did not demonstrate any mutagenicity when using BTC bacteria, which did not contain CYP1A2 (non-induced bacteria, data not shown). This excludes an E. coli i.e. non-CYP1A2 pathway, suggesting an alternative source of electrons for CYP1A2–dependent 2AA activation. Escherichia coli K12, contains an NADPH-ferrodoxin/flavodoxin reductase, which together with flavodoxin, has been shown to sustain, albeit inefficiently, mammalian CYP activity [34–38]. This E. coli reductase is likely responsible for the CYPOR-independent CYP1A2 bioactivation of 2AA. In contrast, this endogenous reductase was not able to sustain CYP1A2-dependent bioactivation of NNK or IQ. Results of the bioactivation of NNK by the different BTC1A2_POR bacteria were more informative in loss of function of the two mutant CYPORs, in that neither mutant CYPOR-containing bacteria demonstrated any mutagenicity, in contrast to the PORwt bacteria.
With both of these ABS-CYPOR variants, it has now been demonstrated both in in vitro reconstitution experiments described in our former study [12] and in the bacterial cellular expression systems that FAD addition can rescue these defects. Therefore, it can be stated that such defects compromise the ability of CYPOR to support, not only steroidogenesis (CYP17A1), but also drug metabolizing and mutagenic activities catalyzed by CYP1A2.
Extending from these results on CYP1A2 as an example, it is highly probable that other major, xenobiotic-metabolizing CYPs may be comprised in their functions by these two ABS-related CYPOR alleles, although caution should be used as particular ABS-CYPOR mutations can exert different effects on CYPs (vida supra). CYP1A2, among others, mediates the elimination of many environmental toxins, as well as many prescribed drugs, to which man is exposed daily [39]. Less effective elimination routes due to reduced CYP activities might lead to severe Adverse Drug Reactions, when patients are administered normal drug dosages. This imbalance in the metabolism of xenobiotics has not been adequately examined in human patients with POR deficiency. Considering the likelihood that ABS patients will undergo surgical procedures to correct the craniofacial defects, as well as treatment with various drugs whose metabolism is dependent upon cytochromes P450, it also is important to understand the basis of these genetic defects and their metabolic consequences in these circumstances. It is also possible that therapeutic approaches to bypass or overcome these genetic deficiencies can be developed, such as the administration of the FAD precursor, riboflavin, in high doses for these particular defects in ABS-afflicted patients as well as during early human gestation should such defects be predictable.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We are grateful for the thoughtful direction and invaluable assistance of Karen McCammon in optimization of protocols for preparation of CYPOR-containing membranes.
Footnotes
This work was supported in part by NIH Grant HL30050 (to B.S.S.M. who is The Robert A. Welch Distinguished Professor in Chemistry, AQ-0012) and grant PTDC/SAU-GMG/71911/2006) of the Fundação para a Ciência e a Tecnologia (Portugal).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Antley R, Bixler D. Birth Defects Orig Artic Ser. 1975;11:397–401. [PubMed] [Google Scholar]
- 2.Chun K, Siegel-Bartelt J, Chitayat D, Phillips J, Ray P. Am J Med Genet. 1998;77:219–224. doi: 10.1002/(sici)1096-8628(19980518)77:3<219::aid-ajmg6>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 3.Flück C, Tajima T, Pandey A, Arlt W, Okuhara K, Verge C, Jabs E, Mendonca B, Fujieda K, Miller W. Nat Genet. 2004;36:228–230. doi: 10.1038/ng1300. [DOI] [PubMed] [Google Scholar]
- 4.Huang N, Pandey A, Agrawal V, Reardon W, Lapunzina P, Mowat D, Jabs E, Van Vliet G, Sack J, Flück C, Miller W. Am J Hum Genet. 2005;76:729–749. doi: 10.1086/429417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guengerich F. In: Cytochrome P450 Structure, Mechanism and Biochemistry. Ortiz de Montellano P, editor. Kluwer Academic/Plenum Publishers; New York: 2005. pp. 377–530. [Google Scholar]
- 6.Arlt W, Walker E, Draper N, Ivison H, Ride J, Hammer F, Chalder S, Borucka-Mankiewicz M, Hauffa B, Malunowicz E, Stewart P, Shackleton C. Lancet. 2004;363:2128–2135. doi: 10.1016/S0140-6736(04)16503-3. [DOI] [PubMed] [Google Scholar]
- 7.Fukami M, Horikawa R, Nagai T, Tanaka T, Naiki Y, Sato N, Okuyama T, Nakai H, Soneda S, Tachibana K, Matsuo N, Sato S, Homma K, Nishimura G, Hasegawa T, Ogata T. J Clin Endocrinol Metab. 2005;90:414–426. doi: 10.1210/jc.2004-0810. [DOI] [PubMed] [Google Scholar]
- 8.Hart S, Wang S, Nakamoto K, Wesselman C, Li Y, Zhong X. Pharmacogenet Genomics. 2008;18:11–24. doi: 10.1097/FPC.0b013e3282f2f121. [DOI] [PubMed] [Google Scholar]
- 9.Han J, Wang S, He X, Liu C, Hong J. Toxicol Sci. 2006;91:42–48. doi: 10.1093/toxsci/kfj139. [DOI] [PubMed] [Google Scholar]
- 10.Wang S, Han J, He X, Wang X, Hong J. Drug Metab Dispos. 2007;35:176–179. doi: 10.1124/dmd.106.011056. [DOI] [PubMed] [Google Scholar]
- 11.Kostrzewa-Nowak D, Paine M, Korytowska A, Serwatka K, Piotrowska S, Wolf C, Tarasiuk J. Cancer Lett. 2007;245:252–262. doi: 10.1016/j.canlet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 12.Marohnic C, Panda S, Martasek P, Masters B. J Biol Chem. 2006;281:35975–35982. doi: 10.1074/jbc.M607095200. [DOI] [PubMed] [Google Scholar]
- 13.Masters BS, Marohnic C. Drug Metab Rev. 2006;38:209–225. doi: 10.1080/03602530600570065. [DOI] [PubMed] [Google Scholar]
- 14.Duarte M, Palma B, Laires A, Oliveira J, Rueff J, Kranendonk M. Mutagenesis. 2005;20:199–208. doi: 10.1093/mutage/gei028. [DOI] [PubMed] [Google Scholar]
- 15.Kranendonk M, Fisher C, Roda R, Carreira F, Theisen P, Laires A, Rueff J, Vermeulen N, Estabrook R. Mutat Res. 1999;439:287–300. doi: 10.1016/s1383-5718(98)00193-4. [DOI] [PubMed] [Google Scholar]
- 16.Duarte M, Palma B, Gilep A, Laires A, Oliveira J, Usanov S, Rueff J, Kranendonk M. Mutagenesis. 2007;22:75–81. doi: 10.1093/mutage/gel054. [DOI] [PubMed] [Google Scholar]
- 17.Dhir V, Ivison H, Krone N, Shackleton C, Doherty A, Stewart P, Arlt W. Mol Endocrinol. 2007;21:1958–1968. doi: 10.1210/me.2007-0066. [DOI] [PubMed] [Google Scholar]
- 18.Pandey A, Kempna P, Hofer G, Mullis P, Flück C. Mol Endocrinol. 2007;21:2579–2595. doi: 10.1210/me.2007-0245. [DOI] [PubMed] [Google Scholar]
- 19.Kranendonk M, Carreira F, Theisen P, Laires A, Fisher C, Rueff J, Estabrook R, Vermeulen N. Mutat Res. 1999;441:73–83. doi: 10.1016/s1383-5718(99)00032-7. [DOI] [PubMed] [Google Scholar]
- 20.Kranendonk M, Mesquita P, Laires A, Vermeulen N, Rueff J. Mutagenesis. 1998;13:263–269. doi: 10.1093/mutage/13.3.263. [DOI] [PubMed] [Google Scholar]
- 21.Fisher C, Caudle D, Martin-Wixtrom C, Quattrochi L, Tukey R, Waterman M, Estabrook R. FASEB J. 1992;6:759–764. doi: 10.1096/fasebj.6.2.1537466. [DOI] [PubMed] [Google Scholar]
- 22.Yim S, Yun C, Ahn T, Jung H, Pan J. J Biochem Mol Biol. 2005;38:366–369. doi: 10.5483/bmbrep.2005.38.3.366. [DOI] [PubMed] [Google Scholar]
- 23.Burke M, Thompson S, Weaver R, Wolf C, Mayer R. Biochem Pharmacol. 1994;48:923–936. doi: 10.1016/0006-2952(94)90363-8. [DOI] [PubMed] [Google Scholar]
- 24.Chauret N, Gauthier A, Nicoll-Griffith D. Drug Metab Dispos. 1998;26:1–4. [PubMed] [Google Scholar]
- 25.Duarte M, Palma B, Gilep A, Laires A, Oliveira J, Usanov S, Rueff J, Kranendonk M. Mutagenesis. 2005;20:93–100. doi: 10.1093/mutage/gei012. [DOI] [PubMed] [Google Scholar]
- 26.Wang M, Roberts D, Paschke R, Shea T, Masters B, Kim J. Proc Natl Acad Sci U S A. 1997;94:8411–8416. doi: 10.1073/pnas.94.16.8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hlavica P, Schulze J, Lewis D. J Inorg Biochem. 2003;96:279–297. doi: 10.1016/s0162-0134(03)00152-1. [DOI] [PubMed] [Google Scholar]
- 28.Pikuleva I, Cao C, Waterman M. J Biol Chem. 1999;274:2045–2052. doi: 10.1074/jbc.274.4.2045. [DOI] [PubMed] [Google Scholar]
- 29.Zhang H, Im S, Waskell L. J Biol Chem. 2007;282:29766–29776. doi: 10.1074/jbc.M703845200. [DOI] [PubMed] [Google Scholar]
- 30.Hughes A, Powell D, Bard M, Eckstein J, Barbuch R, Link A, Espenshade P. Cell Metab. 2007;5:143–149. doi: 10.1016/j.cmet.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 31.Kurzban G, Strobel H. J Chromatogr. 1986;358:296–301. doi: 10.1016/s0021-9673(01)90344-9. [DOI] [PubMed] [Google Scholar]
- 32.Kurzban G, Strobel H. J Biol Chem. 1986;261:7824–7830. [PubMed] [Google Scholar]
- 33.Kurzban G, Howarth J, Palmer G, Strobel H. J Biol Chem. 1990;265:12272–12279. [PubMed] [Google Scholar]
- 34.Jenkins C, Genzor C, Fillat M, Waterman M, Gomez-Moreno C. J Biol Chem. 1997;272:22509–22513. doi: 10.1074/jbc.272.36.22509. [DOI] [PubMed] [Google Scholar]
- 35.Jenkins C, Waterman M. J Biol Chem. 1994;269:27401–27408. [PubMed] [Google Scholar]
- 36.Jenkins C, Waterman M. Biochemistry. 1998;37:6106–6113. doi: 10.1021/bi973076p. [DOI] [PubMed] [Google Scholar]
- 37.Shet M, Fisher C, Estabrook R. Arch Biochem Biophys. 1997;339:218–225. doi: 10.1006/abbi.1996.9868. [DOI] [PubMed] [Google Scholar]
- 38.Waterman M, Jenkins C, Pikuleva I. Toxicol Lett. 1995;82–83:807–813. doi: 10.1016/0378-4274(95)03597-4. [DOI] [PubMed] [Google Scholar]
- 39.Nebert D, Russell D. Lancet. 2002;360:1155–1162. doi: 10.1016/S0140-6736(02)11203-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.