

Abstract
We recently reported that adoptively transferred (AT) exogenous CD4+CD25+ regulatory T cells (Tregs) to wild type (WT) mice can directly act to repress shock/sepsis induced experimental iALI and this is mediated in part by programmed cell death receptor 1 (PD-1). In this study, we further determine whether recipient mouse lacking PD-L1, one of the primary ligands for PD-1, contributes to the manipulation of the Tregs’ capacity to repress lung injury. To do this, Tregs isolated from the spleen of WT mice were AT into PD-L1−/− mice subjected to hemorrhagic shock [Hem] and subsequent to cecal ligation and puncture (CLP) to induce iALI. Samples were collected for analyses 24 hours after CLP. We found that in PD-L1−/− recipient mice, AT WT-Tregs lost the ability to reverse the development of iALI seen in WT recipient mice (i.e., no reduction of lung injury indices assessed by histology and vascular leakage; failure to decrease the lung neutrophil influx [MPO activity] or the rise in lung apoptosis [caspase 3 activity]). Also a significant increase of interlukin-1β (IL-1β) and keratinocyte-derived chemokine (KC), but no changes in IL-6, IL-10 and IL-17A levels in lung tissues were seen in these mice compared with iALI mice without AT of Tregs. Furthermore, we noted that the lung tissue tyrosine phosphatase Src homology region 2 domain-containing phosphatase 1 (SHP-1), but not SHP-2, was activated with the AT of Tregs in PD-L1−/− iALI mice. Finally, through local depletion of CD4+ T cells or CD25+ (Tregs) in the lung, prior to inducing iALI, we found that SHP-1 activation was associated with the loss of Tregs’ protective effects in vivo. Collectively, our data reveal that PD-L1 is a critical modulator of Treg’s ability to suppress iALI and this appears to involve SHP-1 activation.
Keywords: Programmed death receptor-1, neutrophil influx, apoptosis, CTLA-4, SHP-2
Introduction
Acute respiratory distress syndrome (ARDS) is a clinical syndrome defined by acute hypoxemic respiratory failure, bilateral pulmonary infiltrate attributable to edema and normal cardiac filling pressures [1]. With the advances in supportive care, including improvement of gas exchange by extracorporeal membrane oxygenation, many patients do not die at the early stages of ARDS, but rather succumb to the later phases [2]. Injury resolution is an active process [3]; however, the potential mechanisms that drive the repression of inflammation, avoidance of pulmonary fibrosis and remodeling of alveolar epithelium and endothelium are still not well understood.
Currently, the potential role of T-regulatory cells (Tregs), a subset of CD4+ T lymphocytes phenotyped as CD4+CD25+Foxp3+, are being explored in animal models of direct acute lung injury (ALI) induced by intra-tracheal instillation of lipopolysaccharide (LPS) and appear to augment the resolution of ALI/ARDS [4,5,6]. Using a murine model of hemorrhage (shock) in combination with a subsequent septic challenge, which approximates aspects of what is seen in critically injured patients with ARDS, our laboratory has shown that adoptive transfer (AT) of wild type (WT)-Tregs induced a significant repression of the indices of lung injury compared to the vehicle-treated indirect (i)ALI WT recipient mice by a reduction of neutrophil influx to the lung and a decrease of lung cell apoptosis. In addition, these recipient mice had substantially higher concentrations of protein in broncho-alveolar lavage fluid (BAL) and increased interleukin (IL)-10 and IL-17A levels in lung tissues, but significantly decreased levels of lung chemokine keratinocyte-derived chemokine (KC) [7]. Our investigation, together with studies from other laboratories, has thus far demonstrated a therapeutic potential of Tregs in life-threatening complications such as in ALI/ARDS [4,5,6]. However, as a novel approach of cell therapy, growing evidence indicates that instability and plasticity of Tregs remain a major concern for the use of such therapeutic strategies in vivo [8].
Programmed death-1 (PD-1 or CD279) receptor and its ligand, PD-L1 (B7-H1 or CD274) and PD-L2 (B7-DC or CD273) delivers inhibitory signals, which are reported to regulate the balance between T-cell activation, tolerance, and immune-mediated tissue damage (review in [9,10]). PD-1 and PD-L1 are both expressed on Tregs; it is reported that signaling through the PD-L1: PD-1 pathway act in many ways to either promote Treg development or inhibit the expansion/activation of T-effector cells, the cells Tregs interact with; thus, protecting against autoimmunity (review in [9,10]). However, whether and how this pathway modulates the function of Tregs in the resolution of ALI/ARDS is still poorly understood. It is not clear what the role of PD-L1, the primary ligand of PD-1, is in regulating the function of Tregs in these processes.
Mutagenesis studies indicate that the tyrosine within the immunoreceptor tyrosine-based switch motif (ITSM) is essential for PD-1 function in T and B cells [11]. The protein tyrosine phosphatase Src homology region 2 domain-containing phosphatase 1 (SHP-1) and SHP-2 can bind to the ITSM sequence on PD-1 cytoplasmic tail [12]. These SHPs then antagonize T-cell receptor driven activation of the Serine protein kinase (Akt)/phosphatidylinositol-3-kinase (PI3K) pathway, which is thought to underpin the development of the immune suppressed T-effector cell phenotype seen[11,12]. Regarding PD-L1 and/or PD-L2, as these lack an overt ITSM domain, it was assumed they did not produce an intra-cellular signal; however, recent studies suggest this may not wholly be true [13,14]. Unfortunately, the type and significance of this signaling remains to be established.
In this study, we determine whether PD-L1 contributes to the therapeutic effects of Tregs by evaluating the effect of an AT of Tregs derived from the spleens of WT mice transfer into PD-L1−/− mice subjected to hemorrhage (Hem) plus cecal ligation & puncture (CLP). Furthermore, we set out to explore the potential mechanism by which PD-L1: PD-1 ligation contributes to the immune protective/suppressive capacity of Tregs during iALI.
Materials and Methods
Mice
Male WT C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME). PD-L1−/− mice were obtained as a gift of L. Chen (Yale University, New Haven, CT) and maintained in our animal facility. Mice aged 10–12 weeks were used for all experiments. Experiments were performed in accordance with National Institutes of Health guidelines and with approval from the Animal Use Committee of Rhode Island Hospital.
Reagents
Keratinocyte-derived chemokine (KC) and macrophage inflammatory protein-2 (MIP-2) antibodies for ELISA assays were purchased from R&D Systems, Minneapolis, MN. Mouse IL-6, IL-10, tumor necrosis factor-α (TNF-α), monocyte chemoattractant protein (MCP-1), MIP-2 and IL-17A ELISA kits were purchased from BD Bioscience, San Diego, CA. Antibodies for phosphorylated (p)-SHP-1, SHP-1, p-SHP-2, SHP-2, p-Akt and Akt were purchased from Cell Signaling Technology (Danvers, MA). Antibody for beta-actin was purchased from Abcam Company (Cambridge, MA). All other chemicals were of analytical reagent grade and purchased from Sigma Chemical, St Louis, MO.
Rodent Model for iALI (Hemorrhage/Cecal Ligation & Puncture [CLP] Model)
Hemorrhage (Hem)
The non-lethal fix-pressure Hem model used for these experiments has been previously described in our laboratory [15,16,17,18,19]. In brief, mice were anesthetized with isoflurane, restrained in supine position, and catheters were inserted into both femoral arteries. Anesthesia was discontinued, and blood pressure was continuously monitored through one catheter attached to a blood pressure analyzer (BPA; MicroMed, Louisville, KY). When fully awake, as determined by a mean blood pressure of ~95 mmHg, the mice were bled over a 5 to 10 min period to a mean blood pressure of 35 mmHg (±5 mmHg) and kept stable for 90 min. Immediately following Hem, mice were resuscitated with Ringer’s lactate at four times drawn blood volume. After resuscitation, arteries were ligated, catheters removed, and catheter sites bathed with lidocaine and sutured closed. Sham mice were anesthetized, restrained in a supine position and blood vessels were ligated but no blood was drawn [15,16,17,18,19].
Polymicrobial Sepsis/CLP
Twenty four hours after Hem, mice were anesthetized with isoflurane and restrained in supine position. A 1 cm midline incision was made; the cecum was ligated with 5-0 silk thread and punctured twice with a 22-gauge needle. A small amount of fecal material was extruded from the punctured cecum. The cecum was then replaced, and the incision was closed. Mice were resuscitated with 1 ml Ringer’s lactate subcutaneously and returned to their cages. Sham mice were anesthetized, restrained in a supine position for the same duration, and the cecum was exposed, but neither ligated nor punctured [15,16,17,18,19].
Importantly, as we and other reports have previously documented that neither Hem nor CLP alone is capable of inducing substantial increases in the indices of lung injury [15,16,17,18,19], we, therefore, show only Sham-Hem plus Sham-CLP (S/S) as our Sham controls for the study here.
Isolation of T-Regulatory Cells (Tregs) and Adoptive Transfer Protocol
CD4+CD25+ Tregs were isolated from the spleens of WT C57BL/6 mice by the method as previously reported [20]. Splenocytes were obtained by gently grinding the spleens between frosted glass slides and contaminated red blood cells were lysed with a hypotonic solution. The cell suspension was centrifuged at 300 × g for 10 min and the pellet was resuspended in an MACS (Miltenyi Biotec, Auburn, CA) running buffer. This was followed by isolating Tregs, using a two-step process according to the manufacturer’s instructions (CD4+ CD25+T-Regulatory Cell Isolation Kit; Miltenyi Biotec). The CD4+ T cells were pre-enriched by depleting unwanted cells, using a cocktail of antibodies. Then CD25+ cells were positively selected from the enriched CD4+ T-cell fraction. The purity of CD4+CD25+ cells was 90–95% and 80–90% when also co-expressing Foxp3 as a marker by flow cytometric analysis.
For adoptive transfer (AT) groups, 1 × 106 spleen-derived CD4+CD25+ cells from WT mice, in 200 ul sterile DMEM, were intravenously infused into recipient PD-L1−/− mice during resuscitation period of Hem (all recipient mice were PD-L1−/− mice). For the without AT (NOT-AT) control iALI mice, 200 μl sterile DMEM without any cells were intravenously infused into recipient PD-L1−/− mice during Hem resuscitation period. Twenty four hours later, all mice were made septic via CLP as previously described [15,16,17,18,19].
Intratracheal Instillation of Antibodies (Abs)
To examine the effect of the lack of CD4+ and CD25+ T lymphocytes in the lung for some experiments, intratracheal (IT) instillation of functional grade purified rat anti-mouse CD4 Abs (Clone GK1.5, 50 ug; eBioscience, San Diego, CA), anti-mouse CD25 Abs (Clone PC61.5, 50 ug; eBioscience) or isotype control (rat Ig G2b) were given 12 hours prior Hem as previously described [15,20].
Sample Acquisition
Twenty-four hours after CLP, mice were euthanized with an overdose of isoflurane. For the acquisition of BAL, the trachea was exposed via a midline incision and cannulated with a sterile polypropylene 18-gauge catheter. The lungs were lavaged with 0.6 ml of saline two times for an average of 1 ml of lavage fluid recovery per lung. Lavage fluid was centrifuged at 1500 × g for 10 min at 4°C and the supernatant was tested for protein concentrations (index of lung permeability) and cytokine/chemokine levels. For lung histology, the left lobe of the lung was harvested and fixed in 10% formalin, paraffin embedded and tissue sections were prepared as previously described [15,16,17,18,19]. Samples were then stained with hematoxylin and eosin (H & E) and examined by light microscopy. For flow cytometry, The left lobe of the lung underwent an enzymatic digestion as previously described [15,16,17,18,19]. The right lobes of the lung were prepared for myeloperoxidase (MPO) activity or apoptosis (Caspase3 activity) separately.
Lung MPO and Caspase3 Activity Assay
Lung MPO activity, a marker for neutrophil influx, was measured according to the protocols described previously [15,16,17,18,19]. Active Caspase3 activity in lung tissue homogenate was quantified via established protocols [15,16]. Protein concentration was assessed using the Bradford dye-binding procedure (Bio-Rad, Hercules, CA).
Antibodies and Flow Cytometry
Cells in lung tissue digests were phenotyped in combination of PD-1 and cytotoxic T lymphocyte-associatedantigen-4 (CTLA-4) expression by flow cytometry (FACS Array, BD Bioscience, Mountain View, CA) [15,16]. The following monoclonal antibodies (mAbs) conjugated to fluorochromes were used: phycoerythrin (PE)-labeled anti-CD4 (clone GK 1.5), PE-labeled anti-CTLA-4 (UC10-4B9), PE-labeled anti-CD25 (clone PC61.5), allophycocyanin (APC)-labeled anti-granulocyte differentiation antigen 1 (Gr1) (clone RB6-8C5), APC-labeled anti-CTLA-4 (clone UC10-4B9), APC-labeled anti-Foxp3 (clone FJK-16s), APC-labeled anti-CD4 (clone GK 1.5), PE-Cyanine7-labeled anti-PD-1(Clone J43), PE-Cyanine7-labeled anti-CD25 (Clone PC61.5) purchased from eBioscience. Antibodies and their isotype controls were used according to the manufacture’s recommendations. Intracellular staining of Foxp3 was carried out according to the manufacturer’s instructions (eBioscience) as described previously [15,20]. Data were collected on a FACS Array flow cytometer (BD Bioscience) and analyzed using FlowJo software (Tree Star, Ashland, OR).
Quantification of Chemokines and Cytokines
BAL samples and lung tissue homogenate supernatants were analyzed for IL-1β, KC, MIP2, IL-6, IL-10, and IL-17A levels by ELISA (BD Bioscience) as described previously [15,16,17,18,19].
Western Blotting
Forty μg of lung tissue lysate proteins, as determined by a Bradford protein assay were loaded equally onto 10% polyacrylamide gels (Life Technologies Corporation; Carlsbad, CA), and transferred to PVDF membranes (Life Technologies, Corp.). Membranes were blocked for one hour at room temperature in 5% non-fat dry milk in Tris-buffered saline with Tween 20 (TBST), and then probed with anti-mouse phospho (p)-SHP-1 (Tyr564), p-SHP-2 (Tyr580), p-Akt (Thr308) or SHP-1, SHP-2, Akt antibodies (Cell Signaling Technology, Inc., Danvers, MA) overnight at 4°C in TBST containing 5% bovine serum albumin (BSA). After primary antibody incubation, membranes were washed three times in TBST and rabbit anti-mouse secondary antibody (Cell Signaling Technology, Inc.) was added at a concentration of 1:3000 in TBST with 5% BSA. β-actin was blotted as a loading control. Membranes were developed by chemiluminescence using an Amersham prime ECL plus detection system (GE Healthcare Life Sciences; Pittsburgh, PA) and densitometric analyses were performed as previously reported by our laboratory [21].
Statistical Analysis
Data are expressed as mean ± SEM and analyzed using GraphPad Prism 5 statistical analysis and graphing software. Unpaired student t test was used to determine presence of a significant difference between the two groups. Multiple group comparisons were performed using one-way ANOVA with the post hoc test of Tukey’s. Difference at P <0.05 was considered significant.
Results
WT-Tregs lose the ability to suppress shock/sepsis induced iALI in PD-L1−/− mice
In light of our recent demonstration that AT WT-Tregs can directly act to modify the innate immune response induced by experimental iALI in WT mice that did not receive Tregs (they [the mice receiving AT WT-Tregs] suppressed iALI-induced lung injury [as detected by BAL protein and histological changes], repressed neutrophil influx, decreased the rise of iALI-induced tissue cell apoptosis, impeded the rise in lung tissue KC levels, while potentiating local levels of lung IL-10) [7] and Treg expression of PD-1 as a central contributor to lung protection (AT PD-1−/− Tregs attenuated these changes) [7], we hypothesized that a possible role of its primary ligand, PD-L1, is in the modulation of Tregs’ capacity in vivo in the repression of iALI. To address this hypothesis, recipient PD-L1−/− mice were subjected to Hem/CLP to produce iALI and divided into two groups with or without AT of WT-Tregs. Indices of indirect lung injury were measured in the lung 24 hours after CLP.
As protein concentration in the BAL is commonly used to quantify the vascular permeability of lung injury in mice; in the current study, we observed that the protein levels in BAL from PD-L1−/− recipient mice, without AT of Tregs, subjected to Hem plus CLP were significantly increased in comparison with the S/S animals (Fig 1A). This was consistent with our histological findings of an increase in alveolar septal wall thickening associated with marked cellular infiltrates and alveolar congestion/collapse in comparison to S/S animals (Fig 1B–C).
Figure 1. WT-Tregs-AT delivered to PD-L1 deficient iALI mice recapitulates the lung injury induced by shock/sepsis without adoptive transfer.
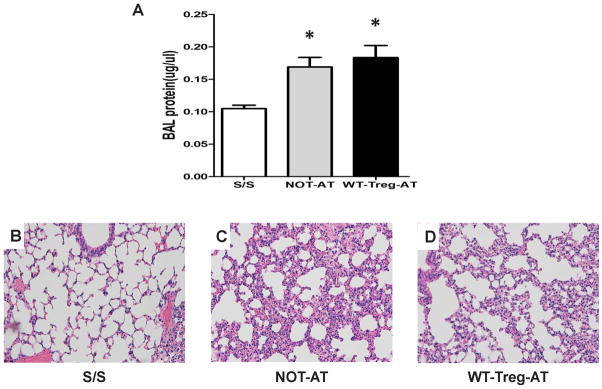
(A) Protein concentrations, as a marker for lung leakage, were measured in the bronchoalveolar lavage fluid (BAL) of S/S (Shem/SCLP) control; NOT-AT (Hem/CLP without adoptive transferring of WT-Tregs) and WT-Tregs-AT (Hem/CLP with adoptive transferring of WT-Tregs) group with PD-L1−/− recipient mice. (n=6–10/group, * p < 0.05 vs. S/S control, determined by one-way ANOVA for multiple group comparisons). All data are expressed as mean ± SEM. (B, C, D) The lungs were fixed, sectioned and stained with H&E. Representative sections are shown for S/S control group (B), NOT-AT group (C) and WT-Tregs-AT group (D) (original magnification, × 200).
We found that there was no change in the BAL protein levels in the WT-Tregs-AT to PD-L1−/− iALI recipient mice compared with the animals without AT Tregs (Fig 1A). Morphologically, sections of lung stained with H&E showed no reduction of the disruption of lung tissue architecture and congestion seen in the WT-Tregs-AT PD-L1−/− group when compared to iALI mice without AT of Tregs (Fig 1C–D).
Previous studies have shown that the influx of neutrophils to the lung play a pivotal role in iALI [15,16,17,18,19]. Here we observed a marked influx of neutrophils into the lung, as measured by an increase of both MPO activity and percentage of Gr1+ cells in the lung tissue of PD-L1−/− mice with iALI (NOT-AT group) when compared to the S/S control animals (Fig 2A–C), which was consistent with the changes seen in WT iALI mice [7,16,18]. However, WT-Tregs-AT to PD-L1−/− iALI mice showed no changes in neutrophil influx compared to iALI mice without AT of WT-Tregs (Fig 2A–C).
Figure 2. Adoptive transfer of WT-Tregs into PD-L1 deficient iALI mice fails to reduce neutrophil influx and apoptosis in the lung.

(A) While both the WT-Tregs-AT and NOT-AT iALI groups had a significant increase in MPO activity compared with S/S group, there was no significant difference of lung tissue MPO activity between the two iALI groups. (B) The lungs were digested and cells were phenotyped for Gr1 expression as a marker for neutrophils. Representative dot plots are shown for expression (percentage) of Gr1+ cells. (C) Consistent with MPO activity, the percentage of Gr1+ cells in lung tissue showed the same trend. (D) While both AT or NOT- AT of WT-Tregs iALI mice had a significant increase in Caspase3 activity comparing with S/S group, no significant difference was noted between the two iALI groups. (n=6–10 group, * p < 0.05 vs. S/S control, determined by one-way ANOVA for multiple group comparisons). All data are expressed as mean ± SEM.
Because our studies have previously shown that pulmonary cell apoptosis is increased after iALI in WT mice [16], we next set out to test if AT of exogenous WT-Tregs could affect lung cell apoptosis in PD-L1−/− recipient mice with iALI. A significant increase of lung cell apoptosis as exhibited by Caspase 3 activity was noted in both Treg-AT and NOT-AT PD-L1−/− mice following Hem/CLP when compared with S/S animals (Fig 2D). By contrast, there was a trend towards an increase, but not significant, in Caspase 3 activity in WT-Tregs-AT to PD-L1−/− mice compared to the iALI animals without Tregs (Fig 2D).
As stated previously, increases in inflammatory cytokines are also thought to be indices of local pulmonary injury/inflammation after the double insult of Hem plus CLP in mice [15,16,17,18,19]. Here we observed that the levels of IL-1β, IL-6 and KC were significantly increased in the lungs of the PD-L1−/− recipient iALI mice as compared with control animals (Fig 3A–C), while AT of WT-Tregs into PD-L1−/− iALI mice caused a significant increase of IL-1β and KC, and no changes of IL-6 concentration in lung tissues when compared with the iALI mice not receiving AT of Tregs (Fig 3A–C). Interestingly, as for IL-10 and IL-17A, there was no significant difference in the levels of these mediators between experimental groups (Fig 3D), which was distinctively different from what we had found in WT-Treg-AT into WT mice [7]. We also observed the same trend for the other important neutrophil chemokine, MIP-2, the pivotal inflammatory cytokines TNF-α and MCP-1 (data not shown). Taken together, this indicates that the exogenous WT-Tregs do not exhibit the beneficial effects of reducing the inflammation and indices of lung injury in recipient PD-L1−/− mice that were exposed to the double insult of Hem followed by CLP.
Figure 3. Adoptive transfer of WT-Tregs into PD-L1 deficient mice differential effects the levels of select chemokines and inflammatory cytokines detected in the lungs following iALI.

While iALI mice (AT or NOT-AT) showed increased levels of IL-1β, KC and IL-6; no changes were seen in IL-10 and IL-17A when compared with S/S group. Alternatively, the WT-Tregs-AT group showed increased in IL-1β (A), KC (B), and had the similar levels of IL-6 (C), IL-10 and IL-17A (D) in lung tissue when compared with the NOT-AT group. (n=6–10 group, * p < 0.05 vs. S/S group; # p < 0.05 vs. NOT-AT group, determined by one-way ANOVA for multiple group comparisons). All data are expressed as mean ± SEM.
Adoptive transfer of WT-Tregs alters the expression of Foxp3, but does not affect the expression of PD-1 or CTLA-4 on Tregs in the lungs of PD-L1−/− recipient mice
Since our studies have previously documented that AT of WT-Tregs, but not AT of PD-1−/−-Tregs had the ability to increase the expression of Foxp3, as well as PD-1 and CTLA-4 on CD4+Foxp3+ cells in the lungs of recipient WT iALI mice [7]. We next attempted to determine the changes of cellular phenotype in the lung after adoptively transferring the WT-Tregs into the PD-L1−/− recipient mice.
In agreement with our prior data [7], we also observed here an increase in the ratio of CD4+Foxp3+ vs. CD4+ Foxp3− T lymphocytes in the lungs of PD-L1−/− iALI mice without Tregs when compared to S/S controls (Fig 4A–B). Furthermore, in iALI mice that received WT-Tregs, a significant increase in this ratio was detected when compared to NOT-AT group (Fig 4A–B). However, no significant difference in the percentage of PD1+CD4+Foxp3+ cells in the lung was noted among all the experimental groups (Fig 4C). Interestingly, we observed a significant up-regulation of the percentage of CTLA-4+CD4+Foxp3+ cells in the PD-L1−/− iALI mice when compared to S/S groups, although no significant differences were noted between the groups with or without exogenous WT-Tregs (Fig 4D). As for the intensity (mean fluorescent intensity, MFI) of PD-1 and CTLA-4 expression on CD4+Foxp3+ cells, all the experimental groups exhibited no significant difference (data not shown). Putting these findings together, this indicates that without PD-L1 on the recipient mouse cells, although the exogenous WT-Tregs have the ability to increase the expression of Foxp3, they lack the ability to increase the expression of PD-1 and CTLA-4 on lung CD4+ Foxp3+ cells during iALI.
Figure 4. Adoptive transfer of WT-Tregs into PD-L1 deficient iALI mice differential effects the ratio of CD4+Foxp3+ to CD4+ cells and the frequency of Tregs expressing PD-1 vs. CTLA-4.

The lungs were digested and cells were phenotyped for CD4+Foxp3+ and CD4+ frequency/expression by flow cytometry. (A) While the ratio of CD4+Foxp3+ vs. total CD4+ T cells was increased in iALI mice when compared with S/S, the AT group was increased further when compared with NOT-AT group. (B) Representative dot plots are shown for the ratio of CD4+Foxp3+ Tregs vs. CD4+ T cells (X axis represents CD4+ T cells and Y axis represents CD4+FoxP3+ T cells, and the square represents ratio of CD4+FoxP3+ Tregs vs. CD4+ T cells). (C) The percentage of lung CD4+Foxp3+ cells expressing PD-1, measured by flow cytometry, was not changed between all groups. (D) While CTLA-4 expression was increased in iALI mice when compared with S/S group, there was no difference between the AT and NOT-AT iALI groups. (n=6–10 group, * p < 0.05 vs. S/S control group; # p < 0.05 vs. NOT-AT group, determined by one-way ANOVA for multiple group comparisons). All data are expressed as mean ± SEM.
PD-L1 deficiency negatively regulate exogenous WT-Tregs’ capacity in the repression of iALI is partially through the recruitment of phosphatase SHP-1 but not SHP-2
As a co-inhibitory receptor bearing ITSM domain, it is believed that the suppression of T-cell function is a result of the recruitment of the phosphatase SHP-1 and/or SHP-2 by PD-1[11]. However, it has been neither documented what the nature of the downstream signaling of the PD-1: PD-L1 pathway nor determined how it affects the Tregs’ function in the development of ALI/ARDS. We next set out to further investigate the potential intra-cellular pathway signaling by which PD-L1/PD-1 may mediate Tregs’ function in vivo. As shown in Fig 5A, we found that p-SHP-1 was undetectable in lung lysates of both PD-L1−/− iALI mice without Tregs and S/S control groups, in contrast, a significantly up-regulated p-SHP-1 level was observed in WT-Tregs recipient PD-L1−/− iALI mouse lungs when compared with iALI mice without Tregs (Fig 5A–B). Unlike the changes in p-SHP-1levels, p-SHP-2 levels, although still not detectable in S/S PD-L1−/− mice, showed a significant increase of p-SHP-2 in the PD-L1−/− iALI mice with or without AT of WT-Tregs (Fig 6A–B). This implies that exogenous WT-Tregs had no effects on activation of SHP-2 in the injured lung of the PD-L1−/− mice (Fig 6A–B). Interestingly, for the WT iALI mice, although we observed the recruitment of both SHP-1 and SHP-2, in the whole lung compared with S/S group, no significant difference in that was noted between the AT or NOT-AT iALI groups (data not shown).
Figure 5. The depletion of either CD4+ or CD25+ cells in the recipient mouse lungs prior to the adoptive transfer of WT-Tregs into PD-L1 deficient iALI mice markedly reduced the extent of activation of SHP-1 (p-SHP) seen in lung tissue.

(A) Representative western blot depicting whole lung tissue lysates probed for phosphorylated-SHP-1(p-SHP-1), total SHP-1 (SHP-1), and β-actin from S/S, NOT-AT and WT-Tregs-AT groups with or without intra-tracheal instillation of CD4 or CD 25 antibody. (B) Semi-quantitation by densitometry and expressed as integrated density (IDT) values of p-SHP-1 relative to IDT values of total SHP-1.
There was a significant increase in p-SHP-1 expression in all three WT-Treg-AT iALIanimals (with or without intra-tracheal instillation of CD4 or CD 25 antibody) vs. S/S and NOT-AT groups. However, a further decline in p-SHP-1 expression was seen in both the local depletion of CD4 T cells and Tregs groups. (n=3–6/group, * p < 0.05 vs. S/S group; # p < 0.05 vs. NOT-AT group; @ p < 0.05 vs. WT-Tregs-AT group without depletion of cells, determined by one-way ANOVA for multiple group comparisons). All data are expressed as mean ± SEM.
Figure 6. Adoptive transfer of WT-Tregs into PD-L1 deficient iALI mice had no effect on the expression of activated/phosphorylated SHP-2 (p-SHP-2) in lung tissue.

(A) Representative western blot depicting lung tissue lysates probed for p-SHP-2, SHP-2, and β-actin from S/S, NOT-AT and WT-Tregs-AT animals (with or without intra-tracheal instillation of CD4 or CD 25 antibody) and (B) Semi- quantitation by densitometry and expressed as integrated density (IDT) values of p-SHP-2 relative to IDT values of SHP-2. While iALI induced an increase in p-SHP-2 expression when compared with S/S control, there was no change in p-SHP-2 expression between NOT-AT, WT-Tregs-AT or antibody treated AT groups.(n=3–6/group, * p < 0.05 vs. S/S group, determined by one-way ANOVA for multiple group comparisons). All data are expressed as mean ± SEM.
To further determine whether the CD4+ T effector-lymphocytes or Tregs mediate the recruitment/activation of SHPs seen in the PD-L1−/− iALI mice with AT of Tregs, local pulmonary CD4+ or CD25+ cells were depleted by IT instillation of anti-mouse CD4 or CD25 monoclonal antibody into PD-L1 −/− mice 12 hours prior to the induction of iALI as previously described [15,20] (a significant decrease in the percentage of lung CD4+ or CD4+CD25+ T cells was confirmed by flow cytometry; data not shown). Following this all the iALI mice then received AT of WT-Tregs. Here we found that the depletion of either CD4+ or CD4+CD25+ T cells significantly decreased the levels of p-SHP-1 in lung tissue when compared with WT-Treg-AT PD-L1−/− iALI mice (Fig 5A–B). Furthermore, depletion of CD4+ cells produced a trend towards a more obvious, but not statistically significant, reduction of the p-SHP-1and/or SHP-1 levels than the local depletion of CD25+ T cells (Fig 5A–B). However, there were no marked differences in the levels of p-SHP-2 and/or SHP-2 seen with or without anti-CD4 or anti-CD25 antibody treatment when compared to the NOT-AT or WT-Treg AT groups (Fig 6A–B).
As a consequence of T lymphocyte stimulation, PD-1 has been reported to inhibit Akt phosphorylation by interfering on CD28-mediated PI3K activation, causing the reduction of cytokine synthesis, blocking T cell proliferation and survival (reviewed in [9,10]). We then decided to determine whether the AT of exogenous Tregs could affect the activation of the downstream protein Akt in the recipient PD-L1−/− iALI mice. Initially, we found that the p-Akt levels were significantly increased in the PD-L1−/− iALI mice when compared to dual sham controls (Fig 7A–B). While with or without the exogenous AT Tregs had no effects on Akt phosphorylation, CD25 depletion, not CD4 depletion, induced a significant increase in the levels of p-Akt in the injured lung (Fig 7A–B) when compared to other iALI mouse groups.
Figure 7. Adoptive transfer of WT-Tregs into PD-L1 deficient iALI mice had no effects on the expression of activated/phosphorylated Akt (p-Akt) in lung tissue, while local depletion of Tregs prior to AT of Tregs augmented the p–Akt levels.

(A) Representative western blot depicting lung tissue lysates probed for p-Akt, Akt, and β-actin from S/S, NOT-AT and WT-Tregs-AT animals (with or without intra-tracheal instillation of CD4 or CD 25 antibody). (B) Semi-quantitation by densitometry and expressed as integrated density (IDT) values of p-Akt relative to IDT values of Akt. While iALI induces an increase in p-Akt expression, there were no changes in p-Akt expression between NOT-AT, WT-Tregs-AT or CD4 antibody treated AT groups. However, CD25 antibody treatment significantly up-regulates p-Akt in the lung when compared with all other groups. (n=3–6/group, * p < 0.05 vs. S/S group; # p < 0.05 vs. NOT-AT group; @ p < 0.05 vs. WT-Tregs-AT group without depletion of cells, determined by one-way ANOVA for multiple group comparisons). All data are expressed as mean ± SEM.
Discussion
Among the B7:CD28 family members, PD-L1 is one of the most widely expressed molecules, which is broadly expressed on a variety of immune cells, including T cells, B cells, dendritic cells, monocytes and non-immune cells including epithelial, endothelial, muscle, hepatocytes, etc. (review in [9,10]). The engagement of PD-L1 with its receptor, PD-1, relays inhibitory signals that lead to suppression of immune responses [22,23]. Previous studies have reported that the interaction between PD-1 and PD-L1 can influence the generation of Tregs, the suppressive capacity of Tregs and the interactions between Tregs with effector T cells [11, 12]. We have recently shown that PD-1 plays an essential role in the protective effects afforded by Tregs against shock/sepsis induced experimental iALI [7]. In the current study, we further demonstrate that PD-L1-mediated signaling is an integral part of the mechanism by which Tregs exert their role in the resolution of iALI.
PD-L1 signaling is known to be an important mediator of Tregs’ action [22]. Amarnath et al. demonstrated PD-L1-mediated protective action of human Tregs in preventing deadly human-into-mouse xenogeneic graft-versus-host disease (xGvHD) [24]. They also showed that not only Tregs, but also PD-L1 expressing effector T cells could completely prevent the xGvHD lethality with a sustained long-lasting effect [25]. Liu et al. [26] further demonstrated that the effects of liver dendritic cells on Treg’s induction and expansion appeared to depend on the PD-L1 signal. A recent in vivo study reported that PD-L1 dysfunction or deficiency abolished Treg-mediated brain protection and neurological improvements after stroke [27]. Consistent with those data, to our knowledge, we show for the first time that transfer of Tregs purified from the WT mouse spleens into the PD-L1 deficient mice lose the beneficial effects of AT Tregs-mediated repression of the development of iALI seen in WT recipient mice. In contrast to an obvious resolution of lung injury and inflammation by the AT of WT-Tregs into the WT iALI recipient mice as demonstrated before [7], here we found that the AT of WT-Tregs into PD-L1 gene deficient iALI recipient mice showed no such reduction of lung injury indices as assessed by histology and vascular leakage. Also, while a significant increase of IL-1β and KC was detected, no changes in IL-6, IL-10 and IL-17A levels in lung tissues of these mice when compared with iALI mice (NOT-AT) that did not receive AT Tregs. In addition, our previous work has shown that the influx of neutrophils to the lung and lung cell apoptosis were suppressed by the AT of WT-Tregs into the WT iALI recipient mice [7]; however, in the present study we observed that when the WT-Tregs were AT to recipient PD-L1 gene deficient mice, they failed to decrease the lung neutrophil influx [MPO activity] or the rise in lung apoptosis. These data confirm a direct need for PD-L1 in the AT Tregs’ capacity to mediate the resolution of lung injury and inflammation in our clinically comparable model of iALI in mice.
Despite the significant knowledge acquired about Tregs over the past few years, very little is known about the potential intra-cellular signaling events by which PD-L1: PD-1 pathway itself may mediate Tregs’ function in vivo. PD-1 contains two tyrosine motifs within its cytoplasmic tail, an immunoreceptor tyrosine-based inhibition motif (ITIM) and an ITSM (review in [9,10,11]), both of which are phosphorylated upon PD-1 engagement with its ligands. It is reported that the suppressive effects of PD-1 ligation are mediated by factors binding the ITSM motif. The recruitment of SHP-2 phosphatase to the phosphorylated Tyr residue in ITSM is thought to be in part responsible for the inhibitory effect exerted by PD-1 through the dephosphorylation of signaling molecules belonging to the TCR or BCR pathways (review in [9,10]). It has also been shown that PD-1 ligation can block CD28-mediated activation of PI3K and the serine-threonine kinase Akt by recruiting SHP-2 (review in [9,10]). A recent report has documented that ligated-PD-1 can form negative costimulatory microclusters that directly inhibit TCR signaling by recruiting phosphatase SHP-2[28]. However, to our knowledge, there are no reports concerning whether PD-L1 recruiting SHP-1 and/or SHP-2 exert immunomodulatory effects on TCR or Tregs in vivo and in vitro. Our data showed that when WT mice were subjected to iALI, there was no significant difference for either p-SHP-1 or p-SHP-2 levels between the NOT-AT and WT-Tregs-AT groups. For the PD-L1-deficient iALI mice, the adoptive transfer of WT-Tregs had the ability to increase the phosphorylation of SHP-1, while no effect on the p-SHP-2 levels in the lung. Through the local specific depletion of Tregs, we further indirectly identified that SHP-1, but not SHP-2, negatively modulated PD-L1-dependent regulation of Tregs’ function in resolving shock/sepsis induced lung injury. Appreciating that one of the main limitations of this indirect depletional approach is that it may not just affect the signal produced by Tregs as the CD25 and CD4 are shared by other cell lineages that might contribute to the PD-1-induced SHP-1/-2 and/or Akt signals assessed here.
SHP-1 is expressed in hematopoietic cells of all lineages including conventional T cells and Tregs. Once phosphorylated and activated, SHP-1 binds to and dephosphorylates its target molecules and terminates the signaling [29,30]. In the absence or decrease of SHP-1 activation, cytokine/growth factor signaling will go unchecked, which may lead to abnormal or pathological responses [29,30]. One recent study demonstrated that SHP-1 could be an endogenous brake and modifier of the suppressive ability of Tregs and loss of SHP-1 activation strongly augments the ability of Tregs to suppress inflammation in a mouse model of delayed-type hypersensitivity [31]. Consistent with these data, in the current study, our findings further confirmed that the loss of Tregs’ function in iALI PD-L1 gene deficient mice is correlated with the SHP-1 activation. Tregs have been shown to require stimulation via TCR to obtain their full functionality [31]. The strength of TCR-initiated signaling within the Tregs directly affects their level of suppressive activity and that SHP-1 appears to function downstream of the TCR in Tregs, thereby, directly modulating their suppressive potential [31]. Furthermore, SHP-1 regulates conjugate formation between Tregs and antigen presenting cells (APCs) that SHP-1-deficient Tregs are more effective in forming tripartite conjugates (T-effectors/APC/Tregs) and thereby limiting the number of bipartite T-effectors-APC conjugates [28]. It has also been reported the PD-L1: PD-1 pathway may control the complicated dynamic interactions among Tregs, T-effectors cells and APCs (review in [9,10]). Yet, the potential mechanisms underpinning to control these interactions remain unclear. In the current study, we indirectly demonstrate that PD-L1 regulates Tregs’ capacity to resolve the shock/sepsis induced iALI by recruiting SHP-1. Future work will be important to understand how PD-L1 and SHP-1 are involved in controlling the interactions among Tregs, T-effector cells and APCs in the development of iALI.
Finally, PD-1 has been shown to alter membrane-proximal signaling events in T cells that can inhibit the induction of PI3K activity and downstream activation of Akt [11]. Both SHP-1 and SHP-2 phosphatases can negatively regulate the activity of Akt [33]. Francisco and colleagues found that PD-L1 played an obligatory role in controlling iTreg (induced Treg) cell development and function through the down-regulation of Akt [34]. In contrast, our data showed that although p-Akt levels were significantly increased in the PD-L1−/− iALI mice when compared to dual sham controls, AT of the exogenous Tregs to these mice had no effects on the Akt phosphorylation in the lung. Furthermore, the augmented activation of Akt was only seen in the lung of Treg depleted mice. As the phosphorylation of Akt occurs at two different sites: Ser473 and Tyr308, which are mediated by different upstream signaling pathways [11,34]. In the current study, we only measured the Tyr308 site. Inasmuch, the effects of PD-L1 on the downstream activation/inhibition of Akt of Tregs warrants further study.
In conclusion, our findings demonstrate the pivotal roles of PD-L1 in Tregs’ function by mediating the repression of double-hit induced iALI. In particular, we show that the inability of exogenous WT Tregs to function in PD-L1 deficient iALI mice is associated with the recruitment of SHP-1 to the injured lung. Along with our previous studies of PD-1’s immunomodulatory role on Tregs, these observations provide a better understanding of the potential mechanism by which Tregs exert their protective role during the resolution of inflammation and injury of iALI. Manipulating PD-L1: PD-1 interactions may provide a novel approach for maintaining and enhancing Tregs’ function. Furthermore, recent studies, 2 phase 1 trials using phosphatase inhibitor sodium stibogluconate, a specific inhibitor targeting SHP-1 activity, have demonstrated safety and targeted inhibition of the anti-tumor response in cancer patients [35,36]. We propose a potential strategy for iALI/ARDS using adoptive Treg cell therapy by combined PD-L1/PD-1 agonists and SHP-1 antagonists.
Acknowledgments
The authors acknowledge L. Chen (Yale University, New Haven, CT) for providing PD-L1−/− mice. We also thank Mr. Paul Monfils at the Core Laboratories Facilities at Rhode Island Hospital for the assistance with the histological preparation/staining done in this study. The research was supported by NIH grants R01GM107149 (to A.A.).
Footnotes
This work was presented as a part of the 37th Annual Conference on Shock as one of the finalist papers read in the New Investigators Award Competition, Charlotte, NC.
Disclosure of Funds:
The research was supported by NIH grants R01GM107149 (to A.A.).
References
- 1.ARDS_Definition Task Force. Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, Camporota L, Slutsky AS. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307(23):2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 2.Ferguson ND, Fan E, Camporota L, Antonelli M, Anzueto A, Beale R, Brochard L, Brower R, Esteban A, Gattinoni L, Rhodes A, Slutsky AS, Vincent JL, Rubenfeld GD, Thompson BT, Ranieri VM. The Berlin definition of ARDS: an expanded rationale, justification, and supplementary material. Intensive Care Med. 2012;38(10):1573–82. doi: 10.1007/s00134-012-2682-1. [DOI] [PubMed] [Google Scholar]
- 3.Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6(12):1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 4.D’Alessio FR, Tsushima K, Aggarwal NR, West EE, Willett MH, Britos MF, Pipeling MR, Brower RG, Tuder RM, McDyer JF, King LS. CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Invest. 2009;119(10):2898–2913. doi: 10.1172/JCI36498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aggarwal NR, D’Alessio FR, Tsushima K, Sidhaye VK, Cheadle C, Grigoryev DN, Barnes KC, King LS. Regulatory T cell-mediated resolution of lung injury: identification of potential target genes via expression profiling. Physiol Genomics. 2010;41(2):109–119. doi: 10.1152/physiolgenomics.00131.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun J, Han ZB, Liao W, Yang SG, Yang Z, Yu J, Meng L, Wu R, Han ZC. Intrapulmonary delivery of human umbilical cord mesenchymal stem cells attenuates acute lung injury by expanding CD4+CD25+ Forkhead Boxp3 (FOXP3)+ regulatory T cells and balancing anti- and pro-inflammatory factors. Cell Physiol Biochem. 2011;27(5):587–596. doi: 10.1159/000329980. [DOI] [PubMed] [Google Scholar]
- 7.Tang L, Bai J, Chung CS, Lomas-Neira J, Chen Y, Huang X, Ayala A. Active players in resolution of shock/sepsis induced indirect lung injury: CD4+CD25+Foxp3+ regulatory T cells (Tregs) and PD-1’s immunomodulatory effects. J Leuko Biol. 2014 doi: 10.1189/jlb.4MA1213-647RR. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, Shah B, Chang SH, Schluns KS, Watowich SS, Feng XH, Jetten AM, Dong C. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29(1):44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gianchecchi E, Delfino DV, Fierabracci A. Recent insights into the role of the PD-1/PD-L1 pathway in immunological tolerance and autoimmunity. Autoimmun Rev. 2013;12(11):1091–1100. doi: 10.1016/j.autrev.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 11.Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173(2):945–954. doi: 10.4049/jimmunol.173.2.945. [DOI] [PubMed] [Google Scholar]
- 12.Kaufmann DE, Walker BD. PD-1 and CTLA-4 inhibitory cosignaling pathways in HIV infection and the potential for therapeutic intervention. J Immunol. 2009;182(10):5891–5897. doi: 10.4049/jimmunol.0803771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin Y, Chauhan SK, El Annan J, Sage PT, Sharpe AH, Dana R. A novel function for programmed death ligand-1 regulation of angiogenesis. Am J Pathol. 2011;178(4):1922–1929. doi: 10.1016/j.ajpath.2010.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Azuma T, Yao S, Zhu G, Flies AS, Flies SJ, Chen L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood. 2008;111(7):3635–3643. doi: 10.1182/blood-2007-11-123141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Venet F, Chung CS, Huang X, Lomas-Neira J, Chen Y, Ayala A. Lymphocytes in the development of lung inflammation: a role for regulatory CD4+ T cells in indirect pulmonary lung injury. J Immunol. 2009;183(5):3472–3480. doi: 10.4049/jimmunol.0804119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monaghan SF, Thakkar RK, Heffernan DS, Huang X, Chung CS, Lomas JL, Cioffi WG, Ayala A. Mechanisms of indirect acute lung injury: a novel role for the coinhibitory receptor, programmed death-1. Ann Surg. 2012;255(1):158–164. doi: 10.1097/SLA.0b013e31823433ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ayala A, Chung CS, Lomas JL, Song GY, Doughty LA, Gregory SH, Cioffi WG, LeBlanc BW, Reichner J, Simms HH, Grutkoski PS. Shock induced neutrophil mediated priming for acute lung injury in mice: divergent effects of TLR-4 and TLR-4/FasL deficiency. Am J Pathol. 2002;161(6):2283–2294. doi: 10.1016/S0002-9440(10)64504-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bai J, Tang L, Lomas JL, Chen Y, McLeish KR, Uriarte SM, Chung CS, Ayala A. TAT-SNAP-23 treatment inhibits the priming of neutrophil functions contributing to shock and/or sepsis-induced extra-pulmonary acute lung injury. Innate Immun. Jan 3; doi: 10.1177/1753425913516524. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lomas JL, Chung CS, Grutkoski PS, LeBlanc BW, Lavigne L, Reichner J, Gregory SH, Doughty LA, Cioffi WG, Ayala A. Differential effects of MIP-2 and KC on hemorrhage induced neutrophil priming for lung inflammation: assessment by adoptive cell transfer in mice. Shock. 2003;19:358–365. doi: 10.1097/00024382-200304000-00011. [DOI] [PubMed] [Google Scholar]
- 20.Wisnoski N, Chung CS, Chen Y, Huang X, Ayala A. The contribution of CD4+ CD25+ T-regulatory-cells to immune suppression in sepsis. Shock. 2007;27(3):251–257. doi: 10.1097/01.shk.0000239780.33398.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hutchins NA, Wang F, Wang Y, Chung CS, Ayala A. Kupffer cells potentiate liver sinusoidal endothelial cell injury in sepsis by ligating programmed cell death ligand-1. J Leukoc Biol. 2013;94(5):963–970. doi: 10.1189/jlb.0113051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8(3):239–245. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- 23.Huang X, Chen Y, Chung CS, Yuan Z, Monaghan SF, Wang F, Ayala A. Identification of B7-H1 as a novel mediator of the innate immune/proinflammatory response as well as a possible myeloid cell prognostic biomarker in sepsis. J Immunol. 2014;192(3):1091–1099. doi: 10.4049/jimmunol.1302252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amarnath S, Mangus CW, Wang JC, Wei F, He A, Kapoor V, Foley JE, Massey PR, Felizardo TC, Riley JL, Levine BL, June CH, Medin JA, Fowler DH. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci Transl Med. 2011;3(111):111–120. doi: 10.1126/scitranslmed.3003130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amarnath S, Costanzo CM, Mariotti J, Ullman JL, Telford WG, Kapoor V, Riley JL, Levine BL, June CH, Fong T, Warner NL, Fowler DH. Regulatory T cells and human myeloid dendritic cells promote tolerance via programmed death ligand-1. Plos Biol. 2010;8(2):e1000302. doi: 10.1371/journal.pbio.1000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu H, Bakthavatsalam R, Meng Z, Li Z, Li W, Perkins JD, Reyes J. PD-L1 signal on liver dendritic cells is critical for Foxp3(+)CD4(+)CD25(+) Treg and liver tolerance induction in mice. Transplant Proc. 2013;45(5):1853–1855. doi: 10.1016/j.transproceed.2013.03.015. [DOI] [PubMed] [Google Scholar]
- 27.Li P, Mao L, Liu X, Gan Y, Zheng J, Thomson AW, Gao Y, Chen J, Hu X. Essential role of program death 1-ligand 1 in regulatory T-cell-afforded protection against blood-brain barrier damage after stroke. Stroke. 2014;45(3):857–864. doi: 10.1161/STROKEAHA.113.004100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 2012;209(6):1201–1217. doi: 10.1084/jem.20112741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J, Somani AK, Siminovitch KA. Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signalling. Semin Immunol. 2000;12(4):361–378. doi: 10.1006/smim.2000.0223. [DOI] [PubMed] [Google Scholar]
- 30.Pao LI, Badour K, Siminovitch KA, Neel BG. Nonreceptor protein-tyrosine phosphatases in immune cell signaling. Annu Rev Immunol. 2007;25:473–523. doi: 10.1146/annurev.immunol.23.021704.115647. [DOI] [PubMed] [Google Scholar]
- 31.Iype T, Sankarshanan M, Mauldin IS, Mullins DW, Lorenz U. The protein tyrosine phosphatase SHP-1 modulates the suppressive activity of regulatory T cells. J Immunol. 2010;185(10):6115–6127. doi: 10.4049/jimmunol.1000622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim JK, Klinger M, Benjamin J, Xiao Y, Erle DJ, Littman DR, Killeen N. Impact of the TCR signal on regulatory T cell homeostasis, function, and trafficking. PLoS One. 2009;4(8):e6580. doi: 10.1371/journal.pone.0006580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Locke NR, Patterson SJ, Hamilton MJ, Sly LM, Krystal G, Levings MK. SHIP regulates the reciprocal development of T regulatory and Th17 cells. J Immunol. 2009;183(2):975–983. doi: 10.4049/jimmunol.0803749. [DOI] [PubMed] [Google Scholar]
- 34.Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, Sharpe AH. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206(13):3015–3029. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yi T, Elson P, Mitsuhashi M, Jacobs B, Hollovary E, Budd TG, Spiro T, Triozzi P, Borden EC. Phosphatase inhibitor, sodium stibogluconate, in combination with interferon (IFN) alpha 2b: phase I trials to identify pharmacodynamic and clinical effects. Oncotarget. 2011;2(12):1155–1164. doi: 10.18632/oncotarget.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naing A, Reuben JM, Camacho LH, Gao H, Lee BN, Cohen EN, Verschraegen C, Stephen S, Aaron J, Hong D, Wheler J, Kurzrock R. Phase I Dose Escalation Study of Sodium Stibogluconate (SSG), a Protein Tyrosine Phosphatase Inhibitor, Combined with Interferon Alpha for Patients with Solid Tumors. J Cancer. 2011;2:81–89. doi: 10.7150/jca.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]