

Abstract
Skeletal muscle microvascular blood flow (MBF) increases in response to physiological hyperinsulinemia. This vascular action of insulin may facilitate glucose uptake. We hypothesized that epoxyeicosatrienoic acids (EETs), a family of arachadonic, acid-derived, endothelium-derived hyperpolarizing factors, are mediators of insulin's microvascular effects. Contrast-enhanced ultrasound (CEU) was performed to quantify skeletal muscle capillary blood volume (CBV) and MBF in wild-type and obese insulin-resistant (db/db) mice after administration of vehicle or trans-4-[4-(3-adamantan-1-ylureido)cyclohexyloxy]benzoic acid (t-AUCB), an inhibitor of soluble epoxide hydrolase that converts EETs to less active dihydroxyeicosatrienoic acids. Similar studies were performed in rats pretreated with l-NAME. CEU was also performed in rats undergoing a euglycemic hyperinsulinemic clamp, half of which were pretreated with the epoxygenase inhibitor MS-PPOH to inhibit EET synthesis. In both wild-type and db/db mice, intravenous t-AUCB produced an increase in CBV (65–100% increase at 30 min, P < 0.05) and in MBF. In db/db mice, t-AUCB also reduced plasma glucose by ∼15%. In rats pretreated with l-NAME, t-AUCB after produced a significant ≈20% increase in CBV, indicating a component of vascular response independent of nitric oxide (NO) production. Hyperinsulinemic clamp produced a time-dependent increase in MBF (19 ± 36 and 76 ± 49% at 90 min, P = 0.026) that was mediated in part by an increase in CBV. Insulin-mediated changes in both CBV and MBF during the clamp were blocked entirely by MS-PPOH. We conclude that EETs are a mediator of insulin-mediated augmentation in skeletal muscle perfusion and are involved in regulating changes in CBV during hyperinsulinemia.
Keywords: epoxyeicosatrienoic acids, contrast ultrasound, insulin, muscle blood flow
acute increases in plasma insulin within and above the normal physiological range produce a dose-dependent increase limb skeletal muscle blood flow (4, 5, 15, 40). This vascular action of insulin is thought to potentiate delivery of glucose to muscle. Techniques that can quantify not only microvascular blood flow (MBF) but also capillary blood volume (CBV) in vivo have demonstrated that capillary recruitment is an important component of microvascular response to insulin (15, 21, 43). A functional increase in CBV increases effective vascular surface area for not only glucose uptake but possibly also insulin transport into tissue (34). Augmentation of skeletal muscle perfusion in response to either insulin or glucose challenge is blunted in insulin resistance (IR) (10, 13, 14, 44), suggesting that microvascular dysfunction contributes to impaired glucose homeostasis. Insulin-mediated capillary recruitment has been shown to involve production of nitric oxide (NO) (40, 41), thereby establishing a potential link between endothelial IR, which is manifested by reduced phosphorylation of endothelial NO synthase (eNOS), and impaired flow response to insulin (24). However, the contributions of other endothelial-derived vasodilators have not been evaluated in detail and may represent a new therapeutic target for treating IR.
Epoxyeicosotrianoic acids (EETs) are a family of regioisomers that are metabolites of arachadonic acid and are formed by 2C and 2J cytochrome P450 epoxygenases (28). There is evidence that EETs are endothelial-derived hyperpolarizing factors (EDHFs) and produce vasodilation directly through calcium-activated potassium channels or the TRPV4 channel and indirectly through NO production (16, 17, 23, 33). This knowledge has led to the development of inhibitors of soluble epoxide hydrolase (sEH), an enzyme responsible for breakdown of EETs to less active dihydroxyeicosatrienoic acids, for a variety of purposes, including as antihypertensive therapy (12, 20). It has been shown that pharmacological interventions that increase EETs also improve insulin-mediated glucose uptake (22, 29, 38). Whether improved insulin-mediated glucose storage is related to the microvascular effects of EETs is untested. We hypothesized that EETs participate in regulating skeletal muscle perfusion and specifically in insulin-mediated augmentation in MBF. To test these hypotheses, we used contrast-enhanced ultrasound (CEU) perfusion imaging of the skeletal muscle microcirculation after inhibition of sEH and during euglycemic hyperinsulinemia after inhibiting production of EETs.
METHODS
Animals.
The study protocol was approved by the Animal Care and Use Committee of the Oregon Health and Science University. We studied 21 male Sprague-Dawley rats weighing 230–270 g (Hilltop Laboratory Animals, Scottdale, PA), 14 wild-type C57Bl/6 mice, and eight insulin-resistant obese db/db mice genetically deficient for the leptin receptor (B6.Cq-m+/+ Leprdb/J; The Jackson Laboratory, Bar Harbor, ME) aged 8–13 wk. Animals were housed with a 12:12-h light-dark cycle and provided with food and water ad libitum. Rats undergoing hyperinsulinemic clamp were studied in the fasting condition.
Animal preparation.
Animals were anesthetized with inhaled isoflurane (1.0–1.5% for mice, 1.5–2.0% for rats), and euthermia was maintained by a heating pad and lamp. In mice, a jugular vein was cannulated for administration of microbubbles and drugs, and a 1.4 French micromanometer-tipped catheter (SPR-671; Millar Instruments) was placed in the right carotid artery for blood pressure measurement in all but three mice. For rats undergoing euglycemic hyperinsulinemic clamp, catheters were placed in the carotid artery for blood sampling and in a jugular vein and the right femoral vein for intravenous infusion of microbubbles, glucose, and insulin. An ultrasonic flow probe (T106; Transonics) was placed on the exposed right femoral artery. In select animals, the micromanometer catheter was placed in the left carotid artery for pressure measurement.
Experimental protocols.
Protocol 1 was designed to characterize changes in perfusion that occur with pharmacological increase in EETs in wild-type and db/db mice. CBV and MBF in the proximal hindlimb skeletal muscle were measured by CEU at baseline and at 15-min intervals for 45 min after administration of the sEH-inhibitor trans-4-[4-(3-adamantan-1-ylureido)cyclohexyloxy]benzoic acid (t-AUCB; 1 mg/kg iv) (provided as a gift from Dr. Bruce Hammock, University of California Davis) in seven wild-type and eight db/db mice or after administration of vehicle [1% dimethylsulfoxide (DMSO) in saline] in six wild-type mice. The dose of t-AUCB was chosen based on pharmacokinetic optimization studies showing that this dose results in plasma concentrations of >100 nM sustained over 1 h without production of hypotension (26). Heart rate (HR), blood pressure (BP), and venous glucose concentration were measured at each interval.
Protocol 2 was designed to characterize changes in perfusion that occur with the pharmacological increase in EETs independent of secondary NO production. In 10 rats, CBV and MBF were measured by hindlimb skeletal muscle CEU at baseline and then 30 min after inhibition of NOS with l-NG-nitroarginine methyl ester (l-NAME; 3 mg/kg, then 50 μg·kg−1·min−1 iv). CEU was then repeated at 30-min intervals for 2 h after administration of the sEH-inhibitor t-AUCB (1 mg/kg iv) or sham controls (n = 5 for each). In an additional four rats, HR and BP were measured continuously after administration of l-NAME and t-AUCB.
Protocol 3 was designed to test the role of EETs in insulin-mediated capillary recruitment. In 11 rats, baseline CEU and femoral artery blood flow measurement and analysis of arterial blood plasma insulin concentration by radioimmunoassay were performed 1 h after surgical preparation to allow steady-state conditions. A euglycemic hyperinsulinemic clamp was then performed by administration of insulin (10 mU·min−1·kg−1) for 90 min. Arterial blood glucose was measured at baseline and at 10-min intervals for the first hour of the clamp and then every 15 min for the remaining hour. Dextrose (30%) was infused at a variable rate to maintain glucose concentration at fasting basal levels. Total body glucose utilization was determined from the dextrose infusion rate required to maintain euglycemia and was expressed normalized to body weight. Hindlimb skeletal muscle CEU, femoral blood flow recording, and arterial plasma insulin measurement were performed at 30 and 90 min after the initiation of the hyperinsulinemic clamp. Upon completion of the clamp, hindlimb muscle samples were obtained for measurement of capillary density by immunohistochemistry. In six of the rats undergoing hyperinsulinemic clamp, EET synthesis was inhibited by N-methylsulfonyl-2-(2-propynyloxy)-benzenehexanamide (MS-PPOH), which was administered by intraperitoneal placement of an osmotic pump (2001D; Durect, Cupertino, CA) 24 h before the clamp. The pump and concentration were designed for a MS-PPOH release rate of 0.21 mg/h, which was similar to doses used to block beneficial effects of sEH inhibitors on rat muscle perfusion (30).
Contrast-enhanced ultrasound.
Lipid-shelled decafluorobutane microbubbles were prepared by sonication of a gas-saturated aqueous suspension of distearoylphosphatidylcholine (2 mg/ml) and polyoxyethylene-40-stearate (1 mg/ml). Microbubble concentration and size distribution were measured by electrozone sensing (Multisizer III; Beckman Coulter, Fullerton, CA). The average diameter of these microbubbles was 1.8–2.0 μm. CEU was performed in the transaxial plane using a linear-array transducer interfaced with an ultrasound imaging system (15L8 transducer, Sequoia 512; Siemens Medical Systems, Mountain View, CA). A contrast-specific multipulse algorithm was used at a transmit frequency of 7 MHz, a mechanical index of 0.18, and a 55-dB dynamic range. Gains were optimized at the beginning of each study to levels that just eliminated background tissue speckle and were kept constant. Blood pool signal (IB) was measured first from the left ventricular cavity at end diastole during an intravenous microbubble infusion rate of 1 × 106 min for mice or 1 × 107 min for rats. The infusion rate was then increased 10-fold, and the proximal hindlimb adductor muscles (adductor magnus and semimembranosus) were imaged midway between the inguinal fold and the knee. Images were acquired at a frame rate of 2 Hz immediately after a brief high-power (MI 1.9) destructive pulse sequence, and time intensity data were fit to the function y = A(1 − e−βt), where y is intensity at time t, A is the plateau intensity, and the rate constant β represents the microvascular flux rate (15, 45). Skeletal muscle CBV was quantified by scaled comparison of plateau intensity to blood pool and calculated by A/(1.06 × IB × F × C), where 1.06 is tissue density (g/cm3), F is the scaling factor that corrected for the different infusion rate for measuring IB to avoid dynamic range saturation, and C is a coefficient to correct for sternal attenuation measured a priori (1.1 for mice, 1.2 for rats) (9). MBF was quantified by the product of CBV and β (15, 45).
Capillary density.
Immunohistochemistry was performed on fixed, paraffin-embedded sections of hindlimb skeletal muscle. For endothelial cell staining, biotinylated griffonia simplicifolia (Vector Laboratories) was used with diaminobenzidine secondary staining. At least 15 random optical fields for each animal were analyzed, blinded to animal identity. Capillary density was determined in transverse muscle sections. Data were expressed as a percentage of the total muscle area.
Statistical analysis.
Comparisons were made by repeated-measures ANOVA for time-dependent data. Post hoc comparisons were made with Bonferroni's corrections for multiple comparisons using either paired Student's t-tests for differences within a subject or non-paired Student's t-tests for comparisons between cohorts. Nonnormally distributed data were compared using either Mann-Whitney or Kruskall-Wallis tests. Changes in blood pressure and heart rate after l-NAME were made using a paired Student's t-test. Correlations were made using linear regression analysis and Pearson product moment. Tests for linear trends for ordinal data were made by a Spearman's correlation coefficient. Data were considered significant at P < 0.05 (two sided).
RESULTS
Functional capillary recruitment mediated by EETs.
In protocol 1, there were no significant changes in either HR or BP after administration of the sEH inhibitor t-AUCB in either wild-type or db/db mice (age 8–13 wk) (Table 1). Baseline venous glucose concentration was on average threefold higher (P < 0.01) for db/db compared with wild-type mice (Table 1). In db/db mice, treatment with t-AUCB resulted in a progressive reduction in venous glucose over 45 min (P = 0.025 for linear trend). On CEU imaging in wild-type mice, t-AUCB produced a rapid increase in CBV and MBF, whereas perfusion did not change in vehicle-treated controls (Figure 1, A and B). Treatment with t-AUCB had no significant effect on microvascular flux rate (β) in wild-type mice. In db/db mice, t-AUCB also produced a significant increase in skeletal muscle CBV and MBF (Fig. 1C). The time to effect tended to be slightly delayed for db/db compared with wild-type mice. In the db/db strain, mouse age was inversely related to baseline MBF (Fig. 1D). However, the peak MBF and CBV achieved after administration of t-AUCB was similar irrespective of age. There was also an anticipated linear relation between age and body mass in db/db mice (range 40–55 g, r2 = 0.74, P = 0.003).
Table 1.
BP, HR, and venous glucose in mice treated with t-AUCB
| Baseline | 15 Min | 30 Min | 45 Min | |
|---|---|---|---|---|
| Wild type | ||||
| Heart rate, beats/min | 390 ± 100 | 421 ± 114 | 425 ± 113 | 417 ± 100 |
| Blood pressure, mmHg | 91 ± 13 | 94 ± 9 | 90 ± 15 | 89 ± 15 |
| Venous glucose, mg/dl | 146 ± 31 | 153 ± 38 | 163 ± 56 | 150 ± 33 |
| db/db | ||||
| HR, beats/min | 472 ± 48 | 490 ± 42 | 509 ± 58 | 521 ± 64 |
| BP, mmHg | 108 ± 14 | 107 ± 18 | 108 ± 17 | 109 ± 19 |
| Venous glucose, mg/dl* | 411 ± 102 | 385 ± 94 | 361 ± 117 | 348 ± 117 |
Values are means ± SE. BP, blood pressure; HR, heart rate; t-AUCB trans-4-[4-(3-adamantan-1-ylureido)cyclohexyloxy]benzoic acid.
P < 0.01 vs. wild type at baseline.
Fig. 1.

A–C: mean (± SE) skeletal muscle capillary blood volume (CBV) in wild-type mice (A), microvascular blood flow (MBF) in wild-type mice (B), and both CBV and MBF in db/db mice (C) at baseline and after administration of trans-4-[4-(3-adamantan-1-ylureido)cyclohexyloxy]benzoic acid (t-AUCB; 1 mg/kg iv) or vehicle (wild-type mice only). *P < 0.05 vs vehicle; †P < 0.05 vs. baseline. D: relation between db/db mouse age and MBF either at baseline or for peak value after administration of t-AUCB. Baseline: y = −9.5X + 126, r2 = 0.71, P = 0.009. t-AUCB: y = −0.2X + 82, r2 < 0.01, P = 0.95.
Direct vs. NO-mediated effects of EETs.
In sEH studies performed in rats pretreated with l-NAME, hemodynamic measurements indicated that l-NAME produced an increase in systolic and diastolic BP and a decrease in HR (Table 2). Administration of t-AUCB 30 min after l-NAME produced only a mild, gradual decrease in blood pressure that was not statistically significant. On CEU imaging, pretreatment of rats with l-NAME produced a mild reduction in CBV (Fig. 2, top). However, MBF did not change because of a concomitant increase in microvascular flux rate (Fig. 2, bottom). Administration of t-AUCB 30 min after l-NAME produced a significant (P < 0.05) increase in CBV that did not occur for l-NAME-treated rats not treated with t-AUCB. However, there were no major changes in MBF between the two groups.
Table 2.
HR and BP in rats treated with l-NAME and t-AUCB
| Baseline | 30 Min | 60 Min | 90 Min | 120 Min | |
|---|---|---|---|---|---|
| Heart rate, beats/min | 381 ± 56 | 347 ± 62* | 329 ± 66 | 314 ± 67 | 309 ± 52 |
| Systolic BP, mmHg | 101 ± 8 | 110 ± 11* | 109 ± 6 | 109 ± 9 | 106 ± 7 |
| Diastolic BP, mmHg | 72 ± 8 | 84 ± 11* | 80 ± 7 | 79 ± 9 | 78 ± 8 |
Values are means ± SE. l-NAME, l-NG-nitroarginine methyl ester.
P < 0.05 vs. baseline.
Fig. 2.
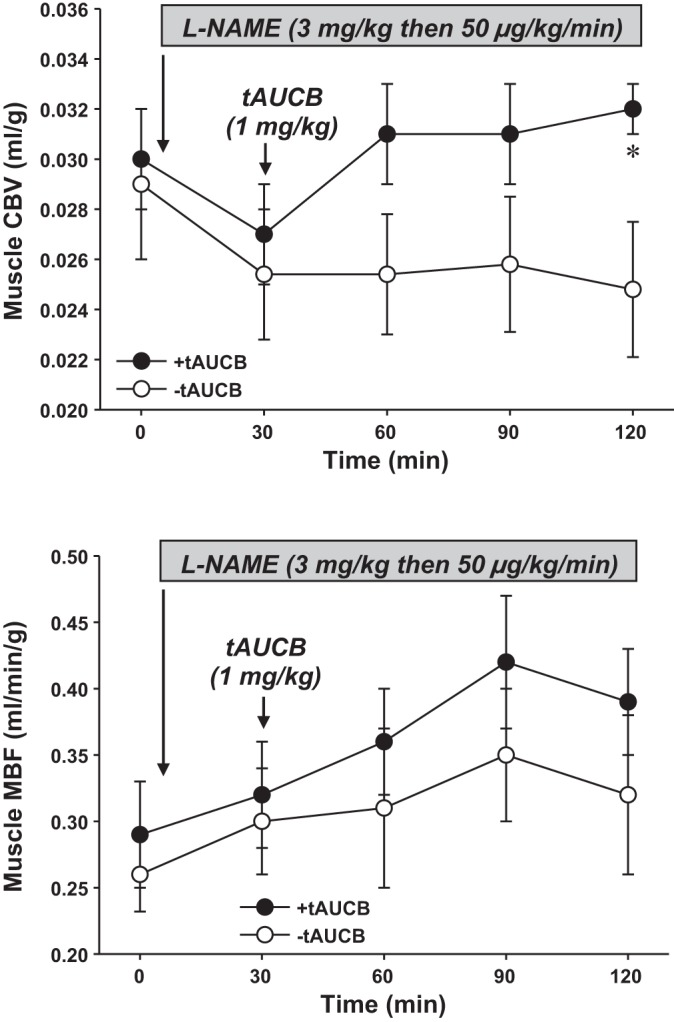
Mean (± SE) skeletal muscle CBV (top) and MBF (bottom) in rats at baseline (t0), after initiation of l-NG-nitroarginine methyl ester (l-NAME), and after administration of t-AUCB (1 mg/kg iv) or sham controls (−t-AUCB) immediately after the t30 study point. *P < 0.05 vs. t30 and vs. −t-AUCB.
EETs in insulin-mediated microvascular response.
In protocol 3, there were no significant differences in basal insulin concentration, arterial or venous glucose concentration, femoral artery blood flow, or limb glucose uptake between control rats and those treated with the EET-inhibitor MS-PPOH (Table 3). Euglycemic hyperinsulinemic clamp produced significant increases in plasma insulin concentration in both groups, although there was a trend toward higher insulin concentrations in the MS-PPOH-treated group at both 30 and 90 min after insulin infusion. The increase in femoral artery blood flow with stable arterial and venous glucose concentrations during hyperinsulinemia indicated that limb glucose uptake increased in both groups. The glucose infusion rate required to maintain euglycemia during the clamp was used as an indicator of total body glucose uptake and was similar between groups (Table 3 and Fig. 3). The glucose infusion rate normalized to plasma insulin concentration, which serves as a measure of insulin sensitivity, was less for MS-PPOH-treated mice, but this difference did not reach statistical significance after correction for multiple comparisons.
Table 3.
Insulin clamp data for untreated and MS-PPOH-treated rats
| Insulin Clamp (n = 6) |
Insulin Clamp + MS-PPOH (n = 5) |
|||||
|---|---|---|---|---|---|---|
| Baseline | 30 min | 90 min | Baseline | 30 min | 90 min | |
| Insulin, ng/ml | 1.27 ± 0.40 | 4.38 ± 2.35† | 5.61 ± 1.64† | 1.21 ± 0.67 | 5.35 ± 1.09† | 6.28 ± 2.34† |
| Arterial glucose, mg/dl | 197 ± 19 | 198 ± 10 | 226 ± 37 | 192 ± 36 | 213 ± 59 | 202 ± 32 |
| Venous glucose, mg/dl | 192 ± 16 | 187 ± 12 | 220 ± 38 | 180 ± 33 | 194 ± 55 | 186 ± 27 |
| Arterial-venous blood glucose, mg/dl | 6 ± 3 | 10 ± 9 | 13 ± 10 | 13 ± 8 | 20 ± 11 | 16 ± 6 |
| GIR, mg·min−1·kg−1 | 0 | 2.4 ± 0.3 | 2.5 ± 0.2 | 0 | 2.2 ± 0.2 | 2.6 ± 0.2 |
| GIR/insulin | 0 | 0.97 ± 1.16 | 0.50 ± 0.19 | 0 | 0.39 ± 0.11 | 0.42 ± 0.12 |
| Heart rate, beats/min | 401 ± 40 | 399 ± 29 | 408 ± 31 | 389 ± 33 | 429 ± 32 | 452 ± 14*† |
| Femoral artery blood flow, ml/min | 0.74 ± 0.11 | 0.85 ± 0.13† | 0.99 ± 0.20† | 0.72 ± 0.06 | 0.95 ± 0.31† | 0.98 ± 0.33† |
Values are means ± SE. MS-PPOH, N-methylsulfonyl-2-(2-propynyloxy)-benzenehexanamide; GIR, glucose infusion rate; GIR/insulin, glucose infusion rate normalized to insulin concentration (103/kg).
P < 0.05 vs. insulin clamp group;
P < 0.05 compared with baseline.
Fig. 3.
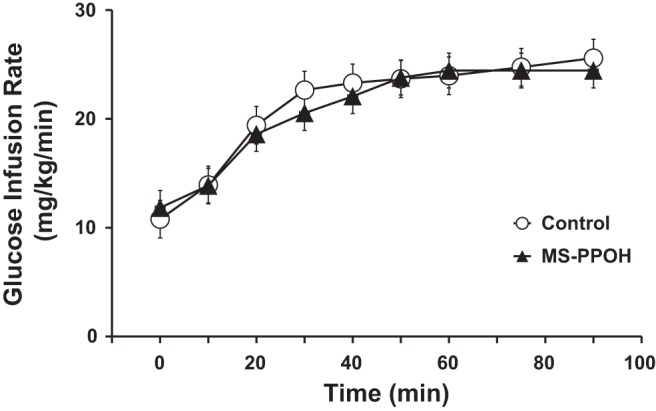
Glucose infusion rate required in rats to maintain euglycemia during the hyperinsulinemic clamp (10 mU·min−1·kg−1). MS-PPOH, N-methylsulfonyl-2-(2-propynyloxy)-benzenehexanamide.
Examples of CEU images and time intensity data at baseline and after 90 min of euglycemic hyperinsulinemia for a control rat and an MS-PPOH-treated rat are shown in Fig. 4. In the control rat (Fig. 4A), an increase in MBF with insulin is manifested by both an increase in the microvascular flux rate (β or rate constant of the exponential function) and an increase in the plateau intensity that when normalized to blood pool represents CBV. For the MS-PPOH-treated rat, insulin did not produce any changes in either the rate constant or plateau intensity. When all animals were analyzed, hyperinsulinemic clamp in control rats produced a small but significant increase in CBV but an even greater, time-dependent increase in MBF (25 ± 31% increase at 30 min, 91 ± 40% increase at 90 min; Fig. 5). In MS-PPOH-treated rats, CBV and MBF were unchanged during the hyperinsulinemic clamp and were not different from saline-treated control rats. Capillary density measured by immunohistochemistry was not significantly different between the groups undergoing the clamp (156 ± 20, 169 ± 30, and 172 ± 43 mm−2 for control, MS-PPOH-treated, and DMSO-treated rats, respectively).
Fig. 4.

Examples of background-subtracted, color-coded, contrast-enhanced ultrasound (CEU) images from rat hindlimb skeletal muscle and corresponding time (pulsing interval) vs. video intensity curves at baseline and after 90 min of euglycemic hyperinsulinemia in a control rat (A) and a MS-PPOH-treated rat (B). See text for details.
Fig. 5.

Mean (± SE) skeletal muscle CBV (A) and MBF (B) in rats undergoing euglycemic hyperinsulinemic clamp (10 mU·min−1·kg−1) or control rats treated with saline. Clamp data are shown for rats with and without MS-PPOH pretreatment. Mean (± SE) %change in CBV (C) and MBF (D) compared with baseline in rats undergoing hyperinsulinemic clamp or saline infusion. *P < 0.05 vs saline; †P < 0.05 vs. insulin without MS-PPOH.
DISCUSSION
The overall aim of this study was to examine the role of EETs in the metabolic regulation of skeletal muscle perfusion. Our studies indicate that treatment with sEH inhibitors produces an increase in skeletal muscle CBV and MBF that is in part independent of NO production. We have also demonstrated that EETs play a role in insulin-mediated augmentation of MBF in skeletal muscle.
There is a growing foundation of evidence supporting the notion that insulin's microvascular effects are permissive for facilitating glucose uptake (5, 15, 40, 42, 43). Animal and human studies have also demonstrated an association between impaired CBV response to insulin and impaired glucose homeostasis, thereby suggesting that impaired vascular responses contribute to IR (13, 14, 39, 44). It has been shown that abnormal capillary responses occur very early in the course of IR in nonhuman primates produced by inactivity and in humans at the stage of obesity and mild to moderate IR (10, 14). These observations have helped to understand how tissue perfusion influences glucose regulation, but they have not yet been translated into new therapies for IR or diabetes mellitus.
Previous studies evaluating the vascular actions of insulin have implicated NO-dependent pathways (11, 35, 40). Insulin at physiological concentrations (100–500 pM) promotes NO production through insulin receptor phosphatidylinositol 3-kinase/Akt signaling of eNOS (32, 42, 47). Skeletal muscle CEU and other nonimaging techniques for assessing the status of the peripheral microcirculation have demonstrated that inhibition of eNOS with l-NAME blunts insulin-mediated augmentation in effective CBV (41, 42). There is also evidence that NO may be exerting an effect through a central nervous system mechanism (6). However, in many studies, inhibition of eNOS does not entirely eliminate insulin-mediated arteriolar vasodilation, suggesting the presence of other mechanisms that can augment glucose uptake through vascular recruitment (6).
In this study, we focused on EETs, which are metabolites of arachadonic acid formed by cytochrome P450 epoxygenases of the 2C and 2J subclasses. These compounds are metabolized to less active dihydroxy eicosanoids by sEH (20, 28, 33). There is strong evidence that, among their varied biochemical effects, EETs are endothelial-derived hyperpolarizing factors and produce vasodilation in many organs, including heart, brain, and skeletal muscle either directly or indirectly through NO (2, 16, 17, 23, 25, 33). Recently, it has been shown that interventions that increase EETs improve insulin sensitivity. In obese and IR hemeoxygenase-deficient mice, all EET isomers were noted to be severely reduced, and inhibition of sEH improved both BP and glucose disposal during insulin challenge (38). In rat and murine models of diet-induced IR, treatment with an sEH inhibitor improved glucose storage after insulin administration or after glucose challenge (1, 22). There have been several other studies confirming that either sEH inhibition or gene-targeted therapies that increase EETs (either CYP-2J3 gene therapy or sEH deletion) lead to improvements in insulin sensitivity (27, 29, 46). Several explanations have been offered for these effects. EETs can increase pancreatic β-cell production of insulin (18, 27), and they have been shown to potentiate insulin's suppressive actions on hepatic gluconeogenesis (37). However, these observations do not explain why peripheral insulin sensitivity is improved by an increase in plasma EETs.
Our results suggest that EETs mediate insulin-mediated capillary recruitment and augmentation of MBF in skeletal muscle. The sEH inhibitor t-AUCB, which selectively increases EETs (36), was used in doses that produce blood levels that have been demonstrated to produce vasodilation (7) and improve fasting blood glucose levels in diabetic mice (49). Using CEU, we showed that t-AUCB produced an increase in muscle CBV and MBF in both wild-type and db/db mice. Rat studies were used to evaluate the effect of t-AUCB in the presence of the eNOS inhibitor l-NAME since hemodynamic effects of l-NAME tend to be more stable in rats than mice and because rats were used for euglycemic hyperinsulinemic clamp studies. In these experiments, t-AUCB still increased CBV after pretreatment with l-NAME. However, MBF was not different between animals treated with l-NAME alone and l-NAME and t-AUCB. When placed in context with the mouse studies, these findings suggest NO-dependent and independent components of the microvascular response to t-AUCB, which is consistent with previous findings that mesenteric arterial dilation to 11,12-EET and 14,15-EET or to sEH inhibitors is only partially inhibited by l-NAME (31, 48). Studies were performed with l-NAME and not in eNOS-deficient mice since these mice have been shown to have a compensatory upregulation of EDHF vasoregulatory response that may have overestimated the vascular response to t-AUCB (19).
Our finding that t-AUCB increased CBV and MBF in obese db/db mice is important since previous studies have suggested that the vasodilatory effects of EETs could be blunted in states of IR (48). Although basal CBV and MBF were substantially lower in db/db than in wild-type mice, the peak CBV and MBF achieved after inhibiting soluble epoxide hydrolase were nearly equal between strains. We chose to study db/db mice over a modest range of age to study a spectrum of mild to moderate IR. There was an inverse relationship between age and either CBV or MBF; however, the peak flow response to t-AUCB was not influenced by age. The increase in muscle perfusion with t-AUCB in db/db mice was also associated with a decreased in plasma glucose, although we cannot necessarily infer that the two were causatively linked.
In our insulin clamp experiments, the selective epoxygenase inhibitor MS-PPOH prevented insulin-mediated increases in CBV and MBF. These data strongly suggest that epoxygenase metabolites that include EETs are involved in insulin-mediated microvascular recruitment in muscle. Although the t-AUCB studies suggested that EETs can mediate flow augmentation, the finding that preinsulin clamp perfusion was not affected by MS-PPOH suggests that EETs may not play a major role in basal control of skeletal muscle perfusion.
There are several limitations of the study that should be mentioned. Although capillary recruitment and MBF augmentation during hyperinsulinemia were prevented by MS-PPOH, measurements of glucose homeostasis were not significantly different between groups. The ratio of glucose infusion to insulin concentration was used as a measurement of IR and was only mildly reduced in the MS-PPOH group. This finding does not necessarily imply that augmentation of muscle perfusion with sEH therapy in IR patients would not be helpful for improving glucose storage, and further studies will be needed to determine whether this is the case. We also have not evaluated the dose response for the sEH inhibitor and instead used concentrations that have been found to have vasodilatory effects. Finally, we did not perform specific assays for EETs and their metabolites because these studies have been performed previously and because of concern that the amount of blood volume required would alter hemodynamics. Finally, although our resting MBF values in mice and rats were similar to previous studies that used microspheres (3, 8), these flows are substantially higher than in humans, which limits extrapolation to human skeletal muscle.
In conclusion, our data suggest that EETs have direct effects on augmenting skeletal muscle perfusion and CBV and that this effect of EETs is an important contributor to insulin's microvascular effects. These findings are important for understanding how drugs that modulate EET metabolism may influence glucose homeostasis through substrate delivery.
GRANTS
J. R. Lindner was supported by National Institutes of Health (NIH) Grants R01-DK-063508, R01-HL-078610, and RC1-HL-100659, and S. Chadderdon was supported by NIH Grant KL2-TR-000152. B. P. Davidson was supported by a Clinical Research Program award (12CRP11890055) from the American Heart Association.
DISCLOSURES
There are no pertinent disclosures for any of the authors.
AUTHOR CONTRIBUTIONS
C.Y.S., S.C., A.X., N.A., B.P.D., and J.R.L. conception and design of research; C.Y.S., S.K., S.C., M.W., Y.Q., A.X., B.P.D., and J.R.L. performed experiments; C.Y.S., S.K., S.C., M.W., Y.Q., A.X., B.P.D., and J.R.L. analyzed data; C.Y.S., S.K., S.C., M.W., Y.Q., A.X., and J.R.L. interpreted results of experiments; C.Y.S., S.K., M.W., and J.R.L. prepared figures; C.Y.S., A.X., and J.R.L. drafted manuscript; C.Y.S., S.K., S.C., M.W., Y.Q., A.X., N.A., B.P.D., and J.R.L. approved final version of manuscript; S.C., N.A., and J.R.L. edited and revised manuscript.
REFERENCES
- 1.Anandan SK, Webb HK, Chen D, Wang YX, Aavula BR, Cases S, Cheng Y, Do ZN, Mehra U, Tran V, Vincelette J, Waszczuk J, White K, Wong KR, Zhang LN, Jones PD, Hammock BD, Patel DV, Whitcomb R, MacIntyre DE, Sabry J, Gless R. 1-(1-acetyl-piperidin-4-yl)-3-adamantan-1-yl-urea (AR9281) as a potent, selective, and orally available soluble epoxide hydrolase inhibitor with efficacy in rodent models of hypertension and dysglycemia. Bioorg Med Chem Lett 21: 983–988, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Archer SL, Gragasin FS, Wu X, Wang S, McMurtry S, Kim DH, Platonov M, Koshal A, Hashimoto K, Campbell WB, Falck JR, Michelakis ED. Endothelium-derived hyperpolarizing factor in human internal mammary artery is 11,12-epoxyeicosatrienoic acid and causes relaxation by activating smooth muscle BK(Ca) channels. Circulation 107: 769–776, 2003. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong RB, Laughlin MH. Blood flows within and among rat muscles as a function of time during high speed treadmill exercise. J Physiol 344: 189–208, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baron AD. Hemodynamic actions of insulin. Am J Physiol Endocrinol Metab 267: E187–E202, 1994. [DOI] [PubMed] [Google Scholar]
- 5.Bonadonna RC, Saccomani MP, Del Prato S, Bonora E, DeFronzo RA, Cobelli C. Role of tissue-specific blood flow and tissue recruitment in insulin-mediated glucose uptake of human skeletal muscle. Circulation 98: 234–241, 1998. [DOI] [PubMed] [Google Scholar]
- 6.Bradley EA, Willson KJ, Choi-Lundberg D, Clark MG, Rattigan S. Effects of central administration of insulin or l-NMMA on rat skeletal muscle microvascular perfusion. Diabetes Obes Metab 12: 900–908, 2010. [DOI] [PubMed] [Google Scholar]
- 7.Bukhari IA, Gauthier KM, Jagadeesh SG, Sangras B, Falck JR, Campbell WB. 14,15-Dihydroxy-eicosa-5(Z)-enoic acid selectively inhibits 14,15-epoxyeicosatrienoic acid-induced relaxations in bovine coronary arteries. J Pharmacol Exp Ther 336: 47–55, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cardinal TR, Hoying JB. A modified fluorescent microsphere-based approach for determining resting and hyperemic blood flows in individual murine skeletal muscles. Vasc Pharmacol 47: 48–56, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carr CL, Qi Y, Davidson B, Chadderdon S, Jayaweera AR, Belcik JT, Benner C, Xie A, Lindner JR. Dysregulated selectin expression and monocyte recruitment during ischemia-related vascular remodeling in diabetes mellitus. Arterioscler Thromb Vasc Biol 31: 2526–2533, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chadderdon SM, Belcik JT, Smith E, Pranger L, Kievit P, Grove KL, Lindner JR. Activity restriction, impaired capillary function, and the development of insulin resistance in lean primates. Am J Physiol Endocrinol Metab 303: E607–E613, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen YL, Messina EJ. Dilation of isolated skeletal muscle arterioles by insulin is endothelium dependent and nitric oxide mediated. Am J Physiol Heart Circ Physiol 270: H2120–H2124, 1996. [DOI] [PubMed] [Google Scholar]
- 12.Chiamvimonvat N, Ho CM, Tsai HJ, Hammock BD. The soluble epoxide hydrolase as a pharmaceutical target for hypertension. J Cardiovasc Pharmacol 50: 225–237, 2007. [DOI] [PubMed] [Google Scholar]
- 13.Clerk LH, Vincent MA, Barrett EJ, Lankford MF, Lindner JR. Skeletal muscle capillary responses to insulin are abnormal in late-stage diabetes and are restored by angiotensin-converting enzyme inhibition. Am J Physiol Endocrinol Metab 293: E1804–E1809, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Clerk LH, Vincent MA, Jahn LA, Liu Z, Lindner JR, Barrett EJ. Obesity blunts insulin-mediated microvascular recruitment in human forearm muscle. Diabetes 55: 1436–1442, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Dawson D, Vincent MA, Barrett EJ, Kaul S, Clark A, Leong-Poi H, Lindner JR. Vascular recruitment in skeletal muscle during exercise and hyperinsulinemia assessed by contrast ultrasound. Am J Physiol Endocrinol Metab 282: E714–E720, 2002. [DOI] [PubMed] [Google Scholar]
- 16.Dimitropoulou C, West L, Field MB, White RE, Reddy LM, Falck JR, Imig JD. Protein phosphatase 2A and Ca2+-activated K+ channels contribute to 11,12-epoxyeicosatrienoic acid analog mediated mesenteric arterial relaxation. Prostaglandins Other Lipid Mediat 83: 50–61, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Earley S, Heppner TJ, Nelson MT, Brayden JE. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ Res 97: 1270–1279, 2005. [DOI] [PubMed] [Google Scholar]
- 18.Falck JR, Manna S, Moltz J, Chacos N, Capdevila J. Epoxyeicosatrienoic acids stimulate glucagon and insulin release from isolated rat pancreatic islets. Biochem Biophys Res Commun 114: 743–749, 1983. [DOI] [PubMed] [Google Scholar]
- 19.Huang A, Sun D, Smith CJ, Connetta JA, Shesely EG, Koller A, Kaley G. In eNOS knockout mice skeletal muscle arteriolar dilation to acetylcholine is mediated by EDHF. Am J Physiol Heart Circ Physiol 278: H762–H768, 2000. [DOI] [PubMed] [Google Scholar]
- 20.Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov 8: 794–805, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inyard AC, Clerk LH, Vincent MA, Barrett EJ. Contraction stimulates nitric oxide independent microvascular recruitment and increases muscle insulin uptake. Diabetes 56: 2194–2200, 2007. [DOI] [PubMed] [Google Scholar]
- 22.Iyer A, Kauter K, Alam MA, Hwang SH, Morisseau C, Hammock BD, Brown L. Pharmacological inhibition of soluble epoxide hydrolase ameliorates diet-induced metabolic syndrome in rats. Exp Diabetes Res 2012: 758614, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katakam PV, Hoenig M, Ujhelyi MR, Miller AW. Cytochrome P450 activity and endothelial dysfunction in insulin resistance. J Vasc Res 37: 426–434, 2000. [DOI] [PubMed] [Google Scholar]
- 24.Kim F, Pham M, Maloney E, Rizzo NO, Morton GJ, Wisse BE, Kirk EA, Chait A, Schwartz MW. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler Thromb Vasc Biol 28: 1982–1988, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li PL, Zhang DX, Ge ZD, Campbell WB. Role of ADP-ribose in 11,12-EET-induced activation of K(Ca) channels in coronary arterial smooth muscle cells. Am J Physiol Heart Circ Physiol 282: H1229–H1236, 2002. [DOI] [PubMed] [Google Scholar]
- 26.Liu JY, Tsai HJ, Hwang SH, Jones PD, Morisseau C, Hammock BD. Pharmacokinetic optimization of four soluble epoxide hydrolase inhibitors for use in a murine model of inflammation. Br J Pharmacol 156: 284–296, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo P, Chang HH, Zhou Y, Zhang S, Hwang SH, Morisseau C, Wang CY, Inscho EW, Hammock BD, Wang MH. Inhibition or deletion of soluble epoxide hydrolase prevents hyperglycemia, promotes insulin secretion, and reduces islet apoptosis. J Pharmacol Exp Ther 334: 430–438, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo P, Wang MH. Eicosanoids, beta-cell function, and diabetes. Prostaglandins Other Lipid Mediat 95: 1–10, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luria A, Bettaieb A, Xi Y, Shieh GJ, Liu HC, Inoue H, Tsai HJ, Imig JD, Haj FG, Hammock BD. Soluble epoxide hydrolase deficiency alters pancreatic islet size and improves glucose homeostasis in a model of insulin resistance. Proc Natl Acad Sci USA 108: 9038–9043, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Merabet N, Bellien J, Glevarec E, Nicol L, Lucas D, Remy-Jouet I, Bounoure F, Dreano Y, Wecker D, Thuillez C, Mulder P. Soluble epoxide hydrolase inhibition improves myocardial perfusion and function in experimental heart failure. J Mol Cell Cardiol 52: 660–666, 2012. [DOI] [PubMed] [Google Scholar]
- 31.Miller AW, Dimitropoulou C, Han G, White RE, Busija DW, Carrier GO. Epoxyeicosatrienoic acid-induced relaxation is impaired in insulin resistance. Am J Physiol Heart Circ Physiol 281: H1524–H1531, 2001. [DOI] [PubMed] [Google Scholar]
- 32.Muniyappa R, Montagnani M, Koh KK, Quon MJ. Cardiovascular actions of insulin. Endocr Rev 28: 463–491, 2007. [DOI] [PubMed] [Google Scholar]
- 33.Mustafa S, Sharma V, McNeill JH. Insulin resistance and endothelial dysfunction: Are epoxyeicosatrienoic acids the link? Exp Clin Cardiol 14: e41–e50, 2009. [PMC free article] [PubMed] [Google Scholar]
- 34.Rattigan S, Clark MG, Barrett EJ. Hemodynamic actions of insulin in rat skeletal muscle: evidence for capillary recruitment. Diabetes 46: 1381–1388, 1997. [DOI] [PubMed] [Google Scholar]
- 35.Scherrer U, Randin D, Vollenweider P, Vollenweider L, Nicod P. Nitric oxide release accounts for insulin's vascular effects in humans. J Clin Invest 94: 2511–2515, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shaik JS, Ahmad M, Li W, Rose ME, Foley LM, Hitchens TK, Graham SH, Hwang SH, Hammock BD, Poloyac SM. Soluble epoxide hydrolase inhibitor trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid is neuroprotective in rat model of ischemic stroke. Am J Physiol Heart Circ Physiol 305: H1605–H1613, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Skepner JE, Shelly LD, Ji C, Reidich B, Luo Y. Chronic treatment with epoxyeicosatrienoic acids modulates insulin signaling and prevents insulin resistance in hepatocytes. Prostaglandins Other Lipid Mediat 94: 3–8, 2011. [DOI] [PubMed] [Google Scholar]
- 38.Sodhi K, Inoue K, Gotlinger KH, Canestraro M, Vanella L, Kim DH, Manthati VL, Koduru SR, Falck JR, Schwartzman ML, Abraham NG. Epoxyeicosatrienoic acid agonist rescues the metabolic syndrome phenotype of HO-2-null mice. J Pharmacol Exp Ther 331: 906–916, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.St-Pierre P, Genders AJ, Keske MA, Richards SM, Rattigan S. Loss of insulin-mediated microvascular perfusion in skeletal muscle is associated with the development of insulin resistance. Diabetes Obes Metab 12: 798–805, 2010. [DOI] [PubMed] [Google Scholar]
- 40.Steinberg HO, Brechtel G, Johnson A, Fineberg N, Baron AD. Insulin-mediated skeletal muscle vasodilation is nitric oxide dependent. A novel action of insulin to increase nitric oxide release. J Clin Invest 94: 1172–1179, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vincent MA, Barrett EJ, Lindner JR, Clark MG, Rattigan S. Inhibiting NOS blocks microvascular recruitment and blunts muscle glucose uptake in response to insulin. Am J Physiol Endocrinol Metab 285: E123–E129, 2003. [DOI] [PubMed] [Google Scholar]
- 42.Vincent MA, Clerk LH, Lindner JR, Klibanov AL, Clark MG, Rattigan S, Barrett EJ. Microvascular recruitment is an early insulin effect that regulates skeletal muscle glucose uptake in vivo. Diabetes 53: 1418–1423, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Vincent MA, Dawson D, Clark AD, Lindner JR, Rattigan S, Clark MG, Barrett EJ. Skeletal muscle microvascular recruitment by physiological hyperinsulinemia precedes increases in total blood flow. Diabetes 51: 42–48, 2002. [DOI] [PubMed] [Google Scholar]
- 44.Wallis MG, Wheatley CM, Rattigan S, Barrett EJ, Clark AD, Clark MG. Insulin-mediated hemodynamic changes are impaired in muscle of Zucker obese rats. Diabetes 51: 3492–3498, 2002. [DOI] [PubMed] [Google Scholar]
- 45.Wei K, Jayaweera AR, Firoozan S, Linka A, Skyba DM, Kaul S. Quantification of myocardial blood flow with ultrasound-induced destruction of microbubbles administered as a constant venous infusion. Circulation 97: 473–483, 1998. [DOI] [PubMed] [Google Scholar]
- 46.Xu X, Zhao CX, Wang L, Tu L, Fang X, Zheng C, Edin ML, Zeldin DC, Wang DW. Increased CYP2J3 expression reduces insulin resistance in fructose-treated rats and db/db mice. Diabetes 59: 997–1005, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zeng G, Nystrom FH, Ravichandran LV, Cong LN, Kirby M, Mostowski H, Quon MJ. Roles for insulin receptor, PI3-kinase, and Akt in insulin-signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation 101: 1539–1545, 2000. [DOI] [PubMed] [Google Scholar]
- 48.Zhang LN, Vincelette J, Chen D, Gless RD, Anandan SK, Rubanyi GM, Webb HK, MacIntyre DE, Wang YX. Inhibition of soluble epoxide hydrolase attenuates endothelial dysfunction in animal models of diabetes, obesity and hypertension. Eur J Pharmacol 654: 68–74, 2011. [DOI] [PubMed] [Google Scholar]
- 49.Zuloaga KL, Krasnow SM, Zhu X, Zhang W, Jouihan SA, Shangraw RE, Alkayed NJ, Marks DL. Mechanism of protection by soluble epoxide hydrolase inhibition in type 2 diabetic stroke. PLoS One 9: e97529, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]