

Abstract
Acute kidney injury is common and has a high mortality rate, and no effective treatment exists other than supportive care. Using cell culture models, we previously demonstrated that exocyst Sec10 overexpression reduced damage to renal tubule cells and speeded recovery and that the protective effect was mediated by higher basal levels of mitogen-activated protein kinase (MAPK) signaling. The exocyst, a highly-conserved eight-protein complex, is known for regulating protein trafficking. Here we show that the exocyst biochemically interacts with the epidermal growth factor receptor (EGFR), which is upstream of MAPK, and Sec10-overexpressing cells express greater levels of phosphorylated (active) ERK, the final step in the MAPK pathway, in response to EGF stimulation. EGFR endocytosis, which has been linked to activation of the MAPK pathway, increases in Sec10-overexpressing cells, and gefitinib, a specific EGFR inhibitor, and Dynasore, a dynamin inhibitor, both reduce EGFR endocytosis. In turn, inhibition of the MAPK pathway reduces ligand-mediated EGFR endocytosis, suggesting a potential feedback of elevated ERK activity on EGFR endocytosis. Gefitinib also decreases MAPK signaling in Sec10-overexpressing cells to levels seen in control cells and, demonstrating a causal role for EGFR, reverses the protective effect of Sec10 overexpression following cell injury in vitro. Finally, using an in vivo zebrafish model of acute kidney injury, morpholino-induced knockdown of sec10 increases renal tubule cell susceptibility to injury. Taken together, these results suggest that the exocyst, acting through EGFR, endocytosis, and the MAPK pathway is a candidate therapeutic target for acute kidney injury.
Keywords: AKI, EGFR, exocyst, MAPK
acute kidney injury is a significant and increasing problem that occurs in 1–7% of hospitalizations and up to 25% of intensive care unit (ICU) admissions. Mortality rates in affected patients, especially in ICUs, can be as high as 50–70% (1, 4, 8, 26, 42, 47). Acute kidney injury is also a significant contributor to the progression of chronic kidney disease (45, 49). Unfortunately, there are still no effective treatments for acute kidney injury other than supportive care. Acute kidney injury most often results from a decrease in effective blood flow to the kidney, such as during an ischemia and reperfusion injury. In this case, reactive oxygen species are generated and renal tubular epithelial cells are severely damaged. The tubular cells lose their ability to form a tight epithelial barrier or maintain polarity, and substantial numbers undergo cell death by apoptosis and/or necrosis. Dead cells are sloughed into the lumen, while dedifferentiation and spreading of remaining viable cells attempt to cover the denuded areas. These remaining tubular cells struggle to proliferate, redifferentiate, and reestablish polarity and a tight epithelial barrier with normal physiological activity (41). To find novel therapeutic targets for acute kidney injury, it seems critical to identify factors that either mediate renal tubule cell damage or stimulate epithelial recovery following injury.
Protein trafficking is essential for such crucial cell functions as delivery of membrane and secreted proteins, establishment and maintenance of polarity, and complex morphogenesis such as tubulogenesis. An essential step in this process involves targeting and docking secretory vesicles carrying polarized proteins, which is mediated by the exocyst complex (24). This is followed by fusion of these vesicles with the plasma membrane and release of the vesicular protein cargo (37). The 750-kDa exocyst complex is comprised of Sec3, Sec5, Sec6, Sec8, Sec10, Sec15, Exo70, and Exo84 (40) and is highly conserved from yeast to humans (16, 30). Sec10 and Sec15 are the most vesicle proximal exocyst components and act as a bridge between Rab GTPases found on the surface of the secretory vesicles and the rest of the exocyst complex in contact with the plasma membrane (12). We and others previously showed that the exocyst traffics primarily basolateral membrane proteins in polarized epithelial cells (10, 22).
The Sec10 subunit is thought to be a central component of the exocyst complex, and modifying levels of Sec10 expression has been shown to result in parallel changes in overall exocyst activity. In yeast, overexpression of a Sec10 NH2-terminal dominant negative displaced the full-length Sec10 from the exocyst complex and resulted in a block of exocytosis and accumulation of secretory vesicles (36). In mammals, overexpression of the NH2-terminal Sec10 fragment acted as a dominant negative and inhibited neurite outgrowth (44). We previously showed that shRNA-mediated knockdown of Sec10 caused protein degradation of other exocyst subunits and decreased exocyst trafficking activity in mammalian epithelial cells (52). Conversely, overexpression of only the Sec10 subunit led to increases in exocyst-mediated trafficking to basolateral surfaces (22) and primary cilia (52). Sec10 overexpression also induced phenotypic changes such as increased tubulogenesis in Madin-Darby canine kidney (MDCK) cell cysts (22).
A well-described in vitro model of the oxidative stress that renal tubule cells incur during acute kidney injury is a brief treatment with H2O2 (39). Following H2O2 treatments, the degree of injury and the time it takes for the injured renal tubule cells to recover their barrier integrity can be measured using transepithelial resistance (TER) as a marker (23, 28, 34). We recently found that stable overexpression of Sec10 in MDCK cells significantly increased phosphorylated (active) ERK, decreased the degree of H2O2 injury, and increased the rate of epithelial barrier recovery following injury. If the MEK inhibitor U0126 was added to prevent ERK phosphorylation, recovery after H2O2 injury was decreased to the same rate as control cells. These data indicated that the faster recovery measured in exocyst-enhanced epithelial cells is mediated through the MAPK pathway (34).
The epidermal growth factor receptor (EGFR) is a member of the ErbB family of receptor tyrosine kinases. Binding of EGF (and other ligands such as transforming growth factor-α) promotes EGFR homo- and heterodimer formation and the autophosphorylation of cytoplasmic regulatory tyrosines that connect the receptor to downstream signaling pathways. Phosphorylated EGFR receptors are internalized primarily via a clathrin-dependent pathway and shuttled to sorting endosomes (20). At this point, phosphorylated EGFR is either sent on to late endosomes, before degradation in the lysosome, or returned to the cell surface (48). EGF binding of EGFR is one of several upstream stimuli that activate the classic MAPK phosphorylation cascade of Raf-MEK-ERK (2).
EGFR gene expression, abundance of EGFR ligands, and EGFR activation have all been shown to increase in a variety of models of experimental acute kidney injury, including ischemia and reperfusion injury (14, 15, 21, 38, 50). Administration of exogenous EGFR ligands has also been shown to accelerate recovery from ischemic acute kidney injury (17, 29). Recently, Chen et al. (3) showed that out of 39 different phosphorylated mouse receptor tyrosine kinases, phospho-EGFR was the most markedly upregulated in response to renal ischemia and reperfusion injury. They went on to show that both administration of erlotinib (a specific EGFR inhibitor) and targeted deletion of the EGFR gene in the renal proximal tubule resulted in markedly delayed structural and functional recovery following ischemia and reperfusion injury (3).
Here we show that the mechanism by which Sec10 overexpression increases ERK phosphorylation and recovery from oxidative injury in renal tubular MDCK cells is EGFR, MAPK, and endocytosis dependent. Furthermore, knockdown of sec10 in vivo in zebrafish results in greater kidney tubule cell damage following photoablation injury.
METHODS
Cell culture and reagents.
All MDCK cell lines used were derived from low passage type II MDCK cells that were obtained from Dr. K. Mostov (University of California, San Francisco, CA) and that were originally cloned by Dr. D. Louvard (European Molecular Biology Laboratory, Heidelberg, Germany). Human Sec10 cDNA with a myc epitope tag added to the COOH terminus was cloned into the pcDNA3 mammalian expression vector, where expression is driven by a CMV promoter (11). This plasmid was stably transfected into type II MDCK cells, and monoclonal lines were selected that stably overexpress Sec10, as described in Lipschutz et al. (22). Stable monoclonal MDCK cell lines with shRNA-mediated Sec10 knockdown were previously generated and characterized by our laboratory (52).
GST pull-downs.
Full-length human Sec10 cDNA was cloned in frame into the plasmid pGEX-4T-1 (Amersham Biosciences, Piscataway, NJ) and transformed into the DE3 strain of Escherichia coli (Stratagene, La Jolla, CA). GST fusion protein expression was induced by adding isopropyl-1-thio-β-d-galactopyranoside to growing cultures and shaking for an additional 3 h at 37°C. Recombinant proteins were purified with glutathione-Sepharose (Amersham Biosciences) following bacterial cell lysis. For pull-down experiments, lysates from wild-type MDCK cells were incubated for 1 h with Sec10-GST, or GST only, bound to glutathione-Sepharose. Pull-downs were washed extensively and resuspended in Laemmli buffer and boiled. Equal amounts were electrophoresed by SDS-PAGE and analysed with Western blotting.
Western blot analysis.
For pull-downs from MDCK cell lysates, immunoblotting was performed with standard methods as previously described (52). Briefly, proteins separated by SDS-PAGE were then transferred to either nitrocellulose or PVDF membranes using a semidry transfer system (Bio-Rad). After being blocked with 5% milk, the membranes were incubated with primary antibodies overnight at 4°C. After being washed, the membrane was then incubated with secondary antibodies either labeled with horseradish peroxidase for chemiluminescent detection or with fluorescence (IRDye; LI-COR Biosciences). The LI-COR Odyssey Imager (LI-COR Biosciences) was used for fluorescent detection of proteins, and the LI-COR Image Studio Lite software was used to quantify band intensities. The primary antibodies used in this study were mouse monoclonal anti-GAPDH (G8795; Sigma, St. Louis, MO), anti-phospho-EGFR (Tyr1068, cat. no. 3777), anti-total-EGFR (cat. no. 4267), anti-phospho-ERK (Thr202/Tyr204, cat. no. 4370), anti-total-ERK (cat. no. 4695) each from Cell Signaling Technology (Beverly, MA), and a rabbit polyclonal anti-Sec10 that we generated and previously characterized (52). Statistical analysis of Western band intensities was performed with Graphpad Prism software, and P values were calculated using either Student's t-test (for two-group comparisons) or one-way ANOVA analysis followed by post hoc Tukey multiple comparisons (for multiple group comparisons).
EGF binding and internalization assay.
Recombinant human EGF (Peprotech, Rocky Hill, NJ) was labeled with 125I (PerkinElmer, Waltham, MA), and the activity of labeled EGF was determined using a phosphotungstic acid precipitation assay. Rate constants of EGF internalization (ke) were measured as previously described with the time scale of measurements being short enough that the effects of recycling were minimal (19, 25). Briefly, serum-starved cells were treated with labeled ligand at 37°C for five different time points from 0 to 7.5 min. Surface-bound and internalized fractions of labeled ligand were collected at five evenly spaced time points by acid-stripping or NaOH solubilization, respectively, and the activities of these fractions were quantified using a 1470 Wizard Gamma Counter (PerkinElmer). From these data, ke was calculated using a simple kinetic model, which included corrections for the effects of nonspecific binding and surface spillover. To determine the effects of inhibition of EGFR, MEK, or dynamin on ke, cells were treated for 1 h with 1 μM gefitinib (LC Labs, Woburn, MA), 10 μM U0126 (LC Labs), or 80 μM Dynasore (EMD Millipore, Billerica, MA) before ke measurement. Stocks of gefitinib, U0126, and Dynasore were prepared in DMSO. Inhibitors were included with 125I-EGF during measurements. After ke values were calculated for each independent experiment, statistical comparisons were performed with Graphpad Prism software using Student's t-test to calculate P values.
Measurement of TER.
Measurement of TER was performed as previously described (34). Briefly, control cells and Sec10-overexpressing cells were grown in triplicate on Transwell filters until TER levels reached steady state (3–5 days postconfluency). For H2O2 injury assays, the filters were incubated with freshly prepared 1 mM H2O2 in complete cell medium for 30 min and then changed back to normal complete cell medium for analysis of recovery. Half the filters were also incubated with 1 μM gefitinib for 1 h before and during the 30-min H2O2 treatment, with continued gefitinib treatment for 7 h afterward. At the indicated time points, TER was measured using an epithelial volt-ohmmeter (model EVOM; World Precision Instruments). Absolute TER values were determined by subtracting the TER of blank filters with medium from all samples, and the unit area resistance was obtained by multiplying the absolute TER by the effective surface area of the filter membrane. Statistical analysis was performed using SPSS software and included direct comparison of measured data at identical timepoints between two groups using Student's t-test. To measure and compare epithelial recovery following injury, additional analysis included linear regressions of recovery times beginning at the nadir of TER (maximum injury) and statistical comparison of the calculated slopes (ohm × cm2/h).
Photoablation of kidney epithelia in zebrafish.
All zebrafish experiments were approved by the Institutional Animal Care and Use Committee at Harvard. The ET(krt8:EGFP)sqet11–9 zebrafish line (henceforth referred to as ET11–9) was used for laser photoablation, which was recently described as a novel technique for studying renal injury in zebrafish (32). This zebrafish line has a part of the proximal tubule and distal tubule labeled with EGFP (6, 33). Embryos were produced by in-crossing heterozygous ET11–9 fish. They were injected at the 1-to-8 cell stage with 28 ng of sec10 morpholino (diluted in 0.5–1 nl of 100 mM KCl, 10 mM HEPES, and 0.1% phenol red at neutral pH), using the Drummond Nanoliter 2000 microinjector (WPI), or with equivalent amounts of standard control morpholino (Genetools). The sec10 morpholinos, the resulting phenotype, and their efficiency at downregulating sec10 have been described in detail in Fogelgren et al. (9). Given variable penetrance of injected morpholinos, sec10 morphant embryos were selected for photoablation injury based on the presence of the described “curly tail up” phenotype and the lack of general toxicity. Embryos were raised in E3 solution until 24 h postfertilization (hpf) and in PTU-containing E3 medium after 24 hpf (0.003% N-phenylthiourea, Sigma).
At 48 hpf, the embryos were anesthetized and immobilized in a 2% low melting point agarose as described by Vasilyev and Drummond (43). The 405-nm laser (100% intensity) of a Zeiss LSM5 laser scanning confocal microscope was used to induce segmental tubule injury within the GFP positive kidney epithelial segment, as described by Palmyre et al. (32). This wavelength allows one to induce injury specifically in the GFP-expressing cells (likely because of the GFP ability to absorb light in this wavelength). The amount of light exposure was adjusted to produce only a focal cell dropout in the control condition [26-μs pixel dwell time, 1 × 8 repeat (8 line average) in a line scan mode]. The amount of immediate injury was estimated by the percentage of GFP intensity reduction at a random cross-section line (by comparing GFP intensity profiles before and immediately after the 405-nm light exposure). The amount of eventual injury was estimated at 3 h postlaser ablation, as this time is generally sufficient for a full extent of kidney injury to develop (32). Confocal maximum intensity projections were generated and the average pixel intensity was measured for the injured segment as well as for the left and right intact epithelia adjacent to the injury site (these were averaged). The ratio of the average pixel intensity in the injured segment to the average pixel intensity in the “bracketing” intact segments was used to calculate the amount of epithelial dropout after photoablation. The measurements of the initial injury were performed in Zeiss Pascal software. The measurements of the eventual injury were made in ImageJ (National Institutes of Health) and analyzed in Excel. Statistical analysis compared injury data from fish with Sec10-injected morpholinos (n = 8) vs. those injected with control morpholinos (n = 11) using a two-tailed t-test, unpaired with unequal variance.
RESULTS
Sec10 biochemically interacts with EGFR, and Sec10 overexpression results in more potent EGFR phosphorylation in response to EGF.
We recently showed that Sec10 overexpression in renal tubule epithelial cells protects against oxidative injury and also enhances recovery of the epithelial barrier. Blocking the MAPK pathway in this system prevented Sec10's protective effects (34). In polarized epithelial cells, the exocyst complex has been mostly associated with basolateral protein delivery (10, 22), although few studies have examined its possible role in receptor tyrosine kinase trafficking. Given that EGFR has been implicated in the recovery from acute kidney injury (3) and that EGF binding to EGFR is a well-described stimulus for activation of the MAPK pathway (2), we first tested for biochemical interactions between the exocyst and EGFR. A Sec10-GST fusion protein was purified on glutathione Sepharose and used as an affinity matrix for the purification of specific binding proteins from MDCK cell lysates. Western blot analysis identified EGFR as a protein pulled down with Sec10-GST, while no EGFR protein was detected in the control GST-only pull-down fraction (Fig. 1A). We also previously showed that Sec10-overexpressing cells have increased basal levels of phosphorylated (active) ERK (p-ERK), which contributed to the protective effects of the exocyst and the enhanced recovery following oxidative injury (34). To test if this could be due to increased EGFR signaling, we stimulated control and Sec10-overexpressing MDCK cells with the same concentration of EGF ligand and examined levels of phosphorylated (active) EGFR (p-EGFR) by analysis of Western blots. In serum-starved MDCK cells treated with EGF for 5 min, we found that p-EGFR levels were significantly higher when Sec10 was overexpressed (Fig. 1B). Despite this increased activation of EGFR, we measured no differences in basal levels of total EGFR in Sec10-overexpressing cells (Fig. 1C) and no differences in the rate of EGFR protein synthesis (Fig. 1D) or membrane delivery (Fig. 1E).
Fig. 1.

Sec10 biochemically interacts with epidermal growth factor receptor (EGFR), and Sec10 overexpression results in more potent EGFR phosphorylation in response to EGF. A: Sec10-GST fusion protein was expressed in Escherichia coli and purified using glutathione-Sepharose beads. After incubation with Madin-Darby canine kidney (MDCK) cell lysate, Sec10-GST, but not GST alone, pulled down EGFR. B: active phosphorylated (p)-EGFR levels were significantly increased in Sec10-overexpressing (OE) compared with control MDCK cells after 2 h of serum starvation followed by 5 min of EGF treatment (100 ng/ml). This experiment was performed in triplicate, and p-EGFR band intensities were measured, normalized to total EGFR (t-EGFR), and quantitatively compared, shown at right. C: EGFR basal protein levels are unchanged in Sec10-overexpressing and control MDCK cells. Sec10-overexpressing and control MDCK cells were grown on Transwell filters for 7 days. The cells were lysed and Western blot was performed for EGFR and GAPDH (a control for protein loading). D: there is no difference in EGFR synthesis in Sec10-overexpressing vs. control MDCK cells. Sec10-overexpressing and control MDCK cells were grown on Transwell filters for several days past confluency (6–7 days). After being washed with PBS, cells were starved for 20 min in MEM medium lacking methionine and then labeled by exposure of the basolateral surface to [35S]methionine for 20 min. The cells were lysed and immunoprecipitation was performed using antibody against EGFR. The immunoprecipitate was then run on an SDS-PAGE gel and analyzed with a phosphorimager. This experiment was repeated 4 times with similar results. E: there is no difference in basolateral membrane delivery of EGFR in Sec10-overexpressing vs. control MDCK cells by pulse chase. Sec10-overexpressing and control MDCK cells were grown on Transwell filters for several days past confluency (6–7 days). After being washed with PBS, cells were starved for 20 min in MEM medium lacking methionine and then were pulsed with [35S]methionine for 20 min, washed extensively, and allowed to chase for 60 min, and sulfo-NHS-biotin was added to the medium in contact with the basolateral surface of the cells. Immunoprecipitation using antibody against EGFR was performed. The antibody-beads-antigen complex was then boiled and the supernatant was reprecipitated with streptavidin beads and the remaining proteins were run on an SDS-PAGE gel and analyzed with a phosphorimager. This experiment was repeated 3 times with similar results.
Increased EGFR phosphorylation is accompanied by increased EGF binding and internalization.
At least one report has implicated the exocyst in endocytosis (31), so we next investigated endocytosis of activated EGFR using radiolabeled EGF (19, 25). Serum-starved MDCK cells were treated with labeled 125I-EGF ligand at 37°C for up to 7.5 min, and both surface-bound and internalized 125I-EGF fractions were collected at regular time points. After calculating the rate constants (ke), we found EGF internalization to be increased in Sec10 overexpressing cells compared with control and Sec10 knockdown (KD) MDCK cells (Fig. 2A). This increase in EGF internalization was prevented by the addition of gefitinib (aka Iressa), an inhibitor of EGFR tyrosine kinase activity (Fig. 2B); the addition of U0126, an inhibitor of MEK (Fig. 2C); as well as the addition of Dynasore, a cell-permeable small molecule inhibitor of dynamin-induced endocytosis (Fig. 2C).
Fig. 2.

EGF endocytosis is increased in Sec10-overexpressing type II MDCK cells in an EGFR- and MEK-dependent manner. Rate constants for EGF endocytosis (ke) were measured by incubating 10 ng/ml of 125I-EGF with cells lines as described in methods. Measurements were made to determine variations in ke as a result of Sec10 overexpression, or knockdown (KD), vs. control (A); EGFR inhibition with gefitinib (B); and MEK or dynamin inhibition using U0126 or Dynasore (C), respectively. Data are represented as average ± SD (n = 3). P values were calculated using an unpaired Student's t-test.
Increased p-ERK in Sec10-overexpressing cells is due to increased signaling by EGFR.
Gefitinib inhibits the EGFR tyrosine kinase by binding to the adenosine triphosphate (ATP)-binding site of the enzyme, preventing downstream activation of the MAPK signal transduction cascade (27). To test if the increased sensitivity of EGFR was the mechanism behind the increased p-ERK levels we had previously reported in Sec10-overexpressing cells (34), we grew control and Sec10-overexpressing MDCK cells for 1 wk on Transwell filters. Under basal conditions, we then added gefitinib at 1 μM for 1 h, which reduced the higher p-ERK levels measured in Sec10-overexpressing cells down to levels equal to control MDCK cells (Fig. 3).
Fig. 3.
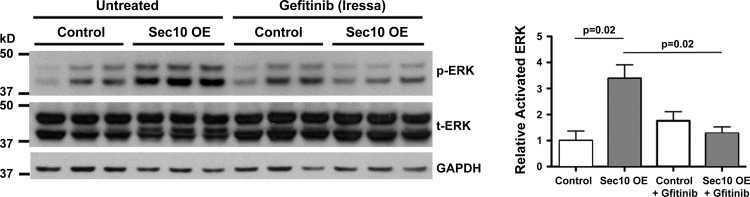
Active p-ERK levels are decreased in Sec10-overexpressing cells following addition of gefitinib. Sec10-overexpressing and control MDCK cells were grown on Transwell filters for 7 days. Under basal conditions, specific inhibition of EGFR kinase activity with gefitinib at 1 μM for 1 h reduced p-ERK levels in Sec10-overexpressing cells to levels seen in control cells. GAPDH was used as a loading control. All cell lines and conditions were run in triplicate. Band intensities were measured, combined for p42 and p44 bands of p-ERK and t-ERK, and the level of p-ERK in each sample was normalized against the level of t-ERK. Quantitative comparisons of triplicates are shown in the graph at right (means ± SE), with P values calculated using one-way ANOVA followed by post hoc Tukey's multiple comparison.
Inhibition of EGFR signaling promotes injury and inhibits recovery of renal tubule cells following H2O2 exposure.
We previously measured the degree of injury following H2O2 treatment and the time it took for the injured epithelial cells to recover their barrier integrity using TER as a marker. We found that overexpression of Sec10 significantly increased active ERK levels, decreased the degree of injury as measured by TER, and increased the rate of epithelial recovery following H2O2-induced injury. If U0126 was added in addition to the H2O2, Sec10-overexpressing cells recovered at the same rate as control cells, indicating that recovery was mediated through the MAPK pathway (34). Here we took a similar approach, but this time used gefitinib to evaluate EGFR's contribution to Sec10's protective role. In the absence of gefitinib, the severity of H2O2-induced injury (the nadir of TER) was greater in the control than the Sec10-overexpressing cells (Fig. 4A). Also, the timing of the nadir of TER was shortened in Sec10-overexpressing cells, indicating epithelial recovery began sooner. These observations were similar to what we previously showed (34). If gefitinib was added in addition to the H2O2, the severity of injury was greater in both cell lines, although Sec10 overexpression was still protective (Fig. 4A). Interestingly, the TER nadir was similar in the gefitinib-treated Sec10-overexpressing and untreated control cells (Fig. 4A), suggesting that EGFR signaling mediates the entire protective effect of Sec10 overexpression. The epithelial recovery rate from the TER nadir to the first point of significant increase was then determined using linear regression. The rate of recovery was decreased in both the Sec10-overexpressing and control cells following the addition of gefitinib (compared with untreated cells), but only reached statistical significance in the control cells (Fig. 4B).
Fig. 4.

Gefitinib increased the degree of H2O2-induced injury and decreased the recovery rate in MDCK cells. Control and Sec10-overexpressing MDCK cells were grown on Transwell filters in triplicate until transepithelial resistance (TER) levels reached steady state. All the filters were treated with 1 mM H2O2 for 30 min, with half the filters incubated with either 1 μM gefitinib or vehicle for 1 h before H2O2 treatment and 7 h afterward. A: TER was measured at the indicated time points. Time 0 represents the first measurement following the H2O2 treatment. *Point in time where the TER was significantly recovered from the nadir (#) experienced during H2O2 treatment. B: rate of TER recovery after the nadir (#) was calculated by linear regression analysis and testing of slopes using SPSS statistical software. Values are means ± SE of 3 separate experiments.
Sec10 inhibition worsens damage in a zebrafish model of acute renal injury.
For in vivo evaluation of Sec10's protective role against injury in renal tubules, we utilized a newly developed zebrafish model of acute kidney injury as described by Palmyre et al. (32). This assay takes advantage of the ET(krt8:EGFP)sqet11-9 zebrafish line, which has a part of the proximal tubule and distal tubule labeled with EGFP. To inhibit Sec10 in these fish, we used microinjections of previously validated antisense morpholinos targeting sec10 (9). The sec10 knockdown embryos were selected based on the presence of tail curvature (9) and the lack of general toxicity. At 48 hpf the embryos were anesthetized and immobilized in agarose (43). With confocal microscopy, a 405-nm laser (100% intensity) was used to induce segmental tubule injury within the GFP-positive kidney epithelial segment. This wavelength allows one to induce injury specifically in GFP-expressing cells (likely because of the GFP ability to absorb light at this wavelength). The amount of immediate injury was estimated by the percentage of GFP intensity reduction at a random cross-section line (by comparing GFP intensity profiles before and immediately after the 405-nm light exposure) and was not significantly different in the Sec10 (n = 8) and control morphants (n = 11) (P = 0.16, data not shown). Additionally, the amount of eventual injury was estimated at 3 h postlaser ablation, a time that is generally sufficient for the full extent of kidney injury to develop (32). For this measurement, we compared the average pixel intensity in the injured segment to the average pixel intensity in the “bracketing” intact segments. This method can be used because injured epithelial cells that undergo necrosis or apoptosis end up permanently losing their GFP fluorescence, as opposed to surviving cells that recover their preinjury fluorescence. After three h, renal tubule cell injury was significantly greater in sec10 morphants compared with control zebrafish (P = 0.034; Fig. 5, A and B). These data supported our in vitro findings that Sec10 expression in renal tubular epithelial cells is protective against cell damage.
Fig. 5.

Renal tubule cell damage after photoablation injury is greater in zebrafish following knockdown of exocyst sec10. Following injection into 1-cell stage embryos of control and splice-site morpholinos (MO) that we had previously shown to efficiently knock down sec10 in zebrafish embryos (9), laser photoablation was performed in EGFP-expressing segments of ET11-9 zebrafish kidney tubule epithelium. The arrows in A point to the boundary between the ablated and intact epithelium. B: Sec10 morphants (n = 8), compared with control morphants (n = 11), showed a significantly greater reduction in GFP positivity and, therefore, greater injury in the photoablated segment at three h following injury (P = 0.034). The %remaining GFP intensity was determined by comparing the average pixel intensity in the injured segment to the average pixel intensity in the “bracketing” intact segments, and statistical comparison was performed via two tail unpaired t-test.
DISCUSSION
Our goal in this study was to determine the mechanism by which exocyst Sec10 overexpression protects against oxidative injury in renal tubule cells. We previously showed that Sec10 overexpression increased MAPK signaling in MDCK cells, which protected the cells against oxidative injury by H2O2 (34). Given the role of the exocyst in trafficking proteins to the basolateral membrane in polarized epithelial cells (10, 22), we hypothesized that the exocyst regulates trafficking of signaling proteins upstream of ERK in the MAPK pathway. We focused on EGFR because of recent studies showing that EGFR is the most upregulated receptor tyrosine kinase in response to renal ischemia and reperfusion injury. Additionally, pharmacologic and genetic inhibition of EGFR in injured kidney proximal tubules markedly delayed cell proliferation and renal structural and functional recovery (3). Indeed, when screening for Sec10-binding proteins with GST pull-down assays from MDCK lysates, we detected a biochemical interaction between EGFR and the exocyst. We then measured levels of EGFR activation in response to EGF and found there was more phosphorylated EGFR in Sec10-overexpressing cells compared with control cells with the same EGF treatments.
At least one study has described a role for the exocyst in endocytosis (31), and EGFR endocytosis upon binding to EGF contributes to subsequent MAPK activation (19). By tracking internalization of radiolabeled EGF ligand, we discovered that the increase in MAPK activation in Sec10-overexpressing cells is likely due to increased EGFR endocytosis. Following the addition of gefitinib, the increase in EGF internalization, the increase in MAPK signaling, and the protective effect on membrane integrity of Sec10 overexpression all returned to levels seen in control MDCK cells.
Like other members of the receptor tyrosine kinase (RTK) superfamily, EGFR dimerizes (or forms heterodimers with other members of the ErbB family) and its kinase is activated upon ligand binding. Also like many other RTKs, the bound EGFR dimer then rapidly undergoes endocytic trafficking to early endosomes. There it either releases its ligand and gets recycled back to the plasma membrane, or it can remain ligand bound and gets degraded after shuttling through the late endosome to the lysosome (48). There is an established connection between EGFR endocytosis and ERK activation. For example in at least some cell settings, expression of a dominant negative form of dynamin (K44A) reduced EGFR endocytosis and significantly impaired EGFR-mediated MAPK activation (19, 46). Similar to what we show here, the exocyst was previously identified as a regulator of endocytosis (31), suggesting that increased EGFR endocytosis is the mechanism for increased ERK phosphorylation in Sec10-overexpressing cells.
A study by Chen et al. (3) showed only inhibition of recovery following pharmacologic and genetic inhibition of proximal tubule EGFR and MAPK signaling. One possible reason that we see both protection and enhanced recovery in Sec10-overexpressing cells is because at the time of injury the EGFR/MAPK signaling pathway is already upregulated in Sec10-overexpressing cells (34). Interestingly, the exocyst component Exo70 was recently shown to be a direct substrate of ERK (35). Exo70 phosphorylation by ERK enhanced the binding of Exo70 to other exocyst components and promoted the assembly of the exocyst complex in response to EGF signaling (35). This sets up the possibility of a positive feedback loop in which increased exocyst expression increases EGFR endocytosis, which would lead to increased ERK signaling and exocyst recruitment.
To support our discovered association between the exocyst and renal cell recovery following injury using an in vitro cell culture model, we performed an in vivo functional study by investigating if sec10 protects against acute kidney injury in zebrafish. Several zebrafish models of acute kidney injury have been previously published (13), although development of the photoablation model by Palmyre et al. (32) represents significant improvement on the previous methods. This model allows a high degree of spatial resolution since laser-targeted GFP-positive epithelial cells lie only within the pronephros tubule, and the timing or injury and recovery have been carefully evaluated (32). Since we have previously demonstrated successful knockdown of zebrafish exocyst sec10 using antisense morpholinos (9), we were able to silence sec10 in this GFP-photoablation zebrafish model and measure the increased damage to kidney tubule cells following injury.
In further support of an in vivo role for the exocyst and MAPK pathway in recovery following acute kidney injury, we recently showed that activation of ERK is required for both the restoration of damaged tubular epithelial cells and the inhibition of fibrosis progression following ischemia/reperfusion injury in mice (18). The MEK inhibitor U0126 inhibited cell proliferation, basolateral relocalization of Na-K-ATPase, and lengthening of primary cilia in tubular epithelial cells. This treatment enhanced the proliferation of interstitial cells and accumulation of extracellular matrix. U0126 also inhibited the normal postinjury increase of Sec10, suggesting that the exocyst may be involved in the repair process following ischemia/reperfusion injury in mice (18). Preclinical or clinical approaches to modifying exocyst activity in targeted tissues require further investigation and development. Unfortunately, no small molecule has been identified that either blocks or enhances exocyst activity or assembly, although gene therapy using adeno-associated viral (AAV) could be a viable approach (7). It is widely believed that cells require fine control of the targeted assembly of the exocyst and its trafficking activity to ensure accuracy of vesicle delivery. This concept is supported by the cell's careful coordination of eight separate protein subunits, with the possibility of subcomplexes for specific functions. Described regulatory mechanisms include activation by small GTPases and kinase phosphorylation of certain subunits (5, 51), and doubtless, more regulatory mechanisms in diverse cell types remain undiscovered. These relationships may allow chemical manipulation of exocyst activity by targeting upstream regulators of the complex assembly. With this goal in mind, it would be of great benefit to initiate screens to identify potential pharmacological modifiers of exocyst assembly and activity.
GRANTS
This work was supported in part by grants from the Veterans Affairs (Merit Award to J. H. Lipschutz), National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-069909 and DK-070980 (to J. H. Lipschutz) and K01-DK-087852 and R0-3DK-100738 (to B. Fogelgren F.), Satellite Healthcare (Norman S. Coplon Extramural Research Grant to J. H. Lipschutz), Hawaii Community Foundation (12ADVC-51347 to B.F.), March of Dimes (Basil O′Connor No. 5-FY14-56 to B. Fogelgren), University of Hawaii RCMI-BRIDGES (5G12MD007601, Pilot Award to B. Fogelgren), Hepato/Renal Fibrocystic Diseases Core Center at the University of Alabama at Birmingham (5P30-DK-074038, Pilot Award to B. Fogelgren), National Science Foundation (Graduate Research Fellowship Award to J. M. Buonato), and the Korean Government (National Research Foundation Grant NRF-2011-220-E00001 to K. Park).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: B.F. and J.H.L. conception and design of research; B.F., X.Z., J.M.B., A.V., J.-I.B., S.Y.C., A.P., N.P., and K.M.P. performed experiments; B.F., X.Z., J.M.B., A.V., A.P., M.J.L., and J.H.L. analyzed data; B.F., X.Z., J.M.B., A.V., M.F.C.-H., A.P., I.A.D., M.J.L., and J.H.L. interpreted results of experiments; B.F., X.Z., J.M.B., A.V., and A.P. prepared figures; B.F., J.-I.B., S.Y.C., M.F.C.-H., N.P., K.M.P., M.J.L., and J.H.L. edited and revised manuscript; M.J.L. and J.H.L. approved final version of manuscript; J.H.L. drafted manuscript.
REFERENCES
- 1.Bagshaw SM, Laupland KB, Doig CJ, Mortis G, Fick GH, Mucenski M, Godinez-Luna T, Svenson LW, Rosenal T. Prognosis for long-term survival and renal recovery in critically ill patients with severe acute renal failure: a population-based study. Crit Care 9: R700–709, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature 410: 37–40, 2001. [DOI] [PubMed] [Google Scholar]
- 3.Chen J, Chen JK, Harris RC. Deletion of the epidermal growth factor receptor in renal proximal tubule epithelial cells delays recovery from acute kidney injury. Kidney Int 82: 45–52, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol 16: 3365–3370, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Choi SY, Chacon-Heszele MF, Huang L, McKenna S, Wilson FP, Zuo X, Lipschutz JH. Cdc42 deficiency causes ciliary abnormalities and cystic kidneys. J Am Soc Nephrol 24: 1435–1450, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choo BG, Kondrichin I, Parinov S, Emelyanov A, Go W, Toh WC, Korzh V. Zebrafish transgenic enhancer TRAP line database (ZETRAP). BMC Dev Biol 6: 5, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chung DC, Fogelgren B, Park KM, Heidenberg J, Zuo X, Huang L, Bennett J, Lipschutz JH. Adeno-associated virus-mediated gene transfer to renal tubule cells via a retrograde ureteral approach. Nephron Extra 1: 217–223, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Mendonca A, Vincent JL, Suter PM, Moreno R, Dearden NM, Antonelli M, Takala J, Sprung C, Cantraine F. Acute renal failure in the ICU: risk factors and outcome evaluated by the SOFA score. Intensive Care Med 26: 915–921, 2000. [DOI] [PubMed] [Google Scholar]
- 9.Fogelgren B, Lin SY, Zuo X, Jaffe KM, Park KM, Reichert RJ, Bell PD, Burdine RD, Lipschutz JH. The exocyst protein Sec10 interacts with polycystin-2 and knockdown causes PKD-phenotypes. PLoS Genet 7: e1001361, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grindstaff KK, Yeaman C, Anandasabapathy N, Hsu S, Rodriguez-Boulan R, Scheller RH, Nelson WJ. Sec6/8 complex is recruited to cell-cell contacts and specifies transport vesicle delivery to the basal-lateral membrane in epithelial cells. Cell 93: 731–740, 1998. [DOI] [PubMed] [Google Scholar]
- 11.Guo W, Roth D, Gatti E, De Camilli P, Novick P. Identification and characterization of homologues of the exocyst component Sec10p. FEBS Lett 404: 135–139, 1997. [DOI] [PubMed] [Google Scholar]
- 12.Guo W, Roth D, Walch-Solimena C, Novick P. The exocyst is an effector for Sec4p, targeting secretory vesicles to sites of exocytosis. EMBO J 18: 1071–1080, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hentschel DM, Park KM, Cilenti L, Zervos AS, Drummond I, Bonventre JV. Acute renal failure in zebrafish: a novel system to study a complex disease. Am J Physiol Renal Physiol 288: F923–F929, 2005. [DOI] [PubMed] [Google Scholar]
- 14.Hise MK, Salmanullah M, Liu L, Drachenberg CI, Papadimitriou JC, Rohan RM. Control of the epidermal growth factor receptor and its ligands during renal injury. Nephron 88: 71–79, 2001. [DOI] [PubMed] [Google Scholar]
- 15.Homma T, Sakai M, Cheng HF, Yasuda T, Coffey RJ, Jr, Harris RC. Induction of heparin-binding epidermal growth factor-like growth factor mRNA in rat kidney after acute injury. J Clin Invest 96: 1018–1025, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsu S, Ting AE, Hazuka CD, Davanger S, Kenny JW, Kee Y, Scheller RH. The mammalian brain rsec6/8 complex. Neuron 17: 1209–1219, 1996. [DOI] [PubMed] [Google Scholar]
- 17.Humes HD, Cieslinski DA, Coimbra TM, Messana JM, Galvao C. Epidermal growth factor enhances renal tubule cell regeneration and repair and accelerates the recovery of renal function in postischemic acute renal failure. J Clin Invest 84: 1757–1761, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jang HS, Han SJ, Kim JI, Lee S, Lipschutz JH, Park KM. Activation of ERK accelerates repair of renal tubular epithelial cells, whereas it inhibits progression of fibrosis following ischemia/reperfusion injury. Biochim Biophys Acta 1832: 1998–2008, 2013. [DOI] [PubMed] [Google Scholar]
- 19.Lazzara MJ, Lane K, Chan R, Jasper PJ, Yaffe MB, Sorger PK, Jacks T, Neel BG, Lauffenburger DA. Impaired SHP2-mediated extracellular signal-regulated kinase activation contributes to gefitinib sensitivity of lung cancer cells with epidermal growth factor receptor-activating mutations. Cancer Res 70: 3843–3850, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 141: 1117–1134, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin JJ, Cybulsky AV, Goodyer PR, Fine RN, Kaskel FJ. Insulin-like growth factor-1 enhances epidermal growth factor receptor activation and renal tubular cell regeneration in postischemic acute renal failure. J Lab Clin Med 125: 724–733, 1995. [PubMed] [Google Scholar]
- 22.Lipschutz JH, Guo W, O'Brien LE, Nguyen YH, Novick P, Mostov KE. Exocyst is involved in cystogenesis and tubulogenesis and acts by modulating synthesis and delivery of basolateral plasma membrane and secretory proteins. Mol Biol Cell 11: 4259–4275, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lipschutz JH, Li S, Arisco A, Balkovetz DF. Extracellular-signal regulated kinases 1/2 control claudin-2 expression in Madin Darby Strain I and II cells. J Biol Chem 280: 3780–3788, 2005. [DOI] [PubMed] [Google Scholar]
- 24.Lipschutz JH, Mostov KE. Exocytosis: the many masters of the exocyst. Curr Biol 12: R212–214, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Lund KA, Opresko LK, Starbuck C, Walsh BJ, Wiley HS. Quantitative analysis of the endocytic system involved in hormone-induced receptor internalization. J Biol Chem 265: 15713–15723, 1990. [PubMed] [Google Scholar]
- 26.Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, Levin A. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care 11: R31, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang JJ, Chewaskulyong B, Jiang H, Duffield EL, Watkins CL, Armour AA, Fukuoka M. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 361: 947–957, 2009. [DOI] [PubMed] [Google Scholar]
- 28.Molitoris BA, Falk SA, Dahl RH. Ischemia-induced loss of epithelial polarity. Role of the tight junction. J Clin Invest 84: 1334–1339, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Norman J, Tsau YK, Bacay A, Fine LG. Epidermal growth factor accelerates functional recovery from ischaemic acute tubular necrosis in the rat: role of the epidermal growth factor receptor. Clin Sci (Lond) 78: 445–450, 1990. [DOI] [PubMed] [Google Scholar]
- 30.Novick P, Field C, Schekman R. Identification of 23 complementation groups required for posttranslational events in the yeast secretory pathway. Cell 21: 205–221, 1980. [DOI] [PubMed] [Google Scholar]
- 31.Oztan A, Silvis M, Weisz OA, Bradbury NA, Hsu SC, Goldenring JR, Yeaman C, Apodaca G. Exocyst requirement for endocytic traffic directed toward the apical and basolateral poles of polarized MDCK cells. Mol Biol Cell 18: 3978–3992, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palmyre A, Lee J, Ryklin G, Camarata T, Selig MK, Duchemin AL, Nowak P, Arnaout MA, Drummond IA, Vasilyev A. Collective epithelial migration drives kidney repair after acute injury. PLoS One 9: e101304, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parinov S, Kondrichin I, Korzh V, Emelyanov A. Tol2 transposon-mediated enhancer trap to identify developmentally regulated zebrafish genes in vivo. Dev Dyn 231: 449–459, 2004. [DOI] [PubMed] [Google Scholar]
- 34.Park KM, Fogelgren B, Zuo X, Kim J, Chung DC, Lipschutz JH. Exocyst Sec10 protects epithelial barrier integrity and enhances recovery following oxidative stress, by activation of the MAPK pathway. Am J Physiol Renal Physiol 298: F818–F826, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ren J, Guo W. ERK1/2 regulate exocytosis through direct phosphorylation of the exocyst component Exo70. Dev Cell 22: 967–978, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roth D, Guo W, Novick P. Dominant negative alleles of SEC10 reveal distinct domains involved in secretion and morphogenesis in yeast. Mol Biol Cell 9: 1725–1739, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rothman JE. Mechanisms of intracellular protein transport. Nature 372: 55–63, 1994. [DOI] [PubMed] [Google Scholar]
- 38.Sakai M, Zhang M, Homma T, Garrick B, Abraham JA, McKanna JA, Harris RC. Production of heparin binding epidermal growth factor-like growth factor in the early phase of regeneration after acute renal injury. Isolation and localization of bioactive molecules. J Clin Invest 99: 2128–2138, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salahudeen AK, Clark EC, Nath KA. Hydrogen peroxide-induced renal injury. A protective role for pyruvate in vitro and in vivo. J Clin Invest 88: 1886–1893, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Terbush DR, Maurice T, Roth D, Novick P. The exocyst is a multiprotein complex required for exocytosis in Saccharomyces cerevisiae. EMBO J 15: 6483–6494, 1996. [PMC free article] [PubMed] [Google Scholar]
- 41.Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med 334: 1448–1460, 1996. [DOI] [PubMed] [Google Scholar]
- 42.Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, Schetz M, Tan I, Bouman C, Macedo E, Gibney N, Tolwani A, Ronco C. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA 294: 813–818, 2005. [DOI] [PubMed] [Google Scholar]
- 43.Vasilyev A, Drummond IA. Live imaging kidney development in zebrafish. Methods Mol Biol 886: 55–70, 2012. [DOI] [PubMed] [Google Scholar]
- 44.Vega IE, Hsu SC. The exocyst complex associates with microtubules to mediate vesicle targeting and neurite outgrowth. J Neurosci 21: 3839–3848, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Venkatachalam MA, Griffin KA, Lan R, Geng H, Saikumar P, Bidani AK. Acute kidney injury: a springboard for progression in chronic kidney disease. Am J Physiol Renal Physiol 298: F1078–F1094, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vieira AV, Lamaze C, Schmid SL. Control of EGF receptor signaling by clathrin-mediated endocytosis. Science 274: 2086–2089, 1996. [DOI] [PubMed] [Google Scholar]
- 47.Waikar SS, Liu KD, Chertow GM. Diagnosis, epidemiology and outcomes of acute kidney injury. Clin J Am Soc Nephrol 3: 844–861, 2008. [DOI] [PubMed] [Google Scholar]
- 48.Wiley HS. Trafficking of the ErbB receptors and its influence on signaling. Exp Cell Res 284: 78–88, 2003. [DOI] [PubMed] [Google Scholar]
- 49.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16: 535–543, 1p following 143, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yano T, Yazima S, Hagiwara K, Ozasa H, Ishizuka S, Horikawa S. Activation of epidermal growth factor receptor in the early phase after renal ischemia-reperfusion in rat. Nephron 81: 230–233, 1999. [DOI] [PubMed] [Google Scholar]
- 51.Zuo X, Fogelgren B, Lipschutz JH. The small GTPase Cdc42 is necessary for primary ciliogenesis in renal tubular epithelial cells. J Biol Chem 286: 22469–22477, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zuo X, Guo W, Lipschutz JH. The exocyst protein Sec10 is necessary for primary ciliogenesis and cystogenesis in vitro. Mol Biol Cell 20: 2522–2529, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]