

Abstract
Hypertension is a serious risk factor for myocardial infarction, heart failure, vascular disease, stroke, and renal failure. Like other complex diseases, hypertension is caused by a combination of genetic and environmental factors. The renin-angiotensin-aldosterone system plays an important role in the regulation of blood pressure. The octapeptide angiotensin II (ANG II) is one of the most active vasopressor agents and is obtained from the precursor molecule, angiotensinogen, by the combined proteolytic action of renin and angiotensin-converting enzyme. ANG II increases the expression of aldosterone synthase (coded by Cyp11B2 gene), which is the rate-limiting enzyme in the biosynthesis of aldosterone. Previous studies have shown that increased expression of aldosterone synthase increases blood pressure and cardiac hypertrophy in transgenic mice. Human Cyp11B2 gene has a T/C polymorphism at −344 positions in its 5′-untranslated region (UTR), and the −344T allele is associated with hypertension. Human Cyp11B2 gene also has an A/G polymorphism at 735 position in its 3′-UTR (rs28491316) that is in linkage disequilibrium with single nucleotide polymorphism at −344. We show here that 1) microRNA (miR)-766 binds to the 735G-allele and not the 735A-allele of the hCyp11B2 gene and 2) transfection of miR-766 reduces the human aldosterone synthase mRNA and protein level in human adrenocortical cells H295R. These studies suggest that miR-766 may downregulate the expression of human aldosterone synthase gene and reduce blood pressure in human subjects containing −344T allele.
Keywords: aldosterone synthase, 3′-untranslated region, single nucleotide polymorphism, miRNA, Cyp11b2, rs28491316, hypertension, blood pressure regulation
hypertension is a serious risk factor for myocardial infarction, heart failure, vascular disease, stroke, and renal failure (10). Hypertension affects 50 million Americans with a prevalence rate of 25–30% in the adult Caucasian population and 1 billion people world-wide (14). The incidence of hypertension and complications due to hypertension is even greater in the African-American population (12). It has been shown that lowering blood pressure has a beneficial effect on the severity of other associated cardiovascular diseases (24). Like other complex diseases, hypertension is caused by the interplay of genetic and environmental factors. However, the molecular mechanisms involved in the pathophysiology of hypertension are not clear (5, 6). In this regard, the renin-angiotensin-aldosterone system plays an important role in the regulation of blood pressure (13, 18, 19). The octapeptide angiotensin II (ANG II) is one of the most potent vasopressor agents and is obtained by the proteolytic cleavage of a larger precursor molecule, angiotensinogen, by renin and angiotensin converting enzyme (7). ANG II acts on the vascular smooth muscle cells to cause vasoconstriction and on the adrenal ZG cells to stimulate aldosterone production. Up to 15% of patients with essential hypertension (22, 28, 34) and 22% of patients with resistant hypertension (2) have an inappropriate excess of aldosterone that leads to an increase in age-related blood pressure and cardiovascular risks. Studies from the Framingham cohort have shown that patients with aldosterone values within the top quartile of the population have a steeper blood pressure rise over a 5 yr period than patients with aldosterone values in the bottom quartile; subjects with aldosterone values between these extremes showed an intermediate rise in blood pressure (9, 33).
Aldosterone synthase, the rate-limiting enzyme in the biosynthesis of aldosterone, is encoded by the Cyp11B2 gene. This gene is expressed mainly in adrenal cortex and to some extent in kidney, brain, and adipose tissue. Makhanova et al. (20) have generated transgenic mice (AShi/hi) that have increased expression of the Cyp11B2 gene. Their experiments show that a modest increase in Cyp11B2 expression did not affect blood pressure in animals fed a normal-salt diet but did make their blood pressure sensitive to high salt and to ANG II infusions on an increased salt diet. These data, together with their previous finding that a decreased level of Cyp11B2 decreases blood pressure in mice on a low-salt diet (21), suggest that Cyp11B2 is a relevant candidate to understand the role of genetics in hypertension. Earlier studies have shown that human Cyp11B2 gene has a T/C polymorphism at −344 positions and −344T allele is associated with hypertension (4, 29). The human Cyp11B2 gene also has an A/G polymorphism at 735 position in its 3′-untranslated region (UTR), which is in linkage disequilibrium with the single nucleotide polymorphism (SNP) at −344. The role of the A/G polymorphism in the 3′-UTR region of the expression of Cyp11B2 gene is not known.
MicroRNAs (miRNAs) are endogenously synthesized short noncoding RNAs of about 20–22 nucleotides that have been shown to play an important role in modulating mammalian gene expression and regulate several key biological functions (15–17). miRNAs normally inhibit the expression of their target genes via posttranscriptional mechanisms (1, 3). It is estimated that there are over 1,000 human miRNAs and that the expression of about 60% of the human protein coding genes can be downregulated by miRNAs. SNPs in the 3′-UTR may create, destroy, or modify the efficiency of miRNA binding to the 3′-UTR of a gene and may modulate its expression (30). The human Cyp11B2 gene has an A/G polymorphism at 735 position in its 3′-UTR. We show here that microRNA (miR)-766 regulates the expression of hCyp11B2 gene by differentially binding to this polymorphic site.
MATERIALS AND METHODS
Cell culture.
Human adrenocortical carcinoma cells (H295R) were routinely cultured as monolayers and maintained in 100 mm tissue culture dishes with “complete media” containing a basal 1:1 mixture of Dulbecco's modified Eagle's and Ham's F12 (DMEM/F12, Gibco-Invitrogen) media, supplemented with 2.5% Nu-Serum IV (BD Biosciences), 1% ITS+ Premix (6.25 μg/ml insulin, 6.25 μg/ml transferrin, 6.25 ng/ml selenium, 1.25 mg/ml bovine serum albumin, 5.35 μg/ml linoleic acid; BD Biosciences), and 0.5% antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin mix, Gibco-Invitrogen). HEK293 cells, purchased from American Type Culture Collection (Manassas, VA), were grown as a monolayer in DMEM supplemented with 10% fetal bovine serum (BenchMark cat. #100-106), 100 units/ml penicillin, and 100 μg/ml streptomycin in a humidified atmosphere of 95% air and 5% CO2 at 37°C in an incubator.
Luciferase reporter constructs.
The 3′-UTR of the human aldosterone synthase (hCyp11B2) gene containing the 735G-allele was PCR-amplified from the human BAC (RP11–304E16) clone using forward 5′-GGA CTA GTA GAC GAT CTT GCT GGC A-3′ and reverse 5′-cga cgc gta ttt ttc caa ggt tta t-3′ primers. These primers amplify the 201–1420 region of the 3′-UTR of the hCyp11B2 gene. The forward primer contains a Spe1 restriction site at the 5′-end, and the reverse primer contains a Mlu1 restriction site at its 3′-end for cloning purpose. The amplified product was treated with restriction enzymes Spe1 and Mlu1 and was directionally cloned in to the multiple cloning site of pMIR-REPORT miRNA expression reporter vector system (Life Technologies). This vector contains the firefly luciferase gene under the control of a mammalian CMV promoter/terminator system and miRNA target-cloning region downstream of the luciferase coding sequence. Plasmid DNA was isolated from recombinant colonies and sequenced to ensure the authenticity and designated as hCyp11B2-pMIR/Luc-735G.
The hCyp11B2-pMIR/Luc-735G plasmid was used as a template to mutate the 735G-allele to the 735A-allele using the QuikChange Lightning Site-Directed Mutagenesis (Agilent Technologies) kit and designated as hCyp11B2-pMIR/Luc-735A. For site-specific mutagenesis, we used forward mutagenic primer (5′-gctgtcccctggaa aaggtcccgagga-3′) and a complementary reverse mutagenic primer (5′-tcctcgggaccttttccaggggacagc-3′) in PCR as per the manufacturer's recommendation. Following PCR, the product was treated with DpnI endonuclease to digest the parental DNA and leaving intact the mutated plasmid DNA. The mutation, 735A, was confirmed by dideoxy-chain termination sequencing. Finally, transformed bacterial cultures were grown, and the reporter constructs were purified by using of a Hi speed Plasmid Medi Kit (Qiagen).
Transfection and luciferase assay.
The H295R and HEK293 cells were seeded at 8 × 104 cells/well in 12-well plates containing 1 ml of complete medium. All the transfections were optimized and performed using siPORT NeoFx transfection reagent (Ambion) as per the manufacturer's recommendations. In brief, 1.5 μl of NeoFX transfection reagent was mixed with 48.5 μl OptiMEM (Invitrogen, 11058021), and the mixture was incubated at room temperature for 10 min. miR-766 mimics or its inhibitors (Ambion) (50 nM) were diluted in 50 μl of OptiMEM and then added into the NeoFX transfection agent. The final mixture was incubated at room temperature for another 10 min and used for transfection. A mutated miR-766 mimic was purchased from Ambion and used as a negative control. Mock transfections were performed in the absence of additional miRNA mimics. The cotransfection experiments were performed with 100 ng of pMIR/Luc reporter constructs. The pMIR-REPORT β-galactosidase reporter control vector was used to normalize the transfection efficiency. After 48 h of incubation, the cells were washed with 1× ice-cold PBS, and the luciferase and β-galactosidase activities were determined by Dual-Light Luciferase & β-galactosidase Reporter Gene Assay System (Life Technologies) as per the manufacturer recommendations.
Real-time PCR.
Total RNA was isolated from miRNA transfected cells with RNeasy Plus minikit (Qiagen). One microgram of RNA was reverse-transcribed into cDNA using a first-strand cDNA synthesis kit (Fermentas). Quantitative real-time RT-PCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems) and Applied Biosystems thermocycler (7500 Fast Real-Time PCR System). Primers for human Cyp11B2 (PPH01239F) and human GAPDH (PPH00150E) were purchased from SuperArray Bioscience Following 95°C incubation for 10 min; 40 cycles of PCR (95°C for 30 s, 60°C for 30 s) were then performed using 2 μl of cDNA, 50 nM PCR primers, and 12.5 μl of SYBR Green PCR Master Mix in 25 μl reactions. Threshold cycles for three replicate reactions were determined, and relative hCyp11B2 gene expression was calculated following normalization with human GAPDH.
miR-766 expression analysis.
The miRNA from H295R human adrenocortical cells was extracted using the mirVana miRNA isolation kit (Ambion). miR-766 quantification was performed using the mirVana quantitative RT-PCR miRNA detection kit (Ambion), as per the manufacturer's recommendations, using the ABI 7500 Fast Real-Time PCR System thermocycler (Applied Biosystems). In brief, total RNA of 20 ng was reverse-transcribed using a TaqMan reverse transcription kit (Applied Biosystems) followed by quantitative PCR performed using TaqMan qPCR assays for miR-766 and U6 snoRNA (Applied Biosystems). The real-time PCR Reaction conditions were 3 min at 94°C followed by 50 cycles of 15 s at 94°C and 30 s at 60°C. Real-time data were obtained during the extension phase, and threshold cycle values were obtained at the log phase of each gene amplification. miRNA-specific signals were normalized using U6 RNA levels. The specificity of the PCR products was confirmed by melting temperature determination of the PCR product and high-resolution electrophoretic analysis in 4% NuSieve 3:1 agarose gels (Cambrex, Rockland, ME) of PCR products.
Immunoblot.
The cell lysates from miRNA-transfected cells were prepared using the Qproteome Mammalian Protein Prep Kit (Qiagen) as per the manufacturer's recommendations. Lysates were cleared by centrifugation, and a total of 30 μg/well protein was resolved by 10% SDS-PAGE and then was electro-blotted onto a polyvinylidene difluoride membrane. The membranes were blocked for 1 h in Odyssey blocking buffer, followed by an overnight incubation at 4°C with 1:2,000 dilution of hCyp11B2 polyclonal antibody raised in goat (Santa Cruz, sc-47655) and then with a 1:10,000 dilution of secondary antibody conjugated with IRDye800 or IRDye700 at room temperature for 60 min, protected from light, along with gentle shaking. The blots were visualized using an Odyssey Imaging System (LI-COR) and were subjected to quantitative analyses using ImageJ. The results were averaged and normalized with β-actin.
Statistical analysis.
All data are expressed as means + SD of at least three independent experiments. Statistical analyses were performed with one-way ANOVA and Tukey-Kramer post hoc analysis. The data were in normal distribution and were checked by the D'Agostino-Pearson omnibus test using GraphPad Prism. Values of P < 0.05 were considered significant.
RESULTS
Human aldosterone synthase +735 G/A polymorphism (rs28491316) occurs in the miR-766 binding site.
The 3′-UTR of the human aldosterone synthase (hCyp11B2) gene contains a G/A polymorphism at the +735 (rs28491316) site. Since miRNAs may bind to nucleotide sequence located in the 3′-UTR of a gene and modulate its expression by posttranscriptional or posttranslational mechanism, we were interested in finding whether any miRNA binds to this region of the hCyp11B2 gene and modulates its expression. TargetScan (Fig. 1A) and Miranda (Fig. 1B) analysis revealed that a SNP at +735 of the hCyp11B2 gene is located in the seed binding sequence of miR-766. This analysis also suggests that the miR-766 has an additional binding site at the 196–203 in the 3′-UTR of the hCyp11B2 gene (Fig. 1D). Computational modeling shows that the nucleotide sequence of the hCyp11B2 gene containing the 735G-allele has perfect Watson-Crick base pairing with miR-766 seed sequence. On the other hand, the presence of the 735A-allele leads to a destabilization of the Watson-Crick base pairing with the seed sequence. Additionally, using 1000 Genomes analysis, we have ascertained that +735A is the minor allele with a frequency of 0.336, whereas +735G is the major allele with a frequency of 0.664.
Fig. 1.

microRNA (miR)-766 binds to the 735G-allele of the hCyp11B2 gene. In silico Target Scan-predicted complementarity between miR-766 and hCyp11B2 3′-untranslated region (UTR) is shown in A, and miRanda predicted complementarity between miR-766 and hCyp11B2 3′-UTR is shown in B. Solid lines represent the Watson-Crick base pairing, and 735 G/A polymorphism is marked in boldface. Nucleotide sequence of mutated miR-766 mimic is shown in C. The 3′-UTR of hCyp11B2 contains another binding site for miR-766 at 296–202, and the alignment with miR-766 is shown in D.
Transient transfection of miR-766 reduces the luciferase activity of 3′-UTR of hCyp11B2 gene containing +735G-allele compared with +735A-allele in HEK293 cells.
To test the hypothesis that miR-766 may regulate the expression of the hCyp11B2 gene, we cloned the nucleotide sequence located between 201 and 1420 of 3′-UTR of the hCyp11B2 gene (containing either +735G-allele or 735A-allele) in an expression vector immediately downstream from the firefly luciferase-coding region. The resulting plasmid constructs were designated as hCyp11B2-pMIR/Luc-735G (containing +735G-allele) and hCyp11B2-pMIR/Luc-735A (containing +735A-allele). To test the effect of miR-766 on the luciferase gene expression, either hCyp11B2-pMIR/Luc-735G or hCyp11B2-pMIR/Luc-735A was cotransfected in HEK293 cells, along with miR-766, and luciferase activity was determined 48 h posttransfection. Control experiments were conducted in the absence of miRNA (mock) or in the presence of mutated miRNA. HEK293 cells are ideal for these experiments because of their ease of transfection and lack of the hCyp11B2 gene. Initial experiments were performed in the presence of 10, 20, 50, and 100 nM miRNA, and since 50 nM miRNA gave the optimum result, this concentration was used in all future experiments. As shown in Fig. 2, luciferase activity remained unchanged in the absence of miRNA (mock) or in the presence of mutated miRNA. On the other hand, luciferase activity of the reporter construct containing the +735G-allele was downregulated by about 65% in the presence of miR-766. However, transfection of miR-766 had no effect on the luciferase activity of reporter construct containing the +735A-allele of the hCyp11B2 gene. These results suggest that the G/A polymorphism at the +735 site of the hCYP11B2 gene has the potential to downregulate its expression in a miRNA-dependent manner.
Fig. 2.
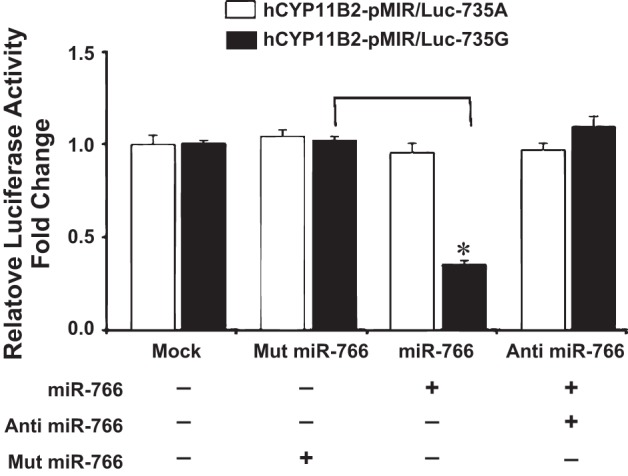
Effect of microRNA (miRNA) mimics miR-766 and anti-miR-766 on the luciferase activity of 3′-UTR of hCyp11B2 gene containing 735G/A polymorphism by dual luciferase assay in HEK293 cells. Human embryonic kidney cells (HEK293) were cotransfected either in the absence of miRNA (mock) or with plasmid construct hCyp11B2-pMIR/Luc-735G or hCyp11B2-pMIR/Luc-735A along with Mut miR-766, miR-766, and anti-miR-766 miRNA mimics (50 nM). Luciferase activity was measured after 48 h of transfection. Firefly luciferase activity was normalized to β-galactosidase expression, and the mean activities ± SE from 3 independent experiments are shown. *P < 0.05. Luc activity of the 735G-allele is shown by dark bar and of the 735A-allele is shown by light bar.
Since the above experiments suggested that miR-766 binds to +735G-allele and downregulates the expression of luciferase gene, we argued that anti-miRNAs should relieve this miRNA-induced downregulation. We therefore examined the effect of anti-miR-766 (antagomir) in the presence of miR-766 by measuring the luciferase activity. Complementary experiments show that 50 nM anti-miR-766 relieves the miR-766-induced downregulation of luciferase activity (Fig. 2). Thus, miR-766-induced downregulation of the luciferase gene is completely attenuated when 50 nM antagomir of miR-766 is used in transient transfection in HEK293 cells.
Transient transfection of miR-766 reduces the luciferase activity of 3′-UTR of hCyp11B2 gene containing +735G-allele compared with +735A-allele in H295R cells.
We next performed a luciferase assay by transient transfection of the above-mentioned reporter constructs in human adrenocortical cells (H295R) since these cells are known to express hCyp11B2 gene. Results of this experiment show that luciferase expression remained the same in the absence of miRNA or presence of mutated miRNA when reporter construct containing the +735G-allele was used (Fig. 3). On the other hand, luciferase expression was downregulated by about 50% in the presence of miR-766 in H295R cells when reporter construct containing the +735G-allele was used in transient transfection (Fig. 3). Cotransfection of the antagomir of miR-766 relieved the downregulation of the luciferase activity in H295R cells, as described above for HEK293 cells (Fig. 2). There was no significant effect of miR-766 or anti-miR-766 on the luciferase activity when reporter construct containing the +735A-allele was used in transfection experiments (Fig. 3). Taken together, results of these experiments show that miR-766 binds to the +735G-allele of the hCyp11B2 gene and downregulates luciferase gene expression either in human kidney or in human adrenocortical cells.
Fig. 3.
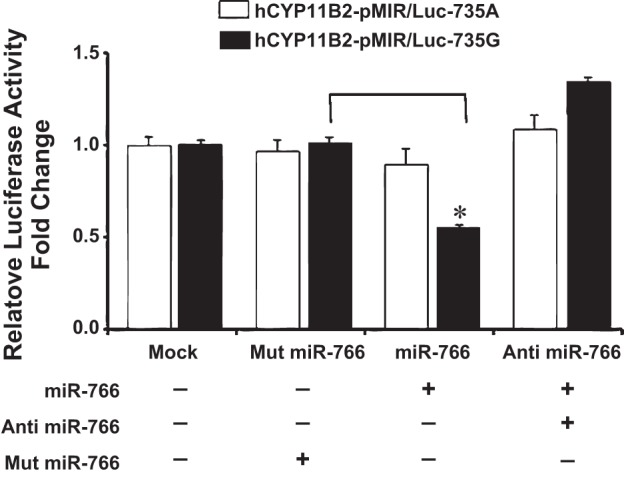
Effect of miRNA mimics miR-766 and anti-miR-766 on the luciferase activity of 3′-UTR of hCyp11B2 gene containing 735G/A polymorphism by dual luciferase assay in H295R cells. H295R cells were cotransfected with hCyp11B2-pMIR/Luc-735G or hCyp11B2-pMIR/Luc-735A along with miR-766, anti-miR-766 miRNA mimics and luciferase activity was determined as described in Fig. 2.
miR-766 reduces hCyp11B2 mRNA level in human adrenocortical (H295R) cells.
Since the above-mentioned experiments suggested that miR-766 selectively binds to +735G-allele in the reporter construct containing 3′-UTR of the hCyp11B2 gene and downregulates luciferase expression, we next wanted to examine whether this miRNA reduces the hCyp11B2 mRNA level in human adrenocortical cells. Before performing this experiment, we determined the nucleotide sequence of 3′-UTR of the hCyp11B2 gene in H295R cells. H295R cells have the +735G-allele of the hCyp11B2 gene (data not shown). Therefore, we transfected these cells with the mock miRNA, mutated-miRNA-766, and miRNA-766. It is important to note that this transfection was made necessary by our inability to detect endogenous miRNA-766 expression in this cell line. Quantitative RT-PCR was performed to analyze hCYP11B2 expression in these settings. As shown in Fig. 4, hCyp11B2 mRNA levels remained same in the absence of miRNA (mock) or presence of mut-miRNA in H295R cells. However, hCyp11B2 mRNA level was reduced by 25% in the presence of 50 nm miR-766 (Fig. 4). One possible reason for this modest reduction in the mRNA level may be that miR-766 has already downregulated the Cyp11B2 mRNA level in H295R cells, since these cells have the +735G-allele.
Fig. 4.
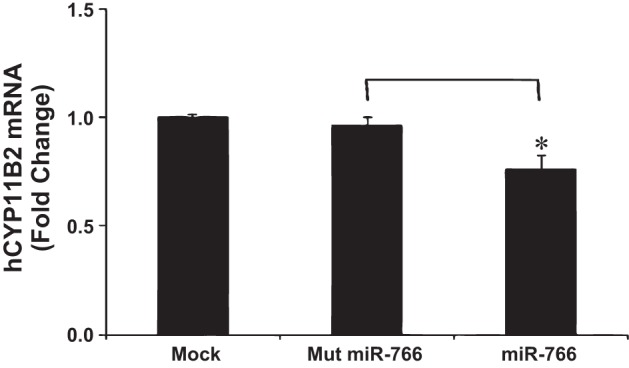
Effect of miR-766 on hCyp11B2 mRNA level in human adrenal (H295R) cells. H295R cells were transfected either in the absence of miRNA (mock) or in the presence of mut miR-766 or miR-766 miRNA mimics (50 nM). After 48 h, transfected cells were washed twice with ice-cold 1× PBS, and total RNA was isolated and reverse-transcribed into cDNA. qRT-PCR was performed; cycle threshold (ct) values for hCyp11B2 were determined and normalized to GAPDH expression. Relative ct values for each group were determined and compared with those for mock transfections taken as 1 for fold change. Each group represents the mean of at least 3 independent experiments. *P < 0.05.
hCYP11B2 expression, in the absence or presence of the miR-766, was confirmed by immunoblotting. As shown in Fig. 5, the aldosterone synthase protein level remained the same when H295R cells were transfected with either the mock miRNA or the mutated miR-766. On the other hand the hCYP11B2 protein level was significantly reduced (P < 0.05) in the presence of miR-766.
Fig. 5.
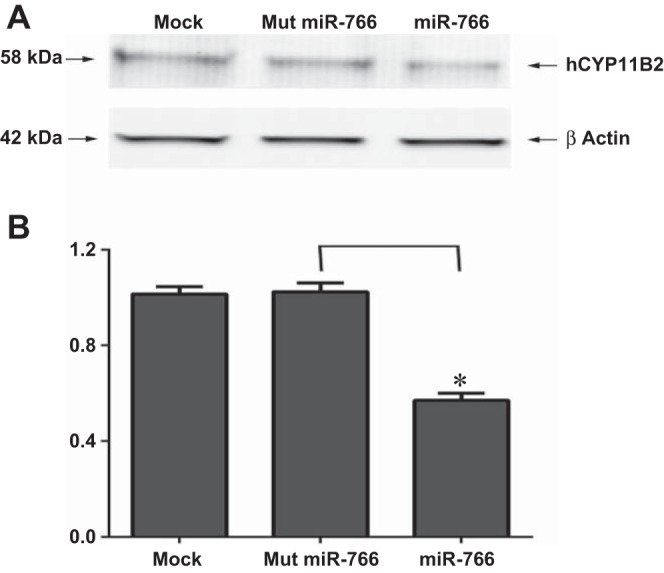
Effect of miR-766 on hCyp11B2 protein level in H295R cells. A: H295R cells were transfected either in the absence of miRNA (mock) or in the presence of Mut miR-766 or miR-766 (50 nM). After 48 h of transfection, cell extract was subjected to SDS-PAGE followed by Western blot analysis using anti-human Cyp11B2 antibody or anti-β-actin antibody. B: hCyp11B2 protein level of each band was measured and normalized to β-actin levels. The downregulation of hCyp11B2 protein after transfection with miR-766 was calculated from 3 independent experiments. *P < 0.05.
DISCUSSION
Hypertension is a complex disease with multiple patho-physiological processes on a backdrop of genetic predisposition. Interindividual variation of blood pressure, up to 45%, can be accounted for by differences in genes regulating the physiological processes governing blood pressure. In this regard, association studies have linked CYP11B2 polymorphisms to human hypertension and cardiovascular diseases (2, 28, 34). In the first key finding of the study, we have identified a G/A polymorphism at +735 in the 3′-UTR of the hCyp11B2 gene (rs28491316) that alters its binding to a particular miRNA. miRNAs are small, noncoding, regulatory RNAs that alter gene expression by transcriptional or posttranscriptional regulation. In silico alignment of the hCyp11B2 3′-UTR sequence by Target Scan and Miranda reveals that the miR-766 binds to the nucleotide sequence around 196–203 and 730–736 in 3′-UTR of the hCyp11B2 gene. The nucleotide sequence of the 3′-UTR of the hCyp11B2 gene harboring the +735G-allele has a perfect Watson-Crick base pair complementary seed sequence of miR-766. On the other hand, if the hCyp11B2 gene has the +735A-allele in its 3′-UTR region, the seed sequence is interrupted, resulting in a thermodynamically less stable complex. As a result, expression of the +735G-allele of the hCyp11B2 gene should be downregulated by posttranscriptional modification of this gene by miR-766. On the other hand, expression of the +735A-allele of the hCyp11B2 gene should not be affected by this miRNA due to a mismatch in the seed sequence. In support of this hypothesis, we show that transfection of miR-766 reduces the human aldosterone synthase mRNA levels in human adrenocortical (H295R) cells.
The second key finding of the study is the negative regulatory effect of the miR-766 on hCYP11B2 expression. The transient transfection experiments allude to increased miRNA binding to the 735G-allele of hCyp11B2 gene in H295R cells. These cells contain the +735G allele of the hCYP11B2 and show significant downregulation of this gene when exposed to miR-766. It is noteworthy that decreased CYP11B2 expression is observed at both mRNA and protein expression level. This suggests that the miR-766 binding to the +735 allele of the hCYP11B2 attenuates the mRNA bioavailability of the gene.
Earlier studies have also shown that the −344T allele located in the 5′-UTR of hCyp11B2 gene is associated with human hypertension (23, 27, 31). Aldosterone is the key regulator of sodium balance via activation of the mineralocorticoid receptors in the principal cells of the cortical collecting tubule. Chronically elevated aldosterone induces cardiac hypertrophy and fibrosis; causes vascular remodeling, including perivascular fibrosis and reduced arterial distensibility; and increases the activity of cellular oxidases and precipitates redox imbalance (11, 25, 26). Complementary clinical studies have demonstrated favorable outcomes in patients with cardiovascular diseases being treated with aldosterone antagonists (SAVE, CONSENSUS). Certain Caucasian and South Asian populations are at an increased risk for salt-sensitive hypertension, and these are the cohorts where −344T has been observed to be associated with high blood pressure (8, 32). We have recently determined (unpublished data) that in transgenic mice the −344T allele leads to increased expression of hCYP11B2 and elevated plasma aldosterone levels. Interestingly, this allele is in linkage disequilibrium with the 735G allele located in the 3′-UTR of the hCyp11B2 gene. Studies presented here show that miR-766 binds to the +735G allele of the hCyp11B2 gene and reduces its mRNA and protein levels in H295R cells. Thus, we are tempted to propose that miR-766 may reduce the expression of hCyp11B2 gene and reduce blood pressure in human subjects containing the −344T allele of this gene. Although aldosterone inhibitors are available to reduce blood pressure, these inhibitors have major side effects. If one can find safe and effective ways to administer miRNAs then they may have useful therapeutic value.
In conclusion, we have identified a novel polymorphism in the 3′-UTR of hCYP11B2 gene that is in linkage disequilibrium with another polymorphism in the 5′-UTR that in turn is associated with hypertension. The polymorphism at the +735 site promotes miR-766-mediated posttranscriptional suppression of the +735G-hCYP11b2 gene. Thus, in patients with the −344T allele of hCYP11B2, who are at increased risk of hypertension, miR-766 could provide therapeutic alternatives by preventing an increase in aldosterone levels.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-81752, HL-105113, and HL-092558.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: S.M., B.M., and M.K.K. performed experiments; B.M. analyzed data; N.P. interpreted results of experiments; N.P. drafted manuscript; N.P. and A.K. edited and revised manuscript; A.K. conception and design of research; A.K. approved final version of manuscript.
ACKNOWLEDGMENTS
We are thankful to other members of the laboratory for help.
REFERENCES
- 1.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calhoun DA, Nishizaka MK, Zaman MA, Thakkar RB, Weissmann P. Hyperaldosteronism among black and white subjects with resistant hypertension. Hypertension 40: 892–896, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell 136: 642–655, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casiglia E, Tikhonoff V, Mazza A, Rynkiewicz A, Limon J, Caffi S, Guglielmi F, Martini B, Basso G, Winnicki M, Pessina AC, Somers VK. C-344T polymorphism of the aldosterone synthase gene and blood pressure in the elderly: a population-based study. J Hypertens 23: 1991–1996, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Chobanian AV. Mixed messages on blood pressure goals. Hypertension 57: 1039–1040, 2011. [DOI] [PubMed] [Google Scholar]
- 6.Coffman TM. Under pressure: the search for the essential mechanisms of hypertension. Nat Med 17: 1402–1409, 2011. [DOI] [PubMed] [Google Scholar]
- 7.Corvol P, Soubrier F, Jeunemaitre X. Molecular genetics of the renin-angiotensin-aldosterone system in human hypertension. Pathol Biol (Paris) 45: 229–239, 1997. [PubMed] [Google Scholar]
- 8.Davies E, Holloway CD, Ingram MC, Inglis GC, Friel EC, Morrison C, Anderson NH, Fraser R, Connell JM. Aldosterone excretion rate and blood pressure in essential hypertension are related to polymorphic differences in the aldosterone synthase gene CYP11B2. Hypertension 33: 703–707, 1999. [DOI] [PubMed] [Google Scholar]
- 9.Funder JW. Primary aldosteronism and low-renin hypertension: a continuum? Nephrol Dial Transplant 28: 1625–1627, 2013. [DOI] [PubMed] [Google Scholar]
- 10.Galis ZS, Thrasher T, Reid DM, Stanley DV, Oh YS. Investing in high blood pressure research: a national institutes of health perspective. Hypertension 61: 757–761, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giacchetti G, Mulatero P, Mantero F, Veglio F, Boscaro M, Fallo F. Primary aldosteronism, a major form of low renin hypertension: from screening to diagnosis. Trends Endocrinol Metab 19: 104–108, 2008. [DOI] [PubMed] [Google Scholar]
- 12.Glasser SP, Judd S, Basile J, Lackland D, Halanych J, Cushman M, Prineas R, Howard V, Howard G. Prehypertension, racial prevalence and its association with risk factors: analysis of the REasons for Geographic And Racial Differences in Stroke (REGARDS) study. Am J Hypertens 24: 194–199, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griendling KK, Murphy TJ, Alexander RW. Molecular biology of the renin-angiotensin system. Circulation 87: 1816–1828, 1993. [DOI] [PubMed] [Google Scholar]
- 14.Lawes CM, Vander HS, Rodgers A. Global burden of blood-pressure-related disease, 2001. Lancet 371: 1513–1518, 2008. [DOI] [PubMed] [Google Scholar]
- 15.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75: 843–854, 1993. [DOI] [PubMed] [Google Scholar]
- 16.Lee Y, Han J, Yeom KH, Jin H, Kim VN. Drosha in primary microRNA processing. Cold Spring Harb Symp Quant Biol 71: 51–57, 2006. [DOI] [PubMed] [Google Scholar]
- 17.Lee Y, Kim M, Han J, Yeom KH, LEES, Baek SH, Kim VN. MicroRNA genes are transcribed by RNA polymerase II. EMBO J 23: 4051–4060, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luft FC. Molecular genetics of human hypertension. J Hypertens 16: 1871–1878, 1998. [DOI] [PubMed] [Google Scholar]
- 19.Luft FC, Mervaala E, Muller DN, Gross V, Schmidt F, Park JK, Schmitz C, Lippoldt A, Breu V, Dechend R, Dragun D, Schneider W, Ganten D, Haller H. Hypertension-induced end-organ damage: a new transgenic approach to an old problem. Hypertension 33: 212–218, 1999. [DOI] [PubMed] [Google Scholar]
- 20.Makhanova N, Hagaman J, Kim HS, Smithies O. Salt-sensitive blood pressure in mice with increased expression of aldosterone synthase. Hypertension 51: 134–140, 2008. [DOI] [PubMed] [Google Scholar]
- 21.Makhanova N, Sequeira-Lopez ML, Gomez RA, Kim HS, Smithies O. Disturbed homeostasis in sodium-restricted mice heterozygous and homozygous for aldosterone synthase gene disruption. Hypertension 48: 1151–1159, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Mulatero P, Stowasser M, Loh KC, Fardella CE, Gordon RD, Mosso L, Gomez-Sanchez CE, Veglio F, Young WF., Jr Increased diagnosis of primary aldosteronism, including surgically correctable forms, in centers from five continents. J Clin Endocrinol Metab 89: 1045–1050, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Munshi A, Sharma V, Kaul S, Rajeshwar K, Babu MS, Shafi G, Anila AN, Balakrishna N, Alladi S, Jyothy A. Association of the -344C/T aldosterone synthase (CYP11B2) gene variant with hypertension and stroke. J Neurol Sci 296: 34–38, 2010. [DOI] [PubMed] [Google Scholar]
- 24.Neal B, MacMahon S, Chapman N. Effects of ACE inhibitors, calcium antagonists, and other blood-pressure-lowering drugs: results of prospectively designed overviews of randomised trials. Blood Pressure Lowering Treatment Trialists' Collaboration. Lancet 356: 1955–1964, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Nogueira AR, Bloch KV. Screening for primary aldosteronism in a cohort of Brazilian patients with resistant hypertension. J Clin Hypertens (Greenwich) 10: 619–623, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ori Y, Chagnac A, Korzets A, Zingerman B, Herman-Edelstein M, Bergman M, Gafter U, Salman H. Regression of left ventricular hypertrophy in patients with primary aldosteronism/low-renin hypertension on low-dose spironolactone. Nephrol Dial Transplant 28: 1787–1793, 2013. [DOI] [PubMed] [Google Scholar]
- 27.Rajan S, Ramu P, Umamaheswaran G, Adithan C. Association of aldosterone synthase (CYP11B2 C-344T) gene polymorphism & susceptibility to essential hypertension in a south Indian Tamil population. Indian J Med Res 132: 379–385, 2010. [PubMed] [Google Scholar]
- 28.Rossi GP, Bernini G, Caliumi C, Desideri G, Fabris B, Ferri C, Ganzaroli C, Giacchetti G, Letizia C, Maccario M, Mallamaci F, Mannelli M, Mattarello MJ, Moretti A, Palumbo G, Parenti G, Porteri E, Semplicini A, Rizzoni D, Rossi E, Boscaro M, Pessina AC, Mantero F. A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. J Am Coll Cardiol 48: 2293–2300, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Russo P, Siani A, Venezia A, Iacone R, Russo O, Barba G, D'Elia L, Cappuccio FP, Strazzullo P. Interaction between the C(-344)T polymorphism of CYP11B2 and age in the regulation of blood pressure and plasma aldosterone levels: cross-sectional and longitudinal findings of the Olivetti Prospective Heart Study. J Hypertens 20: 1785–1792, 2002. [DOI] [PubMed] [Google Scholar]
- 30.Sethupathy P, Collins FS. MicroRNA target site polymorphisms and human disease. Trends Genet 24: 489–497, 2008. [DOI] [PubMed] [Google Scholar]
- 31.Takeuchi F, Yamamoto K, Katsuya T, Sugiyama T, Nabika T, Ohnaka K, Yamaguchi S, Takayanagi R, Ogihara T, Kato N. Reevaluation of the association of seven candidate genes with blood pressure and hypertension: a replication study and meta-analysis with a larger sample size. Hypertens Res 35: 825–831, 2012. [DOI] [PubMed] [Google Scholar]
- 32.Tiago AD, Badenhorst D, Nkeh B, Candy GP, Brooksbank R, Sareli P, Libhaber E, Samani NJ, Woodiwiss AJ, Norton GR. Impact of renin-angiotensin-aldosterone system gene variants on the severity of hypertension in patients with newly diagnosed hypertension. Am J Hypertens 16: 1006–1010, 2003. [DOI] [PubMed] [Google Scholar]
- 33.Vasan RS, Evans JC, Larson MG, Wilson PW, Meigs JB, Rifai N, Benjamin EJ, Levy D. Serum aldosterone and the incidence of hypertension in nonhypertensive persons. N Engl J Med 351: 33–41, 2004. [DOI] [PubMed] [Google Scholar]
- 34.Young WF. Primary aldosteronism: renaissance of a syndrome. Clin Endocrinol (Oxf) 66: 607–618, 2007. [DOI] [PubMed] [Google Scholar]