

Abstract
Stable surface expression of human inhibitory killer cell immunoglobulin-like receptors (KIR) is critical for controlling NK cell function and maintaining NK cell tolerance toward normal MHC-I+ cells. Our recent experiments, however, have found that antibody-bound KIR3DL1 (3DL1) readily leaves the cell surface and undergoes endocytosis to early/recycling endosomes and subsequently to late endosomes. We found that 3DL1 internalization is at least partially mediated by an interaction between the μ2 subunit of the AP-2 clathrin adaptor complex and ITIM tyrosine residues in the cytoplasmic domain of 3DL1. Disruption of the 3DL1/μ2 interaction, either by mutation of the ITIM tyrosines in 3DL1 or mutation of μ2, significantly diminished endocytosis and increased surface expression of 3DL1 in human primary NK cells and cell lines. Furthermore, we found that the 3DL1/AP-2 interaction is diminished upon antibody engagement with the receptor, as compared to untreated cells. Thus, we have identified AP-2-mediated endocytosis as a mechanism regulating the surface levels of inhibitory KIR though their ITIM domains. Based upon our results, we propose a model in which non-engaged KIR are internalized by this mechanism, whereas engagement with MHC-I ligand would diminish AP-2 binding, thereby prolonging stable receptor surface expression and promoting inhibitory function. Furthermore, this ITIM-mediated mechanism may similarly regulate the surface expression of other inhibitory immune receptors.
Introduction
NK cells selectively recognize and kill virus-infected and transformed cells, while remaining tolerant of normal cells (1, 2). Their activation is controlled by a balance of signals from activating (aNKR), adhesion and inhibitory (iNKR) surface receptors (3). Activation is dominantly suppressed upon engagement of iNKRs [especially the human killer cell Ig-like receptors (KIR)] with MHC-I expressed on normal cells. With few exceptions, normal cells elicit NK cell tolerance through their high expression of MHC-I and low expression of ligands for aNKR (4). However, following genotoxic stress (5) or virus infection (6), aNKR ligands can be upregulated and/or MHC-I downregulated on target cells to tip the balance toward NK cell activation and targeted cytotoxicity.
KIR inhibitory function centers around their cytoplasmic ITIMs [(I/V)xYxx(L/V)] (3). KIR engagement with MHC-I ligands results in 1) phosphorylation of ITIM tyrosine residues with subsequent recruitment of SHP-1 and SHP-2 protein tyrosine phosphatases that dominantly suppress aNKR signaling pathways and 2) induced tyrosine phosphorylation of the adaptor Crk, which relocalizes from activating to inhibitory complexes (7–9). These events terminate early NK cell activation signaling and establish tolerance toward normal MHC-I-expressing cells.
The surface levels of KIR or their cognate ligands can directly impact the activation thresholds of NK cells (10, 11), but little is known regarding the mechanisms regulating the surface expression of KIR. Generally, receptor surface expression can be controlled by de novo protein synthesis, endocytosis, recycling back to the cell surface and protein degradation. With respect to KIR, both KIR3DL2 and KIR2DL4 can relocalize from the cell surface to endosomes to mediate intracellular functions (12, 13). Furthermore, polymorphic sequence variants of KIR can exhibit wide disparities in surface expression (14, 15). Also, phosphorylation of serine 394 by PKC appears to stabilize surface expression of KIR3DL1 (3DL1) and other sequence motifs, including the first ITIM tyrosine have been implicated in regulating surface expression (16, 17). These reports demonstrate a need for better mechanistic understanding of KIR endocytosis and intracellular trafficking.
Mammalian cells can internalize receptors constitutively or in response to specific stimuli via either clathrin-dependent or -independent endocytosis (18–20). Clathrin forms a triskelion structure that drives endocytic vesicle formation, but requires adaptors to bind surface receptors. The AP-2 clathrin adaptor is directly implicated in the internalization of many receptors, including transferrin receptor (TfR), LDLR and EGFR (21–23). AP-2 is a heterotetrameric complex composed of α- and β-adaptin that interact with clathrin and the plasma membrane, μ2, which associates with cargo containing tyrosine-based motifs, and σ2, which is involved in binding cargo containing dileucine-based motifs (19, 21). While the mechanism of KIR endocytosis is unknown, the CD94/NKG2A iNKR is reportedly internalized by a macropinocytosis-like pathway, although the sequence elements involved remain undefined (24).
Here we demonstrate that the ITIM sequences of inhibitory KIR, in addition to their role in negative signaling, also provide a handle for 3DL1 internalization. This internalization occurs through interaction with μ2 of the AP-2 clathrin adaptor complex. Our data also suggest that AP-2 association may occur more readily when KIR are not engaged with MHC-I ligand, whereas interaction with MHC-I ligand may reduce AP-2 association, which would promote stable KIR surface expression to prolong inhibitory function.
Materials and Methods
Cells and culture
KHYG-1, NKL, Jurkat, HEK-293T and LentiX 293T (Clontech, Mountain View, CA) cells were cultured as described (8, 25, 26). Healthy volunteer blood donors were recruited by informed consent as approved by the Fox Chase Cancer Center (FCCC) Institutional Review Board. Primary CD56+CD3−KIR3DL1± NK cells were sorted by FACS and cultured in RPMI 1640 medium plus 5% human serum, 10% FBS, and 500–1000 U/ml recombinant human IL-2 (Roche, generously provided by the NCI Biologic Resources Branch, Frederick, MD). Some primary NK cells were restimulated with either irradiated RPMI-8866 cells or irradiated allogeneic PBMC as described (27).
Microscopy
Following attachment to pre-warmed poly-L-lysine slides (BD Pharmingen, San Jose, CA), NKL cells expressing 3DL1-Cherry and EYFP-Rab4 or EGFP-Rab7 (1 day after passage and stimulation with IL-2) were cooled on ice for 15 min and then labeled with DX9-Brilliant Blue 421 (BioLegend, San Diego, CA) for 30 min. Slides were subsequently washed 3x in room temperature (RT) PBS to remove excess mAb, warmed to 37°C for 0–30 min and washed 3x in PBS at RT again. Cells were fixed in pre-warmed PBS containing 3% paraformaldehyde for 15 min at RT. Slides were mounted with 0.16 mm coverslips (Thermo Fisher, Pittsburgh, PA) in ProLong Gold antifade reagent (Life Technologies, Eugene, OR). Images were acquired with EZ-C1 3.80 software on an inverted Nikon TE2000 with C1 confocal scanhead (Nikon, Melville, NY). Z-stacks (0.3 um step size) were collected from at least 25 cells expressing high levels of Rab and 3DL1 for each time point. 3DL1 internalization was analyzed by quantifying the colocalization of DX9 mAb and Rab4 or Rab7. Co-localization was quantified using a rigorous method developed by Manders et al. (28, 29), which measures the degree of overlap of pixels in two separate fluorescent channels independent of their intensity and relative to the total intensity within each channel. Manders’ coefficients were quantified for each 0.3 um slice of each z-stack with the Just Another Co-localization plugin (JACoP) in Image J after thresholding (http://imagej.nih.gov/ij/, JACoP; rsbweb.nih.gov/ij/plugins/track/jacop.html). The resulting coefficients were compared for significance with the Wilcoxon Rank Sum Test using R software (The R Foundation; www.r-project.org). A Manders coefficient of 0 corresponds to non-overlapping distribution, whereas a value of 1 signifies 100% co-localization. To better visualize the co-localization in Figure 1D, 6 of the 0.3 μm slices from the center of representative cells were merged as a max projection to generate the images shown.
Figure 1. 3DL1 localizes to early and late endosomes after antibody engagement.

A. Non-radioactive colorimetric assay comparing the cytotoxicity of either KIR− (Squares; NKL) or 3DL1-Cherry+ NKL cells (Circles; KIR+ NKL) against 721.221 target cells lacking (No KIR ligand) or expressing the 3DL1 ligand HLA-B*051 (KIR ligand) at various NK cell:Target cell (NK:T) ratios. The mean ± S.D. of data from 5 independent experiments is shown. For each experiment, the data were first divided by the “NKL, No KIR ligand” sample (arbitrarily set to 100%) to generate the percentage of total cytotoxicity. Statistical analysis comparing matched samples for each NK cell line was calculated using the Student’s t test with * designating p ≤ 0.05 and ** defining p ≤ 0.01. B. 3DL1 and Rab4/Rab7 localization in representative NKL cells one day after IL-2 stimulation. Following fixation, cells expressing 3DL1-Cherry and either EYFP-Rab4 or EGFP-Rab7, were labeled with Brilliant Blue 421-conjugated DX9 (BV421-DX9). Shown are max-projected images of six 0.3 um slices through the center of representative individual cells. Cherry = 3DL1-Cherry (red); αKIR = Brilliant Blue 421-conjugated DX9 mAb bound to cell surface 3DL1 (blue); Rab4 = EYFP-Rab4 (green); Rab7 = EGFP-Rab7 (green). C. Compilation of data as in B. Thresholded Manders’ coefficients of 3DL1-Cherry colocalized with DX9 mAb (Cell surface), Rab4 (Rab4+ endosome) and Rab7 (Rab7+ endosome). The median of each data set is indicated by a horizontal bar. Data are pooled from 2 independent experiments where each icon represents a separate cell. Black filled icons designate values derived from the representative NK cells shown in panel B. D&E. Intracellular localization of 3DL1 with Rab4 (D) or Rab7 (E) are shown in representative NKL cells after 0–30 min exposure to DX9 mAb. 3DL1-Cherry+ NKL cells were stained with Brilliant Blue 421-conjugated DX9 mAb on ice to label the receptor at the cell surface and kept on ice (time 0) or incubated at 37°C for 15 or 30 min. Shown are max-projected images of six 0.3 μm slices through the center of representative individual cells. F–H. Compilation of data as in panel C. Thresholded Manders’ coefficients of DX9 mAb colocalized with Rab4 or Rab7 (F, top and bottom panels, respectively), 3DL1-Cherry colocalized with Rab4 and Rab7 (G, top and bottom panels, respectively) and DX9 mAb colocalized with 3DL1-Cherry in the same cell populations (H). Black filled icons designate values derived from the representative NK cells shown in panels D or E. Statistical analysis used the Wilcoxon Rank Sum test, where * designates p ≤ 0.05 and ** defines p ≤ 0.01. Data are pooled from 2 independent experiments where each icon represents a separate cell.
Cytotoxicity Assay
The CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega, Madison, WI) was performed according to manufacturer instructions. Briefly, 1×105 721.221 cells [lacking or expressing the 3DL1 ligand (HLA-B*51)] were mixed with NKL cells to achieve NK cell:Target cell ratios (NK:T) of 1.25:1 to 10:1. Cells were incubated together in a U-bottom plate at 37°C in a humidified atmosphere of 7% CO2. After 4 hrs, supernatants were harvested, exposed to substrate and absorbance (490 nm) was measured on a BioTek Microplate Reader EL808 (BioTek, Winooski, VT). Background absorbance from a blank media control was subtracted from all values. Target cell maximum was determined after target cell incubation with kit lysis buffer, while spontaneous values were computed from NK or target cells alone. Specific lysis was calculated as: 100 x (Experimental Absorbance – NK cell spontaneous – Target cell spontaneous)/(Target cell maximum – Target cell spontaneous).
Yeast Two-hybrid Screen
A LexA-based yeast two-hybrid screen was performed as described (25). Bait for the screen was the human 3DL1*0010101 cytoplasmic domain [aa 340-423, http://www.ebi.ac.uk/ipd/kir] fused with LexA-DNA binding domain, which was expressed from the pEG202 plasmid. Control bait constructs encoded the cytoplasmic domains of CD5 (aa 402-495) or murine CD4 (aa 418-457) (30, 31). The 3DL1 ITIM tyrosines (Y) were mutated to alanines (A) (Y377A, Y407A and Y377/407A) using QuikChange II (Stratagene, Santa Clara, CA).
GST pull-down assay
GST-3DL1 fusion proteins [wild type (WT) and Y to A mutant 3DL1 cytoplasmic domains; aa 340-423] were generated from pGEX-4T1 plasmid (GE Healthcare, Piscataway, NJ) in BL21 bacteria, purified on glutathione-agarose (Thermo Scientific, Rockford, IL), incubated with HEK-293T cell lysates, and washed as previously described (16, 32). Adsorbed proteins were separated by SDS-PAGE, transferred to PVDF, and immunoblotted with anti-μ2 Ab (Sigma, St. Louis, MO) or anti-α-adaptin mAb (BD Bioscience, Mountain View, CA).
Immunoprecipitation and Immunoblotting
For experiments in Figure 2, 3DL1+ KHYG-1 cells were lysed in mRIPA buffer [1% IGEPAL CA-630, 0.5% Na deoxycholic acid, 150 mM NaCl, 10 mM Tris, pH 7.5, 0.1% SDS, 2 mM Na orthovanadate, and 1 μg/ml each of aprotinin, leupeptin, and soybean trypsin inhibitor (Sigma)]. 3DL1 was immunoprecipitated with protein G agarose (Millipore, Billerica, MA) pre-coupled with DX9 mAb. Proteins were separated as above, immunoblotted with anti-α-adaptin mAb (BD Transduction Laboratories) and rabbit anti-KIR Ab (16), followed by HRP-conjugated secondary Ab. The resulting blots were visualized ECL (Millipore) and exposure to autoradiography film (Denville, Metuchen, NJ). The intensities of μ2, α-adaptin and KIR bands were quantified using Image J software. The ratio of μ2 or α-adaptin/KIR protein levels for WT was arbitrarily set to 100%. In experiments for Figure 5, KHYG-1 cells (4 days after passage and IL-2 stimulation) were stimulated with 1 mM pervanadate alone, 7.5 ug DX9 alone (a saturating concentration), or both in combination for 10 min on ice prior to lysis, as described (33). These cells were lysed in IP buffer [50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 μg/ml protease inhibitors as above, 2 mM Na orthovanadate, 1 mM NaF, 2 mM EGTA and 0.5% Triton X-100]. 3DL1 was immunoprecipitated from these lysates with 5.133 mAb coupled to cyanogen bromide activated sepharose beads (GE Healthcare). Proteins were separated as above and immunoblotted with anti-α-adaptin pAb (Proteintech, Chicago, IL), 4G10 mAb (Millipore), anti-SHP-1 pAb (Santa Cruz, Dallas, TX) and anti-KIR pAb (#ARP53462; Aviva, San Diego, CA) followed by anti-mouse-IR680 and –rabbit-IR800 (LI-COR Biosciences, Lincoln, NE). Proteins were visualized as described (26). Band intensity was quantified with Image J.
Figure 2. The μ2 subunit of AP-2 interacts with the ITIM tyrosine residues of 3DL1.

A. EGY50 yeast cells were co-transformed with prey plasmid encoding μ2 (aa 146-435) and bait plasmids encoding 3DL1, CD5, or CD4 cytoplasmic domains or empty vector (vector). Yeast were plated on leucine-deficient medium [+ 2% glucose (Glu, Left) or 2% galactose + 0.2% raffinose (Gal, Right)] to induce protein expression. Results are representative of 2 independent transformations. B. Quantitative β-galactosidase assay of EGY50 cells co-transformed with prey plasmid containing μ2 and bait plasmid with cytoplasmic domains of wild type (WT) or alanine mutant 3DL1 (YA, AY or AA). Data are mean ± SD of β-galactosidase production from 3 experiments. C. GST fusion proteins exposed to 293T cell extracts were immunoblotted with anti-μ2 or -α-adaptin Ab. The degree of μ2 and α-adaptin pulldown was determined by quantifying the ratio of μ2 or α-adaptin/KIR protein levels for Y to A mutants relative to WT (set to 100%). Data are representative of 3 independent experiments. D. 3DL1− (lane 1 only) and 3DL1+ KHYG-1 cells were lysed, immunoprecipitated with anti-3DL1 mAb and immunoblotted for α-adaptin, SHP-1 and KIR. L = ladder, WCL = whole cell lysate, E = empty lane. Approximate protein m.w. is indicated on the left-hand side of each immunoblot. Data are representative of 2 independent experiments.
Figure 5. The KIR/AP-2 interaction is regulated by KIR engagement and ITIM phosphorylation.

A. 3DL1 was immunoprecipitated (IP) from unstimulated (Unstim) 3DL1+ KHYG-1 cells or the same cells after treatment for 10 min on ice with pervanadate (PV), DX9 mAb (DX9) or pervanadate and DX9 mAb (PV+DX9). IPs were immunoblotted for phosphotyrosine (pY), α-adaptin, SHP-1 and KIR. B. Compilation of data from 5 experiments performed as in panel A. Band intensities were quantified by Image J and the relative band intensity calculated as a ratio to the intensity of the KIR band in each lane. Each icon represents an independent experiment with the mean value shown as a horizontal bar. The immunoblot shown in panel A was used to generate the band intensity data designated by the gray-filled square icons in panel B. Statistical analysis used the Student’s t test with * designating p ≤ 0.05 and ** defining p ≤ 0.01.
Retroviral and lentiviral constructs and transduction
To disrupt human μ2 (NM_001025205; ATCC, Manassas, VA) binding to tyrosine-based motifs, residues D176 and W420 were mutated to alanine [μ2-dominant negative (DN)]. To visualize KIR localization within the endosomal compartment, 3DL1*001 fused in-frame to mCherry (Genewiz, South Plainfield, NJ) and EYFP-Rab4 or EGFP-Rab7 (generous gift from Dr. Mario Zerial, Max Planck Institute, Dresden, Germany) were subcloned into pBMN-NoGFP to generate retrovirus as described (26, 34). For primary NK cell expression, constructs were subcloned into pCDH-EF1-MCS-T2A-copGFP (System Biosciences, Mountain View, CA) to generate lentivirus by transfecting LentiX 293T cells with pCDH, pMD2.G (VSV-G) and psPAX2 (gag/pol) plasmids (from Dr. Sam Kung, University of Manitoba, Winnipeg). Lentiviral supernatants were harvested 48–72 hr later, filtered and concentrated by ultracentrifugation or PEG precipitation. Viral titers were determined in LentiX 293T cells (35). Primary NK cells were infected on two consecutive days with lentivirus (MOI of 20–40 in 8 μg/ml polybrene). To alleviate compensatory mechanisms resulting from long-term expression of exogenous proteins, cells were assayed 48–72 hr after the second infection.
FACS-based KIR internalization assay
NK cell lines and primary NK cells were always assayed 3 to 4 days after passage into fresh IL-2-containing medium to improve consistency of the results. NK cells were stained with anti-3DL1 (PE-conjugated DX9) or -CD71 (TfR; BioLegend, San Diego, CA) mAbs at 4°C. A sample was left on ice (0 min) and remaining cells were incubated at 37°C for 5–180 min. Cells were moved to ice cold HBSS + 1% FBS + 0.1% NaN3, stained with AlexaFluor 647-conjugated anti-mouse IgG (Invitrogen, Grand Island, NY) at 4°C, and analyzed by flow cytometry on a BD LSR-II. Cell aggregates were gated out by FSH vs. FSA, and viable NK cells were gated by FSH vs. SSC and lack of propidium iodide (Invitrogen) staining. Lentiviral transduced cells were subgated into GFP− (uninfected) and GFP+ (infected) populations. For each sample, % Internalization = 100 – (2° Ab MFIt min/2° Ab MFI 0 min)*100. Jurkat T cells [Fig. 3C] were subjected to an acid wash stripping assay as described (16). Briefly, following internalization, cells were treated with 200 μl of HBSS containing 100 mM glycine and 100 mM NaCl (pH 2.5) at 4°C to remove surface-bound antibody. Cells were washed twice with HBSS plus 1% FBS, and 3DL1 expression was analyzed by FACS.
Figure 3. Mutation of the ITIM tyrosines slows 3DL1 internalization in NK cells.

A. Internalization assay and B. surface expression of 3DL1 (Left panels) and TfR (CD71, Right panels) in NKL cells expressing wild type (WT, ■) or Y377/407A mutant (AA, □) 3DL1. Cells were labeled with PE-conjugated anti-3DL1 or anti-CD71 mAbs at 4°C and incubated for 0–30 min at 37°C. The remaining surface-bound mAb was labeled with AlexaFluor 647-conjugated anti-mouse IgG at 4°C. The percent internalized was quantified by FACS. Surface expression (DX9 mAb MFI) was measured at time 0 min. Results in A and B are from the same 4 experiments, and paired values derived from individual experiments are connected by lines. C. KIR internalization was assessed using an acid-stripping assay in Jurkat T cells (see Methods). The fraction of 3DL1 internalized was quantified over a 2 hr time course (Left panel) and surface expression was assessed at time 0 min (Right panel) in Jurkat T cells that were retrovirally transduced to express 3DL1-WT or -AA. Results are individual determinations from 4 independent experiments, and paired values derived from individual experiments are connected by lines. D. Internalization assay (performed as in A; Left panel) and surface expression at time 0 min (Right panel) of 3DL1-WT or –AA expressed in primary NK cells by lentiviral transduction. 3DL1− NK cells sorted from 3 healthy donors were transduced to express 3DL1-WT or –AA. Presented data are from 7 experiments using cells derived from 6 independent transductions. Paired values derived from individual experiments are connected by lines and values from individual donors are represented by distinct icons. Statistical analysis used the Student’s t test with * designating p ≤ 0.05, ** defining p ≤ 0.01, and n.s. = not significant. E. A representative histogram of 3DL1-WT and 3DL1-AA surface expression from an experiment shown in D.
Results
After antibody engagement, 3DL1 moves to early/recycling and late endosomes
Previously, we provided evidence that 3DL1 internalizes and recycles back to the cell surface in NK cells (16). In that work, we showed that turnover of surface 3DL1 on transduced NK-92 cells was not changed over a 4-hr assay if 50 μg/ml cycloheximide was added to the cells, indicating that: 1) surface expression is quite stable, 2) recycling is occurring, and 3) trafficking to degradation pathways is minimal in the absence of de novo protein synthesis. To better define the subcellular distribution of 3DL1 and determine whether 3DL1 re-localizes to endosomal compartments following internalization from the cell surface, we expressed a fluorescently tagged version of 3DL1 (3DL1-Cherry) in NKL cells. First, we confirmed that the C-terminal tag did not disrupt inhibitory function (Figure 1A), which is consistent with a previous report using 3DL1-EGFP (36). Next, we quantified the amount of 3DL1 expressed on both the cell surface and within the endosomal compartment in fixed NKL cells co-expressing 3DL1-Cherry and either EYFP-Rab4, which marks early/recycling endosomes, or EGFP-Rab7, a late endosomal marker. As expected, a significant fraction of 3DL1-Cherry localized to the plasma membrane (median Manders’ coefficient of 0.3165 and 0.466 in EYFP-Rab4- and EGFP-Rab7-expressing cells, respectively) consistent with its established role in inhibitory signaling (Figure 1B) (3). We also found a significant amount localized to punctuate internal structures, with a small pool coinciding with Rab4+ endosomes (median Manders’ coefficient of 0.0615, Figure 1C, Left) and a more sizeable fraction co-localizing with Rab7+ endosomes (median Manders’ coefficient of 0.222, Figure 1C, Right). Together these data indicate that 3DL1 traffics from the cell surface through the endosomal compartments.
To visualize internalization specifically, we labeled 3DL1 expressed on the cell surface with the DX9 mAb at 4°C and quantified the co-localization of the anti-3DL1 mAb with EYFP-Rab4 (Figure 1D) and EGFP-Rab7 (Figure 1E) at 0–30 min of internalization at 37°C (37). We found a significant increase of anti-3DL1 mAbs co-localizing in Rab4+ endosomes at 15 min, which stabilized to a similar degree at 30 min (Figure 1F, Top). In contrast, significant co-localization of anti-3DL1 mAbs in Rab7+ late endosomes did not occur until the 30 min time point (Figure 1F, Bottom). Similarly, we also observed a significant increase in 3DL1-Cherry co-localized with Rab4+ endosomes at 15 min, and Rab7+ endosomes at 30 min (Figure 1G, Top and Bottom, respectively). We also observed a significant increase in anti-KIR mAbs co-localizing with 3DL1-Cherry at 15 and 30 min (Figures 1H), presumably due to an accumulation of the antibody-bound receptor with denser pools in endosomal compartments after internalization. Taken together, these data are consistent with a slow rate of internalization of antibody-labeled KIR3DL1 moving from the cell surface at time 0 min, to merge with intracellular compartments that include Rab4+ endosomes by 15 min and Rab7+ late endosomes by 30 min.
The μ2 subunit of AP-2 interacts with the cytoplasmic domain of 3DL1
As a means to identify proteins responsible for 3DL1 internalization, we performed a yeast two-hybrid screen using the cytoplasmic domain of 3DL1 as bait. In this screen, we identified 5 clones encoding the μ2 component of the AP-2 clathrin adaptor complex (residues 146 to 435; data not shown). The 3DL1/μ2 interaction was subsequently confirmed in yeast, along with the interaction of μ2 with the cytoplasmic domain of CD5 [which directly interacts with μ2 (38)] but not CD4 [which can only interact indirectly with μ2 through the HIV protein Nef (39)] (Fig. 2A). The μ2 protein interacts with cargo containing tyrosine (Y)-based motifs (Y-X-X-ϕ; X is any amino acid and ϕ is a hydrophobic residue) (19). 3DL1 has two potential μ2 binding sites located within the N- and C-terminal ITIMs, VTY377AQL and ILY407TEL, respectively. We mutated each tyrosine to alanine (A) and assayed their interactions with μ2 in a yeast two-hybrid reporter assay. Individual Y377A and Y407A mutants exhibited significantly decreased interaction with μ2, with the Y377A mutant being most affected (Fig. 2B). Disruption of both tyrosines completely abrogated the μ2/3DL1 interaction.
We next engineered wild type (WT) and mutant 3DL1 cytoplasmic domains as recombinant GST fusion proteins and probed 293T (Fig. 2C) or KHYG-1 cell lysates (data not shown) for interaction with the AP-2 complex. Consistent with the yeast two-hybrid results, 3DL1-WT interacted with both μ2 and α-adaptin of AP-2. In this assay, consistent with the yeast reporter assay, mutation of either Y377 alone or both tyrosines to alanine eliminated interaction with μ2 or α-adaptin, whereas the Y407A mutation only partially disrupted binding (Fig. 2C). Taken together, these in vitro data and the in vivo findings in yeast indicate that the μ2 subunit of AP-2 interacts with the cytoplasmic ITIM tyrosines of 3DL1. While Y377 is crucial for interaction with μ2, Y407 contributes, but is less imperative to binding.
We also tested if AP-2 could be co-immunoprecipitated with full length 3DL1 from NK cells. 3DL1 was isolated from a sorted subset of either 3DL1− or 3DL1+ KHYG-1 cells and probed for AP-2 by immunoblot. Consistent with the GST pulldown data, α-adaptin co-immunoprecipitated with 3DL1 (Fig. 2D).
AP-2 promotes 3DL1 internalization through interaction with ITIM tyrosines
In view of our observations by confocal microscopy that DX9 mAb causes endocytosis of 3DL1, we quantified endocytosis by first labeling cell surface 3DL1 with PE-conjugated-DX9 at 4°C, incubating the cells for various times at 37°C, and then staining with a fluorophore-tagged secondary antibody to determine the amount of DX9 retained on the cell surface (see Methods). To determine whether disruption of the 3DL1/μ2 interaction affects internalization of 3DL1, we compared the endocytic rate of wild type (WT) and Y377/407A (AA) receptor upon antibody binding in NKL cells. The rate of internalization of 3DL1-WT was slow, with less than 25% endocytosed by 30 minutes (Fig. 3A), consistent with our microscopy studies (Fig. 1). In contrast, internalization of the 3DL1-AA mutant was significantly delayed in NKL cells as compared to 3DL1-WT (Fig. 3A). Also, surface expression of 3DL1-AA was consistently higher than 3DL1-WT (Fig. 3B), indicating that disruption of association with the AP-2 clathrin adaptor results in accumulation of 3DL1 on the NK cell surface. In contrast, the rate of endocytosis and surface level of TfR were consistent in these same cells expressing 3DL1-WT or –AA (Fig. 3A and 3B). These data demonstrate that the methodology used to generate KIR-expressing NKL cells (e.g. retroviral transduction) did not globally affect receptor endocytosis and that the 3DL1–AA mutation specifically impacted 3DL1. To confirm that the results represented receptor internalization, rather than dissociation of primary DX9 or TfR antibody, we compared the changes in mean fluorescence intensities (MFI) of differentially fluorophore-conjugated primary and secondary Abs throughout the time course of the assay. Consistent with internalization, we observed a significant decrease in secondary Ab surface staining fluorescence over time, which did not track with a similar decrease in primary Ab fluorescence over the same time course (Suppl. Fig. 1). We also compared the internalization of both 3DL1-WT and -AA in Jurkat T cells using an acid stripping protocol. Again, 3DL1-AA was internalized at a slower rate and expressed at higher surface levels, as compared to 3DL1-WT (Fig. 3C). Unfortunately, primary NK cells and NK cell lines were found to be extremely sensitive to acid wash, which restricted the use of this assay to Jurkat cells.
Next, we compared surface expression and internalization rates in human primary NK cells. We utilized lentiviral transduction and sorting to express 3DL1-WT or –AA in CD3−CD56+3DL1− human primary NK cells. 3DL1-AA internalization was also significantly delayed in primary NK cells and surface expression was significantly elevated compared to 3DL1-WT (Fig. 3D and 3E). Collectively, we conclude that the endocytosis of 3DL1 depends, at least partially, on the cytoplasmic ITIMs, since tyrosine mutation significantly slowed internalization and increased surface expression in cell lines and primary NK cells.
Expression of dominant negative AP-2 reduces 3DL1 internalization
We next tested the impact of expressing a dominant negative (DN) form of the μ2 subunit of AP-2 on 3DL1 surface expression and internalization in primary NK cells. A D176A/W421 mutant of μ2 (designated μ2-DN) disrupts the interaction of μ2 to Y-X-X-ϕbearing cargo (like TfR and KIR) without affecting either the formation of the AP-2 complex or the internalization of dileucine motif-based cargo (21, 40, 41). Since primary NK cells express very low levels of TfR (our unpublished observations), we first showed that μ2-DN expression effectively delayed internalization and increased surface expression of TfR in KHYG-1 cells (Fig. 4A). We also measured surface levels of 3DL1 in NKL cells following expression of μ2-DN. Importantly, μ2-DN expression caused a significantly greater increase in the surface levels of TfR than in KIR surface levels on NKL cells. Furthermore, the impact was transient as the elevation of cell surface expression for both receptors was lost after one week of culture (Suppl. Fig. 2). From these results, we conclude that KIR surface expression levels are more tightly regulated than TfR and compensatory mechanisms rapidly diminish the efficacy of μ2-DN in NK cell lines. To avoid these compensatory mechanisms, we next analyzed the impact of short-term μ2-DN expression on 3DL1 internalization in primary NK cells. To this end, μ2-WT or μ2-DN were next expressed by lentiviral transduction in 3DL1+ human primary NK cells, and the transduced populations were identified by coordinate GFP expression (Fig. 4B). Expression of μ2-DN significantly delayed endocytosis (Figs. 4C and 4D) and increased surface expression of 3DL1 (Fig. 4D) as compared to expression of μ2-WT (Fig. 4C) or control transduction with empty vector (Fig. 4D). In contrast, lentivirus infection and resulting GFP expression alone did not impact 3DL1 internalization or surface expression (Suppl. Fig. 3). These data confirm that the AP-2 clathrin adaptor can significantly contribute to the endocytosis of 3DL1 and thereby influence the levels of receptor surface expression on NK cells.
Figure 4. Expression of dominant-negative μ2 (μ2-DN) delays internalization and increases surface expression of 3DL1 in primary NK cells.
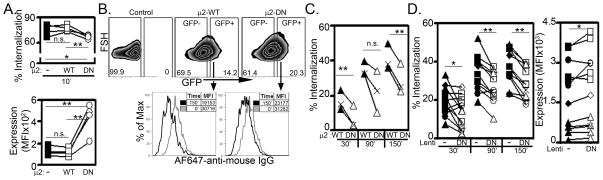
A. TfR internalization (Top panel) and surface expression (Bottom panel) were determined as in Figure 3 in control (−), μ2-WT-expressing, or μ2-DN-expressing KHYG-1 cells. Results are from individual determinations at 10 min (Top panel) or 0 min of internalization (Bottom panel) from 4 independent experiments, with values derived from individual experiments connected by lines. B. Infected primary NK cells are marked by GFP expression following lentiviral transduction and μ2-DN-expressing cells exhibit reduced surface expression of 3DL1. 3DL1+ primary human NK cells were infected with lentivirus containing μ2-WT or μ2-DN. The percentage of GFP− and GFP+ cells for each condition is indicated. Bottom panels: 3DL1 surface expression in the GFP+ populations at time 0 and following 150 min at 37°C with MFI of 3DL1 are shown. C. Data from 3 experiments performed as in B comparing 3DL1+ primary cells infected with lentivirus containing μ2-WT or μ2-DN. Paired values derived from individual experiments are connected by lines, and different donors are represented as distinct icons. D. 3DL1 internalization (Left panel) and surface expression (Right panel) are shown in primary NK cells infected with lentivirus (Lenti) generated with empty vector (−) or μ2-DN (DN) lentivirus. Shown are 12 experiments with NK cells from 5 healthy donors (separate icon/donor), and paired values derived from individual experiments are connected by lines. Statistical analysis used the Student’s t test with * designating p ≤ 0.05 and ** defining p ≤ 0.01.
The KIR/AP-2 interaction is regulated by antibody binding to 3DL1
The ability of KIR to inhibit NK cell cytotoxicity is dependent upon tyrosine phosphorylation of the ITIM tyrosines. Since we have found that AP-2 associates with KIR through these same tyrosines, we next tested if the KIR/AP-2 interaction is regulated by the phosphorylation state of these tyrosines. We hypothesized that since μ2 binds unphosphorylated tyrosines (19), the KIR/AP-2 interaction would be enhanced when the KIR ITIMs are not phosphorylated, but decreased when the ITIM tyrosines are phosphorylated. To test this hypothesis, we immunoprecipitated 3DL1 from unstimulated cells or cells stimulated with 1) pervandate (PV) alone to induce robust and stable tyrosine phosphoryation, 2) DX9 mAb alone, which mimics MHC-I engagement and should transiently increase ITIM phosphorylation (36, 42), or 3) both together. In accordance with previous publications (8, 43), PV-treated cells exhibited a high degree of KIR tyrosine phosphorylation and SHP-1 association. Furthermore, we found that the association of the α-adaptin subunit of AP-2 was significantly diminished following receptor engagement with DX9 in the presence or absence of PV, whereas PV alone reduced α-adaptin association only modestly, which did not reach statistical significance (Fig. 5A and 5B). Collectively these data show that the KIR/AP-2 association is most pronounced in unmanipulated cells, and although tyrosine phosphorylation can reduce the association, antibody engagement seems to further promote AP-2 displacement. In fact, DX9 mAb engagement alone significantly displaced α-adaptin binding, but did not induce tyrosine phosphorylation above baseline in this assay. We were unable to reproducibly observe differences in 3DL1 expression levels on NK cells that had been conjugated with target cells bearing or lacking HLA-B*51 ligand (data not shown). This could be due to inefficiency of ligand engagement under these conditions, however, resulting in only a minor fraction of the total surface 3DL1 being affected, thereby limiting detection of changes in surface levels on a per cell basis. This is consistent with the work of Treanor et al., which showed that only a small fraction of KIR in an immune synapse are phosphorylated in microclusters (44). In contrast, DX9 antibody has the potential to bind all the 3DL1 on the cell surface, and if DX9 binding is consistent with ligand engagement, our data suggest that KIR may be more susceptible to AP-2-dependent internalization when not engaged with ligand, whereas engagement with ligand would displace AP-2 to stabilize the receptor on the surface, where it can mediate prolonged inhibitory signaling to maintain NK cell tolerance.
Discussion
Our results show that 3DL1 can be slowly internalized, first to early/recycling endosomes and subsequently to late endosomes (Fig. 1). Moreover, at least part of this endocytic process is mediated by interaction between 1) the μ2 subunit of the AP-2 clathrin adaptor complex and 2) the ITIM motifs in the KIR cytoplasmic domain (Fig. 2–4). Our findings are consistent with previous report by Chwae et al. that found AP-2 interaction with a chimeric 3DL1 receptor construct in Jurkat T cells, however, an ITIM-mediated basis for the interaction was not defined (45). The same group also provided evidence that the amino-terminal ITIM and several additional sequence elements in the 3DL1 cytoplasmic domain are involved in endocytosis of this chimeric receptor in response to treating the Jurkat cells with PKC agonists (17). We cannot rule out alternative mechanisms that can also mediate KIR endocytosis, but our experiments studied the full-length receptor to characterize the role of AP-2 binding to ITIM tyrosines in primary NK cells. Furthermore, it is important to emphasize that endocytosis of TfR, which is considered to primarily involve AP-2/clathrin (21), was diminished to a similar degree as 3DL1 by expression of μ2-DN in our experiments (Fig. 4). On the other hand, μ2-DN expression resulted in significantly greater elevation of TfR surface expression as compared to 3DL1 (Figs. 4A and Suppl. Fig. 2). Based upon these observations, although AP-2/clathrin can mediate similar degrees of endocytosis of both receptors, we conclude that 3DL1 surface expression is more tightly controlled than TfR, presumably through more efficient recycling of KIR back to the cell surface. The more efficient retention of inhibitory KIR expression on the cell surface is in accordance with the critical role that these receptors play in tolerizing NK cells from attacking normal MHC-I+ cells in the body, whereas the primary function of TfR receptor is to internalize iron.
Following engagement with MHC-I at the immune synapse, KIR are phosphorylated on ITIM tyrosines in aggregated microclusters (44), leading to the recruitment of SHP-1/SHP-2 and inhibitory signaling (3, 7, 8). Since μ2 associates with unphosphorylated tyrosine-based motifs, we expected μ2 to interact with 3DL1 and induce endocytosis only when not engaged with ligand. A similar mechanism has been described for CTLA-4, on which phosphorylation disrupts recruitment of μ2 to a cytoplasmic tyrosine to regulate endocytosis (46, 47). Although we were surprised that pervanadate-induced tyrosine phosphorylation of 3DL1 did not significantly displace α-adaptin binding, it is possible that the pool of 3DL1 associated with AP-2 was not efficiently phosphorylated under these conditions. Instead, we found that the KIR/AP-2 interaction is most profoundly diminished following engagement with DX9 antibody in the presence or absence of pervanadate (Fig. 5). The lack of significant detectable tyrosine phosphorylation of 3DL1 by treatment with DX9 alone suggests that antibody-mediated displacement of AP-2 may result through a mechanism independent of ITIM tyrosine phosphorylation. It is possible that antibody binding induces additional changes in the receptor cytoplasmic domain (in addition to just tyrosine phosphorylation) to more effectively dissociate the clathrin adaptor. Although this mechanism has not been defined, if antibody binding is characteristic of ligand engagement, our results suggest that engaged KIR are maintained at the target cell interface to mediate prolonged inhibitory signaling and sustained self-tolerance toward normal MHC-I-bearing cells. Furthermore, although DX9-engagement for 10 minutes decreased the interaction of KIR with AP-2 (Fig. 5), the antibody-engaged receptor was ultimately slowly internalized by an ITIM/AP-2-dependent process over a longer time course, as shown in our internalization assays (Fig. 3 and 4) and microscopy studies (Fig. 1). In contrast, our data further imply that AP-2-mediated endocytosis of 3DL1 would presumably occur more readily when NK cells are engaged with MHC-I-deficient cells, thereby more efficiently removing the inhibitory KIR from the immune synapse to allow more efficient cytotoxicity. Activating KIR (KIR2DS, KIR3DS) lack full ITIMs and would therefore not be able to directly recruit AP-2. It is possible that the activating KIR can be endocytosed through another mechanism, however, including potential μ2 binding to the ITAM tyrosines of the associated DAP12 adaptor, similar to a recent report of AP-2-mediated BCR internalization through interaction with an ITAM on CD79b (48).
Sequence motifs in KIR that exist outside of the cytoplasmic domain have also been implicated in contributing to endocytosis. Upon binding to CpG oligodeoxynucleotides, KIR3DL2 reportedly relocalizes from the cell surface to early endosomes, thereby transporting the CpG to interact with TLR9 at that location (12). Surprisingly, relocalization in that context was reportedly independent of the cytoplasmic domain, since truncation distal to the transmembrane domain had no impact upon KIR3DL2 internalization. In that report, 3DL1 internalization was also observed upon binding with CpG DNA, although to a lesser extent than KIR3DL2. Furthermore, KIR2DL4, a unique activating receptor that contains a single ITIM (49), can also internalize to early endosomes, where it can mediate intracellular signaling or be degraded following ubiquitylation (13, 25). Published data, however, suggest that internalization of KIR2DL4 is independent of the transmembrane and cytoplasmic domains, since a chimeric receptor consisting of the extracellular domain of KIR2DL4 and transmembrane/cytoplasmic domains of the plasma membrane-localized gp49B receptor was also targeted to endosomes (50). These studies further reinforce the functional relevance of KIR endocytosis and the roles of sequence elements outside of the cytoplasmic domain in mediating internalization. Our work has identified the interaction of the AP-2/clathrin complex with the cytoplasmic ITIMs of 3DL1 as one mechanism shuttling KIR from the cell surface. Although this mechanism is expected to also be operational for other KIR, further analysis is warranted to specifically examine the contributions of ITIM/AP-2 interactions on the endocytosis of KIR3DL2 and KIR2DL4.
Given that KIR-dependent inhibition of NK cell activation is rapid [tyrosine phosphorylation and SHP-1/SHP-2 recruitment occur within minutes of ligand engagement (51)], while the rate of 3DL1 internalization in human primary NK cells is slow (only 19.7 ± 7.25% internalized by 30 min; Fig. 3D, 4C, and 4D), it is unlikely that AP-2-mediated endocytosis of 3DL1 contributes directly to inhibitory function. It is possible, however, that this AP-2-mediated mechanism may also be involved in the KIR-dependent physical transfer of HLA molecules from target cells into NK cells that has previously been reported (52). Alternatively, our data and the consistent expression levels of 3DL1 on the surface of NK cells, suggest that KIR normally undergo constitutive internalization and recycling to maintain inhibitory capacity and tolerance. Although we cannot rule out involvement of other endocytic mechanisms regulating KIR surface expression, our findings define a new functional role for the ITIMs on 3DL1. CTLA4 has been shown to recruit AP-2 through a non-ITIM tyrosine (46, 47, 53), but to our knowledge, our data are the first to demonstrate that AP-2 can internalize an inhibitory receptor through an ITIM binding site. This mechanism may more generally target additional ITIM-bearing receptors for endocytosis.
Supplementary Material
Acknowledgments
We thank Drs. David Wiest and Alana O’Reilly for constructive critique of the manuscript, Drs. Sam Kung and Erica Golemis for reagents and advice, Drs. Alexander MacFarlane IV and Sam Litwin for help with the Wilcoxon rank sum test, and the DNA Sequencing, Flow Cytometry, Bioinformatics & Biostatistics, and Cell Culture Facilities at FCCC for materials and technical support.
Footnotes
This work was supported by NIH grants CA083859 (K.S.C), CA009035 (A.K.P. and D.A.A.-A.), and CA06927 (FCCC) and a Health Research Formula Fund (CURE) grant from the PA Department of Health (K.S.C), which specifically disclaims responsibility for analyses, interpretations or conclusions.
References
- 1.Wallace ME, Smyth MJ. The role of natural killer cells in tumor control--effectors and regulators of adaptive immunity. Springer seminars in immunopathology. 2005;27:49–64. doi: 10.1007/s00281-004-0195-x. [DOI] [PubMed] [Google Scholar]
- 2.Campbell KS, Hasegawa J. Natural killer cell biology: an update and future directions. The Journal of allergy and clinical immunology. 2013;132:536–544. doi: 10.1016/j.jaci.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.MacFarlane AW, 4th, Campbell KS. Signal transduction in natural killer cells. Current topics in microbiology and immunology. 2006;298:23–57. doi: 10.1007/3-540-27743-9_2. [DOI] [PubMed] [Google Scholar]
- 4.Lanier LL. NK cell recognition. Annual review of immunology. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 5.Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436:1186–1190. doi: 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rolle A, Mousavi-Jazi M, Eriksson M, Odeberg J, Soderberg-Naucler C, Cosman D, Karre K, Cerboni C. Effects of human cytomegalovirus infection on ligands for the activating NKG2D receptor of NK cells: up-regulation of UL16-binding protein (ULBP)1 and ULBP2 is counteracted by the viral UL16 protein. Journal of immunology. 2003;171:902–908. doi: 10.4049/jimmunol.171.2.902. [DOI] [PubMed] [Google Scholar]
- 7.Campbell KS, Dessing M, Lopez-Botet M, Cella M, Colonna M. Tyrosine phosphorylation of a human killer inhibitory receptor recruits protein tyrosine phosphatase 1C. The Journal of experimental medicine. 1996;184:93–100. doi: 10.1084/jem.184.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yusa S, Campbell KS. Src homology region 2-containing protein tyrosine phosphatase-2 (SHP-2) can play a direct role in the inhibitory function of killer cell Ig-like receptors in human NK cells. Journal of immunology. 2003;170:4539–4547. doi: 10.4049/jimmunol.170.9.4539. [DOI] [PubMed] [Google Scholar]
- 9.Peterson ME, Long EO. Inhibitory receptor signaling via tyrosine phosphorylation of the adaptor Crk. Immunity. 2008;29:578–588. doi: 10.1016/j.immuni.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anfossi N, Andre P, Guia S, Falk CS, Roetynck S, Stewart CA, Breso V, Frassati C, Reviron D, Middleton D, Romagne F, Ugolini S, Vivier E. Human NK cell education by inhibitory receptors for MHC class I. Immunity. 2006;25:331–342. doi: 10.1016/j.immuni.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 11.Almeida CR, Ashkenazi A, Shahaf G, Kaplan D, Davis DM, Mehr R. Human NK cells differ more in their KIR2DL1-dependent thresholds for HLA-Cw6-mediated inhibition than in their maximal killing capacity. PloS one. 2011;6:e24927. doi: 10.1371/journal.pone.0024927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sivori S, Falco M, Carlomagno S, Romeo E, Soldani C, Bensussan A, Viola A, Moretta L, Moretta A. A novel KIR-associated function: evidence that CpG DNA uptake and shuttling to early endosomes is mediated by KIR3DL2. Blood. 2010;116:1637–1647. doi: 10.1182/blood-2009-12-256586. [DOI] [PubMed] [Google Scholar]
- 13.Rajagopalan S, Bryceson YT, Kuppusamy SP, Geraghty DE, van der Meer A, Joosten I, Long EO. Activation of NK cells by an endocytosed receptor for soluble HLA-G. PLoS biology. 2006;4:e9. doi: 10.1371/journal.pbio.0040009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campbell KS, Purdy AK. Structure/function of human killer cell immunoglobulin-like receptors: lessons from polymorphisms, evolution, crystal structures and mutations. Immunology. 2011;132:315–325. doi: 10.1111/j.1365-2567.2010.03398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parham P. Taking license with natural killer cell maturation and repertoire development. Immunological reviews. 2006;214:155–160. doi: 10.1111/j.1600-065X.2006.00462.x. [DOI] [PubMed] [Google Scholar]
- 16.Alvarez-Arias DA, Campbell KS. Protein kinase C regulates expression and function of inhibitory killer cell Ig-like receptors in NK cells. Journal of immunology. 2007;179:5281–5290. doi: 10.4049/jimmunol.179.8.5281. [DOI] [PubMed] [Google Scholar]
- 17.Chwae YJ, Lee JM, Kim HR, Kim EJ, Lee ST, Soh JW, Kim J. Amino-acid sequence motifs for PKC-mediated membrane trafficking of the inhibitory killer Ig-like receptor. Immunology and cell biology. 2008;86:372–380. doi: 10.1038/icb.2008.5. [DOI] [PubMed] [Google Scholar]
- 18.Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annual review of biochemistry. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- 19.Traub LM. Tickets to ride: selecting cargo for clathrin-regulated internalization. Nature reviews. Molecular cell biology. 2009;10:583–596. doi: 10.1038/nrm2751. [DOI] [PubMed] [Google Scholar]
- 20.Mayor S, Pagano RE. Pathways of clathrin-independent endocytosis. Nature reviews. Molecular cell biology. 2007;8:603–612. doi: 10.1038/nrm2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nesterov A, Carter RE, Sorkina T, Gill GN, Sorkin A. Inhibition of the receptor-binding function of clathrin adaptor protein AP-2 by dominant-negative mutant mu2 subunit and its effects on endocytosis. The EMBO journal. 1999;18:2489–2499. doi: 10.1093/emboj/18.9.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kibbey RG, Rizo J, Gierasch LM, Anderson RG. The LDL receptor clustering motif interacts with the clathrin terminal domain in a reverse turn conformation. The Journal of cell biology. 1998;142:59–67. doi: 10.1083/jcb.142.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grandal MV, Grovdal LM, Henriksen L, Andersen MH, Holst MR, Madshus IH, van Deurs B. Differential Roles of Grb2 and AP-2 in p38 MAPK- and EGF-Induced EGFR Internalization. Traffic. 2011 doi: 10.1111/j.1600-0854.2011.01322.x. [DOI] [PubMed] [Google Scholar]
- 24.Masilamani M, Narayanan S, Prieto M, Borrego F, Coligan JE. Uncommon endocytic and trafficking pathway of the natural killer cell CD94/NKG2A inhibitory receptor. Traffic. 2008;9:1019–1034. doi: 10.1111/j.1600-0854.2008.00738.x. [DOI] [PubMed] [Google Scholar]
- 25.Miah SM, Purdy AK, Rodin NB, MacFarlane AW, 4th, Oshinsky J, Alvarez-Arias DA, Campbell KS. Ubiquitylation of an internalized killer cell Ig-like receptor by Triad3A disrupts sustained NF-kappaB signaling. Journal of immunology. 2011;186:2959–2969. doi: 10.4049/jimmunol.1000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Purdy AK, Campbell KS. SHP-2 expression negatively regulates NK cell function. Journal of immunology. 2009;183:7234–7243. doi: 10.4049/jimmunol.0900088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cella M, Colonna M. Cloning human natural killer cells. Methods in molecular biology. 2000;121:1–4. doi: 10.1385/1-59259-044-6:1. [DOI] [PubMed] [Google Scholar]
- 28.Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. Journal of microscopy. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 29.Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. Journal of cell science. 1992;103(Pt 3):857–862. doi: 10.1242/jcs.103.3.857. [DOI] [PubMed] [Google Scholar]
- 30.Campbell KS, Buder A, Deuschle U. Interactions between the amino-terminal domain of p56lck and cytoplasmic domains of CD4 and CD8 alpha in yeast. European journal of immunology. 1995;25:2408–2412. doi: 10.1002/eji.1830250842. [DOI] [PubMed] [Google Scholar]
- 31.Calvo J, Vilda JM, Places L, Simarro M, Padilla O, Andreu D, Campbell KS, Aussel C, Lozano F. Human CD5 signaling and constitutive phosphorylation of C-terminal serine residues by casein kinase II. Journal of immunology. 1998;161:6022–6029. [PubMed] [Google Scholar]
- 32.Kloeker S, Reed R, McConnell JL, Chang D, Tran K, Westphal RS, Law BK, Colbran RJ, Kamoun M, Campbell KS, Wadzinski BE. Parallel purification of three catalytic subunits of the protein serine/threonine phosphatase 2A family (PP2A(C), PP4(C), and PP6(C)) and analysis of the interaction of PP2A(C) with alpha4 protein. Protein expression and purification. 2003;31:19–33. doi: 10.1016/s1046-5928(03)00141-4. [DOI] [PubMed] [Google Scholar]
- 33.Faure M, Barber DF, Takahashi SM, Jin T, Long EO. Spontaneous clustering and tyrosine phosphorylation of NK cell inhibitory receptor induced by ligand binding. Journal of immunology. 2003;170:6107–6114. doi: 10.4049/jimmunol.170.12.6107. [DOI] [PubMed] [Google Scholar]
- 34.Yusa S, Catina TL, Campbell KS. SHP-1- and phosphotyrosine-independent inhibitory signaling by a killer cell Ig-like receptor cytoplasmic domain in human NK cells. Journal of immunology. 2002;168:5047–5057. doi: 10.4049/jimmunol.168.10.5047. [DOI] [PubMed] [Google Scholar]
- 35.Kung SK. Introduction of shRNAs into primary NK cells with lentivirus. Methods in molecular biology. 2010;612:233–247. doi: 10.1007/978-1-60761-362-6_16. [DOI] [PubMed] [Google Scholar]
- 36.Sharma D, Bastard K, Guethlein LA, Norman PJ, Yawata N, Yawata M, Pando M, Thananchai H, Dong T, Rowland-Jones S, Brodsky FM, Parham P. Dimorphic motifs in D0 and D1+D2 domains of killer cell Ig-like receptor 3DL1 combine to form receptors with high, moderate, and no avidity for the complex of a peptide derived from HIV and HLA-A*2402. Journal of immunology. 2009;183:4569–4582. doi: 10.4049/jimmunol.0901734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Novick P, Zerial M. The diversity of Rab proteins in vesicle transport. Current opinion in cell biology. 1997;9:496–504. doi: 10.1016/s0955-0674(97)80025-7. [DOI] [PubMed] [Google Scholar]
- 38.Lu X, Axtell RC, Collawn JF, Gibson A, Justement LB, Raman C. AP2 adaptor complex-dependent internalization of CD5: differential regulation in T and B cells. Journal of immunology. 2002;168:5612–5620. doi: 10.4049/jimmunol.168.11.5612. [DOI] [PubMed] [Google Scholar]
- 39.Chaudhuri R, Mattera R, Lindwasser OW, Robinson MS, Bonifacino JS. A basic patch on alpha-adaptin is required for binding of human immunodeficiency virus type 1 Nef and cooperative assembly of a CD4-Nef-AP-2 complex. Journal of virology. 2009;83:2518–2530. doi: 10.1128/JVI.02227-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Collawn JF, Stangel M, Kuhn LA, Esekogwu V, Jing SQ, Trowbridge IS, Tainer JA. Transferrin receptor internalization sequence YXRF implicates a tight turn as the structural recognition motif for endocytosis. Cell. 1990;63:1061–1072. doi: 10.1016/0092-8674(90)90509-d. [DOI] [PubMed] [Google Scholar]
- 41.Jing SQ, Spencer T, Miller K, Hopkins C, Trowbridge IS. Role of the human transferrin receptor cytoplasmic domain in endocytosis: localization of a specific signal sequence for internalization. The Journal of cell biology. 1990;110:283–294. doi: 10.1083/jcb.110.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kurago ZB, Lutz CT, Smith KD, Colonna M. NK cell natural cytotoxicity and IFN-gamma production are not always coordinately regulated: engagement of DX9 KIR+ NK cells by HLA-B7 variants and target cells. Journal of immunology. 1998;160:1573–1580. [PubMed] [Google Scholar]
- 43.Yusa S, Catina TL, Campbell KS. KIR2DL5 can inhibit human NK cell activation via recruitment of Src homology region 2-containing protein tyrosine phosphatase-2 (SHP-2) Journal of immunology. 2004;172:7385–7392. doi: 10.4049/jimmunol.172.12.7385. [DOI] [PubMed] [Google Scholar]
- 44.Treanor B, Lanigan PM, Kumar S, Dunsby C, Munro I, Auksorius E, Culley FJ, Purbhoo MA, Phillips D, Neil MA, Burshtyn DN, French PM, Davis DM. Microclusters of inhibitory killer immunoglobulin-like receptor signaling at natural killer cell immunological synapses. The Journal of cell biology. 2006;174:153–161. doi: 10.1083/jcb.200601108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chwae YJ, Lee JM, Kim EJ, Lee ST, Soh JW, Kim J. Activation-induced upregulation of inhibitory killer Ig-like receptors is regulated by protein kinase C. Immunology and cell biology. 2007;85:220–228. doi: 10.1038/sj.icb.7100031. [DOI] [PubMed] [Google Scholar]
- 46.Shiratori T, Miyatake S, Ohno H, Nakaseko C, Isono K, Bonifacino JS, Saito T. Tyrosine phosphorylation controls internalization of CTLA-4 by regulating its interaction with clathrin-associated adaptor complex AP-2. Immunity. 1997;6:583–589. doi: 10.1016/s1074-7613(00)80346-5. [DOI] [PubMed] [Google Scholar]
- 47.Bradshaw JD, Lu P, Leytze G, Rodgers J, Schieven GL, Bennett KL, Linsley PS, Kurtz SE. Interaction of the cytoplasmic tail of CTLA-4 (CD152) with a clathrin-associated protein is negatively regulated by tyrosine phosphorylation. Biochemistry. 1997;36:15975–15982. doi: 10.1021/bi971762i. [DOI] [PubMed] [Google Scholar]
- 48.Busman-Sahay K, Drake L, Sitaram A, Marks M, Drake JR. Cis and trans regulatory mechanisms control AP2-mediated B cell receptor endocytosis via select tyrosine-based motifs. PloS one. 2013;8:e54938. doi: 10.1371/journal.pone.0054938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kikuchi-Maki A, Yusa S, Catina TL, Campbell KS. KIR2DL4 is an IL-2-regulated NK cell receptor that exhibits limited expression in humans but triggers strong IFN-gamma production. Journal of immunology. 2003;171:3415–3425. doi: 10.4049/jimmunol.171.7.3415. [DOI] [PubMed] [Google Scholar]
- 50.Rajagopalan S, Moyle MW, Joosten I, Long EO. DNA-PKcs controls an endosomal signaling pathway for a proinflammatory response by natural killer cells. Science signaling. 2010;3:ra14. doi: 10.1126/scisignal.2000467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abeyweera TP, Merino E, Huse M. Inhibitory signaling blocks activating receptor clustering and induces cytoskeletal retraction in natural killer cells. The Journal of cell biology. 2011;192:675–690. doi: 10.1083/jcb.201009135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carlin LM, Eleme K, McCann FE, Davis DM. Intercellular transfer and supramolecular organization of human leukocyte antigen C at inhibitory natural killer cell immune synapses. The Journal of experimental medicine. 2001;194:1507–1517. doi: 10.1084/jem.194.10.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chuang E, Alegre ML, Duckett CS, Noel PJ, Vander Heiden MG, Thompson CB. Interaction of CTLA-4 with the clathrin-associated protein AP50 results in ligand-independent endocytosis that limits cell surface expression. Journal of immunology. 1997;159:144–151. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.