

Abstract
Recent findings suggest the therapeutic action of relaxin during hypertension is dependent on nitric oxide synthase (NOS) activation; however, the mechanisms underlying the beneficial effects of relaxin on the NOS system have not been fully elucidated. We hypothesized that the protective effects of relaxin include reducing both oxidative stress and the endogenous NOS inhibitor asymmetric dimethylarginine (ADMA). We examined the effect of Serelaxin [human recombinant relaxin-2 (RLX)] in male Sprague-Dawley rats given high-dose angiotensin (ANG) II (400 ng·kg−1·min−1 sc) for 6 wk or shams. RLX was administered (4 μg/h sc) to half of the rats in each group after 2 wk of ANG II for the remaining 4 wk. ANG II induced hypertension and proteinuria, reduced NO oxidation products (NOx), and increased oxidative stress (NADPH oxidase activity, thiobarbituric acid-reactive substances, and 8-isoprostane excretion) and plasma ADMA. While RLX had no effect on sham rats, RLX attenuated the ANG II-dependent hypertension (165 ± 5 vs. 135 ± 13 mmHg, P < 0.05) and proteinuria at 6 wk (62 ± 6 vs. 41 ± 4 mg·day−1·100 g−1, P < 0.05) and normalized oxidative stress and circulating ADMA, in association with restored NOx excretion and kidney cortex NOx. We found that RLX had no impact on the ADMA-regulatory enzymes protein arginine methyltransferase and dimethylarginine-dimethylaminohydrolase (DDAH). Furthermore, RLX treatment did not increase DDAH activity in kidney cortex or liver. These data suggest that benefits of RLX treatment include reduced ADMA levels and increased NO bioavailability, possibly due to its antioxidant effects.
Keywords: nitric oxide, protein arginine methyltransferase-1, dimethylarginine dimethylaminohydrolase, NADPH oxidase
nitric oxide (NO) is an important mediator of vascular tone and renal function, and NO regulates glomerular, vascular, and tubular function in the kidney (12). Therefore, agents that increase NO production and/or bioavailability both systemically and in the kidney may have therapeutic benefit during hypertension. Recent findings suggest a potential therapeutic action of Serelaxin, recombinant human relaxin-2 (RLX), likely via the relaxin peptide family receptor-1 (RXFP-1) (2) during hypertension that is dependent on NO synthase (NOS) activation (22). However, the mechanisms underlying the beneficial effects of RLX on the NOS system have not been fully elucidated.
Regulation of NO biosynthesis is complex, and the endogenous competitive inhibitor of NOS, asymmetric dimethylarginine (ADMA), is one important influence on NO production. We, and others, previously showed that ADMA is increased during chronic ANG II infusion (10, 14, 22). ADMA is generated by protein arginine methyltransferase (PRMT) class 1 enzymes, via methylation of arginine residues in proteins (1), which are then released during proteolysis. Some ADMA is excreted in the urine, but the major route of ADMA elimination is via metabolism by dimethylarginine dimethylaminohydrolase (DDAH) enzymes 1 and 2 (18). The kidney plays an important role in the metabolism of ADMA as the kidney cortex has the highest density of the DDAH enzymes of any organ (20). The DDAH enzymes are also expressed in many other organs, however, and the liver also plays a major role in ADMA catabolism (20).
Reactive oxygen species (ROS) are an important determinant of both NO bioavailability and ADMA levels. Superoxide reacts rapidly with NO to form peroxynitrite, thereby reducing NO bioavailability (31). Oxidative stress also “uncouples” NOS leading to superoxide, rather than NO production (30). In addition, oxidative stress upregulates PRMT-1 gene expression and decreases DDAH activity (27), thus promoting ADMA accumulation. Of note, increased oxidative stress is a feature of angiotensin-dependent hypertension (4, 9, 21).
Because we (22) and others (8, 32) showed that RLX attenuates hypertension and increases NO bioavailability, the purpose of this study was to determine whether the protective effects of RLX include 1) reducing the endogenous NOS inhibitor ADMA in chronic hypertension induced by angiotensin II (ANG II) infusion and 2) whether RLX reduces oxidative stress in ANG II hypertension. In this study, we tested the hypothesis that RLX treatment would improve NO bioavailability and reduce blood pressure in ANG II-dependent hypertension by decreasing oxidative stress and ADMA levels. This model of chronic ANG II infusion involves a “slow pressor” response, where the rise in blood pressure is delayed and in part due to ANG II activation of oxidative stress (9).
MATERIALS AND METHODS
All experiments were performed using male Sprague-Dawley rats (400 to 500 g; Harlan Laboratories) in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved and monitored by the University of Florida Institutional Animal Care and Use Committee. Animals were housed under conditions of constant temperature and humidity and exposed to a 12:12-h light-dark cycle. All rats were given free access to regular rat chow and water.
Rats were randomly assigned to one of four groups: control (CON), serelaxin-treated (RLX), ANG II hypertensive (ANG), and ANG II hypertensive with RLX treatment (ANG+RLX, n = 6–9 per group). ANG II (400 ng·kg−1·min−1; Sigma, St. Louis, MO) or saline vehicle was given via osmotic minipump (Alzet, model 2002, pump replaced every 2 wk) for 6 wk. After 2 wk of ANG II or vehicle treatment, Serelaxin, recombinant human relaxin-2 was then given to half of the rats in each group for the remaining 4 wk. Serelaxin was a gift from Corthera (San Mateo, CA) and was delivered via a separate minipump (Alzet 2ML2, replaced after 2 wk) at a rate of 4 μg/h (7).
At the end of the treatment period, all groups of rats were placed on a low-nitrate diet (custom AIN76C, MP Biomedicals, Solon, OH) for 24 h before urine collection. Then, rats were placed in metabolic cages overnight for the collection of urine. At death, mean arterial pressure (MAP) was measured via the abdominal aorta in anesthetized rats. Our use of the anesthetized terminal MAP measurement for assessment of hypertension in rats was previously validated (23). After MAP measurement, an aortic blood sample was collected and the kidneys were perfused blood free with cold PBS. The right kidney was removed and separated into cortex and medulla, sections of the liver and lung were removed, and the tissues were snap-frozen in liquid nitrogen.
Urine protein concentrations were measured by the Bradford method (BioRad). Total NO production (from NOx = NO3− + NO2−) was measured in urine and kidney cortex homogenates using the Griess reaction (26). Plasma and urine creatinine concentrations were measured by high-performance liquid chromatography as described by us previously (23). Plasma thiobarbituric acid-reactive substances (TBARS) concentrations were measured using the OxiTek TBARS assay kit (Zeptometrix), and urinary 8-isoprostane excretion was measured by EIA after pretreatment of the urine with β-glucuronidase (Oxford Biomedical).
NADPH oxidase-dependent superoxide production was measured by lucigenin (5 μmol/l final concentration, Sigma) chemiluminescence in the presence of NADPH (100 μmol/l final concentration, Sigma) as previously described (24). Kidney cortex samples were homogenized, protein concentrations were measured by the Bradford assay (BioRad), and 35 μg kidney cortex homogenate diluted in 0.9% NaCl were added to wells. Then, 0.9% NaCl or Tempol (10 mmol/l final concentration, Sigma) was added, and the plate was incubated for 30 min at 37°C. Lucigenin and NADPH were added to the wells using an autoinjector, and after a 30-min dark-adapt period, plates were counted on an Orion microplate luminometer. NADPH-stimulated superoxide detection was diminished by Tempol. Enzyme activity was expressed as relative light units (RLU)/μg protein − RLU/μg protein in the presence of Tempol.
ADMA and l-arginine levels were measured in plasma using reverse-phase HPLC with the Waters AccQ-Fluor fluorescent reagent kit using an adaptation of the method of Heresztyn et al. (11) as previously described (23).
Protein abundances were detected using Western blotting as previously described (23). Briefly, samples (200 μg of kidney cortex, 150 μg of liver, 75 μg of lung) were loaded onto 6% (NOS) or 12% (PRMT and DDAH) polyacrylamide gels and separated by electrophoresis. Membranes were incubated overnight with specific antibodies: anti-NOS1 antibody (1:50, to detect alpha isoform, Santa Cruz Biotechnology, Santa Cruz, CA), anti-NOS1 antibody (1:500, to detect beta isoform, Thermo Scientific, Rockford, IL), anti-NOS 3 antibody (1:250, BD Transduction, San Jose, CA), anti-DDAH1 antibody (1:250, Santa Cruz Biotechnology), anti-DDAH2 antibody (1:100, Santa Cruz Biotechnology), anti-PRMT-1 antibody (1:2,000, Upstate, Lake Placid, NY), anti-PRMT-3 antibody (1:250, Abcam, Cambridge, MA). The membranes were then incubated with corresponding secondary antibodies: anti-goat antibody (Santa Cruz Biotechnology), anti-rabbit antibody, or anti-mouse antibody (Bio-Rad, Hercules, CA). Bands of interest were visualized using enhanced chemiluminescence reagent and quantified by densitometry (VersaDoc imaging system and Quantity One Analysis software; Bio-Rad) as integrated optical density (IOD) after subtraction of background. The IOD was factored for Ponceau red staining (Sigma) to correct for any variations in total protein loading and for an internal positive control (PC). The protein abundance was represented as IOD/Ponceau/PC.
DDAH activity was measured by a colorimetric assay measuring the rate of citrulline production as previously described (29). Kidney cortex, liver, and lung were homogenized in sodium phosphate buffer. Tissue homogenate was preincubated with urease for 15 min, and then 100 μl of homogenate containing 2 mg of protein were incubated with 1 mmol/l ADMA for 45 min at 37°C. After deproteinization, the supernatant was incubated with the color mixture at 60°C for 110 min. The absorbance was measured on a Tecan Safire optical system (Tecan) at 466 nm. The DDAH activity was represented as μmol citrulline formation/g protein/min.
Results are presented as means ± SE. Data were analyzed by one-way ANOVA with Newman-Keuls post hoc analysis using SigmaPlot12 software (Systat Software, San Jose, CA). P < 0.05 was considered statistically significant.
RESULTS
RLX treatment alone had no effect on blood pressure, proteinuria, or NO oxidation products (NOx) in the urine or kidney cortex of normotensive rats (Fig. 1). Creatinine clearance was not changed by RLX treatment (1.3 ± 0.2 in CON vs. 1.0 ± 0.2 ml·min−1·100 g body wt−1 in RLX). Six-week infusion of ANG II increased blood pressure and caused significant proteinuria, and ANG II treatment also reduced NOx excretion rate and content in the kidney cortex; however, ANG II had no effect on creatinine clearance (1.0 ± 0.1 ml·min−1·100 g body wt−1). RLX attenuated the effects of ANG II on blood pressure and protein excretion, and normalized kidney cortex and urinary NOx levels, but did not increase creatinine clearance in ANG II-treated rats (0.8 ± 0.1 ml·min−1·100 g body wt−1).
Fig. 1.

Relaxin reduces blood pressure and proteinuria and preserves nitric oxide (NO) production during high-dose ANG II hypertension. Mean arterial pressure (MAP), urinary protein excretion (UProtV), kidney cortex NO metabolite (NOx) content, and urinary NOx excretion rate in control (CON), relaxin-treated (RLX), ANG II hypertensive (ANG), and ANG II hypertensive with relaxin treatment (ANG+RLX) rats; n = 6–9. *P < 0.05 vs. control. +P < 0.05 vs. ANG.
To determine whether RLX restores NOx levels in the renal cortex via an increase in abundance of the NOS enzymes, we examined protein levels of the constitutive NOS isoforms in the kidney cortex (Fig. 2). Neither RLX treatment nor ANG II infusion alone or in combination altered NOS1α (Fig. 2, A and D) or NOS1β (Fig. 2, B and D) in the kidney cortex. ANG II infusion resulted in an increase in NOS3 (Fig. 2, C and D) in the kidney cortex; however, RLX treatment alone did not alter NOS3 enzyme abundance, and RLX treatment in ANG II-treated rats normalized NOS3.
Fig. 2.

Relaxin treatment does not affect renal cortex abundance of NO synthase (NOS) 1 (A and B) or NOS3 (C). Western blot analysis of expression of NOS isoforms in the kidney cortex from control (CON), relaxin-treated (RLX), ANG II hypertensive (ANG), and ANG II hypertensive with relaxin treatment (ANG+RLX) rats; n = 6–9. *P < 0.05 vs. control. D: representative Western blots for the expression of NOS1 and NOS3. MW, molecular weight marker; +, positive control; C, control; R, relaxin-treated; A, ANG II hypertensive; AR, ANG II hypertensive with relaxin treatment.
Because NO levels are also modulated by the presence of ROS, we next measured markers of oxidative stress in rats treated with ANG II and/or RLX. RLX alone had no effect on NADPH oxidase activity in the kidney cortex or urinary excretion of TBARS or 8-isoprostane (Fig. 3). As expected, ANG II hypertension was associated with increases in kidney cortex NADPH oxidase activity and TBARS and 8-isoprostane excretion, and RLX treatment normalized the levels of all three of these markers in hypertensive rats.
Fig. 3.
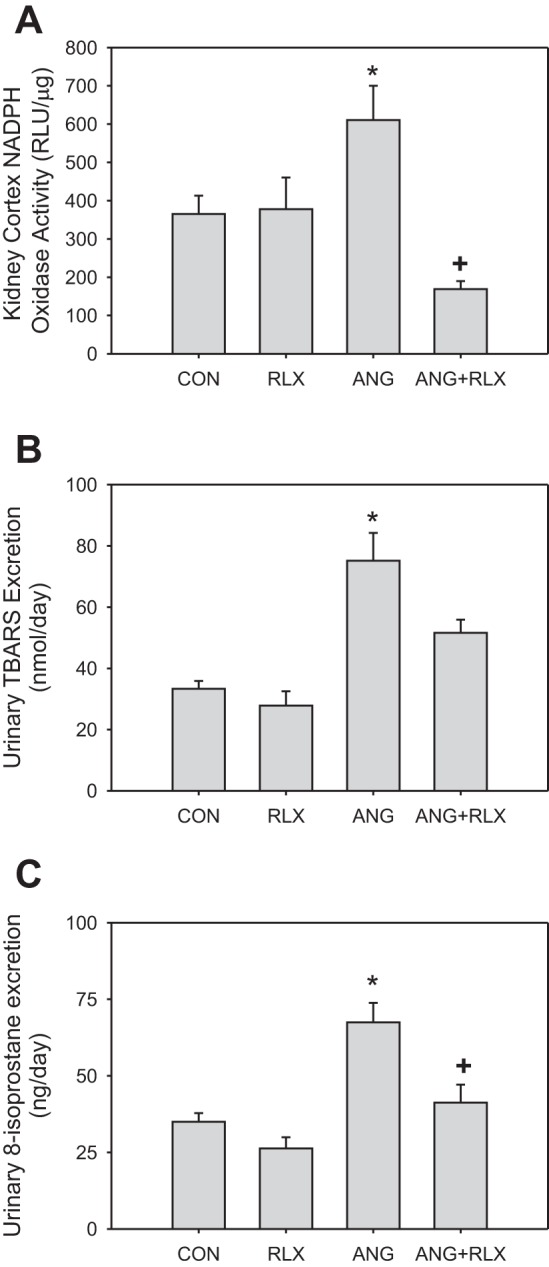
Relaxin reduces oxidative stress during high-dose ANG II infusion. NADPH oxidase activity levels (A) in the kidney cortex and urinary excretion of thiobarbituric acid-reactive substances (TBARS; B) and 8-isoprostane (C) in CON, RLX, ANG, and ANG+RLX rats; n = 6–8. *P < 0.05 vs. control. +P < 0.05 vs. ANG.
We also examined the impact of RLX on the levels of the endogenous NOS inhibitor ADMA and the NOS substrate l-arginine. ANG II treatment increased both ADMA and l-arginine levels (Fig. 4). The arginine-to-ADMA ratio (substrate:inhibitor) is an important determinant of NO production, and this was reduced by ANG II treatment. RLX had no effect on normotensive rats but normalized ADMA and l-arginine levels, thereby restoring the normal l-arginine-to-ADMA ratio in hypertensive rats.
Fig. 4.
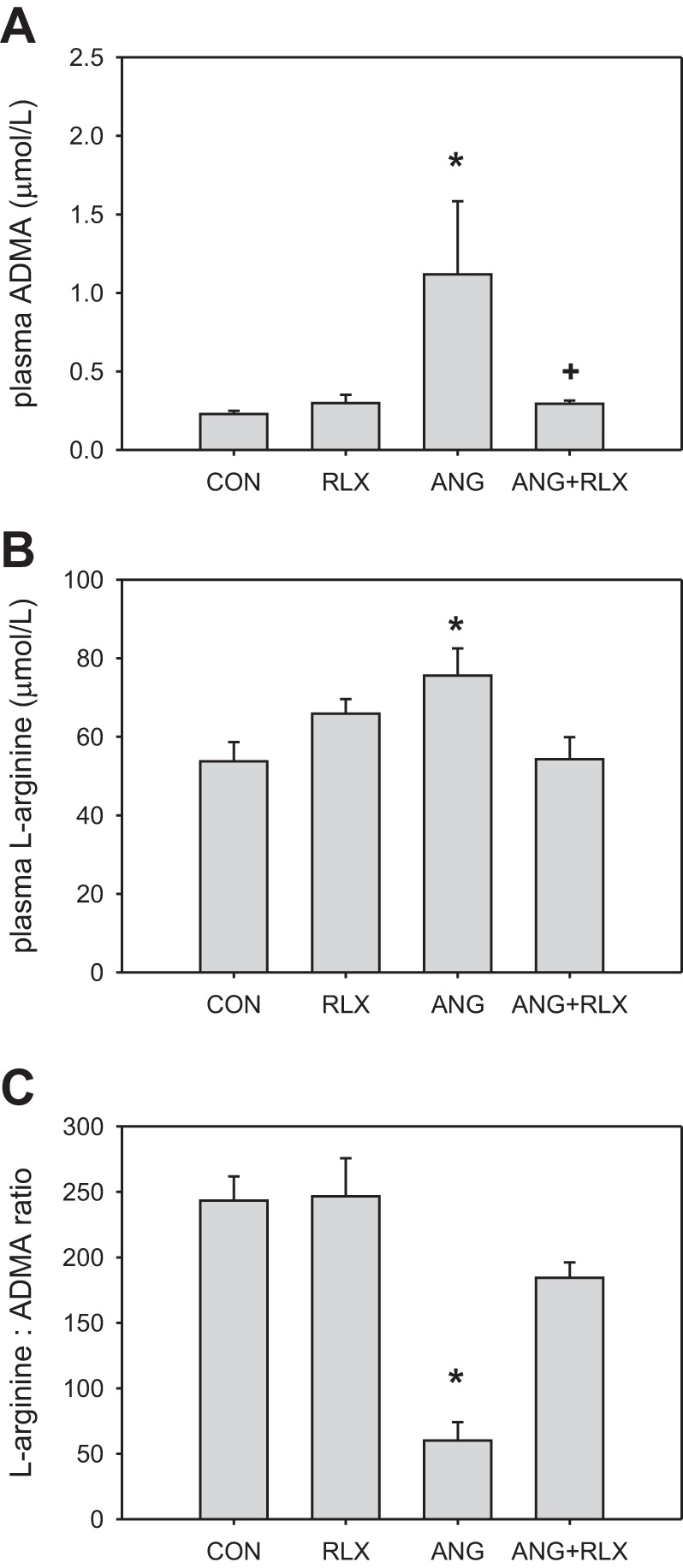
Relaxin reduces plasma asymmetric dimethylarginine (ADMA) levels during high-dose ANG II hypertension. Plasma concentrations of ADMA (A) and l-arginine (B) as measured by HPLC and the ratio of plasma l-arginine-to-plasma ADMA (C) in CON, RLX, ANG, and ANG+RLX rats; n = 6–9. *P < 0.05 vs. control. +P < 0.05 vs. ANG.
We also evaluated the protein abundance of the enzymes that regulate ADMA (PRMT-1 and PRMT-3, DDAH1, and DDAH2) by Western blot. No differences in PRMT-1 or in PRMT-3 abundance were observed among the groups in the kidney cortex, liver, or lung (Fig. 5). No differences were observed in DDAH1 or DDAH2 in either the kidney cortex or liver among groups (Fig. 6, A-D, G). Total DDAH activity, as measured by the production of citrulline, was decreased in the kidney cortex following RLX and/or ANG II treatment; however, activity was similar among groups in the liver.
Fig. 5.

Relaxin has no effect on protein arginine methyltransferase (PRMT) abundance in the kidney cortex, liver, or lung. Western blot analysis of expression of PRMT isoforms in the renal cortex (A-B), liver (C-D), and lung (E-F) of CON, RLX, ANG, and ANG+RLX rats; n = 6–8. G: representative Western blots for the expression of PRMT-1 and PRMT-3.
Fig. 6.

Relaxin does not increase dimethylarginine dimethylaminohydrolase (DDAH) abundance or activity in the kidney cortex or liver. Western blot analysis of expression of DDAH isoforms (A-D) and total DDAH enzymatic activity (E-F) in the kidney cortex and liver from CON, RLX, ANG, and ANG+RLX rats. DDAH activity is expressed as μmol citrulline produced per g of protein per min; n = 6–8. *P < 0.05 vs. control. G: representative Western blots for the expression of DDAH-1 and DDAH-2.
DISCUSSION
In the present study, we report that RLX normalizes MAP, proteinuria, and NO production in rats subjected to chronic ANG II infusion (400 ng·kg−1·min−1) over 6 wk. These findings support and expand on our previous observation that the protective actions of RLX on a lower dose of chronic ANG II (200 ng·kg−1·min−1) for 3 wk are mediated by restoration of NO production (22). The novel findings in the present study are that 1) RLX is effective in preventing ANG II-induced oxidative stress, indicated by reductions in NADPH oxidase activity and measures of lipid peroxidation (TBARS and 8-isoprostane), and 2) RLX prevents the ANG II-mediated increases in plasma ADMA, restoring the l-arginine:ADMA ratio in rats with ANG II-dependent hypertension. Thus, this work extends the current understanding of the cardiorenal effects of RLX by demonstrating the ability of RLX to attenuate oxidative stress and ADMA levels in chronic ANG II-induced hypertension.
NO plays an important role in the regulation of blood pressure and the maintenance of renal health. There is increasing evidence that some actions of RLX are mediated through increased abundance or activity of the NOS enzymes. RLX activates different isoforms of NOS in various tissues (for a review, see Ref. 25), and McGuane and colleagues (17) reported that endothelial NOS phosphorylation contributes to RLX-induced vasodilation. We previously reported that the anti-hypertensive effects of RLX during 3 wk of ANG II infusion are dependent on activation of NOS and that RLX had no protective effect on blood pressure or renal injury during hypertension induced by chronic NOS inhibition (22). In the current study, RLX treatment during 6 wk of chronic ANG II-induced hypertension was associated with restoration of the NOx levels in the kidney cortex and in the urine. Based on the Western blot data, however, this restoration of NO production by RLX was not due to increased NOS1 or NOS3 protein abundance. The major renal NOS in the normal kidney is NOS1α, and neither RLX treatment nor ANG II infusion alone or in combination altered levels of this protein. NOS1β is present in very low abundance in the normal kidney but is activated in some models of chronic kidney disease that feature significant glomerulosclerosis (3). However, the chronic ANG II infusion model is associated with only mild glomerulosclerosis (13, 23) despite the presence of significant proteinuria, macrophage infiltration, and interstitial fibrosis (19). In the current study, ANG II treatment had no effect on renal cortex NOS1β; RLX was also without effect on this protein. Chronic ANG II administration resulted in an increase in NOS3 protein in the kidney cortex which in the context of the concurrent increase in oxidative stress could be maladaptive. In the presence of superoxide, increased NOS3 could contribute to further ROS production via NOS uncoupling or generation of peroxynitrite (30). While RLX alone did not alter NOS3 enzyme abundance, the RLX treatment in ANG II-treated rats reduced NOS3 protein levels to normal. These findings are in contrast to the effects of RLX in the Dahl salt-sensitive rats where porcine RLX increases both NOS1 and NOS3 in kidney (32). While it is unclear why the opposite results were observed in these two models of experimental hypertension, it should be noted that there are differences in the source of RLX (recombinant human vs. porcine) and that the Dahl salt-sensitive rat is a known model of NO deficiency and low NOS expression (5, 28).
The regulation of NOS activity depends on much more than simply abundance of the enzyme. There is considerable posttranslational modification of NOS activity and one major influence is via oxidative stress. Oxidation of the essential NOS cofactor tetrahydrobioptern (BH4) to BH2, and the subsequent BH4 deficiency, uncouples NOS and converts it to a superoxide generator (30). Superoxide also directly inactivates NO by combining to form toxic peroxynitrite which in turn produces the vasoconstrictor isoprostanes (21). Chronic ANG II infusion is known to cause widespread oxidative stress, indicated by increased vascular and renal cortex NADPH oxidase activity (4, 9). Chronic ANG II infusion also leads to elevations in 8-isoprostane production (21), which taken together implicate increases in superoxide production during chronic ANG II infusion. In the present study, we confirmed increased renal cortical NADPH oxidase activity with 6 wk chronic ANG II as well as increased urinary excretion of both TBARS and 8-isoprostane. The novel finding in our study is that RLX restores all these oxidative stress markers to normal, implying that RLX inhibits the ANG II stimulation of superoxide. Although this pathway has not been studied in other hypertensive models, previous studies in models of ischemia-reperfusion have demonstrated antioxidant effects of RLX treatment. RLX decreased lipid peroxidation and 8-hydroxy-2′-deoxyguanosine levels and increased the expression of manganese superoxide dismutase in ileal tissue from animals subjected to splanchnic artery occlusion and also decreased lipid peroxidation in myocardial tissue following ischemia-reperfusion injury (15, 16). The effects of RLX to either inhibit ROS production or enhance the neutralization of ROS should be further explored in hypertensive models. Recent work by Chow et al. (6) also demonstrated that the renal anti-fibrotic actions of RLX are dependent on heterodimerization of the RLX receptor, RXFP-1, and the angiotensin type 2 receptor, AT2. It is possible that this interaction may inhibit activation of the AT1 receptor, thereby providing a potential mechanism by which RLX treatment reduces oxidative stress during ANG II-dependent hypertension.
Thus, the RLX-induced inhibition of ANG II-stimulated ROS certainly contributes to the restoration of normal NO production in the present study. We also examined the impact of RLX on the levels of the endogenous NOS inhibitor ADMA in the present study. We and others previously demonstrated that ANG II is associated with increased ADMA levels (10, 14, 23); however, the effects of RLX on regulation of ADMA, PRMT, and DDAH activity have not previously been examined. In the present study, ANG II treatment increased both ADMA and l-arginine levels but with a more marked effect on ADMA; hence, the arginine-to-ADMA ratio was reduced by ANG II treatment which promotes falls in NO production. RLX had no effect on normotensive rats, but in chronic ANG II-infused rats RLX restored the normal l-arginine-to-ADMA ratio, which facilitates increased NO production.
RLX could reduce ADMA levels via a reduction in ADMA synthesis/release and/or increased metabolic degradation by DDAH. There were no differences in the PRMT-1 and PRMT-3 enzymes in kidney cortex, liver, or lung. Since concomitant RLX did not reduce PRMT levels, but did return plasma ADMA to normal, this is unlikely to be the cause of the increased ADMA with chronic ANG II. We have no measures of PRMT activity in these studies, and it is possible that increased activity occurred in some locations in the body, in the absence of increased protein abundance. Another possibility is that the inflammatory state induced by chronic ANG II promoted proteolysis with increased release of free ADMA. Indeed, chronic ANG II infusion in mice leads to skeletal muscle breakdown via activation of inflammatory pathways (33).
Plasma ADMA levels are also regulated by degradation by the DDAH enzymes, and DDAH activity in the kidney and liver accounts for the majority of DDAH catabolism (20). We found no impact of chronic ANG II, RLX, or ANG II + RLX on the abundance of DDAH1 and DDAH2 in kidney cortex or liver. The “in vitro” DDAH activity was similar among groups in the liver but decreased in the kidney cortex during chronic ANG II. However, similar falls in kidney cortex DDAH were also seen with RLX alone or RLX + ANG II treatment and thus cannot account for the ANG II-mediated rise in plasma ADMA. It is reasonable to hypothesize that RLX may have effects on other target organs, possibly by modulating DDAH activity in the vasculature.
In conclusion, these data reinforce previous findings that agents that stimulate the RLX pathway may be a therapeutic target for the treatment of hypertension. Understanding the pathways by which RLX preserves NO bioavailability could be an important key unlocking the potential to target this pathway for hypertension and the associated end-organ damage.
GRANTS
This work was supported by National Institutes of Health (NIH) Grant K01 DK095018 to J. M. Sasser and NIH Grant R01 DK56843 to C. Baylis.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.M.S. and C.B. conception and design of research; J.M.S. and M.W.C. performed experiments; J.M.S. and M.W.C. analyzed data; J.M.S., M.W.C., and C.B. interpreted results of experiments; J.M.S. and M.W.C. prepared figures; J.M.S. drafted manuscript; J.M.S., M.W.C., and C.B. edited and revised manuscript; J.M.S. and C.B. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Harold Snellen for expert technical assistance and Dennis Stewart and Corthera for providing recombinant human relaxin-2.
REFERENCES
- 1.Anthony S, Leiper J, Vallance P. Endogenous production of nitric oxide synthase inhibitors. Vasc Med 10: 3–9, 2005. [DOI] [PubMed] [Google Scholar]
- 2.Bathgate RD, Halls ML, Van der Westhuizen ET, Callander GE, Kocan M, Summers RJ. Relaxin family peptides and their receptors. Physiol Rev 93: 405–480, 2013. [DOI] [PubMed] [Google Scholar]
- 3.Baylis C. Nitric oxide deficiency in chronic kidney disease. Am J Physiol Renal Physiol 294: F1–F9, 2008. [DOI] [PubMed] [Google Scholar]
- 4.Chabrashvili T, Kitiyakara C, Blau J, Karber A, Aslam S, Welch WJ, Wilcox CS. Effects of ANG II type 1 and 2 receptors on oxidative stress, renal NADPH oxidase, and SOD expression. Am J Physiol Regul Integr Comp Physiol 285: R117–R124, 2003. [DOI] [PubMed] [Google Scholar]
- 5.Chen PY, Sanders PW. l-arginine abrogates salt-sensitive hypertension in Dahl/Rapp rats. J Clin Invest 88: 1559–1567, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chow BSM, Kocan M, Bosnyak S, Sarwar M, Wigg B, Jones ES, Widdop RE, Summers RJ, Bathgate RAD, Hewitson TD, Samuel CS. Relaxin requires the angiotensin II type 2 receptor to abrogate renal interstitial fibrosis. Kidney Int 86: 75–85, 2014. [DOI] [PubMed] [Google Scholar]
- 7.Danielson L, Sherwood OD, Conrad KP. Relaxin is a potent renal vasodilator in conscious rats. J Clin Invest 103: 525–533, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fallowfield J, Hayden AL, Snowdon VK, Aucott RL, Stutchfield BM, Mole DJ, Pellicoro A, Gordon-Walker TT, Henke A, Schrader J, Trivedi PJ, Princivalle M, Forbes SJ, Collins JE, Iredale JP. Relaxin modulates human and rat hepatic myofibroblast function and ameliorates portal hypertension in vivo. Hepatology 59: 1492–1504, 2014. [DOI] [PubMed] [Google Scholar]
- 9.Fukui T, Ishizaka N, Rajagopalan S, Laursen JB, Capers Q, Taylor WR, Harrison DG, De Leon H, Wilcox JN, Griendling KK. p22phox mRNA expression and NADPH oxidase activity are increased in aortas from hypertensive rats. Circ Res 80: 45–51, 1997. [DOI] [PubMed] [Google Scholar]
- 10.Hasegawa K, Wakino S, Tatematsu S, Yoshioka K, Homma K, Sugano N, Kimoto M, Hayashi K, Itoh H. Role of asymmetric dimethylarginine in vascular injury in transgenic mice overexpressing dimethylarginie dimethylaminohydrolase 2. Circ Res 101: e2–e10, 2007. [DOI] [PubMed] [Google Scholar]
- 11.Heresztyn T, Worthley MI, Horowitz JD. Determination of l-arginine and NG, NG- and NG, NG′-dimethyl-l-arginine in plasma by liquid chromatography as AccQ-Fluor fluorescent derivatives. J Chromatogr B Analyt Technol Biomed Life Sci 805: 325–329, 2004. [DOI] [PubMed] [Google Scholar]
- 12.Kone BC, Baylis C. Biosynthesis and homeostatic roles of nitric oxide in the normal kidney. Am J Physiol Renal Physiol 272: F561–F578, 1997. [DOI] [PubMed] [Google Scholar]
- 13.Lombardi D, Gordon KL, Polinsky P, Suga S, Schwartz SM, Johnson RJ. Salt-sensitive hypertension develops after short-term exposure to angiotensin II. Hypertension 33: 1013–1019, 1999. [DOI] [PubMed] [Google Scholar]
- 14.Luo Z, Teerlink T, Griendling K, Aslam S, Welch WJ, Wilcox CS. Angiotensin II and NADPH oxidase increase ADMA in vascular smooth muscle cells. Hypertension 56: 498–504, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Masini E, Bani D, Bello MG, Bigazzi M, Mannaioni PF, Sacchi TB. Relaxin counteracts myocardial damage induced by ischemia-reperfusion in isolated guinea pig hearts: evidence for an involvement of nitric oxide. Endocrinology 138: 4713–4720, 1997. [DOI] [PubMed] [Google Scholar]
- 16.Masini E, Cuzzocrea S, Mazzon E, Muià C, Vannacci A, Fabrizi F, Bani D. Protective effects of relaxin in ischemia/reperfusion-induced intestinal injury due to splanchnic artery occlusion. Br J Pharmacol 148: 1124–1132, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGuane JT, Debrah JE, Sautina L, Jarajapu YPR, Novak J, Rubin JP, Grant MB, Segal M, Conrad KP. Relaxin induces rapid dilation of rodent small renal and human subcutaneous arteries via PI3 kinase and nitric oxide. Endocrinology 152: 2786–2796, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ogawa T, Kimoto M, Sasaoka K. Occurrence of a new enzyme catalyzing the direct conversion of NG,NG-dimethyl-l-arginine to l-citrulline in rats. Biochem Biophys Res Commun 148: 671–677, 1987. [DOI] [PubMed] [Google Scholar]
- 19.Ozawa Y, Kobori H, Suzaki Y, Navar LG. Sustained renal interstitial macrophage infiltration following chronic angiotensin II infusions. Am J Physiol Renal Physiol 292: F330–F339, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palm F, Onozato ML, Luo Z, Wilcox CS. Dimethylarginine dimethylaminohydrolase (DDAH): expression, regulation, and function in the cardiovascular and renal systems. Am J Physiol Heart Circ Physiol 293: H3227–H3245, 2007. [DOI] [PubMed] [Google Scholar]
- 21.Reckelhoff JF, Romero JC. Role of oxidative stress in angiotensin-induced hypertension. Am J Physiol Regul Integr Comp Physiol 284: R893–R912, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Sasser JM, Molnar M, Baylis C. Relaxin ameliorates hypertension and increases nitric oxide metabolite excretion in angiotensin II but not N(ω)-nitro-l-arginine methyl ester hypertensive rats. Hypertension 58: 197–204, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sasser JM, Moningka NC, Cunningham MW, Croker B, Baylis C. Asymmetric dimethylarginine in angiotensin II-induced hypertension. Am J Physiol Regul Integr Comp Physiol 298: R740–R746, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sasser JM, Sullivan JC, Elmarakby AA, Kemp BE, Pollock DM, Pollock JS. Reduced NOS3 phosphorylation mediates reduced NO/cGMP signaling in mesenteric arteries of deoxycorticosterone acetate-salt hypertensive rats. Hypertension 43: 1080–1085, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Sasser JM. The emerging role of relaxin as a novel therapeutic pathway in the treatment of chronic kidney disease. Am J Physiol Regul Integr Comp Physiol 305: R559–R565, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sütö T, Losonczy G, Qiu C, Hill C, Samsell L, Ruby J, Charon N, Venuto R, Baylis C. Acute changes in urinary excretion of nitrite + nitrate do not necessarily predict renal vascular NO production. Kidney Int 48: 1272–1277, 1995. [DOI] [PubMed] [Google Scholar]
- 27.Sydow K, Münzel T. ADMA and oxidative stress. Atheroscler Suppl 4: 41–51, 2003. [DOI] [PubMed] [Google Scholar]
- 28.Szentiványi M, Zou AP, Mattson DL, Soares P, Moreno C, Roman RJ, Cowley AW. Renal medullary nitric oxide deficit of Dahl S rats enhances hypertensive actions of angiotensin II. Am J Physiol Regul Integr Comp Physiol 283: R266–R272, 2002. [DOI] [PubMed] [Google Scholar]
- 29.Tain YL, Baylis C. Determination of dimethylarginine dimethylaminohydrolase activity in the kidney. Kidney Int 72: 886–889, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vásquez-Vivar J, Kalyanaraman B, Martásek P. The role of tetrahydrobiopterin in superoxide generation from eNOS: enzymology and physiological implications. Free Radic Res 37: 121–127, 2003. [DOI] [PubMed] [Google Scholar]
- 31.Wilcox CS. Oxidative stress and nitric oxide deficiency in the kidney: a critical link to hypertension? Am J Physiol Regul Integr Comp Physiol 289: R913–R935, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Yoshida T, Kumagai H, Suzuki A, Kobayashi N, Ohkawa S, Odamaki M, Kohsaka T, Yamamoto T, Ikegaya N. Relaxin ameliorates salt-sensitive hypertension and renal fibrosis. Nephrol Dial Transplant 27: 2190–2197, 2012. [DOI] [PubMed] [Google Scholar]
- 33.Zhang L, Du J, Hu Z, Han G, Delafontaine P, Garcia G, Mitch WE. IL-6 and serum amyloid A synergy mediates angiotensin II-induced muscle wasting. J Am Soc Nephrol 20: 604–612, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]