

Abstract
Microvascular barrier integrity is dependent on bioavailable nitric oxide (NO) produced locally by endothelial NO synthase (eNOS). Under conditions of limited substrate or cofactor availability or by enzymatic modification, eNOS may become uncoupled, producing superoxide in lieu of NO. This study was designed to investigate how eNOS-dependent superoxide production contributes to endothelial barrier dysfunction in inflammatory lung injury and its regulation. C57BL/6J mice were challenged with intratracheal LPS. Bronchoalveolar lavage fluid was analyzed for protein accumulation, and lung tissue homogenate was assayed for endothelial NOS content and function. Human lung microvascular endothelial cell (HLMVEC) monolayers were exposed to LPS in vitro, and barrier integrity and superoxide production were measured. Biopterin species were quantified, and coimmunoprecipitation (Co-IP) assays were performed to identify protein interactions with eNOS that putatively drive uncoupling. Mice exposed to LPS demonstrated eNOS-dependent increased alveolar permeability without evidence for altered canonical NO signaling. LPS-induced superoxide production and permeability in HLMVEC were inhibited by the NOS inhibitor nitro-l-arginine methyl ester, eNOS-targeted siRNA, the eNOS cofactor tetrahydrobiopterin, and superoxide dismutase. Co-IP indicated that LPS stimulated the association of eNOS with NADPH oxidase 2 (Nox2), which correlated with augmented eNOS S-glutathionylation both in vitro and in vivo. In vitro, Nox2-specific inhibition prevented LPS-induced eNOS modification and increases in both superoxide production and permeability. These data indicate that eNOS uncoupling contributes to superoxide production and barrier dysfunction in the lung microvasculature after exposure to LPS. Furthermore, the results implicate Nox2-mediated eNOS-S-glutathionylation as a mechanism underlying LPS-induced eNOS uncoupling in the lung microvasculature.
Keywords: acute respiratory distress syndrome, endothelial nitric oxide synthase uncoupling, endothelial barrier function, nitric oxide, NADPH oxidase
increasing age imparts greater risk for developing acute respiratory distress syndrome (ARDS) and is associated with worse short- and long-term outcomes (14, 19, 22, 27, 30). Both systemic and pulmonary vascular function are known to deteriorate as a consequence of age (15, 16, 29, 41). The pulmonary vascular endothelium is central to the development of ARDS, as it plays a key role in regulating inflammatory cell recruitment, maintaining alveolar barrier integrity, and regulating regional blood flow to match alveolar ventilation. Pulmonary vasomotor dysfunction has been associated with poor outcome from critical illness and specifically ARDS (10, 48), and we have demonstrated that promotion of lung capillary barrier integrity reduces regional edema accumulation and improves gas exchange in animal models of ARDS (37, 42, 49). The mechanisms underlying pulmonary endothelial barrier dysfunction and the development of novel therapeutics targeting the endothelium have been active areas of investigation for years without significant impact on treatment of ARDS, and the effect of aging on pulmonary microvascular function remains poorly defined (29).
Endothelial function is critically dependent on endothelial nitric oxide synthase (eNOS)-derived local production of NO, which reduces leukocyte and platelet adhesion to the endothelium and contributes to regulation of acute inflammation and microvascular barrier integrity in both systemic (8) and pulmonary vascular beds (43, 57). The role eNOS plays in the development of lung injury, however, remains unclear. Early investigation suggested that eNOS expression had no effect on LPS-induced acute lung injury (ALI) (47). On the other hand, despite electron microscopic evidence identifying paracellular gaps in the lung microvasculature of eNOS-deficient mice (43), both pharmacological inhibition and deficiency of eNOS have recently been demonstrated to be protective against lung edema formation in the setting of increased left atrial pressure (31) and in response to high tidal volume mechanical ventilation (45).
Reactive oxygen species (ROS) are thought to play a role in physiological aging (34) and are known to promote endothelial permeability and contribute to lung injury. Oxidative signaling has been linked to activation of Rho GTPase proteins (55), endothelial cytoskeletal reorganization, paracellular gap formation (28), and permeability (46). NADPH oxidase (Nox), xanthine oxidase, and mitochondria, all significant producers of ROS, participate in the development of endothelial permeability and ALI (1, 6, 9, 11, 44). In recent years, dysfunctional NOS has emerged as an important source of ROS, and it has been associated with the development of ventilator-induced lung injury (52); however, its role in vascular dysfunction in inflammatory ARDS remains to be explored.
Under conditions of limited substrate, cofactor oxidation (32) or posttranslational thiol modification (12) eNOS has been shown to become uncoupled, whereby, rather than oxidize arginine to produce citrulline and NO, electrons transfer univalently to oxygen to produce superoxide. Endothelial NOS uncoupling contributes to endothelial dysfunction in hypertension and atherosclerosis (53, 56), to vascular dysfunction with aging (7, 33), and has been associated with the development of ventilator-induced lung injury in mice (52). Similarly, tetrahydrobiopterin (BH4) supplementation has been demonstrated to protect rats from organ dysfunction and mortality associated with endotoxemia (4, 17), and arginase activity is increased in animal models of sepsis (5). We hypothesized that eNOS uncoupling plays a key pathophysiological role in the evolution of microvascular permeability and vasomotor dysfunction in inflammatory acute lung injury and that ROS formation in the inflamed lung may oxidize BH4 or modify eNOS itself, serving to uncouple eNOS and promote “feed-forward” ROS generation and subsequent endothelial barrier dysfunction.
Nox are a major source of O2·− in vascular and inflammatory cells. In contrast to inflammatory cells, vascular Nox enzymes display a low-level constitutive activity and respond to stimuli on the order of minutes to hours as opposed to instantaneously (24). Vascular Nox2 plays a role in lung endothelial ROS generation in response to ischemia (2), Nox expression is increased in the lung in response to inflammation (18, 24), and various isoforms have been linked to the development of lung injury (11, 40, 44, 54). Given that Nox2 is expressed by pulmonary endothelium (2, 25), we hypothesized that Nox2-dependent superoxide production in pulmonary endothelium leads to eNOS uncoupling and subsequent endothelial barrier dysfunction in inflammatory lung injury.
We present data demonstrating that eNOS deficiency is protective against LPS-induced lung permeability edema formation in mice. Exploring the mechanism underlying this protection in vitro, we identified a novel association between Nox2 and eNOS in lung microvascular endothelial cells stimulated with LPS. In doing so, Nox2 promotes direct oxidative posttranslational modification of eNOS by S-glutathionylation and sulfenic acid formation. These eNOS modifications drive ongoing superoxide production and endothelial monolayer barrier dysfunction, which is prevented by eNOS inhibition or silencing, and are present in lung tissue from mice exposed to intratracheal LPS.
MATERIALS AND METHODS
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh.
Materials.
LPS from Escherichia coli (E. coli) 0111:B4 (L-2630), Nω-Nitro-l-arginine methyl ester hydrochloride (l-NAME), ascorbate, and superoxide dismutase (SOD) were purchased from Sigma-Aldrich; BH4 and 7,8-dihydro-l-biopterin (BH2) were obtained from Cayman Chemical; Nox2 ds-tat peptide was synthesized at the Tufts University Core Facility (http://www.tucf.org/peptidesynthesis-f.html) as previously described (43a). Anti-eNOS (sc-654), anti-inducible NOS (iNOS) (sc-8310), and anti-Nox2 (sc-74514) were purchased from Santa Cruz Biotechnology; anti-phospho-eNOS S1177 and T495 were obtained from BD Biosciences (612392 and 612706, respectively); anti-glutathione (GSH) (101-A) and anti-cysteine sulfenic acid (SOH, 07-2139) were obtained from Virogen and EMD Millipore, respectively, and anti-GTP cyclohydrolase 1 (ab69962) and β-actin (ab6276) were obtained from Abcam. Silencing RNA, scramble RNA, and transfection reagent (Oligofectamine) were purchased from Dharmacon.
In vivo protocols.
Male C57BL/6J (Jackson Laboratories Bar Harbor) and endothelial NOS deficient (eNOS−/−) mice (12 and 24 wk old) were anesthetized with 3% isoflurane and exposed to vehicle or 2–3 mg/kg E. coli endotoxin (E. coli O55:B5, List Biologicals) dissolved in sterile water via supraglottic placement and aspiration. Subsequently, they were allowed to recover from anesthesia and had free access to water and chow for 6 or 24 h in a warmed chamber before death. Mice were killed by intraperitoneal overdose of pentobarbital. The left lung was clamped at the hilum, resected, and immediately flash frozen in liquid nitrogen for storage at −80°C. Bronchoalveolar lavage (BAL) was performed on the right lung with serial instillation of three aliquots (0.6, 0.5, 0.5 ml) of warmed sterile saline + 0.6 mM EDTA. Aliquots were pooled and centrifuged at 13,000 g for 5 min, and supernatant was retrieved and frozen at −80°C for subsequent analysis. BAL fluid protein concentration was determined as a measure of alveolar permeability by standard bicinchoninic acid assay (Fisher Thermo Scientific) as previously reported (37, 42, 49).
Cell culture techniques.
Human lung microvascular endothelial cells (HLMVEC; PromoCell, Lonza) were grown to confluence in endothelial basic medium-2 (EBM-2; Lonza) supplemented with 10% FBS, human recombinant epidermal growth factor, human recombinant insulin-like growth factor-1, human basic fibroblast growth factor, vascular endothelial growth factor, hydrocortisone, ascorbic acid, heparin, gentamicin, and amphotericin B. For experiments, endothelial cells (passages 5–10) were seeded onto 12-well plates and serum starved (EBM-2 + 0.5% FBS) overnight. The cells were then stimulated with LPS (1–50 μg/ml for 0–6 h) and/or in the presence or absence of various pharmacological agents [l-NAME, 100 μM; pegylated SOD (PEG-SOD), 50 U/ml; BH4, 50 μM; BH2, 50 μM; ascorbate, 500 μM; Nox2ds-tat, 10 μM, Scrmb-tat 10 μM] in a medium consisting of EBM-2 with 5% FBS containing LPS-binding protein and soluble CD14 (sCD14) to promote LPS action according to Goldblum and colleagues (23).
NOS activity assays.
Cyclic GMP levels were determined in lung tissue homogenates using ELISA according to kit manufacturer instructions (no. 581021, Cayman Chemical) (39, 51). NOS activity was measured in tissue homogenates by quantifying the conversion of [3H]-arginine to citrulline in the presence and absence of l-NAME according to kit manufacturer instructions (no. 781001, Cayman Chemical) (3, 13).
Biopterin quantification.
BH4 levels were measured by high-performance liquid chromatography with florescence detection, as described previously (50). Briefly, lung tissues or HLMVEC were homogenized in ice-cold extract buffer (50 mM Tris·HCl, pH 7.4, 1 mM dithiothreitol, 1 mM EDTA) and centrifuged at 16,000 g for 15 min at 4°C. The supernatant was subjected to protein assay and BH4 detection. Protein was removed from the supernatant by adding 10 μl of a 1:1 mixture of 1.5 M HClO4 and 2 M H3PO4 to 90-μl extracts, followed by centrifugation at 13,000 g for 3 min at 4°C. To determine total biopterin [BH4, BH2, and oxidized biopterin (B)] by acidic oxidation, 10 μl of 1% iodine and 2% KI in 1 M HCl solution was added to 90 pl of protein-free supernatant. To determine BH2 + B by alkaline oxidation, 10 μl of 1 M NaOH was added to 80 μl of extract, then 10 μl of 1% iodine and 2% KI in 1 M NaOH solution was added. After 1-h incubation at room temperature in the dark, 20 μl of 1 M H3PO4 was added to acidify alkaline-oxidation samples followed by 5 μl of fresh ascorbic acid (20 mg/ml in water) to reduce iodine. After centrifugation at 13,000 g for 10 min at 4°C, 50 μl of supernatant was injected into a 250 mm × 4.6 mm × 5 μm Spherisorb ODS-1 column (Alltech Associates) and eluted with a methanol and water (5:95, vol/vol) mobile phase running at a flow rate of 1 ml/min. Fluorescence was detected at 450 nm emission wavelength after excitation at 350 nm (RF10AXL; Shimadzu). Tetrahydrobiopterin concentration, expressed as picomoles per milligram of protein, was calculated by subtracting BH2 + B from total biopterin.
ROS measurement.
Superoxide (O2·−) production was measured by L-012 chemiluminescence assay in lung tissue homogenates from C57BL/6J mice 6 or 24 h after LPS exposure (2–3 mg/kg intratracheally, n ≥ 4). Tissue homogenates were incubated with the luminol derivative L-012 (500 μM) for 15 min. Chemiluminescence was quantified over time using a Biotek Synergy 4 Microplate reader (BioTek). The production of O2·− was confirmed by the addition of PEG-SOD (200 U/ml), and NOS dependence was determined by supplementing the homogenate mixture with l-NAME (100 μM) (13).
HLMVEC superoxide production was measured in cell media by SOD-inhibitable dichlorofluorescein (DCF) fluorescence and/or electron paramagnetic resonance (EPR) spectroscopy. Endothelial monolayers following treatment with LPS and/or drugs in 12-well plates were washed with PBS and incubated 30 min with H2DCF diacetate (10 μM, Sigma-Aldrich) with or without SOD (400 U/ml). Cells were washed twice with PBS and incubated with 2 mM calcium in PBS (0.5 ml) for 1 h. Supernatant fluorescence was measured using the Biotek Synergy 4 Microplate (excitation and emission wavelengths 485 and 530 nm, respectively) (26). Superoxide production was determined by calculating the fluorescence difference between comparable wells with and without SOD. For EPR, cells were harvested in the absence of proteases by mechanical dissociation and resuspended at 3 × 106 cells/ml in Chelex-treated PBS pH 7.4. O2·− formation was assessed using the EPR spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine·HCl (CMH) (200 μM) ± l-NAME (100 μM) or SOD (400 U/ml) for 10 min at 37°C as previously described (35). Spectra represent CM· intensity consisting of five signal-averaged scans from t = 9 to t = 10 min.
NO measurement.
HLMVEC NO production was determined by l-NAME inhibitable diaminofluoroscein 2 (DAF-2) fluorescence. Endothelial monolayers (6-well plates) following treatment with LPS in the presence or absence of various pharmaceuticals were washed twice with PBS and incubated 1 h with DAF-2 diacetate (1 μM, Cayman Chemical) in 100 μM l-arginine or 1 mM l-NAME in PBS. Supernatant fluorescence was measured at excitation and emission wavelengths of 485 and 530 nm, respectively. NO production was determined by calculating the difference in fluorescence between l-arginine- and l-NAME-treated cells.
Western blot.
Lung tissue from LPS-injured and control mice was homogenized in protein lysis buffer (20 mM Tris·HCl, pH 7.0, 1% NP-40, 137 mM NaCl, 1 mM PMSF, 1 μM pepstatin, and 1 μM aprotinin) at 4°C, sonicated on ice, and centrifuged at 13,000 g for 5 minutes at 4°C. Supernatant was collected and frozen at −80°C for future analysis. Western blot was performed after SDS-PAGE separation of 30 mg of protein on an 8% Tris-glycine gel and transfer to PVDF membrane. Alternatively, HLMVEC were lysed in Laemmli sample buffer, and proteins were separated in 12% SDS gel and transferred to nitrocellulose membranes. Membranes were blotted for iNOS, eNOS, phosphorylated eNOS (pS1177, pT495), Nox2, or β-actin using standard techniques. Briefly, membranes were blocked in Tris-buffered saline supplemented with 5% nonfat milk (or 5% BSA for antiphosphorylated protein antibodies) and 0.1% Tween 20. Membranes were incubated with primary antibody overnight at 4°C followed by serial washing and secondary incubation with species-appropriate horseradish peroxidase-coupled antibody at room temperature for 1 h. Protein detection and quantification were performed using standard chemiluminescence detection techniques (Kodak In-Vivo Imaging System). Membranes were subsequently probed with β-actin antibody for normalization of protein loading.
Coimmunoprecipitation.
Endothelial cells grown in 10-cm plates were washed with ice-cold PBS and incubated on ice for 15 min with 500 μl lysis buffer (20 mM Tris·HCl, pH 7.0, 1% NP-40, 137 mM NaCl, 1 mM PMSF, 1 μM pepstatin, and 1 μM aprotinin). The cells were scrape-harvested, and nuclear and cellular debris were removed by centrifugation for 10 min at 16,000 g at 4°C. Alternatively, in a subset of mice exposed to intratracheal LPS, lung tissue was homogenized in lysis buffer (50 mM Tris·HCl, 0.5% NP-40, 10% glycerol, 137 mM NaCl, 2 mM EDTA, 1 mM PMSF, pH 7.5, and 1× protease inhibitor mixture; Roche), and the concentration of protein was determined by BCA assay (Thermo Scientific).
Cell supernatants or lung tissue homogenates were incubated with primary antibody for 1 h at 4°C, followed by addition of 20 μl protein A-G agarose beads (50% slurry) for 1 h at 4°C. Immunoprecipitates were washed three times with ice-cold lysis buffer, separated by centrifugation for 30 s at 16,000 g, and then heated with Laemmli sample buffer for electrophoresis and Western blot analysis. For detection of GSH conjugated with eNOS, eNOS immunoprecipitates were heated with Laemmli sample buffer without 2-mercaptoethanol. For detection of SOH conjugated with eNOS, the lysis buffer was supplemented with 1 mM dimedone, and the Laemmli sample buffer contained 100 mM maleimide without 2-mercaptoethanol (36).
Endothelial monolayer permeability.
To determine microvascular barrier integrity, HLMVEC were plated on gelatin-coated culture inserts (3-μm pore size) in 12-well companion plates and grown to confluence in the growth medium. Monolayers were serum-starved overnight and then stimulated with LPS (1–50 μg/ml) in the presence or absence of various pharmacological agents (l-NAME, 100 μM; PEG-SOD, 50 U/ml; BH4, 50 μM; BH2, 50 μM; ascorbate, 500 μM; Nox2ds-tat, 10 μM, Scrmb-tat 10 μM) for 4 h in EBM-2 containing 5% FBS. Subsequently, FITC-labeled dextran (40 kDa, Sigma) was added to the upper chamber (0.1 mg/ml), and PBS was added to the lower chamber (to prevent the formation of an oncotic pressure gradient) for 30 min. Medium was collected from the lower chamber, and the fluorescence was measured using a BioTek fluorimeter (485-nm excitation, 530-nm emission). The fold change in FITC-dextran fluorescence intensity over controls was used as a measure of monolayer paracellular permeability.
siRNA.
Endothelial cells were seeded in 12-well plates and allowed to grow for 24 h in antibiotic-free EBM-2 containing 10% serum and supplements. Next, each well was incubated with 80 pmol of eNOS or Nox2 siRNA (small interfering ribonucleic acid) or scramble RNA and 3.15 μl of Oligofectamine reagent in 1 ml of antibiotic-free EBM-2 containing 10% FBS and supplements for 24 h. The medium was then changed to regular growth medium, and the cells were cultured for another 24 h. Transfected cells were seeded to inserts for permeability assay or treated with LPS and/or drugs for experiments as described above.
Statistical analysis.
Primary statistical analyses were performed by Student's t-tests or one-way ANOVA where appropriate with post hoc analyses using the Bonferroni test for multiple comparisons.
RESULTS
eNOS contributes to LPS-induced lung injury in aged mice.
To determine the role of eNOS in LPS-induced lung injury in vivo, young (10–12 wk, n = 29) and old (24 wk, n = 10) C57BL/6J and eNOS-deficient (eNOS−/−, 23 young and 10 old) mice were exposed to intratracheal LPS (2–3 mg/kg in 100 μl) or vehicle (sterile water, 100 μl) and allowed to recover for 6 h. Subsequently, mice were killed, and BAL was performed for protein assay. Intratracheal LPS precipitated lung microvascular permeability in young mice (88 ± 8 μg/ml vehicle vs. 288 ± 36 μg/ml LPS, P = 0.0001), which was accentuated in aged animals (200 ± 41 μg/ml vehicle vs. 1,014 ± 84 μg/ml LPS, P < 0.0001). Whereas young eNOS−/− mice demonstrated a nonsignificant trend toward reduced alveolar permeability, aged eNOS−/− mice were protected against lung microvascular permeability in response to LPS in vivo [BAL protein following LPS = 208 ± 39 μg/ml in young mice (P = 0.2) and 565 ± 83 μg/ml in old mice (P = 0.002), Fig. 1]. Measures of canonical eNOS signaling including eNOS phosphorylation, cGMP levels, and arginine-to-citrulline turnover were not altered in lung tissue from young wild-type mice exposed to LPS compared with vehicle (Fig. 2, A and B, n ≥ 3 each, P = NS). As a surrogate measure of eNOS uncoupling in lung tissue, biopterins were quantified in tissue homogenates (50). Lung tissue BH2 was significantly increased in young mouse lungs after LPS exposure compared with vehicle [11.1 ± 1.0 vs. 15.6 ± 0.9 pmol/mg protein in vehicle and LPS-treated animals, respectively (n ≥7, P = 0.005), Fig. 2B], suggesting the possibility that eNOS is uncoupled in mouse lungs following LPS exposure. To determine whether uncoupled eNOS may be a source of ROS in the lung, tissue homogenates were prepared from C57BL/6J mice (n = 8) exposed to LPS (2.5 mg/kg in 100 μl sterile water) for 24 h. BAL protein was increased (2,263 ± 233 μg/ml) 24 h after LPS suggesting significant lung injury at this time point. Lung tissue from mice exposed to LPS demonstrated significant l-NAME-dependent superoxide production by L-012 chemiluminescence (0.29 ± 0.05 vs. 0.17 ± 0.02 O2·− Abs U/g protein per minute in the absence or presence of l-NAME, respectively; n = 5, P = 0.05) (Fig. 2C), suggestive of NOS dependent superoxide production in LPS-exposed lungs.
Fig. 1.
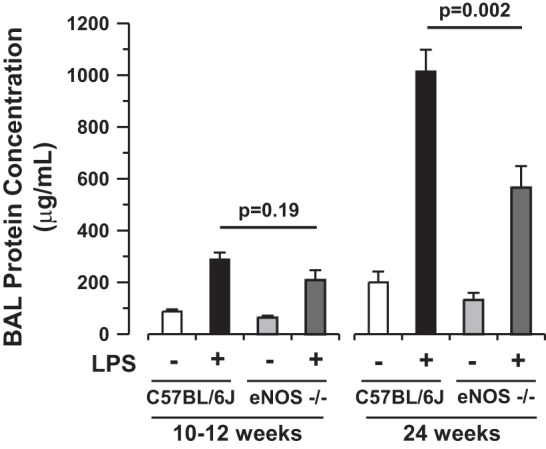
Endothelial nitric oxide synthase (eNOS) deficiency is protective against lung injury induced by intratracheal LPS. Depicted are bronchoalveolar lavage (BAL) fluid protein analyses from young (10–12 wk, n ≥ 9 each, mean ± SE) and old (24 wk, n ≥ 3 each) C57BL6/J or eNOS-deficient (eNOS−/−) mice exposed to intratracheal vehicle (100 μl, open or light shaded bars) or endotoxin (LPS, 2–3 mg/kg in 100 μl sterile water, solid or dark shaded bars) for 6 h. LPS-induced BAL protein accumulation is attenuated by eNOS deficiency attenuated by eNOS deficiency in aged mice (P = 0.002 by 1-way ANOVA).
Fig. 2.

Intratracheal LPS leads to eNOS uncoupling in mouse lung tissue. A: Western blot analysis of phosphorylated eNOS protein in lung tissue homogenates from mice exposed to intratracheal vehicle (100 μl) or LPS (2–3 mg/kg in 100 μl sterile water, n = 3 each) for 6 h. B: arginine-to-citrulline turnover and cyclic GMP production in lung tissue homogenates from mice exposed to intratracheal vehicle (100 μl, open bars) or LPS (2–3 mg/kg in 100 μl sterile water, solid bars, n ≥ 3 each) for 6 h. Taken together, these data suggest that canonical signaling is unchanged in mouse lungs exposed to LPS compared with controls. C: lung tissue biopterin expression from mice exposed to intratracheal vehicle (100 μl, open bars) or LPS (2–3 mg/kg in 100 μl sterile water, solid bars) for 6 h. 7,8-Dihydro-l-biopterin (BH2) is increased in lung tissue exposed to LPS compared with controls (n ≥ 7, P < 0.01). BH4, tetrahydrobiopterin. D: L-012 chemiluminescence (superoxide production) in mouse lung tissue homogenate 24 h following intratracheal LPS exposure (2.5 mg/kg) in the presence (gray line, bar) or absence (black line, bar) of the eNOS inhibitor nitro-l-arginine methyl ester (l-NAME) (100 μM, n = 5 each). l-NAME-dependent superoxide production is the sine qua non of eNOS uncoupling. RLU, relative light units.
Uncoupled eNOS contributes to endothelial permeability in response to LPS in vitro.
To further explore the role of eNOS uncoupling in the regulation of alveolar barrier function in response to an inflammatory stimulus, we shifted to an in vitro model. HLMVEC were exposed to LPS (1–50 μg/ml) for up to 6 h, and then O2·− production and monolayer permeability were measured. LPS-induced HLMVEC superoxide production, measured by DCF oxidation, peaked 4 h after stimulation (1.7 ± 0.3-fold increase in response to 1 μg/ml, Fig. 3A), was independent of dose, and correlated with an increase in monolayer dextran permeability (1.6 ± 0.1-fold increase after 4 h compared with baseline, Fig. 3B). O2·− production after high-dose LPS treatment (50 μg/ml) was confirmed by EPR using the cyclic hydroxylamine CMH, which demonstrated a 65% increase in signal over untreated HLMVEC (Fig. 3C, top) and correlated with increased monolayer permeability (data not shown). LPS-induced (1 μg/ml) superoxide production and dextran permeability were attenuated by administration of the nonspecific antioxidants ascorbate and BH4 (Fig. 4, A and B), both of which may serve to recouple eNOS function.
Fig. 3.
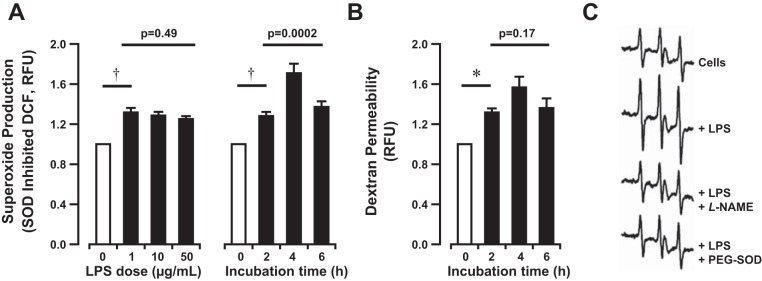
Bacterial LPS induces superoxide production and barrier dysfunction in human lung microvascular endothelial cells (HLMVEC) in vitro. A: superoxide dismutase (SOD)-inhibitable dichlorofluorescein (DCF) fluorescence in media extracted from cultured HLMVEC following bacterial LPS exposure (0–50 μg/ml) for 0–6 h in vitro. B: fluorescent dextran accumulation in basal media after apical exposure of HLMVEC to bacterial LPS (1 μg/ml) for 0–6 h in vitro. LPS induces dose-independent superoxide production and increased permeability to fluorescent dextran peaking at 4 h after stimulation. Bars represent the means ± SE of n = 3 separate experiments for each dose and n ≥ 8 at each time point. †P < 0.0001 and *P < 0.01 by 1-sided paired t-test. 1-way ANOVA used for comparison of dose or time dependence. RFU, relative fluorescence units. C: HLMVEC exposed to LPS (50 μg/ml) ± pegylated SOD (PEG-SOD) (25 U/ml) for 12 h, harvested in the absence of proteases by mechanical dissociation, and resuspended at 3 × 106 cells/ml in Chelex-treated PBS pH 7.4. Superoxide formation was monitored with the electron paramagnetic resonance (EPR) spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine·HCl (CMH) (200 μM) ± l-NAME (100 μM) for 10 min at 37°C. Spectra represent CM· intensity consisting of 5 signal-averaged scans from t = 9 to t = 10 min. Tracings displayed from top to bottom are as follows: untreated, LPS treated, LPS treated (+PEG-SOD), LPS treated (+l-NAME). LPS increased the CMH EPR signature amplitude by 65% over untreated cells. Both l-NAME and SOD attenuated the signal to levels comparable to untreated cells.
Fig. 4.
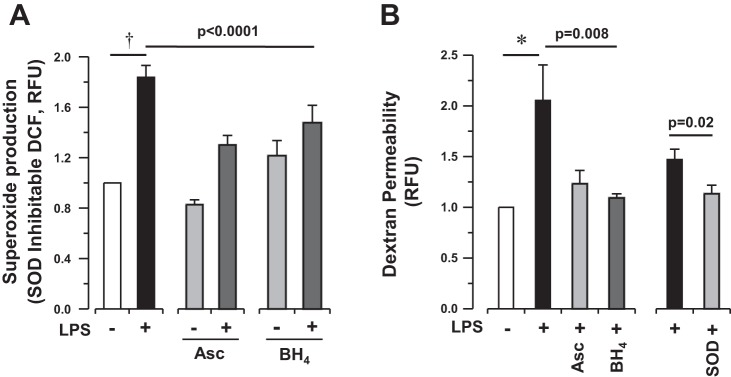
Bacterial LPS-induced superoxide production is associated with the development of barrier dysfunction in vitro. A: SOD-inhibitable DCF fluorescence in media extracted from HLMVEC following LPS exposure (1 μg/ml for 4 h) in the presence of ascorbate (500 μM) (Asc) or BH4 (50 μM). B: fluorescent dextran accumulation in basal media after apical exposure of HLMVEC to LPS (1 μg/ml for 4 h) in the presence of ascorbate (500 μM), BH4 (50 μM), or SOD (50 U/ml). SOD experiment is shown separately, as it represents separate experiment with its own control cells. LPS-induced permeability is attenuated in the presence of antioxidants or superoxide-specific SOD. Bars represent means ± SE of n ≥ 12 individual experiments. Statistical analysis by 1-way ANOVA (Asc and BH4) and Student's t-test (SOD). Post hoc analysis by Bonferroni test for superoxide production (A, Asc P = 0.002, BH4 P = 0.11) and for permeability (B, Asc P = 0.04, and BH4 P = 0.01). †P < 0.0001 and *P < 0.01 by 1-sided paired t-test.
Canonical eNOS signaling was assessed in HLMVEC exposed to LPS for 4 h. Interestingly, iNOS was not detectable in cultured HLMVEC (Fig. 5A). Neither iNOS nor eNOS protein expression was altered in HLMVEC in response to LPS stimulation. Similarly, canonical eNOS phosphorylation was unchanged in LPS-exposed HLMVEC (Fig. 5A). Measurement of NO production showed a significant decrease in HLMVEC 4 h after LPS exposure to levels similar to those of eNOS-silenced cells (Fig. 5, B and C).
Fig. 5.

Bacterial LPS induces eNOS-dependent superoxide production and monolayer permeability in HLMVEC in vitro. A: Western blot analysis of inducible NOS (iNOS), eNOS, and phosphorylated eNOS protein in HLMVEC lysates after exposure to vehicle or LPS (1 μg/ml for 4 h). LPS does not induce iNOS or eNOS expression or activation of eNOS as represented by enzyme phosphorylation. B: Western blot analysis of eNOS expression in the presence or absence of LPS (1 μg/ml for 4 h) after siRNA incubation. C: diaminofluoroscein (DAF) fluorescence in media extracted from HLMVEC following LPS exposure (1 μg/ml for 4 h) in the presence of eNOS siRNA or scramble RNA. LPS exposure (1 μg/ml for 4 h) inhibits NO production in HLMVEC in vitro to a degree similar to eNOS silencing. D and E: eNOS-dependent superoxide production (DCF fluorescence) and permeability (basilar dextran fluorescence), respectively, in HLMVEC exposed to LPS (1 μg/ml for 4 h) in vitro. Bars represent mean data from n ≥ 12 individual experiments. †P < 0.0001 and *P < 0.01 by 1-sided paired t-test. 1-way ANOVA reported for treatment response compared with LPS response. Post hoc analysis by Bonferroni test (P = 0.03 for LPS vs. LPS + l-NAME and P < 0.001 for LPS vs. LPS + siRNA). Permeability in eNOS silenced cells normalized to isolate the LPS effect, as eNOS silencing altered baseline dextran fluorescence in vitro. The change in dextran permeability induced by LPS in eNOS-deficient cells is significantly lower than the change in control cells (P = 0.04 by Student's t-test).
To determine whether uncoupled eNOS was in part responsible for LPS-induced O2·− production, H2DCFDA oxidation to DCF was measured after NOS inhibition. Both pharmacological inhibition of NOS by l-NAME (100 μM) and eNOS silencing prevented HLMVEC superoxide production induced by low-dose LPS (1 μg/ml) stimulation (l-NAME + LPS 1.2 ± 0.3-fold vs. LPS 1.7 ± 0.4-fold, eNOS siRNA + LPS 0.9 ± 0.1-fold vs. eNOS scrRNA + LPS 1.8 ± 0.2-fold compared with baseline, Fig. 5D). Consistent with this finding, both l-NAME and SOD attenuated the CM· EPR signal induced by high-dose LPS (50 μg/ml) to levels comparable to untreated HLMVEC (Fig. 3C, middle bottom and bottom). Taken together, these data suggest that eNOS provides a source for superoxide production in HLMVEC stimulated with LPS.
Endothelial monolayer permeability increased ∼1.7-fold following LPS stimulation (1 μg/ml for 4 h). Similar to the effect on superoxide production, treatment with l-NAME (100 μM) abrogated the permeability increase (1.2 ± 0.1-fold increase compared with baseline control, Fig. 5E). Similar to results published by Predescu and colleagues (43), eNOS silencing increased basal permeability in HLMVEC monolayers (1.6 ± 0.1-fold increase compared with eNOS scramble RNA control), suggesting a role for endogenous NO production in the maintenance of barrier integrity. Confirming the effects of l-NAME, eNOS silencing also abrogated the increase in monolayer permeability induced by LPS in vitro (63 ± 13% increase after LPS in scramble RNA controls vs. 14 ± 11% increase after LPS in silencing RNA experiments, P = 0.04 for difference in change, Fig. 5E). These data suggest that eNOS uncoupling contributes to LPS-induced monolayer barrier dysfunction in HLMVEC.
Nox2 contributes to endothelial permeability in response to LPS in vitro.
As previously indicated, vascular Nox2 plays a role in lung endothelial ROS generation in response to ischemia (2), and increased Nox expression has been described in the lung in response to inflammation (18, 24). In our model system, endothelial Nox2 expression was increased following LPS exposure (Fig. 6A). As with eNOS silencing, Nox2 silencing slightly increased basal permeability in HLMVEC monolayers (1.2 ± 0.1-fold increase compared with Nox2 scramble RNA control, P = 0.1). Inhibition of Nox2 using the specific peptide inhibitor (Nox2ds-tat) and silencing of Nox2 using a specific siRNA strategy (Fig. 6A) prevented both LPS-induced superoxide production [LPS + Scrmb-tat 1.7 ± 0.1-fold increase vs. LPS + Nox2ds-tat 1.2 ± 0.1-fold increase (P = 0.005), LPS + Nox2 scrRNA 1.5 ± 0.1-fold increase vs. LPS + Nox2 siRNA 1.1 ± 0.1-fold increase (P = 0.0006), Fig. 6B] and monolayer permeability (48 ± 11% increase after LPS in scramble RNA controls vs. 5 ± 7% increase in silencing RNA experiments, P = 0.004 for difference in change, Fig. 6C) in vitro. These data suggest that, in addition to eNOS uncoupling, Nox2-dependent superoxide production contributes to LPS-induced monolayer barrier dysfunction in HLMVEC.
Fig. 6.

Bacterial LPS induces NADPH oxidase 2 (Nox2)-dependent superoxide production and monolayer permeability in HLMVEC in vitro. A: Western blot analysis of Nox2 expression in the presence or absence of LPS (1 μg/ml for 4 h) after siRNA incubation. B and C: Nox2-dependent superoxide production (DCF fluorescence) and permeability (basilar dextran fluorescence), respectively, in HLMVEC exposed to LPS (1 μg/ml for 4 h) in vitro in the presence or absence of Nox2 inhibition by specific peptide (Nox2ds-tat, 10 μM) or RNA silencing. Bars represent mean data from n ≥ 11 individual experiments. Statistical analysis performed by 1-way ANOVA. Permeability in Nox2-silenced cells was normalized to isolate the LPS effect, as Nox2 silencing altered baseline dextran fluorescence in vitro. The change in dextran permeability induced by LPS in Nox2-deficient cells is significantly lower than the change in control cells (*P = 0.04 by Student's t-test).
Nox2-dependent oxidation of eNOS drives uncoupling in LPS-stimulated lung endothelium.
As noted previously, biopterin oxidation or oxidative modification of the enzyme directly may lead to the uncoupling of eNOS enzymatic function and the production of O2·− in lieu of NO. Our in vivo data suggested that biopterin oxidation contributed to eNOS uncoupling in the inflammatory lung injury (Fig. 2B). Given that Nox2 contributes to superoxide production in HLMVEC exposed to LPS, we explored the possibility that biopterin oxidation leads to subsequent eNOS uncoupling in LPS-exposed cells. In contrast to LPS-exposed mouse lungs, biopterin levels were unchanged in HLMVEC treated with LPS, and BH2 supplementation did not affect monolayer barrier integrity (dextran increase from baseline in LPS 1.5 ± 0.1-fold vs. LPS + BH2 1.8 ± 0.4-fold, P = NS, Fig. 7), suggesting that this is not the mechanism underlying eNOS uncoupling in lung microvascular endothelial cells.
Fig. 7.
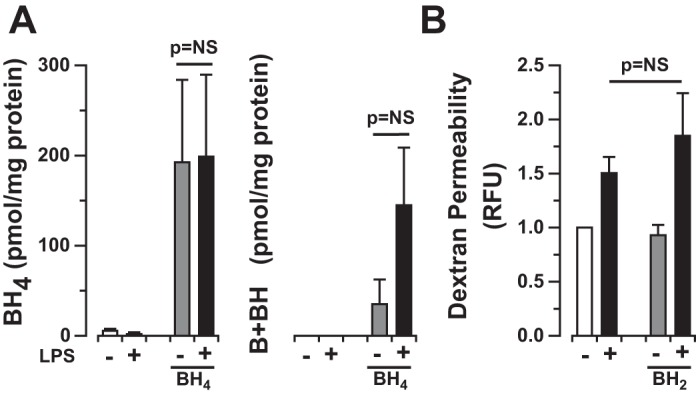
Dihydrobiopterin expression does not contribute to LPS-induced permeability in HLMVEC in vitro. A: biopterin expression in HLMVEC following LPS (1 μg/ml for 4 h) exposure in the presence or absence of BH4 (50 μM). Bars represent means ± SE from n = 4 individual experiments, P = NS. Biopterin expression is minimal in HLMVEC at baseline and following LPS exposure. B: HLMVEC permeability (basal dextran fluorescence) following LPS (1 μg/ml for 4 h) exposure in the presence or absence of BH2 (50 μM). BH2 exposure has no effect on HLMVEC monolayer permeability at baseline or following LPS exposure. Bars represent means ± SE from n = 8 individual experiments, P = NS.
We next explored the possibility that Nox2-dependent superoxide production modifies the eNOS protein leading downstream to uncoupling of the enzyme. Coimmunoprecipitation revealed that eNOS associates with Nox2 in HLMVEC following LPS stimulation (Fig. 8A). Similarly, LPS induces S-glutathionylation of eNOS protein (eNOS-GSH) and eNOS cysteine oxidation to sulfenic acid (eNOS-SOH) that is inhibited in the presence of the Nox2ds-tat peptide inhibitor (Fig. 8, B and C). Taken together, these observations suggest that Nox2-dependent superoxide production leads to posttranslational oxidative modifications of the eNOS protein that promote uncoupling of enzymatic function and eNOS-dependent superoxide production.
Fig. 8.

Bacterial LPS induces eNOS association with Nox2 and subsequent oxidative modification of eNOS enzyme in HLMVEC in vitro. A: coimmunoprecipitation (co-IP) of eNOS and Nox2 protein in HLMVEC following LPS (1 μg/ml for 4 h) exposure. Bacterial endotoxin induces an association of eNOS with Nox2 in HLMVEC in vitro. IB, immunoblotting. B: representative co-IP of eNOS protein with S-glutathionyl (GSH) and sulfenic acid (SOH) moieties following LPS (1 μg/ml for 4 h) exposure in the presence or absence of the Nox2-specific peptide inhibitor Nox2ds-tat (10 μM). C: densitometry quantification of n ≥ 3 individual co-IP experiments presented in B. Statistical analysis performed by 1-way ANOVA. LPS-induced oxidative modification of eNOS protein (S-glutathionylation and sulfenic acid formation) is dependent on Nox2.
Finally, we returned to the murine model to determine whether Nox2 associates with eNOS and induces S-glutathionylation of eNOS protein (eNOS-GSH) in lungs exposed to LPS in an age-dependent manner. Coimmunoprecipitation of mouse lung tissue homogenates 6 h following intratracheal LPS administration reveals that eNOS does in fact associate with Nox2 in the injured lung and that eNOS protein is glutathionylated to a greater degree in aged mice compared with young mice (Fig. 9). Furthermore, Nox2 association with eNOS and subsequent S-glutathionylation are associated with a nonsignificant trend toward increased O2·− production in old mice compared with young mice as determined by L-012 chemiluminescence [0.36 ± 0.02 (aged) vs. 0.29 ± 0.02 (young) Abs/mg lung tissue per min, n = 4, P = 0.07 by two-tailed t-test, data not shown] without significant difference in the l-NAME-dependent component.
Fig. 9.
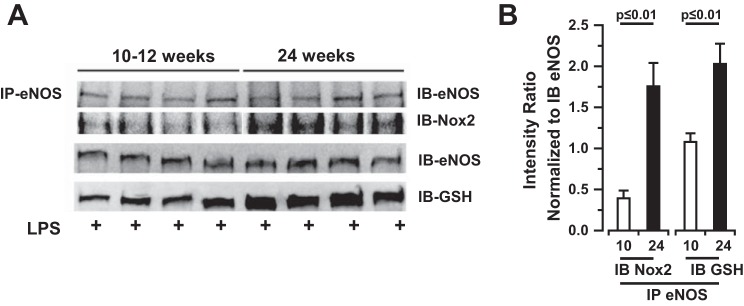
Bacterial LPS induces eNOS association with Nox2 and subsequent oxidative modification of eNOS enzyme in mouse lungs in vivo. A: co-IP of eNOS and Nox2 protein and S-glutathionyl moieties in mouse lung tissue homogenates 6 h following LPS (2 mg/kg) exposure. Bacterial endotoxin induces an association of eNOS with Nox2 and with GSH in murine lungs in vivo. B: densitometry quantification of n = 4 individual co-IP experiments presented in A for young (10 wk old) and old (24 wk old) mice. Statistical analysis performed by 2-sample t-test.
DISCUSSION
Pulmonary endothelial barrier integrity is dependent on local NO bioavailability (43), which also serves to regulate local inflammation, reducing leukocyte and platelet adhesion to the endothelium. eNOS is the key enzyme regulating local NO production at the level of the endothelium, but the role eNOS plays in the development of lung injury remains unclear. When uncoupled, eNOS produces superoxide in lieu of NO (56), thereby reducing NO bioavailability and producing a potentially toxic product known to contribute to endothelial cytoskeletal reorganization and paracellular gap formation (28), both key mechanisms underlying the development of endothelial permeability (21, 46). eNOS uncoupling has recently been associated with the development of lung injury in the setting of high tidal volume mechanical ventilation (52). Our data both in vivo and in vitro support the hypothesis that eNOS uncoupling contributes to lung microvascular endothelial barrier dysfunction in the setting of inflammatory lung injury. The mechanisms by which O2·− contributes to lung endothelial permeability, however, remain unanswered by this work and represent one focus of ongoing work in the laboratory.
From a mechanistic perspective, our study provides novel insight into how eNOS may become uncoupled in inflammatory states. Three potential mechanisms have been demonstrated to uncouple NOS: 1) substrate limitation, 2) oxidation of the necessary cofactor, BH4 (32), and 3) posttranslational oxidative modification of the enzyme (12). Nox are important sources of superoxide production in inflammatory cells as well as the microvasculature of the lung (2, 11, 18, 24, 25, 40, 44, 54), and deficiency of Nox2 has been demonstrated to be protective against lung edema formation in a murine model of Gram-negative sepsis (20). We hypothesized that Nox2-dependent superoxide production and/or downstream reactive metabolites would contribute to lung microvascular endothelial barrier dysfunction in response to LPS through oxidation of BH4 or the eNOS enzyme itself. Indeed, our data demonstrate that specific inhibition of Nox2 reduces superoxide production by lung microvascular endothelial cells and prevents the barrier disruption that follows endotoxin exposure in vitro (Fig. 6).
Our data in vivo suggest that BH4 oxidation may contribute to NOS uncoupling in inflammatory lung injury with BH2 being increased in lung tissue from mice exposed to intratracheal LPS (Fig. 2). However, our in vitro experiments indicate that the source of biopterin in the intact lung is not the microvascular endothelial cells. Neither BH4 nor BH2 were detectable in HLMVEC lysates in the presence or absence of LPS (Fig. 7), and treatment of the endothelium with BH2 did not contribute to barrier disruption in vitro. Taken together, these data suggest that this mechanism is not particularly important for regulation of eNOS function in the lung microvasculature. It is possible that conduit endothelium, inflammatory cells, or epithelial cells contribute to the biopterin pool in the intact lung. Further study is required to elucidate the source of biopterin in the lung and the contribution of biopterin oxidation to eNOS uncoupling in vivo.
As oxidative modification of eNOS, e.g., S-glutathionylation, promotes uncoupling of enzymatic function (12), we explored the hypothesis that the barrier protection afforded by Nox2 inhibition was potentially mediated through regulation of eNOS oxidation. Our data demonstrate that eNOS protein associates with Nox2 in lung microvascular endothelium after exposure to LPS. This close approximation may enable low-level Nox2-catalyzed superoxide and/or reactive metabolites of superoxide to modify the structure of eNOS, thereby driving uncoupling and accentuating superoxide production (Fig. 10). To answer this question, we examined whether LPS exposure results in oxidative modification of the eNOS protein. Our data clearly show that LPS causes both S-glutathionylation of the eNOS protein as well as eNOS SOH formation in a Nox2-dependent fashion in vitro (Fig. 8). Furthermore, our data suggest that oxidative modification of eNOS occurs in vivo in response to endotoxin exposure in an age-dependent manner (Fig. 9). Although this particular experiment was underpowered to detect a significant difference in O2·− production in vivo, aged mice exhibited a trend toward increased ROS production after LPS. Further study is required to validate 1) age-dependent differences in O2·− production, 2) define specific enzymatic source(s) in this model, and 3) identify factors that may alter the identity of downstream metabolites of O2·− requisite for oxidative modification eNOS. The association of Nox2 with eNOS following inflammatory stimulation represents a previously undescribed mechanism underlying barrier dysregulation in the lung microvascular endothelium, and this mechanism of cross talk between redox enzymes to promote uncoupling of eNOS has not previously been described in the literature. How these enzymes come together at the cell surface remains yet to be explored although both enzymes have been previously associated with caveolae (38).
Fig. 10.
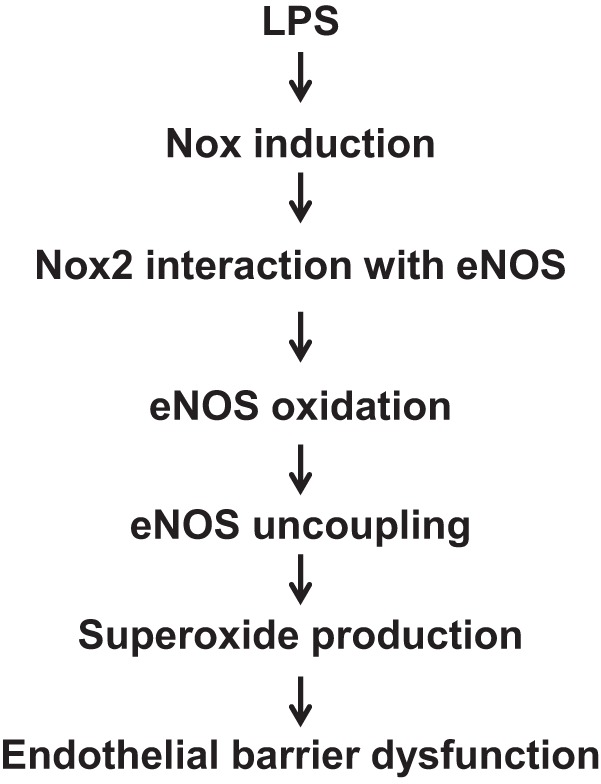
Conceptual model for mechanisms by which LPS induces eNOS uncoupling in HLMVEC by Nox2-dependent oxidative modification of eNOS protein.
The population admitted to intensive care units across the country and around the world is aging (22). From a translational perspective, eNOS uncoupling has been associated with vascular dysfunction with aging (7, 29, 33), and it is well described that vascular endothelial dysfunction plays a key role in the development and consequences of ARDS (10, 21, 37, 42, 45). Furthermore, the risk of developing ARDS increases with age, and both short- and long-term outcomes are worse in elderly patients (14, 19, 22, 27, 30). Similar to data in humans, our data suggest that older mice develop worse lung injury in response to LPS compared with their younger counterparts, and deficiency of eNOS is protective against lung edema formation in aged mice (Fig. 1). Taken together, these data suggest a potentially important role for eNOS uncoupling in the development of barrier dysfunction with age. One potential explanation for these phenomena is that NOS becomes uncoupled with age, reducing NO bioavailability and predisposing to lung microvascular barrier dysfunction at baseline, which is then exacerbated with inflammatory stimulation. Although we have demonstrated that eNOS associates with Nox2 and undergoes S-glutathionylation in an age-dependent manner after stimulation with LPS in vivo (Fig. 9), more work is necessary to further validate our findings in vivo and to determine the time course and relative importance of age-dependent NOS uncoupling to the development of lung injury. As antioxidant therapies have not proven successful in treating lung injury in all work to date, further work is necessary to explore the therapeutic potential of strategies aimed at recoupling NOS function and restoring NO bioavailability for the treatment of inflammatory lung injury in patients demonstrating a vascular phenotype of disease. Perhaps these strategies may prove beneficial in aging populations.
GRANTS
This project was supported by the NHLBI (K08-HL83101 to B. McVerry), the American Heart Association (12GRNT11820019 to B. McVerry, 10SDG3560005 to E. Kelley), and the University of Pittsburgh Vascular Medicine Institute in collaboration with the Institute for Transfusion Medicine (B. McVerry).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: F.W., W.S.S., E.E.K., M.T.G., and B.J.M. conception and design of research; F.W., W.S.S., S.S., H.L., Yinna Wang, L.W., Ying Wang, E.E.K., and B.J.M. performed experiments; F.W., W.S.S., S.S., E.E.K., A.F.C., M.T.G., and B.J.M. analyzed data; F.W., W.S.S., S.S., E.E.K., A.F.C., M.T.G., and B.J.M. interpreted results of experiments; F.W. and B.J.M. prepared figures; F.W. and B.J.M. drafted manuscript; F.W., W.S.S., S.S., E.E.K., A.F.C., M.T.G., and B.J.M. edited and revised manuscript; F.W., W.S.S., S.S., H.L., Yinna Wang, L.W., Ying Wang, E.E.K., A.F.C., M.T.G., and B.J.M. approved final version of manuscript.
REFERENCES
- 1.Abdulnour RE, Peng X, Finigan JH, Han EJ, Hasan EJ, Birukov KG, Reddy SP, Watkins JE, 3rd, Kayyali US, Garcia JG, Tuder RM, Hassoun PM. Mechanical stress activates xanthine oxidoreductase through MAP kinase-dependent pathways. Am J Physiol Lung Cell Mol Physiol 291: L345–L353, 2006. [DOI] [PubMed] [Google Scholar]
- 2.Al-Mehdi AB, Zhao G, Dodia C, Tozawa K, Costa K, Muzykantov V, Ross C, Blecha F, Dinauer M, Fisher AB. Endothelial NADPH oxidase as the source of oxidants in lungs exposed to ischemia or high K+. Circ Res 83: 730–737, 1998. [DOI] [PubMed] [Google Scholar]
- 3.Alef MJ, Vallabhaneni R, Carchman E, Morris SM, Jr, Shiva S, Wang Y, Kelley EE, Tarpey MM, Gladwin MT, Tzeng E, Zuckerbraun BS. Nitrite-generated NO circumvents dysregulated arginine/NOS signaling to protect against intimal hyperplasia in Sprague-Dawley rats. J Clin Invest 121: 1646–1656, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bahrami S, Fitzal F, Peichl G, Gasser H, Fuerst W, Banerjee A, Strohmaier W, Redl H, Werner-Felmayer G, Werner ER. Protection against endotoxemia in rats by a novel tetrahydrobiopterin analogue. Shock 13: 386–391, 2000. [DOI] [PubMed] [Google Scholar]
- 5.Bansal V, Ochoa JB. Arginine availability, arginase, and the immune response. Curr Opin Clin Nutr Metab Care 6: 223–228, 2003. [DOI] [PubMed] [Google Scholar]
- 6.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87: 245–313, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Belik J, Jerkic M, McIntyre BA, Pan J, Leen J, Yu LX, Henkelman RM, Toporsian M, Letarte M. Age-dependent endothelial nitric oxide synthase uncoupling in pulmonary arteries of endoglin heterozygous mice. Am J Physiol Lung Cell Mol Physiol 297: L1170–L1178, 2009. [DOI] [PubMed] [Google Scholar]
- 8.Bucci M, Roviezzo F, Posadas I, Yu J, Parente L, Sessa WC, Ignarro LJ, Cirino G. Endothelial nitric oxide synthase activation is critical for vascular leakage during acute inflammation in vivo. Proc Natl Acad Sci USA 102: 904–908, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Budinger GR, Mutlu GM, Urich D, Soberanes S, Buccellato LJ, Hawkins K, Chiarella SE, Radigan KA, Eisenbart J, Agrawal H, Berkelhamer S, Hekimi S, Zhang J, Perlman H, Schumacker PT, Jain M, Chandel NS. Epithelial cell death is an important contributor to oxidant-mediated acute lung injury. Am J Respir Crit Care Med 183: 1043–1054, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bull TM, Clark B, McFann K, Moss M. Pulmonary vascular dysfunction is associated with poor outcomes in patients with acute lung injury. Am J Respir Crit Care Med 182: 1123–1128, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carnesecchi S, Deffert C, Pagano A, Garrido-Urbani S, Metrailler-Ruchonnet I, Schappi M, Donati Y, Matthay MA, Krause KH, Barazzone Argiroffo C. NADPH oxidase-1 plays a crucial role in hyperoxia-induced acute lung injury in mice. Am J Respir Crit Care Med 180: 972–981, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen CA, Lin CH, Druhan LJ, Wang TY, Chen YR, Zweier JL. Superoxide induces endothelial nitric-oxide synthase protein thiyl radical formation, a novel mechanism regulating eNOS function and coupling. J Biol Chem 286: 29098–29107, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cruz JA, Bauer EM, Rodriguez AI, Gangopadhyay A, Zeineh NS, Wang Y, Shiva S, Champion HC, Bauer PM. Chronic hypoxia induces right heart failure in caveolin-1-/- mice. Am J Physiol Heart Circ Physiol 302: H2518–H2527, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davidson TA, Rubenfeld GD, Caldwell ES, Hudson LD, Steinberg KP. The effect of acute respiratory distress syndrome on long-term survival. Am J Respir Crit Care Med 160: 1838–1842, 1999. [DOI] [PubMed] [Google Scholar]
- 15.Dzau VJ, Antman EM, Black HR, Hayes DL, Manson JE, Plutzky J, Popma JJ, Stevenson W. The cardiovascular disease continuum validated: clinical evidence of improved patient outcomes. I. Pathophysiology and clinical trial evidence (risk factors through stable coronary artery disease). Circulation 114: 2850–2870, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Dzau VJ, Antman EM, Black HR, Hayes DL, Manson JE, Plutzky J, Popma JJ, Stevenson W. The cardiovascular disease continuum validated: clinical evidence of improved patient outcomes. II. Clinical trial evidence (acute coronary syndromes through renal disease) and future directions. Circulation 114: 2871–2891, 2006. [DOI] [PubMed] [Google Scholar]
- 17.Fitzal F, Redl H, Strohmaier W, Werner ER, Bahrami S. A 4-amino analogue of tetrahydrobiopterin attenuates endotoxin-induced hemodynamic alterations and organ injury in rats. Shock 18: 158–162, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Frey RS, Ushio-Fukai M, Malik AB. NADPH oxidase-dependent signaling in endothelial cells: role in physiology and pathophysiology. Antioxid Redox Signal 11: 791–810, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gajic O, Afessa B, Thompson BT, Frutos-Vivar F, Malinchoc M, Rubenfeld GD, Esteban A, Anzueto A, Hubmayr RD, Second International Study of Mechanical Ventilation and ARDS-net Investigators. Prediction of death and prolonged mechanical ventilation in acute lung injury. Crit Care 11: R53, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao XP, Standiford TJ, Rahman A, Newstead M, Holland SM, Dinauer MC, Liu QH, Malik AB. Role of NADPH oxidase in the mechanism of lung neutrophil sequestration and microvessel injury induced by Gram-negative sepsis: studies in p47phox-/- and gp91phox-/- mice. J Immunol 168: 3974–3982, 2002. [DOI] [PubMed] [Google Scholar]
- 21.Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, English D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest 108: 689–701, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garland A, Olafson K, Ramsey CD, Yogendran M, Fransoo R. Epidemiology of critically ill patients in intensive care units: a population-based observational study. Crit Care 17: R212, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldblum SE, Brann TW, Ding X, Pugin J, Tobias PS. Lipopolysaccharide (LPS)-binding protein and soluble CD14 function as accessory molecules for LPS-induced changes in endothelial barrier function, in vitro. J Clin Invest 93: 692–702, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res 86: 494–501, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Griffith B, Pendyala S, Hecker L, Lee PJ, Natarajan V, Thannickal VJ. NOX enzymes and pulmonary disease. Antioxid Redox Signal 11: 2505–2516, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han M, Pendem S, Teh SL, Sukumaran DK, Wu F, Wilson JX. Ascorbate protects endothelial barrier function during septic insult: Role of protein phosphatase type 2A. Free Radic Biol Med 48: 128–135, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herridge MS, Tansey CM, Matte A, Tomlinson G, Diaz-Granados N, Cooper A, Guest CB, Mazer CD, Mehta S, Stewart TE, Kudlow P, Cook D, Slutsky AS, Cheung AM, Canadian Critical Care Trials Group. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med 364: 1293–1304, 2011. [DOI] [PubMed] [Google Scholar]
- 28.Hinshaw DB, Burger JM, Armstrong BC, Hyslop PA. Mechanism of endothelial cell shape change in oxidant injury. J Surg Res 46: 339–349, 1989. [DOI] [PubMed] [Google Scholar]
- 29.Jane-Wit D, Chun HJ. Mechanisms of dysfunction in senescent pulmonary endothelium. J Gerontol Biol A Sci Med Sci 67: 236–241, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnston CJ, Rubenfeld GD, Hudson LD. Effect of age on the development of ARDS in trauma patients. Chest 124: 653–659, 2003. [DOI] [PubMed] [Google Scholar]
- 31.Kaestle SM, Reich CA, Yin N, Habazettl H, Weimann J, Kuebler WM. Nitric oxide-dependent inhibition of alveolar fluid clearance in hydrostatic lung edema. Am J Physiol Lung Cell Mol Physiol 293: L859–L869, 2007. [DOI] [PubMed] [Google Scholar]
- 32.Karuppiah K, Druhan LJ, Chen CA, Smith T, Zweier JL, Sessa WC, Cardounel AJ. Suppression of eNOS-derived superoxide by caveolin-1: a biopterin-dependent mechanism. Am J Physiol Heart Circ Physiol 301: H903–H911, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim JH, Bugaj LJ, Oh YJ, Bivalacqua TJ, Ryoo S, Soucy KG, Santhanam L, Webb A, Camara A, Sikka G, Nyhan D, Shoukas AA, Ilies M, Christianson DW, Champion HC, Berkowitz DE. Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats. J Appl Physiol 107: 1249–1257, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liochev SI. Reactive oxygen species and the free radical theory of aging. Free Radic Biol Med 60: 1–4, 2013. [DOI] [PubMed] [Google Scholar]
- 35.Malik UZ, Hundley NJ, Romero G, Radi R, Freeman BA, Tarpey MM, Kelley EE. Febuxostat inhibition of endothelial-bound XO: implications for targeting vascular ROS production. Free Radic Biol Med 51: 179–184, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maller C, Schroder E, Eaton P. Glyceraldehyde 3-phosphate dehydrogenase is unlikely to mediate hydrogen peroxide signaling: studies with a novel anti-dimedone sulfenic acid antibody. Antioxid Redox Signal 14: 49–60, 2011. [DOI] [PubMed] [Google Scholar]
- 37.McVerry BJ, Peng X, Hassoun PM, Sammani S, Simon BA, Garcia JG. Sphingosine 1-phosphate reduces vascular leak in murine and canine models of acute lung injury. Am J Respir Crit Care Med 170: 987–993, 2004. [DOI] [PubMed] [Google Scholar]
- 38.Milovanova T, Chatterjee S, Hawkins BJ, Hong N, Sorokina EM, Debolt K, Moore JS, Madesh M, Fisher AB. Caveolae are an essential component of the pathway for endothelial cell signaling associated with abrupt reduction of shear stress. Biochim Biophys Acta 1783: 1866–1875, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mo L, Wang Y, Geary L, Corey C, Alef MJ, Beer-Stolz D, Zuckerbraun BS, Shiva S. Nitrite activates AMP kinase to stimulate mitochondrial biogenesis independent of soluble guanylate cyclase. Free Radic Biol Med 53: 1440–1450, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muzaffar S, Shukla N, Angelini GD, Jeremy JY. Acute hypoxia simultaneously induces the expression of gp91phox and endothelial nitric oxide synthase in the porcine pulmonary artery. Thorax 60: 305–313, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O'Rourke MF, Safar ME, Dzau V. The Cardiovascular Continuum extended: aging effects on the aorta and microvasculature. Vasc Med 15: 461–468, 2010. [DOI] [PubMed] [Google Scholar]
- 42.Peng X, Hassoun PM, Sammani S, McVerry BJ, Burne MJ, Rabb H, Pearse D, Tuder RM, Garcia JG. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med 169: 1245–1251, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Predescu D, Predescu S, Shimizu J, Miyawaki-Shimizu K, Malik AB. Constitutive eNOS-derived nitric oxide is a determinant of endothelial junctional integrity. Am J Physiol Lung Cell Mol Physiol 289: L371–L381, 2005. [DOI] [PubMed] [Google Scholar]
- 43a.Rey FE, Cifuentes ME, Kirash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O2- and systolic blood pressure in mice. Circ Res 89: 408–414, 2001. [DOI] [PubMed] [Google Scholar]
- 44.Sato K, Kadiiska MB, Ghio AJ, Corbett J, Fann YC, Holland SM, Thurman RG, Mason RP. In vivo lipid-derived free radical formation by NADPH oxidase in acute lung injury induced by lipopolysaccharide: a model for ARDS. FASEB J 16: 1713–1720, 2002. [DOI] [PubMed] [Google Scholar]
- 45.Schmidt EP, Damarla M, Rentsendorj O, Servinsky LE, Zhu B, Moldobaeva A, Gonzalez A, Hassoun PM, Pearse DB. Soluble guanylyl cyclase contributes to ventilator-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol 295: L1056–L1065, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shasby DM, Lind SE, Shasby SS, Goldsmith JC, Hunninghake GW. Reversible oxidant-induced increases in albumin transfer across cultured endothelium: alterations in cell shape and calcium homeostasis. Blood 65: 605–614, 1985. [PubMed] [Google Scholar]
- 47.Speyer CL, Neff TA, Warner RL, Guo RF, Sarma JV, Riedemann NC, Murphy ME, Murphy HS, Ward PA. Regulatory effects of iNOS on acute lung inflammatory responses in mice. Am J Pathol 163: 2319–2328, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stamm J, McVerry BJ, Mathier M, Donahoe M, Saul M, Gladwin MT. Doppler-defined pulmonary hypertension in medical intensive care unit patients: Retrospective investigation of risk factors and impact on mortality. Pulm Circ 1: 95–102, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szczepaniak WS, Zhang Y, Hagerty S, Crow MT, Kesari P, Garcia JG, Choi AM, Simon BA, McVerry BJ. Sphingosine 1-phosphate rescues canine LPS-induced acute lung injury and alters systemic inflammatory cytokine production in vivo. Transl Res 152: 213–224, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tie L, Li XJ, Wang X, Channon KM, Chen AF. Endothelium-specific GTP cyclohydrolase I overexpression accelerates refractory wound healing by suppressing oxidative stress in diabetes. Am J Physiol Endocrinol Metab 296: E1423–E1429, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Totzeck M, Hendgen-Cotta UB, Luedike P, Berenbrink M, Klare JP, Steinhoff HJ, Semmler D, Shiva S, Williams D, Kipar A, Gladwin MT, Schrader J, Kelm M, Cossins AR, Rassaf T. Nitrite regulates hypoxic vasodilation via myoglobin-dependent nitric oxide generation. Circulation 126: 325–334, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vaporidi K, Francis RC, Bloch KD, Zapol WM. Nitric oxide synthase 3 contributes to ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol 299: L150–L159, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA., Jr Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci USA 95: 9220–9225, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang W, Suzuki Y, Tanigaki T, Rank DR, Raffin TA. Effect of the NADPH oxidase inhibitor apocynin on septic lung injury in guinea pigs. Am J Respir Crit Care Med 150: 1449–1452, 1994. [DOI] [PubMed] [Google Scholar]
- 55.Wojciak-Stothard B, Tsang LY, Paleolog E, Hall SM, Haworth SG. Rac1 and RhoA as regulators of endothelial phenotype and barrier function in hypoxia-induced neonatal pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 290: L1173–L1182, 2006. [DOI] [PubMed] [Google Scholar]
- 56.Xia Y, Tsai AL, Berka V, Zweier JL. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J Biol Chem 273: 25804–25808, 1998. [DOI] [PubMed] [Google Scholar]
- 57.Yang Y, Yin J, Baumgartner W, Samapati R, Solymosi EA, Reppien E, Kuebler WM, Uhlig S. Platelet-activating factor reduces endothelial nitric oxide production: role of acid sphingomyelinase. Eur Respir J 36: 417–427, 2010. [DOI] [PubMed] [Google Scholar]