

Abstract
Animals with a history of sodium depletions exhibit increases in salt intake, a phenomenon described as the sensitization of sodium appetite. Using a novel experimental design, the present experiments investigated whether putative molecular markers of neural plasticity and changes in the message for components of the brain renin-angiotensin-aldosterone-system (RAAS) accompany the sensitization of sodium appetite. An initial set of experiments examined whether the glutamatergic N-methyl-d-aspartate receptor antagonist MK-801 would attenuate sodium appetite sensitization and prevent changes in mRNA expression associated with sensitization. Rats with repeated sodium depletions exhibited enhanced sodium appetite and mRNA expression for components of the RAAS in areas along the lamina terminalis (LT), a region of the brain that is important for the regulation of body fluid homeostasis, and these effects were significantly attenuated by MK-801 pretreatment. A second set of experiments investigated whether successive sodium depletions would elevate sodium intake and induce a pattern of fos-B staining consistent with the Δfos-B isoform in areas along the LT. The pattern of fos-B staining in the subfornical organ was consistent with the characteristics of Δfos-B expression. Specifically, fos-B/Δfos-B expression was increased 4 days after the last of a series of sodium depletions, fos-B/Δfos-B expression was nearly absent in control rats, and the quantity of fos-B/Δfos-B staining was directly associated with a history of sodium depletions. These findings demonstrate that the sensitization of sodium appetite is associated with sustained molecular alterations in the LT that are indicative of neural plasticity and upregulation of the central RAAS.
Keywords: salt appetite, sensitization, neural plasticity, sodium, Δfos-B
when omnivores and herbivores become sodium-deficient, they exhibit sodium appetite, which entails seeking and ingesting salty substances. Sodium appetite is commonly assessed by measuring intakes of hypertonic saline solutions (1.5–3.0% wt/vol NaCl) in rats. The expression of sodium appetite is mediated through the actions of the peptide ANG II and the steroid aldosterone. ANG II and aldosterone act as hormones synthesized in the periphery [i.e., the circulating renin-angiotensin-aldosterone-system (RAAS)] and also as neurotransmitters/neuromodulators generated within the central nervous system (i.e., the central RAAS; see Refs. 11, 12, 15, and 20). Forebrain nuclei, including the subfornical organ (SFO), organum vasculosum of the lamina terminalis, and median preoptic nucleus, are located along the lamina terminalis (LT) and are critical for maintaining body fluid homeostasis; collectively, they monitor and process information related to circulating ANG II, aldosterone, and plasma osmolality (25, 26). Structures of the LT also house components of the central RAAS, including the synthetic enzymes and precursors of ANG II and aldosterone, as well as receptors for both the steroid and peptide (24, 25, 41). When rats are repeatedly depleted of sodium, they display elevated sodium intake (10, 14, 22, 39, 43). Rats exhibit progressive (3, 39, 44) or rapid (19, 22, 43) increases in sodium intake after one to three episodes of sodium deficiency depending on the depletion protocol employed. This increase in salt intake operationally defines sodium appetite sensitization.
Previous studies have demonstrated that the development of sodium appetite sensitization is dependent on ANG II, aldosterone, and N-methyl-d-aspartate receptor (NMDAR) signaling in the central nervous system. Antagonism of ANG II type 1 receptors (AT1R) (39) or blockade of ANG II synthesis concurrent with central mineralocorticoid receptor (MR) antagonism (43) prevents the sensitization of sodium appetite. Administration of central ANG II along with systemic aldosterone induces sensitization (43). Additionally, blockade of NMDARs prior to sodium depletion prevents sensitization of sodium appetite (22). It is likely that sodium depletion induces neuroplasticity in the central nervous system through the coordinated actions of ANG II, aldosterone, and glutamate, which are responsible for the elevation of sodium intake. Such plasticity has been related to long-lasting changes in gene transcription and mRNA translation, neuronal structure, receptor expression, among many other alterations in neural or glial molecular biology and function (37, 54, 58).
Many sodium depletion protocols require a long latency between the induction of sodium loss and the actual expression of sodium appetite (generally between 8 h to several days) (23). Coadministration of the diuretic furosemide along with the antihypertensive drug captopril (furo/cap) causes an extracellular dehydration concomitant with a slight drop in blood pressure (31, 50). These effects result in a rapid onset of thirst and sodium appetite with a latency of ∼1 h (17, 31, 50). Rats repeatedly treated with furo/cap reliably exhibit sensitization of sodium appetite when the order of water and sodium access is controlled by the experimenter (22). Specifically, when water is offered for 90 min before sodium access (the water-first/sodium-second iteration of the furo/cap protocol) rats exhibit a progressive increase in sodium intake over successive furo/cap treatments. It is currently unclear as to whether NMDAR antagonism affects the progressive increase in sodium intake observed in this protocol.
One goal of the present study was to extend previous findings on the effect of NMDAR blockade on the sensitization of sodium appetite in the water-first/sodium-second iteration of the furo/cap protocol. A second aim was to examine changes in the expression of molecular components indicative of sustained alterations in activity of the central RAAS and putative markers of neural plasticity in the LT. In an initial experiment, we tested whether NMDAR antagonism would attenuate the sensitization of sodium appetite and whether repeated sodium depletions would result in an NMDAR-dependent increase in mRNA expression for components of the RAAS and a MR-related molecular marker of plasticity, serum- and glucocorticoid-induced kinase (SGK), in the LT. In a second set of experiments, it was hypothesized that sodium depletion would induce a pattern of fos-B expression in areas along the LT that would be consistent with the characteristics of the Δfos-B isoform, which is associated with neural plasticity (38). Consistent with our hypotheses, repeated sodium depletions induced increased expression of molecular markers related to both the brain RAAS and neural plasticity that correlated with sodium intake. Such changes lasted for several days after sodium depletion, and NMDAR antagonism prevented many of them. These findings lend further support to the hypothesis that neural plasticity within the LT contributes to the sensitization of sodium appetite.
MATERIALS AND METHODS
Subjects.
All experiments were conducted in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (36) and were approved by The University of Iowa Animal Care and Use Committee. Male Sprague-Dawley rats (Harlan Teklad, Indianapolis, IN) weighing between 275 and 300 g upon arrival were used as subjects. Rats were maintained on a 12:12-h light-dark cycle and housed in translucent (28.5 × 28.5 × 17.5 cm) or wire mesh suspended (24 × 17.2 × 17.0 cm) cages in a temperature- and humidity-controlled room. Unless noted otherwise, the animals had ad libitum access to filtered tap water, 1.8% wt/vol hypertonic saline (1.8% NaCl), and NIH-31-irradiated modified open formula mouse/rat diet. Rats had at least 3 days of ad libitum 1.8% NaCl access prior to experimentation. The studies used 1.8% NaCl intake to assess sodium appetite and the consumption of deionized water to assess thirst.
Extracellular dehydration protocol.
Extracellular dehydration was induced by subcutaneous injection of furosemide (10 mg/kg; Hospirca Lake Forest, IL) along with the antihypertensive drug captopril (5 mg/kg; Sigma Aldrich, St. Louis, MO), which results in a slight drop in blood pressure (∼10 mmHg) (31, 50). Water was offered immediately after furo/cap treatment and 90 min later 1.8% NaCl access was added.
Effect of MK-801 on sodium appetite sensitization and mRNA expression in the LT.
We previously found that administration of the NMDAR antagonist MK-801 prevented sodium appetite sensitization in the furo/cap model when sodium access was offered prior to water (22). One purpose of our studies was to extend the generalizability of this finding by using a water-first/sodium-second protocol. Rats (n = 12 in the vehicle group and 13 in the MK-801 group) were pretreated with vehicle (95% saline, 5% DMSO) or MK-801 (0.15 mg/kg) 20 min prior to receiving furo/cap treatment. Immediately after furo/cap treatment, they were allowed access to water for 90 min, and intakes were recorded. At the end of the 90-min water access period, sodium access was allowed, and fluid intakes were recorded for an additional 90 min. Rats were treated 3 times with this protocol with each treatment separated by 4 days.
Separate groups of rats were used to investigate whether repeated sodium depletions induce changes in mRNA expression in the LT. Three experimental groups (n = 4) were employed: a vehicle-pretreated and sham-depleted group, a vehicle pretreated and furo/cap-depleted group, and an MK-801 (0.15 mg/kg) pretreated and furo/cap depleted group. Each group received vehicle or MK-801 20 min prior to furo/cap or sham depletion. Rats received a total of 3 furo/cap or sham treatments each separated by 4 days. Five days after the treatments, rats were euthanized by decapitation, the LT was dissected by hand, and mRNA expression for AT1R, angiotensin type 2 receptors (AT2R), angiotensin-converting enzyme (ACE) 1 and 2, MR, and SGK was measured using real-time RT-PCR.
RT-PCR was performed as described previously (56, 57). Briefly, mRNA was isolated using TRIzol (Invitrogen). mRNA transcription was accomplished using random hexamers under the manufacturer's instructions (Applied Biosystems, Foster City, CA). Primers are listed in Table 1. cDNA amplification was performed by a C1000 thermocycler system (Bio-Rad). mRNA levels were normalized to GAPDH levels and calculated using the ΔΔCt method. Each assay was run in triplicate to ensure reliability. Results are expressed as relative fold change compared with sham-depleted rats.
Table 1.
Primers used for reverse transcription real-time PCR
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| ACE1 | GTGTTGTGGAACGAATACGC | CCTTCTTTATGATCCGCTTGA |
| ACE2 | TTAAGCCACCTTACGAGCCTC | GCCAATGTCCATGGAGTCAT |
| AT1R | CTCAAGCCTGTCTACGAAAATGAG | GTGAATGGTCCTTTGGTCGT |
| AT2R | ACCTTTTGAACATGGTGCTTTG | TTTCCTATGCCAGTGTGCAG |
| MR | GCCCGGCAAATCTCAACAACTCAA | TTAGGGAAAGGAACGTCGTGAGCA |
| SGK | AATGGCGGAGAGCTGTTCTA | TGTGCTCGATGTTCTCCTTG |
Effect of repeated sodium depletions on sensitization of sodium appetite and fos-B/Δfos-B expression in the LT.
To examine the effect of furo/cap on the sensitization of sodium appetite, three groups of rats were used (n = 11). One group of rats received four sham depletions, a second received three sham depletions prior to a single furo/cap depletion, and a third received 4 furo/cap depletions. Rats were treated with a depletion or sham depletion every 4 days, and fluid access was offered in accordance with the water-first/sodium-second experimental protocol. Water and 1.8% NaCl intakes were recorded on the 4th depletion or sham depletion. Immediately following furo/cap treatment, water was offered for 90 min, and then sodium access was allowed. During the 4th depletion, intakes of water and sodium were recorded for 180 min at 90-min intervals.
A subset of rats (n = 4) were killed 4 days after the last depletion or sham depletion and perfused transcardially with PBS followed by 4% paraformaldehyde (PFA). To control for variability in staining efficiency, all cycles of tissue collection and staining were performed in cohorts, such that an equal number of sham, single, and repeatedly depleted animals were run in parallel. Brains were postfixed for 4–6 h in 4% PFA and then transferred to vials containing 20% sucrose dissolved in PBS. Brains were left in sucrose overnight at 4°C, and the next morning, a series of three nonconsecutive 40-μm slices were sectioned with a cryostat and stored in cryoprotectant at −20°C. Tissue was removed from storage and washed 3 times in PBS, blocked in normal rabbit serum for 1 h, and then incubated with a goat-raised anti-fos-B antibody (1:2,000–1:4,000; sc-48-G; Santa Cruz Biotechnology, Dallas, TX) for 48 h at 4°C. This antibody detects both fos-B and the truncated isoform of fos-B known as Δfos-B. Tissue was then washed three times and incubated with rabbit-raised biotinylated anti-goat antibody for 1 h (1:200; Santa Cruz), washed three times, and incubated with avidin-biotin complex (Vector Labs Elite Kit, Burlingame, CA) for 1 h. Tissue was washed 3 times and exposed to DAB and hydrogen peroxide for ∼4 min to visualize sites of protein expression. Sections were mounted and coverslipped. A digital image of the SFO was taken at approximately equivalent rostral-caudal coordinates, and cell counts were performed manually by an experimenter blind to the treatment conditions using ImageJ (NIH, version 1.46r).
Statistics.
All results were analyzed using SigmaPlot build 12.0.0.182 (Systat Software, Chicago, IL). Statistical significance was set at P < 0.05. In studies examining the effect of MK-801 on sensitization, the difference in sodium intake between the first and third depletion was used as the dependent variable, and these data were analyzed with Student's t-test. Depletion, fos-B, and RT-PCR data were analyzed using a one-way ANOVA, and when a significant main effect was observed, Newman-Keul's post hoc tests were employed to probe group differences.
RESULTS
Effect of MK-801 on sodium appetite sensitization and mRNA expression in the LT.
MK-801 pretreatment significantly attenuated sensitization of sodium appetite, such that the increase in total salt intake between the first and third depletions was reduced in the MK-801-pretreated group t(23) = 2.336; P < 0.05 (Fig. 1). No differences in salt intake during the first depletion were observed between vehicle and MK-801-pretreated rats t(23) = 0.265; P = 0.79. No effects of MK-801 on water intake were observed (data not shown). Analyses revealed a main effect of treatment on mRNA expression for AT1R F(2,9) = 4.938; P < 0.05, MR F(2,9) = 8.372; P < 0.01, and SGK F(2,9) = 5.056; P < 0.05. No treatment effects were observed in mRNA expression for AT2R F(2,9) = 1.179; P = 0.35, ACE1 F(2,9) = 2.037; P = 0.18, and ACE2 F(2,9) = 2.458; P = 0.14. mRNA data are displayed in Fig. 2. Compared with sham-treated animals, repeated furo/cap treatments elevated mRNA expression for AT1R, MR, and SGK. MK-801 treatment prevented these changes, such that mRNA expression for AT1R, MR, and SGK did not increase over sham-treated values.
Fig. 1.
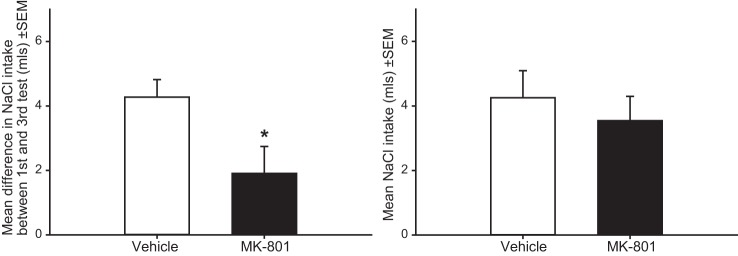
Effect of MK-801 pretreatment on sodium appetite sensitization in the water-first/sodium-second protocol. Rats pretreated with MK-801 displayed an attenuated sensitization of sodium appetite (left; *P < 0.05); however, MK-801 has no effect on sodium intake during the first furo/cap treatment (right).
Fig. 2.

Effect of repeated furo/cap treatments and MK-801 pretreatment on mRNA expression in the lamina terminalis. mRNA expression relative to control levels is graphed on the y-axis. Compared with sham-depleted rats, rats with a history of furo/cap treatments expressed greater levels of mRNA for the AT1R, MR, and SGK (*P < 0.05 vs. 3 times sham-treated rats). Rats pretreated with MK-801 failed to show an elevation of message for AT1R and expressed significantly less message for MR and SGK compared with rats with a history of furo/cap treatments (#P < 0.05 vs. 3 times furo/cap). No significant differences in AT2R, ACE1, or ACE2 mRNA expression were observed. All data are expressed as arbitrary units.
Effect of repeated sodium depletions on sensitization of sodium appetite and fos-B/Δfos-B expression in the LT.
Analyses revealed a main effect of depletion number, F(2,30) = 23.819; P < 0.001, in which rats with a history of sodium depletions exhibited greater salt intake than rats with a single depletion, and both groups of depleted rats exhibited greater salt intake than sham-depleted rats (Fig. 3A). A main effect of depletion number on water intake was also found F(2,30) = 7.576; P < 0.005. Sodium-depleted rats exhibited greater water intake compared with sham-treated rats (Fig. 3B), but no significant differences were observed between rats that received a single sodium depletion or repeated sodium depletions. A main effect of depletion number on total fluid intake was observed F(2,30) = 14.683; P < 0.001. Rats that received extracellular dehydration exhibited greater total fluid intake, but there were no differences between rats that received a single depletion or repeated depletions (Fig. 3C). Finally, a greater proportion of total fluid intake consisted of sodium intake in rats with a history of sodium depletions compared with rats receiving a single depletion t(20) = 3.354; P < 0.005 (Fig. 3D).
Fig. 3.

Sensitization of sodium appetite. A: rats with a history of sodium depletions display greater 1.8% NaCl intake than rats with a single depletion. A and B: depleted rats drank more 1.8% NaCl and water than sham-treated rats. C: combined water and sodium intake was elevated in single and repeatedly depleted rats compared with sham-depleted rats. D: a greater proportion of total fluid intake consisted of sodium intake in repeatedly depleted rats compared with rats with a single depletion. *P < 0.05 vs. sham. #P < 0.05 vs. single depleted rats.
A survey of brains for immunohistochemical evidence of increased protein levels indicated that the SFO was the only region in the brain that exhibited consistent differences in fos-B/Δfos-B expression across treatments. Therefore, all fos-B/Δfos-B analyses were performed on the SFO. These analyses revealed a main effect of depletion number on fos-B/Δfos-B expression F(2,9) = 12.4; P < 0.005. Post hoc tests identified that sham-depleted rats exhibited very low levels of fos-B/Δfos-B staining, rats with a single depletion exhibited significantly increased fos-B/Δfos-B expression than seen in sham-depleted rats, and animals with a history of four depletions exhibited greater levels of fos-B/Δfos-B than sham- and single-depleted rats (Figs. 4 and 5).
Fig. 4.
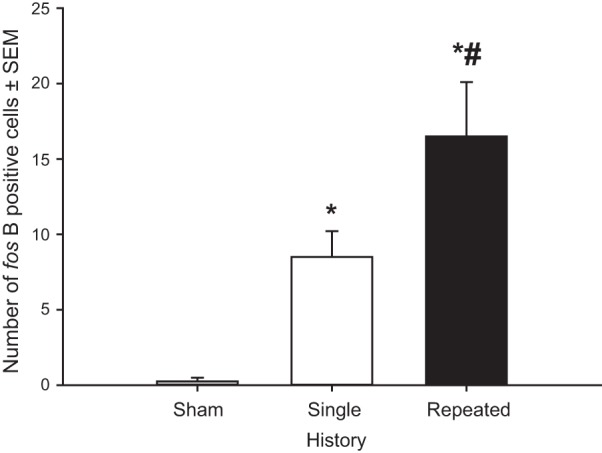
Effect of sodium depletions on fos-B/Δfos-B expression in the SFO. Sham-pretreated rats exhibited virtually no fos-B/Δfos-B staining, rats with a single sodium depletion exhibited moderate amounts of fos-B/Δfos-B expression, and rats with repeated sodium depletions exhibited robust expression of fos-B/Δfos-B. *P < 0.05 vs. sham-treated rats. #P < 0.05 vs. single-depletion rats.
Fig. 5.
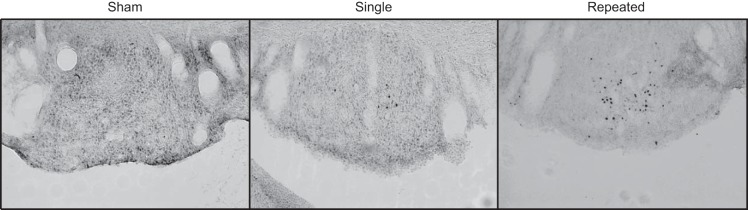
Representative fos-B/Δfos-B staining in the SFO. ×10 images were taken of the posterior portion of the SFO ∼1.4 mm caudal from bregma.
DISCUSSION
The primary findings from the present experiments are that 1) MK-801 pretreatment attenuated the sensitization of sodium appetite in the water-first/sodium-second protocol, 2) sodium appetite sensitization is associated with maintained elevation of mRNA expression of key components of the brain RAAS in the LT region, and 3) salt preference during sodium depletion is associated with a sustained elevation of fos-B/Δfos-B in the SFO. A list of the molecular markers investigated in the present study and their significance is presented in Table 2.
Table 2.
List of molecular markers examined in the present study
| Abbreviation | Description |
|---|---|
| ACE1 | Angiotensin-converting enyzme type 1–An enzyme that converts ANG I into ANG II |
| ACE2 | Angiotensin converting enzyme type 2–A converting enzyme that appears to promote effects opposite of ACE1 by deactivating ANG II and converting ANG I into ANG 1–9 (4) |
| AT1R | Angiotensin type 1 receptor–The primary receptor that mediates the actions of ANG II on water and sodium intake and vasoconstriction |
| AT2R | Angiotensin type 2 receptor–Receptor for ANG II that may serve counter-regulatory functions |
| MR | Mineralocorticoid receptor–Receptor for aldosterone that promotes sodium intake, vasoconstriction, and sodium retention |
| SGK | Serum- and glucuocorticoid-induced kinase–Second messenger induced by mineralocorticoid receptor activation that appears to be involved in some forms of neural plasticity (29) |
| fos-B | An immediate early gene that is upregulated after neurons become activated |
| Δfos-B | A truncated isoform of fos-B that lacks the catalytic moeity, allowing it to remain elevated for weeks after induction. Δfos-B appears to be involved in some forms of neural plasticity (37, 40) |
The behavioral results from these experiments are consistent with prior reports of enhanced salt appetite as a function of episodic sodium depletions (14, 22, 43, 44). It was found that when using the water-first/sodium-second protocol, rats with a history of sodium depletions drank more 1.8% NaCl than rats with a single depletion, and they drank a greater proportion of sodium relative to water. Importantly, pretreatment with 0.15 mg/kg MK-801 attenuated sensitization of sodium appetite in this protocol but did not influence salt intake on the first depletion. The observed attenuation in sodium appetite is consistent with the idea that sensitization of sodium appetite is dependent on signaling involving NMDARs, which are critical for the initiation of many forms of neural plasticity (7).
Rats with a history of furo/cap treatments displayed elevated message for components of the central RAAS. Specifically, AT1R and MR mRNA were upregulated 5 days after the last sodium depletion. Importantly, this effect was blocked by MK-801 pretreatment. To our knowledge, this is the first evidence that sodium depletions can have long-lasting NMDAR-dependent effects on the central RAAS. Others have found that treatments that affect ANG II or MR levels can have long-lasting effects on the central expression of ANG II receptors (6, 45) and administration of exogenous deoxycorticosterone acetate, aldosterone, or ANG II elevates angiotensin receptor expression in the diencephalon (27, 52, 53, 55). In the present study, no significant differences were observed in components of the RAAS that have been found to serve counterregulatory functions, such as ACE2 or AT2R expression (4, 5).
SGK mRNA was increased after sodium depletion and NMDAR antagonism prevented this increase. SGK is a MR-dependent kinase (35, 46) that has been linked to neural plasticity (28, 49, 51). Rats transfected with a dominant negative isoform of SGK in the hippocampus display impaired expression of hippocampal long-term potentiation, fear conditioning, novel object recognition, and spatial recognition (28, 29, 51). It is possible that SGK expression is increased after sodium depletion due to aldosterone action at the MR and that SGK may play an important role in shaping neural plasticity within components of the LT to sensitize sodium intake.
Expression of fos-B/Δfos-B staining in the SFO was directly associated with salt preference. Rats with a history of sodium depletions exhibited a greater preference for sodium relative to water in addition to total sodium intake. Rats with a history of sodium depletion also exhibited greater fos-B/Δfos-B expression relative to rats that were sodium-depleted for the first time. fos-B is an immediate early gene that is expressed during neuronal activity (37, 38). Δfos-B is an isoform of fos-B. Δfos-B lacks the catalytic domain present in fos-B, which results in it remaining elevated for weeks after induction (37, 38). As such, Δfos-B expression follows a characteristic pattern. Baseline expression of Δfos-B is nearly absent in brain nuclei. However, in response to treatments associated with the induction of neural plasticity (e.g., repeated stress and administration of addictive drugs) Δfos-B is expressed and remains elevated for weeks, and it accumulates over repeated treatments (37, 38). The pattern of fos-B/Δfos-B staining observed in the present study follows the characteristics of the Δfos-B isoform. A near-complete absence of staining was observed in sham-depleted rats, whereas rats that had experienced a single depletion exhibited a moderate amount of staining, and a greater amount of staining was observed in rats depleted repeatedly. Therefore, it is likely that sodium depletion induced increased expression of Δfos-B in the SFO and that Δfos-B accumulated over the course of repeated depletions.
It is possible that the expression of fos-B/Δfos-B observed after sodium depletion was due to the ingestion of sodium (i.e., expressed due to associative learning). This seems to be unlikely, as previous studies have suggested that the sensitization of sodium appetite is a nonassociative phenomenon (13). Rats will exhibit increased salt intake, even when salt is replenished through means independent of ingestion (13). It is also unlikely that the expression of fos-B/Δfos-B was due to a possible change in circulating ANG II or aldosterone caused by sodium depletion as repeated sodium depletions do not appear to change circulating levels of either hormone (44).
The present experiments have potential limitations that need to be acknowledged. The fos-B/Δfos-B and mRNA data are correlative in nature, and the functional significance of these changes remains to be tested. mRNA was collected from the entirety of the LT, and we cannot pinpoint an exact structure or set of structures, where these molecular changes may be occurring. Functional studies employing viral vectors could be used to elevate or eliminate AT1R or MR receptors in key brain areas in the LT to provide insight into the importance of receptor expression and the sensitization of sodium appetite. Additionally, there are dominant-negative forms of SGK and Δfos-B that allow for the possibility of testing the functional importance of both of these molecules in sodium appetite sensitization.
Together, the present findings indicate that a history of sodium depletions elevates sodium intake in addition to inducing a long-lasting elevation of molecular markers related to neural plasticity (SGK and a pattern of staining that appears to be Δfos-B) and those related to the central RAAS (AT1R and MR). Sensitization of sodium appetite and enhanced expression of AT1R, MR, and SGK mRNA were dependent on intact NMDA receptor signaling. On the basis of this and previous findings, it is possible that the coordinated actions of glutamate (22), ANG II (3, 56), and aldosterone (11, 18, 43, 55, 57) produce long-lasting changes in neural structures containing components of the RAAS. The amount of fos-B/Δfos-B staining in the SFO was associated with sodium depletion history and may contribute to sodium appetite sensitization. Additional evidence implicates the SFO in sensitization of sodium appetite as the immediate early gene c-fos is elevated in the SFO in sodium-deficient rats with a history of depletion relative to rats with no prior history of sodium depletion (34). Some of the molecular changes observed in the current findings have been shown to promote glutamatergic AMPA receptor insertion, which is critically involved in producing long-term potentiation (30). Specifically, SGK induces surface expression of AMPA receptors (49), and Δfos-B has been linked to neural plasticity with respect to drugs of abuse (it aids in the insertion of GluA2-containing AMPA receptors into the neuronal membrane) (37, 38, 40). These findings add to a growing body of evidence that supports the occurrence of neural plasticity in body fluid homeostasis neurocircuitry (22, 34, 42, 56), and they provide insight into important molecular changes that are likely to drive sensitization of sodium appetite.
Perspectives and Significance
ANG II and aldosterone impinge upon or are released in central nervous system areas involved in body fluid homeostasis to initiate long-lasting neuroplastic changes that cause an animal to seek out and ingest greater quantities of sodium. This is an example of allostasis, or the change in operating levels of physiological endpoints produced by repeated or sustained physiological or environmental challenges to homeostasis (23, 33, 48). Many animals experience threats to sodium homeostasis, including surviving in environments bereft of sodium, sickness that evokes emesis and diarrhea, excess sweating, pregnancy, and trauma-induced hemorrhage. To help restore and protect sodium balance, it is in all likelihood adaptive for physiological systems to undergo plasticity that results in enhanced sodium intake. This provides a buffer against future loss, increases the likelihood that a sodium-replete animal will consume sodium and learn the location of sodium in the environment, and allows an animal to replenish more quickly depleted sodium stores and restore extracellular volume (16, 23, 44). However, when animals are in environments with a surplus of sodium, such as humans in Western societies, the increased drive to seek out and ingest sodium may have negative health consequences for some individuals (1, 2, 8, 9, 21, 23, 47). Sodium appetite sensitization and the excess sodium intake associated with sodium appetite sensitization are examples of allostasis and allostatic load, respectively (32, 33). In other words, the experience of sodium depletion heightens the sensitivity or activity of neural systems involved in maintaining body fluid homeostasis, and this increases the chance of developing pathologies, such as hypertension. Preclinical work aiming to understand the biological mechanisms mediating excess sodium intake may result in treatments that help ameliorate excess salt intake in Western societies.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: S.W.H. and A.K.J. conception and design of research; S.W.H., Z.Z., T.G.B., and B.X. performed experiments; S.W.H. and Z.Z. analyzed data; S.W.H., Z.Z., and A.K.J. interpreted results of experiments; S.W.H. prepared figures; S.W.H. drafted manuscript; S.W.H., Z.Z., T.G.B., B.X., and A.K.J. edited and revised manuscript; S.W.H., Z.Z., T.G.B., B.X., and A.K.J. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Tom Nordstrom and Nora Bruhn for their technical assistance and Marilyn Dennis for comments on the manuscript. This research was supported by National Institutes of Health Grants HL-14388, HL-098207, and MH-08241. The authors have no disclosures to report.
REFERENCES
- 1.Bibbins-Domingo K, Chertow GM, Coxson PG, Moran A, Lightwood JM, Pletcher MJ, Goldman L. Projected effect of dietary salt reductions on future cardiovascular disease. N Engl J Med 362: 590–599, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown IJ, Tzoulaki I, Candeias V, Elliott P. Salt intakes around the world: implications for public health. Int J Epidemiol 38: 3: 791–813, 2009. [DOI] [PubMed] [Google Scholar]
- 3.Bryant R, Epstein A, Fitzsimons J, Fluharty S. Arousal of a specific and persistent sodium appetite in the rat with continuous intracerebroventricular infusion of angiotensin II. J Physiol 301: 365–382, 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burrell LM, Johnston CI, Tikellis C, Cooper ME. ACE2, a new regulator of the renin-angiotensin system. Trends Endocrinol Metab 15: 166–169, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carey RM, Jin X, Siragy HM. Role of the angiotensin AT2 receptor in blood pressure regulation and therapeutic implications. Am J Hypertens 14: 98S–102S, 2001. [DOI] [PubMed] [Google Scholar]
- 6.Castrén E, Saavedra JM. Angiotensin II receptors in paraventricular nucleus, subfornical organ, and pituitary gland of hypophysectomized, adrenalectomized, and vasopressin-deficient rats. Proc Natl Acad Sci USA 86: 725–729, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cotman CW, Monaghan DT, Ganong AH. Excitatory amino acid neurotransmission: NMDA receptors and Hebb-type synaptic plasticity. Annu Rev Neurosci 11: 61–80, 1988. [DOI] [PubMed] [Google Scholar]
- 8.Dahl LK. Salt and hypertension. Am J Clin Nutr 25: 231–244, 1972. [DOI] [PubMed] [Google Scholar]
- 9.Dahl LK, Knudsen KD, Heine MA, Leitl GJ. Effects of chronic excess salt ingestion: modification of experimental hypertension in the rat by variations in the diet. Circ Res 22: 11–18, 1968. [DOI] [PubMed] [Google Scholar]
- 10.De Luca LA, Pereira-Derderian DTB, Vendramini RC, David RB, Menani JV. Water deprivation-induced sodium appetite. Physiol Behav 100: 535–544, 2010. [DOI] [PubMed] [Google Scholar]
- 11.Epstein AN. Mineralocorticoids and cerebral angiotensin may act together to produce sodium appetite. Peptides 3: 493–494, 1982. [DOI] [PubMed] [Google Scholar]
- 12.Epstein AN. Neurohormonal control of salt intake in the rat. Brain Res Bull 27: 315–320, 1991. [DOI] [PubMed] [Google Scholar]
- 13.Falk JL. Serial sodium depletion and NaCl solution intake. Physiol Behav 1: 75–77, 1966. [Google Scholar]
- 14.Falk JL. Water intake and NaCl appetite in sodium depletion. Psychol Rep 16: 315–325, 1965. [DOI] [PubMed] [Google Scholar]
- 15.Ferguson AV, Washburn DL, Latchford KJ. Hormonal and neurotransmitter roles for angiotensin in the regulation of central autonomic function. Exp Biol Med (Maywood) 226: 85–96, 2001. [DOI] [PubMed] [Google Scholar]
- 16.Fessler D. An evolutionary explanation of the plasticity of salt preferences: prophylaxis against sudden dehydration. Med Hypotheses 3: 412–415, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Fitts DA, Masson DB. Forebrain sites of action for drinking and salt appetite to angiotensin or captopril. Behav Neurosci 103: 865–872, 1989. [DOI] [PubMed] [Google Scholar]
- 18.Fluharty SJ, Epstein AN. Sodium appetite elicited by intracerebroventricular infusion of angiotensin II in the rat: II. Synergistic interaction with systemic mineralocorticoids. Behav Neurosci 97: 746–758, 1983. [DOI] [PubMed] [Google Scholar]
- 19.Frankmann SP, Dorsa DM, Sakai RR, Simpson JB. A single experience with hyperoncotic colloid dialysis persistently alters water and sodium intake. The Physiology of Thirst and Sodium Appetite. NATO ASI Series. New York: Plenum Press, 1986. [Google Scholar]
- 20.Gomez-Sanchez CE, Zhou MY, Cozza EN, Morita H, Foecking MF, Gomez-Sanchez EP. Aldosterone biosynthesis in the rat brain. Endocrinology 138: 3369–3373, 1997. [DOI] [PubMed] [Google Scholar]
- 21.Henry JP. Stress, salt and hypertension. Social Sci Med 26: 293–302, 1988. [DOI] [PubMed] [Google Scholar]
- 22.Hurley SW, Johnson AK. Dissociation of thirst and sodium appetite in the furo/cap model of extracellular dehydration and a role for N-methyl-d-aspartate receptors in the sensitization of sodium appetite. Behav Neurosci 127: 890–898, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hurley SW, Thunhorst RL, Johnson AK. Sodium appetite sensitization. In: Neurobiology of Body Fluid Homeostasis: Transduction and Integration (Series IV: Frontiers in Neuroscience), edited by De L, uca LA, Johnson AK, Menani JV. Boca Raton, FL: Taylor and Francis, 2013, p. 279–301. [Google Scholar]
- 24.Johnson AK. The sensory psychobiology of thirst and salt appetite. Med Sci Sports Exerc 39: 1388–1400, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Johnson A, Thunhorst R. The neuroendocrinology, neurochemistry and molecular biology of thirst and salt appetite. In: Handbook of Neurochemistry and Molecular Neurobiology: Behavioral Neurochemistry, Neuroendocrinology and Molecular Neurobiology 3rd ed, New York: Springer, 2007, p. 641–687. [Google Scholar]
- 26.Johnson A, Thunhorst R. The neuroendocrinology of thirst and salt appetite: visceral sensory signals and mechanisms of central integration. Front Neuroendocrinol 18: 292–353, 1997. [DOI] [PubMed] [Google Scholar]
- 27.King SJ, Harding JW, Moe KE. Elevated salt appetite and brain binding of angiotensin II in mineralocorticoid-treated rats. Brain Res 448: 140–149, 1988. [DOI] [PubMed] [Google Scholar]
- 28.Lee CT, Tyan SW, Ma YL, Tsai MC, Yang YC, Lee EH. Serum- and glucocorticoid-inducible kinase (SGK) is a target of the MAPK/ERK signaling pathway that mediates memory formation in rats. Eur J Neurosci 23: 1311–1320, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Lee EH, Hsu WL, Ma YL, Lee PJ, Chao CC. Enrichment enhances the expression of sgk, a glucocorticoid-induced gene, and facilitates spatial learning through glutamate AMPA receptor mediation. Eur J Neurosci 18: 2842–2852, 2003. [DOI] [PubMed] [Google Scholar]
- 30.Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25: 103–126, 2002. [DOI] [PubMed] [Google Scholar]
- 31.Masson DB, Fitts DA. Subfornical organ connectivity and drinking to captopril or carbachol in rats. Behav Neurosci 103: 873–880, 1989. [DOI] [PubMed] [Google Scholar]
- 32.McEwen BS. Seminars in Medicine of the Beth Israel Deaconess Medical Center: Protective and damaging effects of stress mediators. N Engl J Med 338: 171–189, 1998. [DOI] [PubMed] [Google Scholar]
- 33.McEwen BS, Stellar E. Stress and the individual: mechanisms leading to disease. Arch Intern Med 153: 2093–2101, 1993. [PubMed] [Google Scholar]
- 34.Na ES, Morris MJ, Johnson RF, Beltz TG, Johnson AK. The neural substrates of enhanced salt appetite after repeated sodium depletions. Brain Res 1171: 104–110, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naray-Fejes-Toth A., Fejes-Toth G. The sgk, an aldosterone-induced gene in mineralocorticoid target cells, regulates the epithelial sodium channel. Kidney Int 57: 1290–1294, 2000. [DOI] [PubMed] [Google Scholar]
- 36.National Research Council (U.S.), Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Institute for Laboratory Animal Research, and National Academies Press. Guide for the Care and Use of Laboratory Animals. Washington, D.C.: National Academies Press, 2011, p. 220. [Google Scholar]
- 37.Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci 2: 119–128, 2001. [DOI] [PubMed] [Google Scholar]
- 38.Nestler EJ, Barrot M, Self DW. DeltaFosB: a sustained molecular switch for addiction. Proc Natl Acad Sci USA 98: 11042–11046, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pereira DTB, Menani JV, De Luca LAJ. FURO/CAP: A protocol for sodium intake sensitization. Physiol Behav 99: 472–481, 2010. [DOI] [PubMed] [Google Scholar]
- 40.Perrotti LI, Hadeishi Y, Ulery PG, Barrot M, Monteggia L, Duman RS, Nestler EJ. Induction of ΔFosB in reward-related brain structures after chronic stress. J Neurosci 24: 10594–10602, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phillips MI. Functions of angiotensin in the central nervous system. Annu Rev Physiol 49: 413–435, 1987. [DOI] [PubMed] [Google Scholar]
- 42.Roitman MF, Na E, Anderson G, Jones TA, Bernstein IL. Induction of a salt appetite alters dendritic morphology in nucleus accumbens and sensitizes rats to amphetamine. J Neurosci 22: RC225, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakai RR, Fine WB, Epstein AN, Frankmann SP. Salt appetite is enhanced by one prior episode of sodium depletion in the rat. Behav Neurosci 101: 724–731, 1987. [DOI] [PubMed] [Google Scholar]
- 44.Sakai RR, Frankmann SP, Fine WB, Epstein AN. Prior episodes of sodium depletion increase the need-free sodium intake of the rat. Behav Neurosci 103: 186–192, 1989. [DOI] [PubMed] [Google Scholar]
- 45.Shelat SG, Fluharty SJ, Flanagan-Cato LM. Adrenal steroid regulation of central angiotensin II receptor subtypes and oxytocin receptors in rat brain. Brain Res 807: 135–146, 1998. [DOI] [PubMed] [Google Scholar]
- 46.Shigaev A, Asher C, Latter H, Garty H, Reuveny E. Regulation of sgk by aldosterone and its effects on the epithelial Na+ channel. Am J Physiol Renal Physiol 278: F613–F619, 2000. [DOI] [PubMed] [Google Scholar]
- 47.Stamler J. The INTERSALT Study: background, methods, findings, and implications. Am J Clin Nutr 65: 2 Suppl: 626S–642S, 1997. [DOI] [PubMed] [Google Scholar]
- 48.Sterling P, Eyer J. Allostasis: a new paradigm to explain arousal pathology. In: Handbook of Life Stress, edited by Fisher S, Reason J. 1988, p. 629–649. [Google Scholar]
- 49.Strutz-Seebohm N, Seebohm G, Mack AF, Wagner HJ, Just L, Skutella T, Lang UE, Henke G, Striegel M, Hollmann M, Rouach N, Nicoll RA, McCormick JA, Wang J, Pearce D, Lang F. Regulation of GluR1 abundance in murine hippocampal neurones by serum- and glucocorticoid-inducible kinase 3. J Physiol 565: 381–390, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thunhorst RL, Johnson A. Renin-angiotensin, arterial blood pressure, and salt appetite in rats. Am J Physiol Regul Integr Comp Physiol 266: R458–R465, 1994. [DOI] [PubMed] [Google Scholar]
- 51.Tsai KJ, Chen SK, Ma YL, Hsu WL, Lee EH. Sgk, a primary glucocorticoid-induced gene, facilitates memory consolidation of spatial learning in rats. Proc Natl Acad Sci USA 99: 3990–3995, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilson KM, Sumners C, Hathaway S, Fregly MJ. Mineralocorticoids modulate central angiotensin II receptors in rats. Brain Res 382: 87–96, 1986. [DOI] [PubMed] [Google Scholar]
- 53.Wilson KM, Sumners C, Fregly MJ. Effects of increased circulating angiotensin II (AII) on fluid exchange and binding of All in the brain. Brain Res Bull 20: 493–501, 1988. [DOI] [PubMed] [Google Scholar]
- 54.Wright JW, Harding JW. The brain angiotensin system and extracellular matrix molecules in neural plasticity, learning, and memory. Prog Neurobiol 72: 263–293, 2004. [DOI] [PubMed] [Google Scholar]
- 55.Xue B, Beltz TG, Yu Y, Guo F, Gomez-Sanchez CE, Hay M, Johnson AK. Central interactions of aldosterone and angiotensin II in aldosterone- and angiotensin II-induced hypertension. Am J Physiol Heart Circ Physiol 300: H555–H564, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xue B, Zhang Z, Johnson RF, Johnson AK. Sensitization of slow pressor angiotensin II (Ang II)-initiated hypertension: induction of sensitization by prior Ang II treatment. Hypertension 59: 459–466, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xue B, Zhang Z, Roncari CF, Guo F, Johnson AK. Aldosterone acting through the central nervous system sensitizes angiotensin II-induced hypertension. Hypertension 60: 1023–1030, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci 24: 1071–1089, 2001. [DOI] [PubMed] [Google Scholar]