

Abstract
Precise regulation of the kinetics and magnitude of Ca2+ signaling enables this signal to mediate diverse responses, such as cell migration, differentiation, vesicular trafficking, and cell death. Here, we showed that the Ca2+-binding protein calmodulin (CaM) acted in a positive feedback loop to potentiate Ca2+ signaling downstream of the Tec kinase family member Itk. Using NMR (nuclear magnetic resonance), we mapped CaM binding to two loops adjacent to the lipid-binding pocket within the Itk pleckstrin homology (PH) domain. The Itk PH domain bound synergistically to Ca2+/CaM and the lipid phosphatidylinositol-3,4,5-trisphosphate [PI(3,4,5)P3], such that binding to Ca2+/CaM enhanced the binding to PI(3,4,5)P3 and vice versa. Disruption of CaM binding attenuated Itk recruitment to the membrane and diminished release of Ca2+ from the endoplasmic reticulum. Moreover, disruption of this feedback loop abrogated Itk-dependent production of the proinflammatory cytokine IL-17A (interleukin-17A) by CD4+ T cells. Additionally, we found that CaM associated with PH domains from other proteins, indicating that CaM may regulate other PH domain–containing proteins.
Introduction
The pleckstrin homology (PH) domain is a beta barrel formed by two antiparallel β sheets and a C-terminal amphipathic helix and was initially identified over 20 years ago as a repeated domain in the protein Pleckstrin (1). Since its discovery, PH domains have been recognized in proteins from bacteria to mammals (2, 3). Several hundred mammalian proteins that participate in diverse cellular functions contain one or multiple PH domains. PH domains are generally recognized as membrane-targeting domains (4, 5); although these domains may have other functions as well. Lipid-binding PH domains have positively charged residues in the loops between specific β strands (β1/β2, β3/β4, and β6/β7) and these charged residues differentially interact with negatively charged lipids, including phosphoinositides, such as phosphatidylinositol-4-phosphate [PI4P], phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2], phosphatidylinositol-3,4-bisphosphate [PI(3,4)P2], and phosphatidylinositol-3,4,5-trisphosphate [PI(3,4,5)P3] (1). Although most lipid-binding PH domains interact weakly or promiscuously with a range of lipid targets, a few PH domains have high specificity and submicromolar affinity for specific phospholipids (1). Among these, the PH domains of Akt and Tec family kinases interact with PI(3,4,5)P3, a plasma membrane phospholipid that is generated by phosphatidylinositol -3 kinase (PI3K) following receptor activation (6). The Akt family of serine/threonine kinases promotes survival and proliferation in most cell types. The Tec family of tyrosine kinases is critical for the development and activation of immune cells. Mutations in the PH domain of the Tec kinase Bruton’s tyrosine kinase (Btk) that disrupt PI(3,4,5)P3 binding result in defective B cell responses, causing a primary immunodeficiency disease known as X-linked agammaglobulinaemia (XLA) in humans and X-linked immunodeficiency (Xid) in mice (7, 8). In contrast, a Btk mutant (E41K) that increases the association of the PH domain with phospholipids is constitutively active and has cellular transforming activity (9).
Mutation in humans or gene targeting in mice of the Tec kinase IL-2 inducible tyrosine kinase (Itk) disrupts T cell function, resulting in primary immunodeficiency disease (10, 11). Itk-deficient mice fail to mount a protective T helper type 2 (TH2) response to parasites, including Nippostrongylus brasiliensis, Schistosoma mansoni, and Leishmania major (12, 13). Itk activity is required for optimal T cell activation and expansion; Itk-deficient T cells ultimately fail to provide immune protection due to insufficient production of the cytokine interleukin 4 (IL-4) due to reduced activation of the transcription factor NFATc (12). Similarly, NFATc-dependent production of the pro-inflammatory cytokine IL-17A is also disrupted in Itk-deficient T cells (14). Optimal IL-17A depends on maximal signaling by the T cell receptor (TCR) and Itk activation in particular (14). Given the importance of IL-17A in promoting contact hypersensitivity, collagen-induced arthritis, and experimental autoimmune encephalomyelitis (15), targeting Itk pharmacologically may ameliorate some T cell-mediated autoimmune diseases.
Tec kinases promote cellular responses by activating phospholipase Cγ (PLCγ) to generate the second messengers, diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3) (10, 11). IP3 production triggers Ca2+ signaling (16, 17). Cytosolic Ca2+ concentrations in resting cells are maintained at a low concentration, typically 100 nM, by actively pumping free Ca2+ into the endoplasmic reticulum (ER) and into the extracellular space where Ca2+ concentrations are in the mM range (16). IP3 binding to IP3 receptors on the ER triggers release of ER-stored Ca2+ and subsequent Ca2+ influx from the extracellular environment through store-operated plasma membrane channels. The large Ca2+ concentration differential present in resting cells provides a powerful and rapid mechanism to activate cellular responses through regulation of cytosolic Ca2+ concentrations.
Ca2+ directly alters the activity of proteins by binding to C2 and EF hand domains (16). Ca2+ also indirectly activates effectors by regulating the conformation of the evolutionarily conserved allosteric regulator calmodulin (CaM). Ca2+ binding to the four EF hands of CaM (defined as Ca2+/CaM) promotes its association with many cellular enzymes and ion channels, leading to their activation or deactivation (16). Ca2+/CaM-dependent effectors include myosin light chain kinase (MLCK), CaM kinases, and calcineurin, which induce myosin-dependent cellular contraction, cellular differentiation, and NFAT-dependent gene transcription, respectively.
Here, we investigated whether CaM bound and regulated the PI(3,4,5)P3 -binding PH domains of the Tec family kinases. We found that Ca2+/CaM binds to Itk, but not the related Tec kinase Btk. To examine the interaction between Ca2+/CaM and Itk further, we used nuclear magnetic resonance (NMR) spectroscopy to map CaM binding to two loops within the Itk PH domain that are adjacent to the lipid-binding pocket. Ca2+/CaM and PI(3,4,5)P3 cooperated to enhance binding to either ligand. Disruption of Ca2+/CaM binding attenuated Itk recruitment to the membrane and subsequent activation of PLCγ1, indicating that Ca2+/CaM binding to the Itk PH domain acts in a positive feedback loop to potentiate and sustain Ca2+ signaling. Disruption of this feedback loop abrogated Itk-dependent production of the pro-inflammatory cytokine IL-17A by CD4+ T cells. Moreover, we present data that Ca2+/CaM may be a general binding partner and potential regulator of other proteins with PH domains.
Results
Ca2+/CaM binds to the Itk PH domain but not the Btk PH domain
To test whether CaM interacts with the Tec kinases Itk and Btk, we incubated mouse splenocyte lysates with CaM-coated beads and assessed the presence of endogenous Itk or Btk in the precipitated samples by Western blot(Fig. 1A). As a positive control, we coprecipitated both kinases using PI(3,4,5)P3-coated beads. Both Tec kinases bound to the phospholipid ligand; however, only Itk interacted with CaM and more Itk was present in the precipitates as the concentration of Ca2+ was increased in the precipitation buffer. Coprecipitation experiments with yellow fluorescent protein (YFP) fused to either full-length Itk or the with the PH domain of Itk showed that the PH domain of Itk mediated the interaction with CaM (fig. S1). We further characterized the interaction using recombinant Itk PH domain generated and purified from bacteria and CaM. The isolated Itk PH domain bound directly to CaM in a Ca2+-dependent manner; a Ca2+concentration of 1 μM was used to mimic cytosolic Ca2+ concentrations in activated T cells (Fig. 1B). The presence of the Ca2+-chelator EGTA reduced the interaction between CaM and the Itk PH domain (Fig. 1B).
Fig 1. Itk association with CaM is Ca2+ dependent and enhanced by PI(3,4,5)P3 binding.

(A) CaM coprecipitation of endogenous Itk or Btk from mouse splenocyte lysates was perfomed with the indicated concentrations of Ca2+ in the precipitation buffer. Affinity purification with PI(3,4,5)P3-coated beads served as a positive control. (B) Direct comparison of recombinant Itk PH domain binding to PI(3,4,5)P3, Apo-CaM (EGTA), and Ca2+/CaM (Ca2+). (C) Effect of PIP3, IP4 and PIP2 addition on the coprecipitation of Itk or calcineuron with CaM from primary T cell lysates. (D) Dose-dependent enhancement of Itk binding to CaM by addition of PI(3,4,5)P3. All data are representative of 3 experiments. PIP2, PI(4,5)P2; PIP3, PI(3,4,5)P3; IP4, Ins(1,3,4,5)P4; ppt, precipitation; rItkPH, recombinant Itk PH domain
PI(3,4,5)P3 promotes CaM binding to the Itk PH domain
Because Itk activation requires PH domain-mediated recruitment to the membrane by PI(3,4,5)P3, we investigated the effect of PI(3,4,5)P3 binding on the Itk PH domain interaction with CaM by adding soluble phosphatidylinositides to the cellular lysates during CaM precipitation. Remarkably, PI(3,4,5)P3, but not Ins(1,3,4,5)P4 [a physiological mimic of the PI(3,4,5)P3 head group] or its membrane precursor PI(4,5)P2, enhanced CaM binding to endogenous Itk in a dose-dependent manner (Fig. 1C and D). These data indicated that both Ca2+ and PI(3,4,5)P3 enhanced the binding of the Itk PH domain to CaM.
Structural characterization of the Itk PH-CaM binding interface
We used NMR spectroscopy to identify the specific residues in both CaM and the Itk PH domain that mediate the interaction. We measured the resonance frequencies of each amide N-H group in the protein (either CaM or the C96E/T110I Itk PH domain variant, which facilitates PH domain solubilization (18)) using the 1H-15N Heteronuclear Single Quantum Correlation (HSQC) spectrum and assigned the backbone N-H resonances of CaM using data from the Biological Magnetic Resonance Data Bank (BMRB) and assigned the backbone N-H resonances for the Itk PH domain using the standard suite of triple-resonance NMR experiments (see Materials and Methods).
We then added the unlabeled Itk PH domain to 15N-labeled CaM (Fig. 2A) or unlabeled CaM to the 15N-labeled Itk PH domain (Fig. 2B). The spectral changes in both titrations revealed extensive line broadening upon addition of increasing concentrations of binding partner (Fig. 2A and B). Such line broadening can be ascribed to the size of the CaM-Itk PH complex, can indicate a protein-protein interaction that is undergoing intermediate exchange on the NMR timescale, or can result from both. A subset of the CaM and Itk PH domain resonances showed pronounced spectral changes upon addition of small amounts of binding partner (Fig. 2A and B) consistent with formation of a specific complex. Mapping of the residues that correspond to these resonances onto a structure of Ca2+/CaM (19) and a model of the Itk PH domain (see Materials and Methods) revealed that these residues cluster to contiguous regions on the tertiary structure of each domain (Fig. 3A and B). In Ca2+/CaM, the residues showing the largest spectral shift occur in both the N- and C-domains (Fig. 2A and 3A). The region of the Itk PH domain involved in the interaction with Ca2+/CaM includes the β3/β4 and β5/β6 loops, the adjacent β-strands 2, 3 and 4 and portions of β1 and β5 (Fig. 3B). These regions of the Itk PH domain are adjacent to and not overlapping with the PI(3,4,5)P3 binding pocket (Fig. 3B and C), consistent with an allosteric mechanism for PI(3,4,5)P3 enhancement of CaM binding (Fig. 1C and D).
Fig 2. Structural characterization of the binding interface between the Itk PH domain and CaM.

(A) (top) Overlay of 1H-15N-HSQC spectra of 150 μM 15N-Ca2+/CaM with unlabeled Itk PH domain titrated at indicated molar ratios (red to green); residues with the largest spectral changes upon binding are labeled. (bottom) Representative regions of the 1H-15N-HSQC spectra for the first three points in the titration showing line-broadening of the selected CaM resonances (labeled in red and boxed). Resonances that show only partial line broadening are labeled in black. (B) (top) Overlay of 1H-15N-HSQC spectra of 300 μM 15N-ItkPH with unlabeled Ca2+/CaM titrated at indicated molar ratios (red to green); residues showing significant spectral changes are labeled. (bottom) Representative regions of the 1H-15N-HSQC spectra for the first three points in the titration showing line-broadening of the selected Itk PH domain resonances (labeled in red). Resonances that show modest line broadening (presumably due to increased molecular weight of the complex rather than direct interaction with CaM) are labeled in black. Asterisk (*) indicates resonances that could not be unequivocally assigned.
Fig 3. Structural models of the binding interface between the Itk PH domain and CaM.
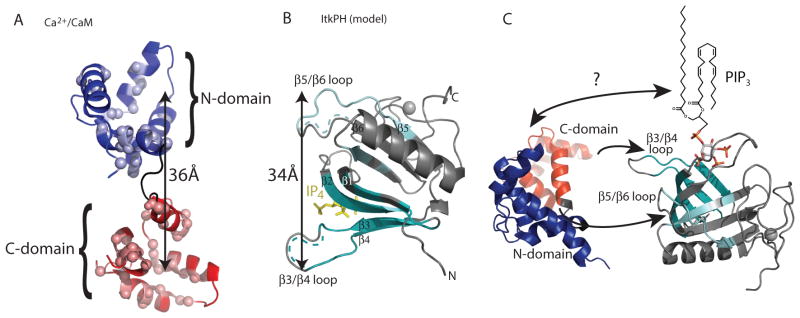
(A) Spectral changes induced by addition of the Itk PH domain mapped onto the Ca2+/CaM structure (PDB entry 2KDU). Blue spheres indicate N-domain residues perturbed on binding of the Itk PH domain to Ca2+/CaM and red spheres indicate C-domain residues affected by binding. The doubled headed arrow indicates the 36Å distance between the two lobes in this extended structure of Ca2+/CaM. (B) Spectral changes are induced by addition of CaM mapped onto a structural model of the Itk PH domain bound to IP4. IP4 is in yellow, the gray ball is a bound zinc ion, and the regions of the PH domain for which the NMR resonances were affected upon addition of Ca2+/CaM are indicated in cyan. For both the β3/β4 loop and the β5/β6 loop the dotted lines indicate regions for which NMR assignments could not be completed. Because chemical shift mapping suggest that the β5 strand and the β5/β6 loop along with the β4 strand and the β3/β4 loop are the CaM target sites, the 34Å distance between the sites is indicated for comparison to the distance between the N- and C-domains of CaM shown in panel (A). (C) Alternative view of the Itk PH domain with the IP4-binding site at the top and the lipid chain present in PI(3,4,5)P3 added to indicate the possible location of the membrane relative to the CaM-binding site on the Itk PH domain. Another representative structure of Ca2+-CaM (PDB entry 2MGU) is shown in an orientation that would allow the N- and C-domains to contact the PH domain β5/β6 and β3/β4 loops, respectively. The arrow between CaM and PI(3,4,5)P3 indicates the possibility for additional contacts that may contribute to the observed cooperativity. PIP3, PI(3,4,5)P3
The Itk PH domain also binds apo-CaM (Ca2+ free calmodulin), albeit much more weakly (Fig. 4A). Compared to the NMR titration with Ca2+/CaM (Fig. 2A), addition of the unlabeled Itk PH domain to 15N-labeled apo-CaM produced less extensive line broadening even at much higher concentrations, and spectral changes were observed for only a small subset of CaM resonances, the majority of which localize to the CaM C-domain when mapped onto the structure (20) (Fig. 4B). Consistent with this observation, the NMR analysis indicated that the same residues that showed the largest shifts in the Ca2+/CaM C-domain fragment in the presence of the Itk PH domain (Fig. 4C) also shifted, albeit to a lesser extent, in the apo form of the CaM C-domain bound to the Itk PH domain (Fig. 4D).
Fig 4. Binding of apo-CaM and the CaM C-domain to the Itk PH domain.

(A) Overlay of 1H-15N-HSQC spectra of 150 μM 15N-labeled apo-CaM with increasing amounts of unlabeled Itk PH domain titrated at the indicated molar ratios (red to green); resonances that exhibit the largest spectral changes throughout the titration are labeled with the corresponding residue. (B) Residues exhibiting the most change mapped onto the structure of apo-CaM (PDB entry 1DMO) are represented as spheres. (C, D) Unlabeled Ca2+/CaM-C (C) or apo-CaM-C (D) was titrated into 300 μM 15N-labeled Itk PH domain at molar ratios identical to that used in Figure 2C ([CaM] increases from red to green). Selected regions of the HSQC spectra from the titration are shown; resonances showing the largest chemical shift changes are labeled in (C) and the same resonances are indicated in (D).
The spectral changes that occurred upon addition of the isolated CaM C-domain (Ca2+ bound or apo) to the Itk PH domain were restricted to the β-strands 2, 3, and 4 and the β3/β4 loop in the Itk PH domain (Fig. 3B). The region of the Itk PH domain that encompasses the β5/β6 loop and β-strand 5 only showed spectral changes on titration of Ca2+/CaM. Regardless of the presence or absence of Ca2+, the CaM N-domain fragment by itself and the Itk PH domain did not interact (fig. S2). Thus, the CaM C-domain and the region surrounding the β3/β4 loop in the Itk PH domain seem to be most critical in mediating the interaction between CaM and the Itk PH domain.
CaM promotes Itk activity and amplifies Ca2+ signaling through a positive feedback loop
TCR stimulation promotes increased association of endogenous Itk from cellular lysates with PI(3,4,5)P3-coated beads in vitro (21) and recruitment to the immune synapse in T cells (22). We, therefore, asked whether CaM is necessary for TCR-induced Itk binding to PI(3,4,5)P3. We stimulated purified T cells with antibodies that crosslink surface CD3 and CD4 receptors in the presence or absence of W-7, an inhibitor of Ca2+/CaM (23), and compared the binding of endogenous Itk from cell lysates to PI(3,4,5)P3–coated beads. Pharmacologic inhibition of CaM with W-7 reduced basal and TCR-induced binding of endogenous Itk from T cell lysates to PI(3,4,5)P3–coated beads, indicating that CaM enhanced PI(3,4,5)P3 binding (Fig. 5A). Consistent with decreased Itk membrane recruitment and activity, Itk-mediated phosphorylation of PLCγ1 was also decreased by W-7 (Fig. 5A). However, activity of the upstream kinase Lck was unaffected by CaM inhibition (Fig. 5A). Together, these data indicated that CaM and PI(3,4,5)P3 cooperatively promoted Itk-mediated PLCγ1 phosphorylation in response to TCR stimulation.
Fig 5. CaM promotes Itk activity and amplifies Ca2+ signaling in a positive feedback loop.

(A) Effect of CaM inhibition (W-7) on TCR-stimulated Itk binding to PI(3,4,5)P3 and Itk-mediated PLCγ1 phosphorylation. Thymocytes were stimulated with antibodies recognizing the CD3 and CD4 (anti-CD3 & 4) subunits of the TCR for 1 minute. TCR stimulation was performed in the presence of vehicle (MeOH ctrl) or W-7 (30μM). Itk association with PI(3,4,5)P3-coated beads and TCR-induced Lck and PLC γ1 phosphorylation at the indicated residues was assessed by Western blot analysis. (B) TCR-induced or ionomycin-induced cytosolic calcium accumulation in thymocytes exposed to vehicle (MeOH ctrl), W-7, or its inactive analog W-12. Ca2+ release from the ER was measured first in the absence of extracellular Ca2+ followed by Ca2+ entry through the plasma membrane (PM) by addition of 1 mM Ca2+ to the sample buffer. TCR stimulation was performed as in (A) in the presence or absence of W-7 (30μM) or W-12 (30μM). (C) Effect of depletion of Ca2+ from the ER by BAPTA-AM (10μM) and EGTA (5 mM) or inhibition of PLCγ1 catalytic activity (U73122, 5μM) on Itk binding to PI(3,4,5)P3 and subsequent Itk-mediated PLCγ1 phosphorylation. Thymocytes were pretreated with indicated reagents for 30 minutes prior to stimulation with biotin-conjugated antibodies to CD3 and CD4 and streptavidin in warm PBS. Samples were separated by SDS-PAGE and analyzed by Western blot analysis with the indicated antibodies. (D) Effect of Thapsigargin (Thap)-mediated increase of cytosolic Ca2+ on Itk-dependent PLCγ1 phosphorylation. Thymocytes were stimulated for 1 minute with biotin-conjugated antibody to CD3 and where indicated CD4 in the presence of 0 – 1 μM Thap. Samples were separated by SDS-PAGE and analyzed by Western blot analysis with the indicated antibodies. All data are representative of 3 experiments.
By hydrolyzing PI(4,5)P2, PLCγ1 generates the second messengers, DAG and IP3. IP3 stimulates Ca2+ release from the ER by binding to IP3 receptors, which are ligand-activated Ca2+ channels. To determine whether optimal Itk-dependent Ca2+ release requires CaM, we measured cytosolic Ca2+ signals with a ratiometric Ca2+-indicator dye in primary mouse thymocytes exposed to W-7. W-7, but not vehicle or the nonfunctional analog W-12, diminished TCR-induced Ca2+ release from the ER (Fig. 5B). As a control for equal loading of the Ca2+-indicator dye and equivalent Ca2+ levels in the stores, thymocytes were treated with the ionophore, Ionomycin (Ion). Vehicle, W-7 and W-12 treatment had little effect on Ionomycin-induced Ca2+ release from the ER stores (Fig. 5B). These data support a positive feedback role for Ca2+/CaM in promoting Itk membrane recruitment and triggering of downstream Ca2+ signals.
To further evaluate the positive feedback of Ca2+ on Itk activity, we assessed the effect of depleting total intracellular Ca2+ with BAPTA-AM treatment and inhibiting PLCγ1 activity on Itk activity. BAPTA-mediated Ca2+ depletion reduced Itk binding to PI(3,4,5)P3 and PLCγ1 phosphorylation (Fig. 5C). Inhibition of PLCγ1 enzymatic activity with U73122, which is downstream of Itk, also reduced Itk binding to PI(3,4,5)P3 (Fig. 5C). Conversely, increasing cytosolic Ca2+ concentration by inhibiting the ER Ca2+ pump with Thapsigargin promoted Itk-mediated PLCγ1 phosphorylation. However, TCR-induced activation of the upstream kinase Zap70 was unaffected by increased cytosolic Ca2+ levels (Fig. 5D). Together, these data indicate the presence of Ca2+-dependent positive feedback on Itk activity.
Disruption of CaM binding abrogates Itk recruitment to the immune synapse and IL-17A production
Because pharmacologic inhibition of CaM can disrupt many signaling processes, we generated loop-swap Itk (LS-Itk) with mutations in the PH domain that selectively disrupted binding to CaM but not binding to PI(3,4,5)P3. We used the following criteria to select the residues to mutate. (i)NMR analysis identified the Itk PH domain β-strands 2, 3, and 4, and the β3/β4 and β5/β6 loops as the principal regions involved in CaM binding (Fig. 3B and C). (ii) We focused on the β3/β4 and β5/β6 loops, because these disordered regions may adopt a helical structure when CaM binds (24, 25); CaM binding to its targets typically involves association with α-helical structures (26). (iii) The closely related Btk PH domain does not bind CaM (Fig. 1A) and the amino acid sequences of both the β3/β4 and β5/β6 loops of the Btk PH domain differ from the corresponding regions in Itk. (iv) We focused on the smaller β3/β4 loop region, the site of the interaction with the CaM C-domain in the presence or absence of Ca2+ (Fig. 4C and D). Thus, we swapped the 5-amino acid β3/β4 loop in Itk with the corresponding 7 amino acids in Btk to produce LS-Itk (Fig. 6A). When expressed in 293 epithelial cells, LS-Itk exhibited reduced co-precipitation with CaM than did wild-type Itk (Fig. 6B). However, the association of LS-Itk with PI3,4,5P3-coated beads was indistinguishable from that of wild-type Itk, suggesting that mutation in the β3/β4 loop does not alter the fold of the PH domain. In contrast to LS-Itk, numerous mutations within the β-strands of the Itk PH domain either did not disrupt CaM binding or disrupted both CaM and PI(3,4,5)P3 binding (table S1, fig. S3).
Fig. 6. A Itk mutant that cannot bind CaM is not recruited to the immune synapse in Jurkat T cells, and Itk-deficient CD4 T cells reconstituted with mutant exhibit reduced Ca2+ signaling and IL-17A production.

(A) LS)-Itk was generated by replacing the 5 amino-acid β3/β4 loop in Itk PH domain with the corresponding 7-amino acid loop from Btk. (B) Ability of LS-Itk to bind CaM and PI(3,4,5)P3 in vitro. Cell lysates from HEK293 cells transfected with WT or LS-Itk were assessed for Itk binding to Ca2+/CaM or PI(3,4,5)P3 by co-precipitation assay. Data are representative of 3 experiments. (C) Jurkat T cells expressing pRuby-LifeAct and WT- or LS-Itk-YFP fusion proteins were conjugated to Daudi (Turquoise-labeled) B cells in the presence of SEE super-antigen. Actin (red) and YFP (green) colocalization was assessed by confocal microscopy and quantified. (D) Primary Itk-deficient CD4+ T cells reconstituted with retrovirus expressing WT-Itk or LS-Itk and bicistronic GFP were loaded with Indo-1 and stimulated with antibodies against TCR subunits to detect changes in cytosolic Ca2+ amounts, which are plotted at the right as a ratio of Indo-1 Violet to Indo-1 Blue over time. GFP− and GFP+ T cells represent nontransduced and transduced cells, respectively. Data are representative of 3 experiments. (E) Primary Itk-deficient CD4+ T cells reconstituted with retrovirus expressing WT- or LS-Itk were induced to differentiate into TH17 cells in culture and assessed for IL-17A and IFNγ production. Left plots show the distribution of GFP-negative and -positive CD4+ T cells with GFP gates for middle and right plots indicated by the boxed regions. Middle and right plots show the abundance of IL-17A-positive cells in GFP-negative (non-reconstituted Itk−/− cell) and GFP-positive (WT-Itk- or LS-Itk-reconstituted Itk−/− cell) populations respectively that produce low amounts of IFNγ, thereby defining the percent of TH17 cells. Data are representative of 3 experiments. DIC, Differential interference contrast; FSC, Foward scatter; MFI, Mean fluorescence intensity
To investigate the role of CaM in recruiting Itk to the membrane of T cells, we assessed localization of Itk to the immunological synapse where PI(3,4,5)P3 generation occurs using Jurkat T cells, which respond to CD3 stimulation by activating Akt and Itk-dependent PLCγ1 (fig. S4). Although a fusion between wild-type Itk and YFP (WT-Itk-YFP) efficiently localized to actin-rich synapses formed between Jurkat T cells and Daudi B cells, the LS-Itk-YFP did not (Fig. 6C and fig. S5), indicating that Itk recruitment to the synapse required the presence of the β3-β4 loop native to Itk, not that of Btk. This implies that CaM is important for Itk membrane recruitment and subsequent Itk activation. To assess Ca2+ signaling, we reconstituted primary Itk-deficient CD4+ T cells with either WT-Itk or LS-Itk, stimulated the TCR, and monitored the amount of cytosolic Ca2+. As previously reported for Itk-deficient T cells (11, 12), Ca2+ did not change in response to TCR stimulation, as detected by the ratiometric Ca2+ indicator dye Indo-1 (Fig. 6D). However, TCR stimulation produced increased cytosolic Ca2+ in Itk-deficient T cells retrovirally reconstituted with WT-Itk, but not in those reconstituted with LS-Itk (Fig. 6D). Together, these data support a model in which initial increases in Ca2+ promote CaM-mediated enhancement of Itk membrane recruitment and activation to further amplify Ca2+ signaling.
We also evaluated the importance of CaM binding to Itk on T cell functions that depend on Itk and Ca2+ signaling. Itk is required for NFAT-dependent IL-17A production by TH17 cells (14), an inflammatory T cell subset the deregulation of which has been implicated in mediating autoimmune diseases (15). Compared to cells reconstituted with WT-Itk, retroviral reconstitution of primary Itk-deficient CD4+ T cells with LS-Itk failed to rescue IL-17A production (Fig. 6E), suggesting that positive feedback of Itk activity through Ca2+/CaM influences the efficiency of pro-inflammatory T cell responses.
CaM is a putative protein ligand for multiple PH domains
NMR analysis revealed that the β3/β4 and β5/β6 loops of the Itk PH domain interacted with CaM. Given the general conservation of PH domain structure and loop positioning, we assessed the potential for CaM to bind other PH domains. We analyzed all annotated mouse PH domains in the UniProt database (27) for CaM binding potential using a prediction algorithm based on known CaM-binding proteins (28). Consistent with our experimental findings, the Itk PH domain was predicted to be a CaM-binding protein. In addition to Itk, we found that 49% of 236 PH domains are predicted to bind to CaM, with 28% and 18% predicted to bind with intermediate and high affinities, respectively (Fig. 7A, table S3). To assess the accuracy of this prediction, we cloned ten PH domains from proteins of diverse function and tissue expression that were predicted to bind with low to high affinity and two PH domains with no predicted affinity (Fig. 7B). Coprecipitation assays with cell lysates containing PH domain-YFP fusion proteins showed that six of the predicted PH domains bound CaM in a Ca2+-dependent manner and that of the four predicted CaM-binding PH domains and both PH that were not predicted to bind CaM did not associate (Fig. 7B). Although further characterization of CaM binding to individual PH domains is required to substantiate these predictions, the data indicate that many PH domains may be regulated by Ca2+/CaM, greatly expanding the number of potential effector proteins downstream of the second messenger Ca2+ and its binding partner CaM
Fig. 7. CaM is a putative protein ligand for multiple PH domains.

(A) Calmodulin target prediction for mouse PH domain-containing proteins annotated in Uniprot predicts that >50% of PH domains bind CaM. Predicted relative affinities are based on the number of consecutive amino acids (aa) scoring 8 in the algorithm: low (1–7 consecutive aa), medium (8–14 aa), and high affinity (15–21 aa). See table S3 for the results of the analysis of the 236 PH domains tested (B) PH domains with indicated predicted affinities and the PH domain of Itk were assessed in precipitation assays with apo-CaM and Ca2+/CaM (+/− Ca2+). For some PH domain-YFP fusions, smaller fragments likely derived from internal translational start sites were observed.
Discussion
Ca2+ is an important second messenger that regulates multiple cellular behaviors, including migration, differentiation, and death, and cellular processes, such as vesicular trafficking and enzyme activation. Precise control of the kinetics and magnitude of intracellular Ca2+ concentrations helps to activate the appropriate Ca2+-dependent response. In T cells, the TCR activates the Tec kinase Itk to induce Ca2+ signaling following recruitment of Itk to the membrane by PI(3,4,5)P3. Here, we identified a positive feedback mechanism by which Ca2+/CaM cooperates with PI(3,4,5)P3 for binding to the Itk PH domain and potentiates further Itk recruitment and activation. We mapped key interacting residues in both CaM and Itk and demonstrated that in the Itk PH domain the CaM-binding β3/β4 loop that adjoins the PI(3,4,5)P3 binding pocket is required for Itk-dependent amplification of production of the proinflammatory cytokine IL-17A. Lastly, we propose that Ca2+/CaM regulation may extend to other PH domains.
Distinct intracellular Ca2+ patterns contribute to different T cell differentiation programs. During T cell development, thymocytes with appropriate TCR affinities are positively selected to mature into CD4+ helper or CD8+ cytotoxic T cells, while cells with potentially autoreactive TCRs die by negative selection. Ca2+ patterns differ between thymocytes receiving maturation versus death signals. Sustained intermediate Ca2+ concentrations are induced during positive selection and high but transient Ca2+ concentrations are induced during negative selection of CD4+ cells (29). Peripheral CD4+ T cells also exhibit different patterns of Ca2+ signaling that are likely required for their differentiation and effector function. Although TH1, TH2, and TH17 cells all respond to TCR stimulation by inducing a rapid spike in intracellular Ca2+ concentration, TH1 and to a lesser extent TH17 cells show sustained oscillatory Ca2+ signals (30). In contrast, Ca2+ concentrations in TH2 cells rapidly decrease following stimulation. Decreasing Ca2+ patterns translate into decreased amounts of nuclear NFAT (30). These studies emphasize the importance of tuning Itk-dependent Ca2+ signals to the appropriate degree to induce protective T cell responses. Interestingly, a report has identified a role for Itk in controlling the tissue infiltration of autoreactive T cells (31). Thus, targeting the Ca2+/CaM feedback loop that controls Itk activity identified here may present a new strategy for preventing tissue infiltration of autoimmune T cells, as well as therapeutic treatment of autoimmunity by limiting Itk-dependent IL-17A production.
Our data also support the changing perception of PH domains as multifunctional regulatory domains rather than simply membrane targeting domains (4). Indeed, less than a quarter of all mammalian PH domains bind lipids, and of those that do, less than 10% bind phosphoinositides with high specificity and affinity (1). Several PH domains bind directly to small guanosine triphosphatases (GTPases), including Cdc42, Rho, and Arf1 (ADP-ribosylation factor 1), through residues within the intervening β loops of the PH domain (32–35). Here, the we found that the β3/β4- and β5/β6-intervening loops of the Itk PH domain interact with CaM and are required for enhanced Itk association with PI(3,4,5)P3. CaM also bind the Akt PH domain (36). Analysis of overlapping peptide fragments from the Akt PH domain localized CaM binding to a region within the β barrel, representing an unconventional interaction. Interestingly, CaM functionally prevents the PH domain of Akt from binding PI(3,4,5)P3, suggesting that CaM inhibits Akt activity (36). However, a separate study supports a positive role for CaM on Akt because CaM inhibitors reduce Akt-dependent cell growth of breast tumor cell lines (37). Further structural and biochemical analyses as provided here for Itk will be required to determine the effect of CaM on Akt activation.
The finding that CaM interacts with the Itk PH domain and PH domains from a subset of other proteins suggests that these PH domain-containing proteins are putative effectors of Ca2+ signaling. CaM may allosterically regulate protein activity directly, as occurs with conventional CaM-binding effectors, or indirectly by integrating Ca2+ signals with those provided by GTPases or membrane phospholipids. For PH domains without lipid-binding potential or with uncharacterized function, lipid-binding potential should be (re-)evaluated in the presence of Ca2+/CaM in addition to assessing lipid-independent activities.
The precise mechanism by which the Itk PH domain, Ca2+/CaM, and PI(3,4,5)P3 work together to control and fine tune signals emanating from the TCR has yet to be firmly established. Several possibilities are emerging. Like the PH domains of certain guanine exchange factors (GEFs) for Rho (RhoGEFs) and the PH domain of Akt (38–41), the Itk PH domain may serve an autoinhibitory role. For Itk, association with Ca2+/CaM may prevent formation of an autoinhibitory structure, resulting in release of the Itk catalytic machinery and exposure of the lipid-binding pocket on the Itk PH domain for membrane anchoring. However, the closely related kinase Btk, which is present in B cells, mast cells, and myeloid cells, does not appear to be co-regulated by CaM, suggesting that if Ca2+ responses in these cell types are amplified by positive feedback that it is not through the same mechanism we found for Itk in T cells.
Our NMR data and in vitro binding assays suggest a model for how CaM engages the Itk PH domain and how PI(3,4,5)P3 (and not IP4) might cooperatively enhance the interaction between CaM and Itk. The emerging picture has parallels with the binding of CaM to the NMDA receptor NR1C0 site (42) and to melittin (43). In these complexes, the C-domain of apo-CaM binds the target with moderate affinity, and the N-domain does not interact measurably. A rise in Ca2+, producing Ca2+/CaM causes both the C-domaind and N-domain to bind the target tightly. The apparent noncontiguous nature of the Itk PH domain as a CaM target is reminiscent of several other CaM-controlled systems; the ryanodine receptor, a protein that binds each domain of CaM through noncontiguous sites (44), the water-channel protein aquaporin-0 engages CaM through two disparate regions (45), and voltage-gated sodium channels wherein CaM is thought to bridge a C-terminal motif and a linker region that is distant in primary sequence (46). The noncontiguous Itk PH domain residues targeted by Ca2+/CaM, in particular the β3/β4 and β5/β6 loops (Fig. 3B and C), may mediate binding to full-length CaM in a manner that requires CaM to maintain a semi-extended conformation rather than the collapsed conformation typical of many CaM-mediated interactions. Precedence for CaM engaging its targets in an extended fashion include the synaptic vesicle priming protein, Munc13 (19), and the structure of CaM bound to the matrix (MA) domain of the HIV-1 Gag protein (47). In both cases, the distance between the centers of the two CaM-binding pockets (C-domain and N-domain) is approximately 36Å, which corresponds to the 34Å distance from the β3/β4 loop to the β5/β6 loop in the Itk PH domain. The CaM-MA complex structure also reveals that CaM binding modulates the fold of the MA domain (47), which we anticipate could also occur for the Itk PH domain leading to the induction of helix formation of the large β3/β4 and β5/β6 loops for optimal CaM binding.
Although our NMR titration data did not include PI(3,4,5)P3, the Itk PH domain ligand, binding data suggested a role for PI(3,4,5)P3 in stabilizing the CaM-PH domain interaction (Fig. 1D). The emerging model of an extended CaM protein binding the β3/β4 and β5/β6 loops of Itk PH does not appear mutually exclusive with PI(3,4,5)P3 binding to the same PH domain (Fig. 3C). Interactions between CaM and the membrane, CaM-mediated conformational changes in the Itk PH domain that enhance lipid binding, or even structural changes in the membrane itself to fully accommodate a CaM-Itk-PI(3,4,5)P3 complex are all possibilities. Indeed, CaM interactions with myristoylated proteins have been described (48–51), providing evidence for direct interaction between CaM and hydrophobic lipid-like structures.
Previous findings have suggested that soluble IP4 enhances Itk binding to PI(3,4,5)P3 (21). In that case, Itk dimerization and allosteric communication across the protein-protein interface was invoked to explain how IP4 both enhances PI(3.,4,5)P3 binding to the PH domain of Itk and binds to the same location as PI(3,4,5)P3 on the PH domain. The extent to which Itk dimerization or oligomerization plays a role in the ability of CaM to enhance the association of Itk with PI(3,4,5)P3 deserves further attention. However, we did not find an effect of IP4 on the CaM-Itk interaction. The previous findings were based in part on in-vitro binding experiments conducted in the absence of Ca2+ (21), precluding Ca2+/CaM interactions with Itk. Moreover, the physiological relevance of IP4 on Itk activation has only been explored in developing T cells in the thymus; IP4-deficient mice have a defect in T cell development (21, 52, 53). The contribution of IP4 and its effect on Ca2+/CaM and PI(3,4,5)P3 binding to Itk, and Itk-dependent T cell functions, such as TH2 and TH17 responses, remain unexplored in peripheral T cells. The possibility that IP4 and CaM independently promote Itk activity in a cell-stage or cell-type specific manner requires future assessment.
As further experiments clarify the mechanistic details for Ca2+/CaM-regulation of Itk-mediated signaling, we will gain a clearer understanding of how Itk activity is fine tuned to promote distinct T cell functions. The apparent complexity in Itk regulation is likely a reflection of how important controlling the magnitude and kinetics of Ca2+ responses is for balancing T cell responses to prevent immunodeficiency and autoimmunity, pathologies that may occur as a result of too little or too much TCR and Itk signaling.
Materials and Methods
Mice, cell lines, plasmids
All mice were housed under specific-pathogen-free conditions in the Washington University School of Medicine animal facilities in accordance with institutional guidelines. Lymphoid organs were harvested from 6–10 week old H2-Ab1tm1Gru B2mtm1Jae (MHC−/−, Taconic Farms, Model 4080) or Itk−/− mice on the C57BL/6 background.
Wild-type and LS-mutant Itk were cloned into the pFLRu-YFP vector and were used to generate Jurkat stable cell lines as previously described (54). The human B cell line Daudi was stably transfected with pFLRu-Turquoise. Wild-type and LS-Itk were also cloned into a MSCV-based retrovirus (pCMV2.1) expressing GFP bicistronically as previously described (21) and used to transduce murine CD4+ T cells.
Cloning and analysis of mouse PH domains
RNA was prepared from various mouse tissues from C57BL/6 mice with TRIzol (Invitrogen). cDNA was synthesized with Supercript III reverse transcriptase (Invitrogen). Sequences for PH domains were cloned into pcDNA3.1 and tagged with YFP by introducing a BamHI (GGATCC) site within the reverse primer. A flexible linker (Gly-Gly-Gly-Gly-Ser-Gly-Gly-Gly-Gly-Ser), which lacks a CaM-binding site, was introduced by PCR between the PH domain and YFP. The Cdc42bpa-PH domain, which contains an internal BamHI site, was cloned using a BglII (AGATCT) site with a flexible linker (Gly-Gly-Gly-Arg-Ser-Gly-Gly-Gly-Gly-Ser). The Itk PH domain was cloned as a structural unit with the Tec homology domain. The boundaries for PH domains of unknown function were defined by combining the Uniprot annotations with domain predictions generated by Protein Homology/analogY Recognition Engine (Phyre) (55). Cloned PH domains preserved the C-terminal α helix without extraneous sequence extensions. LS-Itk was generated by bridge PCR mutation. Primers used for generating LS-Itk and Itk PH domain-YFP fusions are listed in table S2.
PH domain-YFP fusion proteins were expressed in 293 epithelial cells, and cell lysates were incubated with Apo-CaM- (with 1 mM EGTA) or Ca2+/CaM- (with 100 nM CaCl2) coated beads (Sigma) for 1.5 hours at 4°C. Beads were washed 3–5 times with 1x lysis buffer (1% Triton X-100, 60 mM octylglucoside, 150 mM NaCl, 25 mM Tris pH 7.5) containing protease (Mini Complete, EDTA-free Protease Inhibitor Cocktail, Roche) and phosphatase inhibitors (PhosSTOP, phosphatase inhibitor cocktail, Roche) and then denatured in 1x NuPAGE sample buffer (Life Technologies) at 99°C for 10 minutes prior to SDS-PAGE. Nitrocellulose membranes were probed overnight at 4°C with primary antibodies and 30–45 min. with secondary antibodies anti-rabbit or anti-mouse conjugated to horseradish peroxidase. PH domain-YFP fusion proteins were detected by Western blot using GFP-specific antibody (JL-8, cross-reactive with YFP, Clontech).
Protein expression and purification for NMR studies
Recombinant Itk PH domain used in this study contains the double mutation, C96E/T110I, that has been previously reported to facilitate production of soluble PH domain that retains PI(3,4,5)P3 binding (18).
Itk PH domain C96E/T110I (amino acids 1–154, mus. musculus) was expressed and purified as previously described (18). Briefly, a modified pET20b vector was used to express Itk PH domain with an N-terminal His6-GB1 tag in (DE3)BL21 cells. Protein was purified using Ni-NTA chromatography, followed by Factor Xa cleavage of the His6-GB1 tag and size-exclusion chromatography. The following rat Calmodulin constructs were expressed and purified as described previously (56): CaM-FL (1–148), CaM-C (76–148), CaM-N75 (1–75), CaM-N80 (1–80). For NMR titrations, proteins were dialyzed into 50 mM HEPES pH 7.4, 150 mM NaCl, 2 mM DTT, 0.02% NaN3 (and 1 mM CaCl2 for Ca2+/CaM experiments). For apo-CaM titrations, CaM was treated with EGTA or EDTA and then dialyzed into calcium-free NMR buffer. The Itk PH domain has an extended region at its C-terminus that binds a Zn2+ ion that is likely necessary for the proper fold of this domain; hence, the dialysis was necessary to remove EDTA or EGTA prior to performing the experiments. For Ca2+/CaM experiments, a five-fold excess of CaCl2 was added to the purified CaM, which was then dialyzed into 1 mM CaCl2 NMR buffer.
NMR Spectroscopy and 1H-15N Backbone chemical shift assignments of ItkPH
All NMR spectra were collected on a Bruker AVII 700 spectrometer with a 5 mm HCN z-gradient cryoprobe operating at a 1H frequency 700.13 MHz, with a sample temperature of 298K. We assigned 75% of the backbone 1H/15N chemical shifts using the Sparky (57) and MARS (58) software programs, utilizing the following pairs of triple-resonance experiments: HNCA and HN(CO)CA, HNCO and HN(CA)CO, and CBCA(CO)NH and CBCANH. Spectra are referenced to DSS, directly in the 1H dimension and indirectly for the 13C and 15N dimensions, according to standard procedures. NMRPipe (59) and NMRViewJ (60) were also used for data processing, visualization, and analysis. 1H-15N backbone assignments for CaM were obtained from the BMRB (61) (entry 6541 for 15N-Ca2+/CaM).
NMR titrations
NMR titrations were carried out as described previously (62). For each titration, unlabeled ligand (either Itk PH or CaM) was added to 15N-labeled protein and 1H-15N HSQC spectra were acquired for the indicated molar ratios. The concentration of 15N-Ca2+/CaM was diluted from 150 μM to 123 μM over the course of the titration. For 15N-Itk PH, the starting concentration of 300 μM was diluted to 248 μM by the final point of the titration.
Structural model of the Itk PH domain
The structural model of Itk PH domain used to interpret the NMR data was constructed with I-TASSER (63) and MODELLER (64), using the available Btk PH domain structures (PDB entries 1BTK and 1B55) as templates.
Cell stimulations
MHC−/− thymocytes were rested at 37°C for 20–30 minutes prior to stimulation. Thymocytes (2 x 107/sample) were stained in phosphate-buffered saline (PBS) with biotin-conjugated antibodies against CD3 and antibodies against CD4 for 15 minutes at 4°C. Following two washes in PBS, cells were stimulated with 1 μg/ml streptavidin in pre-warmed PBS at 37°C. Where indicated, thymocytes were pretreated with vehicle control, 30 μM of W-7 [N-(6-Aminohexyl)-5-chloro-1-naphthalenesulfonamide hydrochloride, Sigma A3281, MLCK (IC50 = 51 μM)] or W-12 [N-(4-Aminobutyl)-2-naphthalenesulfonamide hydrochloride, Sigma A3168, MLCK (IC50 = 300 μM))] 10 μM of BAPTA-AM (Sigma) with 5 mM EGTA, or 5 μM of U73122 (Cayman Chemical Company) prior to stimulation.
Jurkat T cells were rested in RPMI-1640 with 1% fetal bovine serum (FBS) overnight and then on ice for 1 hr. Cells (106) in PBS were rested on ice for 30 min and then stimulated with 0.5 μg/ml antibody against CD3 (OKT3, eBioscience) in pre-warmed PBS at 37°C.
Cells were lysed directly with 4x lysis buffer (4% Triton X-100, 240 mM octylglucoside, 600 mM NaCl, 100 mM Tris pH 7.5, 4 mM EDTA) containing protease (Mini Complete, EDTA-free Protease Inhibitor Cocktail, Roche) and phosphatase inhibitors (PhosSTOP, phosphatase inhibitor cocktail, Roche). Lysates were cleared of cellular debris by centrifugation at 13,200 x g for 15 minutes at 4°C.
Precipitation and immunoblot analyses
Cell lysates or purified Itk PH domain were incubated with beads coated with PI(3,4,5)P3 (Echelon Biosciences), Apo-CaM (with 1 mM EGTA) or Ca2+/CaM (with 100 nM CaCl2) CaM (Sigma) for 1.5 hours at 4°C. PI(3,4,5)P3, IP4, or PI(4,5)P2 were added to the precipitation system to study their effects on CaM binding to Itk. Beads were washed 3–5 times with 1x lysis buffer and then denatured in 1x sample buffer at 99°C for 10 minutes prior to SDS-PAGE. Nitrocellulose membranes were probed overnight at 4°C with primary antibodies and 30–45 min. with secondary antibodies anti-rabbit or anti-mouse conjugated to horseradish peroxidase. Protein abundance was detected by chemiluminescence. Calmodulin (EP799Y)-specific antibody was from Abcam. Calcineurin-, Btk-, phosphorylated PLCγ1 (Try783)-, phosphorylated Src (Tyr416)- and phosphorylated ZAP-70 (Tyr319)-specific antibodies were from Cell Signaling Technologies. Itk-specific antibody was from BD Bioscience. GFP (JL-8)-specific antibody was from Clontech, and the GAPDH-HRP antibody was from Sigma.
Calcium mobilization measurements
Thymocytes were loaded with 1 μg/ml Fura-2-AM and 0.02% Pluronic (Invitrogen) in Ca2+-containing buffer (1 mM CaCl2, 135 mM NaCl, 5 mM KCl, 1 mM MgCl2, 5.6 mM glucose, 10 mM HEPES pH 7.4, 0.1% BSA) at 37°C for 30 min. After two washes, cells were resuspended in Ca2+-free buffer (135 mM NaCl, 5 mM KCl, 1 mM MgCl2, 5.6 mM glucose, 10 mM HEPES pH 7.4, 0.1% BSA) and transferred into a poly-L-lysine-treated 96 well-assay plate at 5 X 105 cells/well. Calcium response was measured on the FlexStation (Molecular Devices) at 37°C as previously described (65). 5 μg/ml of biotin-conjugated antibody against CD3 (2C11, Biolegend) and 1 μg/ml of biotin-conjugated antibody against CD4 (GK1.5, Biolegend) together with 15 μg/ml of streptavidin were used for TCR stimulation. 5 μg/ml of Ionomycin (Sigma) were used for inducing release of Ca2+ from ER in Ca2+-free extracellular buffer.
Wild-type and LS-mutant reconstituted Itk−/− murine CD4+ T cells were rested without CD3 and CD28 stimulation for 48 hours before measuring calcium response. Cells were loaded with 1 μg/ml Indo-1-AM and 0.02% Pluronic (Invitrogen) in RPMI (10% FBS) at 37°C for 30 min. Cells were washed twice and resuspended in RPMI (1% FBS) and kept at room temperature. Cells were pre-warmed for 5 min at 37°C before experiments. Calcium response was measured on an LSRII flow cytometer (BD) by adding 10 μg/ml antibody against CD3 and 2.5 μg/ml antibody against CD4 followed by crosslinking with 25 μg/ml streptavidin. Calcium responses were recorded for 4 min after addition of streptavidin. The ratio of Indo-1 Violet to Indo-1 Blue was plotted over time for GFP+ (reconstituted) and GFP− (non-transduced) cells.
Immune synapse analyses
Jurkat cells expressing either wild-type- or LS-mutant Itk-YFP fusion proteins were transfected with pRuby-LifeAct. Jurkat-Daudi conjugates were made as previously described (66). For quantification, conjugates were chosen randomly and manually scored for colocalization of actin and Itk by a blinded individual.
TH17 polarization and retroviral transduction
Naïve CD4+CD62L+CD25−CD44−/low cells were purified from Itk−/− mice as previously described (14). Cells were cultured in plates coated with antibodies against CD3 (2C11, 10 μg/ml) and CD28 (37.51, 5 μg/ml) under TH0 condition (see below) for 48 hr and then under TH17 conditions for an additional 48 hr. TH17 cells were stimulated with 50 ng/ml of Phorbol 12-myristate 13-acetate (PMA, Sigma) and 1 μg/ml of ionomycin (Sigma) in presence of Brefeldin A (Biolegend) for 4 hr. Cells were fixed and permeabilized with Cytofix/Cytoperm kit (BD Biosciences) and stained for surface CD4 and intracellular IL-17A and IFN-γ. TH0 condition: 10 μg/ml each of antibodies against IL-4 (11B11), IFN-γ (H22), and IL-12 (17.8) (Biolegend) in IMDM supplemented with 10% FBS (HyClone), 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine, and 55 μM 2-mercaptoethanol (Invitrogen); TH17 condition: TH0 condition supplemented with 20 ng/ml of IL-6 (Peprotech), 5 ng/ml of TGFβ1 (R&D), 10 ng/ml of IL-1β (Peprotech), and 10 ng/ml of IL-23 (R&D).
Itk-encoding retroviruses were packaged by transfection of PlatE cells (67) with Fugene 6 (Roche). Retroviral supernatants were collected at 48 hrs after transfection, filtered through 0.45 μM filters, and used to spinoculate T cells on day 2 of culture at 2500 rpm for 1.5 hr at room temperature with 8 μg/ml of polybrene (Sigma).
Supplementary Material
Fig. S1. CaM binding maps to the Itk PH domain.
Fig. S2. The N-domain of CaM does not bind to the Itk PH domain.
Fig. S3. Structural model of the Itk PH domain showing the location of the mutated residues tested.
Fig. S4. TCR stimulation promotes Akt activation and PLCγ1 phosphorylation in Jurkat E6.1 cells.
Fig. S5. Kinetic analysis of recruitment of WT-Itk or LS-Itk to the immune synapse
Table S3. Calmodulin binding predictions for 236 mouse PH domains listed in the Uniprot database.
Table S1. Effect of Itk PH domain point mutations on protein abundance, PI(3,4,5)P3 binding, and CaM binding
Table S2. Oligo primers for cloning PH domains and YFP
Acknowledgments
We thank N. Mathis for technical assistance, Y. Feng for pFLRu and pRuby-LifeAct constructs, and M. Ikura for discussion on CaM binding prediction. This study was supported by the National Institutes of Health grants GM057001 (M.A.S), AI043957 (A.H.A.) and AI089805 (Y.H.H).
Footnotes
Author contributions: X.W. and Y.H.H. conceived the study. X.W. performed the biochemical and functional experiments. S.E.B. performed and analyzed the NMR studies. M.A.S. and A.H.A. analyze the NMR studies. J.H. and A.S.S. helped with imaging. X.X. analyzed confocal micrographs. X.W., S.E.B., M.A.S., A.H.A. and Y.H.H. prepared the manuscript.
Competing interests: The authors declare no competing interests.
References
- 1.Lemmon MA. Pleckstrin homology (PH) domains and phosphoinositides. Biochem Soc Symp. 2007:81–93. doi: 10.1042/BSS0740081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu Q, Bateman A, Finn RD, Abdubek P, Astakhova T, Axelrod HL, Bakolitsa C, Carlton D, Chen C, Chiu HJ, Chiu M, Clayton T, Das D, Deller MC, Duan L, Ellrott K, Ernst D, Farr CL, Feuerhelm J, Grant JC, Grzechnik A, Han GW, Jaroszewski L, Jin KK, Klock HE, Knuth MW, Kozbial P, Krishna SS, Kumar A, Marciano D, McMullan D, Miller MD, Morse AT, Nigoghossian E, Nopakun A, Okach L, Puckett C, Reyes R, Rife CL, Sefcovic N, Tien HJ, Trame CB, van den Bedem H, Weekes D, Wooten T, Hodgson KO, Wooley J, Elsliger MA, Deacon AM, Godzik A, Lesley SA, Wilson IA. Bacterial pleckstrin homology domains: a prokaryotic origin for the PH domain. J Mol Biol. 2010;396:31–46. doi: 10.1016/j.jmb.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu JW, Mendrola JM, Audhya A, Singh S, Keleti D, DeWald DB, Murray D, Emr SD, Lemmon MA. Genome-wide analysis of membrane targeting by S. cerevisiae pleckstrin homology domains. Mol Cell. 2004;13:677–688. doi: 10.1016/s1097-2765(04)00083-8. [DOI] [PubMed] [Google Scholar]
- 4.Moravcevic K, Oxley CL, Lemmon MA. Conditional peripheral membrane proteins: facing up to limited specificity. Structure. 2012;20:15–27. doi: 10.1016/j.str.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harlan JE, Hajduk PJ, Yoon HS, Fesik SW. Pleckstrin homology domains bind to phosphatidylinositol-4,5-bisphosphate. Nature. 1994;371:168–170. doi: 10.1038/371168a0. [DOI] [PubMed] [Google Scholar]
- 6.Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13:195–203. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- 7.Rawlings DJ, Saffran DC, Tsukada S, Largaespada DA, Grimaldi JC, Cohen L, Mohr RN, Bazan JF, Howard M, Copeland NG, et al. Mutation of unique region of Bruton’s tyrosine kinase in immunodeficient XID mice. Science. 1993;261:358–361. doi: 10.1126/science.8332901. [DOI] [PubMed] [Google Scholar]
- 8.Thomas JD, Sideras P, Smith CI, Vorechovsky I, Chapman V, Paul WE. Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science. 1993;261:355–358. doi: 10.1126/science.8332900. [DOI] [PubMed] [Google Scholar]
- 9.Li T, Tsukada S, Satterthwaite A, Havlik MH, Park H, Takatsu K, Witte ON. Activation of Bruton’s tyrosine kinase (BTK) by a point mutation in its pleckstrin homology (PH) domain. Immunity. 1995;2:451–460. doi: 10.1016/1074-7613(95)90026-8. [DOI] [PubMed] [Google Scholar]
- 10.Andreotti AH, Schwartzberg PL, Joseph RE, Berg LJ. T-cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harb Perspect Biol. 2010;2:a002287. doi: 10.1101/cshperspect.a002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu KQ, Bunnell SC, Gurniak CB, Berg LJ. T cell receptor-initiated calcium release is uncoupled from capacitative calcium entry in Itk-deficient T cells. J Exp Med. 1998;187:1721–1727. doi: 10.1084/jem.187.10.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fowell DJ, Shinkai K, Liao XC, Beebe AM, Coffman RL, Littman DR, Locksley RM. Impaired NFATc translocation and failure of Th2 development in Itk-deficient CD4+ T cells. Immunity. 1999;11:399–409. doi: 10.1016/s1074-7613(00)80115-6. [DOI] [PubMed] [Google Scholar]
- 13.Schaeffer EM, Yap GS, Lewis CM, Czar MJ, McVicar DW, Cheever AW, Sher A, Schwartzberg PL. Mutation of Tec family kinases alters T helper cell differentiation. Nat Immunol. 2001;2:1183–1188. doi: 10.1038/ni734. [DOI] [PubMed] [Google Scholar]
- 14.Gomez-Rodriguez J, Sahu N, Handon R, Davidson TS, Anderson SM, Kirby MR, August A, Schwartzberg PL. Differential expression of interleukin-17A and -17F is coupled to T cell receptor signaling via inducible T cell kinase. Immunity. 2009;31:587–597. doi: 10.1016/j.immuni.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maddur MS, Miossec P, Kaveri SV, Bayry J. Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am J Pathol. 2012;181:8–18. doi: 10.1016/j.ajpath.2012.03.044. [DOI] [PubMed] [Google Scholar]
- 16.Clapham DE. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 17.Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol. 2007;7:690–702. doi: 10.1038/nri2152. [DOI] [PubMed] [Google Scholar]
- 18.Boyken SE, Fulton DB, Andreotti AH. Rescue of the aggregation prone Itk Pleckstrin Homology domain by two mutations derived from the related kinases, Btk and Tec. Protein Sci. 2012;21:1288–1297. doi: 10.1002/pro.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodriguez-Castaneda F, Maestre-Martinez M, Coudevylle N, Dimova K, Junge H, Lipstein N, Lee D, Becker S, Brose N, Jahn O, Carlomagno T, Griesinger C. Modular architecture of Munc13/calmodulin complexes: dual regulation by Ca2+ and possible function in short-term synaptic plasticity. EMBO J. 2010;29:680–691. doi: 10.1038/emboj.2009.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang M, Tanaka T, Ikura M. Calcium-induced conformational transition revealed by the solution structure of apo calmodulin. Nature structural biology. 1995;2:758–767. doi: 10.1038/nsb0995-758. [DOI] [PubMed] [Google Scholar]
- 21.Huang YH, Grasis JA, Miller AT, Xu R, Soonthornvacharin S, Andreotti AH, Tsoukas CD, Cooke MP, Sauer K. Positive regulation of Itk PH domain function by soluble IP4. Science. 2007;316:886–889. doi: 10.1126/science.1138684. [DOI] [PubMed] [Google Scholar]
- 22.Singleton KL, Gosh M, Dandekar RD, Au-Yeung BB, Ksionda O, Tybulewicz VL, Altman A, Fowell DJ, Wulfing C. Itk controls the spatiotemporal organization of T cell activation. Sci Signal. 2011;4:ra66. doi: 10.1126/scisignal.2001821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Osawa M, Swindells MB, Tanikawa J, Tanaka T, Mase T, Furuya T, Ikura M. Solution structure of calmodulin-W-7 complex: the basis of diversity in molecular recognition. J Mol Biol. 1998;276:165–176. doi: 10.1006/jmbi.1997.1524. [DOI] [PubMed] [Google Scholar]
- 24.Radivojac P, Vucetic S, O’Connor TR, Uversky VN, Obradovic Z, Dunker AK. Calmodulin signaling: analysis and prediction of a disorder-dependent molecular recognition. Proteins. 2006;63:398–410. doi: 10.1002/prot.20873. [DOI] [PubMed] [Google Scholar]
- 25.Nagulapalli M, Parigi G, Yuan J, Gsponer J, Deraos G, Bamm VV, Harauz G, Matsoukas J, de Planque MR, Gerothanassis IP, Babu MM, Luchinat C, Tzakos AG. Recognition pliability is coupled to structural heterogeneity: a calmodulin intrinsically disordered binding region complex. Structure. 2012;20:522–533. doi: 10.1016/j.str.2012.01.021. [DOI] [PubMed] [Google Scholar]
- 26.Crivici A, Ikura M. Molecular and structural basis of target recognition by calmodulin. Annual review of biophysics and biomolecular structure. 1995;24:85–116. doi: 10.1146/annurev.bb.24.060195.000505. [DOI] [PubMed] [Google Scholar]
- 27.Consortium U. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40:D71–75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yap KL, Kim J, Truong K, Sherman M, Yuan T, Ikura M. Calmodulin target database. J Struct Funct Genomics. 2000;1:8–14. doi: 10.1023/a:1011320027914. [DOI] [PubMed] [Google Scholar]
- 29.Lo WL, Donermeyer DL, Allen PM. A voltage-gated sodium channel is essential for the positive selection of CD4(+) T cells. Nat Immunol. 2012;13:880–887. doi: 10.1038/ni.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weber KS, Miller MJ, Allen PM. Th17 cells exhibit a distinct calcium profile from Th1 and Th2 cells and have Th1-like motility and NF-AT nuclear localization. J Immunol. 2008;180:1442–1450. doi: 10.4049/jimmunol.180.3.1442. [DOI] [PubMed] [Google Scholar]
- 31.Jain N, Miu B, Jiang JK, McKinstry KK, Prince A, Swain SL, Greiner DL, Thomas CJ, Sanderson MJ, Berg LJ, Kang J. CD28 and ITK signals regulate autoreactive T cell trafficking. Nature medicine. 2013 doi: 10.1038/nm.3393. [DOI] [PMC free article] [PubMed]
- 32.Chen Z, Medina F, Liu MY, Thomas C, Sprang SR, Sternweis PC. Activated RhoA binds to the pleckstrin homology (PH) domain of PDZ-RhoGEF, a potential site for autoregulation. J Biol Chem. 2010;285:21070–21081. doi: 10.1074/jbc.M110.122549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chhatriwala MK, Betts L, Worthylake DK, Sondek J. The DH and PH domains of Trio coordinately engage Rho GTPases for their efficient activation. J Mol Biol. 2007;368:1307–1320. doi: 10.1016/j.jmb.2007.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Godi A, Di Campli A, Konstantakopoulos A, Di Tullio G, Alessi DR, Kular GS, Daniele T, Marra P, Lucocq JM, De Matteis MA. FAPPs control Golgi-to-cell-surface membrane traffic by binding to ARF and PtdIns(4)P. Nat Cell Biol. 2004;6:393–404. doi: 10.1038/ncb1119. [DOI] [PubMed] [Google Scholar]
- 35.Snyder JT, Worthylake DK, Rossman KL, Betts L, Pruitt WM, Siderovski DP, Der CJ, Sondek J. Structural basis for the selective activation of Rho GTPases by Dbl exchange factors. Nature structural biology. 2002;9:468–475. doi: 10.1038/nsb796. [DOI] [PubMed] [Google Scholar]
- 36.Dong B, Valencia CA, Liu R. Ca(2+)/calmodulin directly interacts with the pleckstrin homology domain of AKT1. J Biol Chem. 2007;282:25131–25140. doi: 10.1074/jbc.M702123200. [DOI] [PubMed] [Google Scholar]
- 37.Coticchia CM, Revankar CM, Deb TB, Dickson RB, Johnson MD. Calmodulin modulates Akt activity in human breast cancer cell lines. Breast cancer research and treatment. 2009;115:545–560. doi: 10.1007/s10549-008-0097-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bi F, Debreceni B, Zhu K, Salani B, Eva A, Zheng Y. Autoinhibition mechanism of proto-Dbl. Mol Cell Biol. 2001;21:1463–1474. doi: 10.1128/MCB.21.5.1463-1474.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calleja V, Alcor D, Laguerre M, Park J, Vojnovic B, Hemmings BA, Downward J, Parker PJ, Larijani B. Intramolecular and intermolecular interactions of protein kinase B define its activation in vivo. PLoS biology. 2007;5:e95. doi: 10.1371/journal.pbio.0050095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calleja V, Laguerre M, Parker PJ, Larijani B. Role of a novel PH-kinase domain interface in PKB/Akt regulation: structural mechanism for allosteric inhibition. PLoS biology. 2009;7:e17. doi: 10.1371/journal.pbio.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Das B, Shu X, Day GJ, Han J, Krishna UM, Falck JR, Broek D. Control of intramolecular interactions between the pleckstrin homology and Dbl homology domains of Vav and Sos1 regulates Rac binding. J Biol Chem. 2000;275:15074–15081. doi: 10.1074/jbc.M907269199. [DOI] [PubMed] [Google Scholar]
- 42.Akyol Z, Bartos JA, Merrill MA, Faga LA, Jaren OR, Shea MA, Hell JW. Apo-calmodulin binds with its C-terminal domain to the N-methyl-D-aspartate receptor NR1 C0 region. J Biol Chem. 2004;279:2166–2175. doi: 10.1074/jbc.M302542200. [DOI] [PubMed] [Google Scholar]
- 43.Newman RA, Van Scyoc WS, Sorensen BR, Jaren OR, Shea MA. Interdomain cooperativity of calmodulin bound to melittin preferentially increases calcium affinity of sites I and II. Proteins. 2008;71:1792–1812. doi: 10.1002/prot.21861. [DOI] [PubMed] [Google Scholar]
- 44.Xiong LW, Newman RA, Rodney GG, Thomas O, Zhang JZ, Persechini A, Shea MA, Hamilton SL. Lobe-dependent regulation of ryanodine receptor type 1 by calmodulin. J Biol Chem. 2002;277:40862–40870. doi: 10.1074/jbc.M206763200. [DOI] [PubMed] [Google Scholar]
- 45.Reichow SL, Clemens DM, Freites JA, Nemeth-Cahalan KL, Heyden M, Tobias DJ, Hall JE, Gonen T. Allosteric mechanism of water-channel gating by Ca2+-calmodulin. Nat Struct Mol Biol. 2013;20:1085–1092. doi: 10.1038/nsmb.2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sarhan MF, Tung CC, Van Petegem F, Ahern CA. Crystallographic basis for calcium regulation of sodium channels. Proc Natl Acad Sci U S A. 2012;109:3558–3563. doi: 10.1073/pnas.1114748109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vlach J, Samal AB, Saad JS. Solution structure of calmodulin bound to the binding domain of the HIV-1 matrix protein. J Biol Chem. 2014;289:8697–8705. doi: 10.1074/jbc.M113.543694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hayashi N, Matsubara M, Jinbo Y, Titani K, Izumi Y, Matsushima N. Nef of HIV-1 interacts directly with calcium-bound calmodulin. Protein Sci. 2002;11:529–537. doi: 10.1110/ps.23702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsubara M, Jing T, Kawamura K, Shimojo N, Titani K, Hashimoto K, Hayashi N. Myristoyl moiety of HIV Nef is involved in regulation of the interaction with calmodulin in vivo. Protein Sci. 2005;14:494–503. doi: 10.1110/ps.04969605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matsubara M, Nakatsu T, Kato H, Taniguchi H. Crystal structure of a myristoylated CAP-23/NAP-22 N-terminal domain complexed with Ca2+/calmodulin. EMBO J. 2004;23:712–718. doi: 10.1038/sj.emboj.7600093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matsubara M, Titani K, Taniguchi H, Hayashi N. Direct involvement of protein myristoylation in myristoylated alanine-rich C kinase substrate (MARCKS)-calmodulin interaction. J Biol Chem. 2003;278:48898–48902. doi: 10.1074/jbc.M305488200. [DOI] [PubMed] [Google Scholar]
- 52.Pouillon V, Hascakova-Bartova R, Pajak B, Adam E, Bex F, Dewaste V, Van Lint C, Leo O, Erneux C, Schurmans S. Inositol 1,3,4,5-tetrakisphosphate is essential for T lymphocyte development. Nat Immunol. 2003;4:1136–1143. doi: 10.1038/ni980. [DOI] [PubMed] [Google Scholar]
- 53.Wen BG, Pletcher MT, Warashina M, Choe SH, Ziaee N, Wiltshire T, Sauer K, Cooke MP. Inositol (1,4,5) trisphosphate 3 kinase B controls positive selection of T cells and modulates Erk activity. Proc Natl Acad Sci U S A. 2004;101:5604–5609. doi: 10.1073/pnas.0306907101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feng Y, Nie L, Thakur MD, Su Q, Chi Z, Zhao Y, Longmore GD. A multifunctional lentiviral-based gene knockdown with concurrent rescue that controls for off-target effects of RNAi. Genomics Proteomics Bioinformatics. 2010;8:238–245. doi: 10.1016/S1672-0229(10)60025-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 56.Sorensen BR, Faga LA, Hultman R, Shea MA. An interdomain linker increases the thermostability and decreases the calcium affinity of the calmodulin N-domain. Biochemistry. 2002;41:15–20. doi: 10.1021/bi011718+. [DOI] [PubMed] [Google Scholar]
- 57.Goddard TD. University of California; San Francisco: [Google Scholar]
- 58.Jung YS, Zweckstetter M. Mars -- robust automatic backbone assignment of proteins. Journal of biomolecular NMR. 2004;30:11–23. doi: 10.1023/B:JNMR.0000042954.99056.ad. [DOI] [PubMed] [Google Scholar]
- 59.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. Journal of biomolecular NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 60.Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 61.Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J, Livny M, Mading S, Maziuk D, Miller Z, Nakatani E, Schulte CF, Tolmie DE, Kent Wenger R, Yao H, Markley JL. BioMagResBank. Nucleic Acids Res. 2008;36:D402–408. doi: 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Severin A, Joseph RE, Boyken S, Fulton DB, Andreotti AH. Proline isomerization preorganizes the Itk SH2 domain for binding to the Itk SH3 domain. J Mol Biol. 2009;387:726–743. doi: 10.1016/j.jmb.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 65.Marshall IC, Boyfield I, McNulty S. Ratiometric Ca(2)(+) measurements using the FlexStation((R))Scanning Fluorometer. Methods Mol Biol. 2013;937:95–101. doi: 10.1007/978-1-62703-086-1_4. [DOI] [PubMed] [Google Scholar]
- 66.Lin J, Weiss A. The tyrosine phosphatase CD148 is excluded from the immunologic synapse and down-regulates prolonged T cell signaling. J Cell Biol. 2003;162:673–682. doi: 10.1083/jcb.200303040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000;7:1063–1066. doi: 10.1038/sj.gt.3301206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. CaM binding maps to the Itk PH domain.
Fig. S2. The N-domain of CaM does not bind to the Itk PH domain.
Fig. S3. Structural model of the Itk PH domain showing the location of the mutated residues tested.
Fig. S4. TCR stimulation promotes Akt activation and PLCγ1 phosphorylation in Jurkat E6.1 cells.
Fig. S5. Kinetic analysis of recruitment of WT-Itk or LS-Itk to the immune synapse
Table S3. Calmodulin binding predictions for 236 mouse PH domains listed in the Uniprot database.
Table S1. Effect of Itk PH domain point mutations on protein abundance, PI(3,4,5)P3 binding, and CaM binding
Table S2. Oligo primers for cloning PH domains and YFP