

Abstract
Discordant myocardial growth and angiogenesis can explain left ventricular (LV) hypertrophy progressing toward heart failure with aging. Sirtuin 1 expression declines with age; therefore we explored the role played by angiogenesis and Sirtuin 1 in the development of cardiomyopathy. We compared the cardiac function of 10- to 15-wk-old (wo), 30–40 wo, and 61–70 wo endothelial Sirtuin 1-deleted (Sirt1endo−/−) mice and their corresponding knockout controls (Sirt1Flox/Flox). After 30–40 wk, Sirt1endo−/− animals exhibited diastolic dysfunction (DD), decreased mRNA expression of Serca2a in the LV, and decreased capillary density compared with control animals despite a similar VEGFa mRNA expression. However, LV fibrosis and hypoxia-inducible factor (HIF)1α expression were not different. The creation of a transverse aortic constriction (TAC) provoked more severe DD and LV fibrosis in Sirt1endo−/− compared with control TAC animals. Although the VEGFa mRNA expression was not different and the protein expression of HIF1α was higher in the Sirt1endo−/− TAC animals, capillary density remained reduced. In cultured endothelial cells administration of Sirtuin 1 inhibitor decreased mRNA expression of VEGF receptors FLT 1 and FLK 1. Ex vivo capillary sprouting from aortic explants showed impaired angiogenic response to VEGF in the Sirt1endo−/− mice. In conclusion, the data demonstrate 1) a defect in angiogenesis preceding development of DD; 2) dispensability of endothelial Sirtuin 1 under unstressed conditions and during normal aging; and 3) impaired angiogenic adaptation and aggravated DD in Sirt1endo−/− mice challenged with LV overload.
Keywords: hypoxia, echocardiography, VEGF, angiogenesis, Adriamycin cardiomyopathy, transverse aortic constriction
the development of left ventricular hypertrophy (LVH) during pathological situations (hypertension, myocardial ischemia) is considered to be an adaptive response to the increased wall stress exerted on the left ventricle (LV). Although normal cardiomyocytes grow during childhood or exercise, and such growth is not associated with heart failure, in aging LVH becomes a high risk factor for heart failure. The reasons why cardiac growth can progress toward heart failure are incompletely understood. However, cardiac function has been shown to depend on the angiogenic capacity of the cardiac vessels. The disruption of coordinated myocardial growth and matching angiogenesis leads to the progression to heart failure (27). With aging the angiogenic capacity decreases, which could explain at least in part why LVH in older patients is associated with cardiac failure (16). In aging animals, angiogenic competence is decreased. Mouse aortic rings obtained from these animals show 40% reduction in vascular sprouting induced by VEGF compared with young animals (22). A similar defect occurs in prematurely senescent Klotho mice (26). Of note, a caloric restriction regimen rescues angiogenic competence of aortic rings obtained from 24-mo-old rats by 45–63% (9). One of the downstream targets of caloric restriction, Sirtuin 1, is robustly expressed in endothelial cells and tends to decline with age and after application of cardiovascular stressors (4).
Sirtuin 1 is a NAD+-dependent deacetylase residing in the nucleus and the cytoplasm of mammalian cells. It has been demonstrated that SIRT1 plays an essential role in responses to stressors, like reactive oxygen species (10). SIRT1 knockout mice are not viable; on the other hand, SIRT1 transgenic mice show phenotypes resembling calorie restriction. Deletion of SIRT1 in hepatocytes leads to hepatic steatosis and inflammation (10). The role of SIRT1 in cell fate and metabolic regulation is based on its effects augmenting FOXO-regulated stress resistance, interaction with p53, E2F1, PGC1a, peroxisome proliferator-activated receptor-γ (PPARγ), and endothelial nitric oxide synthase (eNOS), to name a few (12), thus underlining its function as a sensor of the metabolic and cellular energy state of cells, deacetylating many key proteins and per se regulating a wide range of functions (i.e., senescence, apoptosis, longevity). Angiogenesis is one of these functions regulated by Sirtuin 1 (21). The loss of Sirtuin 1 expression in endothelial cells leads to impaired angiogenesis, premature senescence, and downregulation of several proangiogenic genes (4).
Therefore, the objective of the present study was to better understand the role played by angiogenesis and one of its key regulators, Sirtuin 1, in the development of cardiomyopathy; hence, we conducted a study to explore the cardiac consequences of the inactivation of Sirtuin 1 in coronary endothelial cells.
MATERIALS AND METHODS
Mice with endothelial Sirt1 deletion.
The animal study protocol was in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and was approved by the Institutional Animal Care and Use Committee of New York Medical College.
The endothelial Sirt1-deleted mouse model was created by crossbreeding of B6;129-Sirt1tm1Ygu/J mice (homozygous for targeted allele Sirt1co/co, viable and fertile, containing a loxP-flanked neomycin cassette upstream and downstream of exon 4 of the targeted gene) (5) with Tie2-Cre transgenic mice [B6.cg-tg(tek-cre)1ywa/J] expressing Cre recombinase in vascular endothelial cells (both from Jackson Lab) (14). The resulting Sirt1endo+/− mice were mated with Sirt1Flox/Flox mice to obtain endothelial-deleted Sirt1 mutant mice. Endothelial Sirt1endo−/− knockout (and corresponding knockout control Sirt1Flox/Flox) mice were obtained at Mendelian ratios and used in the experiments. Genotyping was performed by tail DNA PCR analysis. Tail DNA was isolated with the REDExtract-N-Amp Tissue PCR Kit (Sigma-Aldrich, St Louis, MO). Primer sequences used for genotyping floxed Sirt1 allele were as follows: SIRT1 forward 5′GGT TGA CTT AGG TCT TGT CTG3′, SIRT1 reverse 5′CGT CCC TTG TAA TGT TTC CC3′, and for SIRT1 null allele 5′AGG CGG ATT TCT GAG TTC GA3′. Tie2 transgene was detected with the following primers: forward 5′GCG GTC TGG CAG TAA AAA CTA TC3′ and reverse 5′ GTG AAA CAG CAT TGC TGT CAC TT3′. PCR products were analyzed on 1.5% Tris-acetate-EDTA (TAE) acrylamide gels. Male and female mice were used in the same proportion in the presented experiments.
Consequences of aging for Sirt1endo−/− cardiac functions.
We compared three groups of young (10–15 wk old; n = 10), middle-aged (30–40 wk old; n = 15), and old (61–70 wk old; n = 11) Sirt1endo−/− and control (Sirt1Flox/Flox) animals. Each group was examined by echocardiography followed by exsanguination under ketamine-xylazine anesthesia. The heart was washed with cold phosphate-buffered saline (PBS) in situ and then carefully harvested, and sections of LV were used for histology, Western blot, and quantitative PCR (qPCR) studies.
Left ventricular pressure overload in Sirt1endo−/− mice.
We compared two groups composed of 30- to 40-wk-old control (Sirt1Flox/Flox; n = 7) and Sirt1endo−/− (n = 5) animals exposed to a LV pressure overload and one group of same-age control animals without surgery (n = 5). The pressure overload was created by transverse aortic constriction (TAC) under general anesthesia 7 wk before euthanasia. The procedure for TAC has been described elsewhere (7). Briefly, under general anesthesia with isoflurane, oral intubation, and ventilation, the animal's sternum was opened at the first two ribs and the aortic arch was exposed. A 27½-gauge needle was placed aside the aortic arch, and a suture (silk suture 6/0) was ligated around the aorta and the needle. The needle was removed, and then chest and skin were closed with autoresorbent sutures. Transthoracic echocardiography (TTE) was performed before and 1 wk and 7 wk after the surgery. After the last TTE, the animals were euthanized by exsanguination under ketamine-xylazine anesthesia. The heart was washed with cold PBS in situ and then carefully harvested, and sections of LV were subjected to histological studies, Western blotting, and qPCR studies.
Adriamycin cardiomyopathy in Sirt1endo−/− mice.
Similar studies were conducted in mice with Adriamycin cardiotoxicity, a model of dystrophic myocardial injury. Thirty-week-old Sirt1endo−/− (n = 5) and control (n = 5) mice were treated with doxorubicin hydrochloride (Sigma Chemical, St. Louis, MO) according to a procedure described previously (2). Doxorubicin was dissolved in saline and injected intraperitoneally at 4 mg/kg (10 ml/kg body wt) twice a week (every Monday and Thursday) for a total of 10 injections. Animals were not treated for 2 wk between the first four injections and the last six injections. TTE was performed at baseline, 5 wk after the initiation of treatment, and 1 wk before euthanasia (9 wk after the beginning of treatment). Animals were euthanized after the last injection (10 wk after the beginning of treatment). The heart was washed with cold PBS in situ and then carefully harvested, and sections of LV were subjected to histological studies.
Echocardiography.
TTE examinations were performed under isoflurane inhalation general anesthesia with a heart rate of ∼400–450 beats per minute (bpm). Isoflurane was administered with a vaporizer. After induction with 3.5–4.5% isoflurane in an isolated chamber, anesthesia was maintained with 1–1.5% isoflurane delivered through a small nose cone. TTE measurements were performed with a commercially available echocardiograph (770, Visualsonic) with a 30-MHz transducer. M-mode echocardiography of the LV was performed in order to measure the following parameters: LV end-diastolic diameter (LVEDD), LV end-systolic diameter (LVESD), diastolic posterior wall thickness (PW), and diastolic septal wall thickness (SW), according to American Society of Echocardiography guidelines (24). LV mass was calculated with the following formula: LV mass = 1.04 × [(LVEDD + PW + SW)3 − LVEDD3]. The shortening fraction (SF) was calculated as the difference between the LVEDD and the LVESD divided by the LVEDD. Mitral pulsed Doppler was recorded at the tip of the mitral valve with an apical view, in order to analyze the isovolumic relaxation time (IVRT), the mitral flow duration, the duration of diastole, and the ejection time. The Tei index was calculated as the difference between the duration of diastole and the mitral flow duration divided by the ejection time (29).
Western blotting.
Expression of proteins of interest was quantified with Western blot analysis. Briefly, LV tissues were homogenized twice for 10 s with a Polytron homogenizer (Pro Scientific) at 4°C in RIPA buffer [50 mM Tris·HCl (pH 7.4), 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM Na3VO4, 1 mM NaF, and a protease inhibitor cocktail tablet]. Homogenates were centrifuged at 14,000 rpm for 15 min at 4°C, and the supernatants were assayed. The protein concentration was determined in a Bradford assay (Bio-Rad). Samples were denatured in Laemmli buffer (125 mM Tris·HCl, 2% SDS, 1% β-mercaptoethanol, 10% glycerol, 0.005% bromophenol blue, pH 6.8) and heated at 95°C for 5 min. Twenty micrograms of total protein per sample was separated on 4–20% acrylamide/bisacrylamide SDS-PAGE under constant-current conditions. Proteins were then transferred to polyvinylidene difluoride (PVDF) 0.45-μm membranes (Millipore) in a transfer buffer at 90 V for 90 min. Membranes were then blocked with 5% nonfat milk in Tris-buffered saline (TBS) with 0.1% Tween 20 for 1 h at room temperature and immunoblotted with the desired primary antibody in TBS BSA 0.1% overnight at 4°C. The source and concentration of hypoxia-inducible factor (HIF)1α antibody were Millipore and 1:1,000. After several washings in TBS-0.1% Tween 20, membranes were incubated with horseradish peroxidase (HRP)-conjugated goat anti-mouse antibody (1:5,000). Each secondary antibody was incubated for 1 h at room temperature prior to development with an enhanced chemiluminescence detection kit (Millipore). Bands were visualized and quantified with ImageJ software (version 1.45; NIH). Protein levels were normalized against endogenous β-tubulin levels.
Immunoprecipitation.
One hundred micrograms of total LV protein extract prepared for Western blot analyses was used for immunoprecipitation. Proteins were incubated with HIF1α antibody (Millipore, 1:500) overnight at 4°C and then for 2 h at 4°C with 20 μl of protein A/G plus-agarose immunoprecipitation reagent (Santa Cruz Biotechnologies). Immune complexes were collected by centrifugation and washed three times with ice-cold PBS. After a final wash, the supernatant was discarded and the pellet was resuspended in SDS lysis buffer and then boiled in 5× SDS loading dye for 5 min. Protein was separated by SDS-PAGE and transferred onto PVDF membranes. Immunoprecipitated proteins were then detected with anti-acetyl lysine antibody (1:1,000, Abcam) or HIF1α antibody (Millipore, 1:800) overnight at 4°C. Bands were visualized and quantified with ImageJ software (version 1.45; NIH).
Quantitative PCR.
Total RNA was extracted from LVs with a RNA purification kit (Denville Scientific), and total RNA (1 μg) was transcribed into complementary DNA with the High Capacity cDNA Reverse Transcription Kit according to the manufacturer's protocol (Applied Biosystems). The following primers were used: Serca2a (Forward TGA ATC TGA CCC AGT GGC TGA, Reverse ACT CCA GTA TTG CAG GCT CCA), PGC1a (Forward AGC CTA TGA GCA CGA AAG GCT CAA, Reverse TGG CCC TTT CAG ACT CCC GCT), Sirtuin 3 (Forward TCA CAA CCC CAA GCC CTT TT, Reverse GTG GGC TTC AAC CAG CTT TG), Sirtuin 6 (Forward GAC CTG ATG CTC GCT GAT GA, Reverse CTG GCG GTC ATG TTT TGT GG), short endoglin (Forward TGA GTA TCC CAA GCC TCC ACC CCA T, Reverse CTG AGG GGC GTG GGT GAA GGT CAG), long endoglin (Forward GCA CTC TGG TAC ATC TAT TCT CAC ACA CGT GG, Reverse GGG CAC TAC GCC ATG CTG CTG GTG G), Collagen 1 (Forward CTG CTG GCA AAG ATG GAG A, Reverse ACC AGG AAG ACC CTG GAA TC), VEGFA (Forward TCC ACC ATG CCA AGT GGT CCC AGG CTG CAC CCA, Reverse GAC GTG GGC ACG CAC TCC AGG), and 18S (Forward AAG GAG ACT CTG GCA TGC TAA C, Reverse CAG ACA TCT AAG GGC ATC ACA GAC). SYBR Green chemistry was used to perform quantitative determinations of the mRNAs. The cDNAs were amplified with a Stratagene Mx3005P (Agilent Technologies) sequence detection system (Applied Biosystems). 18S was used to normalize differences in RNA isolation, RNA degradation, and the efficiency of the reverse transcription reaction. Changes in mRNA expression were determined with the comparative threshold method (17). RNA expression was also expressed in relation to the levels observed in control mice.
Histochemical studies.
Hearts were fixed in 4% PFA for 24 h, embedded in paraffin, and sectioned (8 μm). The heart sections were stained with hematoxylin and eosin, Masson's trichrome, and anti-CD31 (Abcam ab28364).
Analysis of five to eight randomly selected fields (×400 magnification) was conducted. Masson's trichrome images were quantified with a digital method by Photoshop software described previously (6). This technique allows measurement of the percentage of trichrome-positive pixels (blue) within the total number of tissue pixels (blue and red).
For CD31 staining, sections were first deparaffinized, rehydrated, and then exposed to an antigen retrieval solution (Target retrieval solution, Dako). Slides were stained by the streptavidin biotin procedure with the LSAB system HRP (Dako). Briefly, slides were incubated with the primary antibody (CD31, Abcam, 1:100 dilution) overnight at 4°C after peroxidase blocking. Slides were then exposed to the streptavidin peroxidase solution followed by chromogen solution. Counterstain was performed with hematoxylin.
The number of CD31-positive structures per image (×400 magnification) were counted and normalized to the cardiomyocyte area. The cardiomyocyte area was quantified per image with Adobe Photoshop software v8.0 (Adobe Systems, San Jose, CA). First, each picture was adjusted to obtain the same pixel dimensions per image. Then the area of blank pixel (interstitial spaces) was automatically measured. The cardiomyocyte area was calculated as the total area of the image minus the blank areas (interstitial spaces) (31).
Cell culture.
Immortalized human umbilical vein endothelial cells (HUVECs) (ATCC, Manassas, VA), were maintained in DMEM supplemented with 10% FBS and 1% penicillin under conditions of 37°C and 5% CO2. The cells were cultured in 35-mm dishes. Half of the dishes were treated daily with Sirtuin 1 inhibitor III (Calbiochem, Millipore) at 10 μmol/well for 5 days. At day 5, the cells were harvested and total cell RNA was isolated with the RNA Purification kit (Denville scientific) for qPCR according to the same procedure described above. The following primers were used: human FLT1 (Forward GCA CCA TAC CTC CTG CGA AA, Reverse TGG TGG CTT TGC AGT GAT AGA), human FLK1 (Forward CGT GTC TTT GTG GTG CAC TG, Reverse GGT TTC CTG TGA TCG TGG GT), and human 18S (Forward GAG GAT GAG GTG GAA CGT GT, Reverse TCT TCA GTC GCT CCA GGT CT).
Aortic sprouting angiogenesis assay.
Thoracic aortas were obtained from 12-wk-old control and Sirt1endo−/− mice. Aortic rings sectioned with a 1-mm interval were embedded in 3D Matrigel in a 96-well plate and cultured at 37°C in EGM2 without VEGF or supplemented with VEGF. Newly formed capillary cords in explant cultures were imaged after 6 days with a Nikon TE2000 microscope equipped with a CCD camera (Hamamatsu Photonics) at a magnification of ×40. Quantitative angiogenesis assays were performed according to a previously published protocol (3).
Statistical analyses.
All experiments were repeated at least three times. Values are given as means ± SE. Data were analyzed with a Mann-Whitney U-test for comparison of two groups or ANOVA (with or without repeated measurements), with post hoc analysis for multiple group comparisons by the Bonferroni method. A P value of <0.05 was considered statistically significant.
RESULTS
Characterization of the Sirt1endo−/− mouse model.
Our strategy to delete SIRT1 in endothelial cells, similar to that used by Potente et al. (21), has been previously reported (4, 32). We crossbred B6;129-Sirt1tm1Ygu/J mice (homozygous for targeted allele Sirt1co/co, viable and fertile, containing a loxP-flanked neomycin cassette upstream and downstream of exon 4 of the targeted gene) with Tie2-Cre transgenic mice. The offspring thrive as their wild-type littermates (Mendelian distribution), are normotensive, and show normal blood counts (data not shown). Endothelial and endothelial progenitor cells isolated from Sirt1endo−/− mice, however, showed an increased proportion of senescent and apoptotic cells under basal conditions and reduced resistance to stress, as shown by our group (4). En face aortic preparations stained for the expression of senescence-associated β-galactosidase revealed a dramatic increase in the frequency of senescent endothelial cells already at the age of 12 wk (4).
Consequences of aging for Sirt1endo−/− cardiac functions.
The body weight of the animals increased with age but remained similar between the control and Sirt1endo−/− animals (Table 1). On echocardiography, the Sirt1endo−/− animals, after 30–40 wk of age, progressively developed a diastolic dysfunction with a higher IVRT and Tei index. LV mass and systolic function remained similar (Fig. 1). The diastolic dysfunction was in accord with the decreased mRNA expression of Serca2a in LV (Fig. 2). We also found a lower capillary density (number of CD31-positive structures per cardiomyocyte) in the Sirt1endo−/− group compared with the control animals (Fig. 3), whereas the quantification of Masson's trichrome staining in the LV and the mRNA expression of Collagen 1 were similar between the control and Sirt1endo−/− groups (Fig. 4).
Table 1.
Body weight and left ventricular echocardiographic parameters in mice of different ages
| 10–15 wo |
30–40 wo |
60–70 wo |
||||
|---|---|---|---|---|---|---|
| Control | Sirt1endo−/− | Control | Sirt1endo−/− | Control | Sirt1endo−/− | |
| Body weight, g | 19.1 ± 1.5 | 19.5 ± 1.6 | 24.1 ± 1.2 | 25 ± 1.4 | 34.1 ± 1.6 | 32 ± 3 |
| LVEDD, mm | 3.67 ± 0.15 | 3.8 ± 0.19 | 4 ± 0.14 | 3.9 ± 0.17 | 4.28 ± 0.1 | 4.17 ± 0.1 |
| LVESD, mm | 2.53 ± 0.16 | 2.74 ± 0.24 | 2.81 ± 0.12 | 2.77 ± 0.19 | 3 ± 0.1 | 3 ± 0.11 |
| IVS, mm | 0.63 ± 0.05 | 0.63 ± 0.04 | 0.65 ± 0.02 | 0.7 ± 0.04 | 0.74 ± 0.03 | 0.66 ± 0.04 |
| PW, mm | 0.58 ± 0.03 | 0.59 ± 0.05 | 0.63 ± 0.02 | 0.68 ± 0.04 | 0.8 ± 0.03 | 0.76 ± 0.04 |
Values are means ± SE. LVEDD, left ventricular end-diastolic diameter; LVESD, left ventricular end-systolic diameter; IVS, interventricular septum; PW, posterior wall; wo, weeks of age.
Fig. 1.

Cardiac consequences of endothelial Sirtuin 1 inactivation at different ages: comparisons of left ventricular (LV) weight/body weight, shortening fraction, isovolumic relaxation time (IVRT), and Tei index at different ages between control and Sirt1endo−/− mice measured by echocardiography. Echocardiography shows a diastolic dysfunction after 30–40 wk of age (wo) with increased IVRT and Tei index. Shortening fraction and LV weight/body weight were similar between the groups of experimental animals.
Fig. 2.
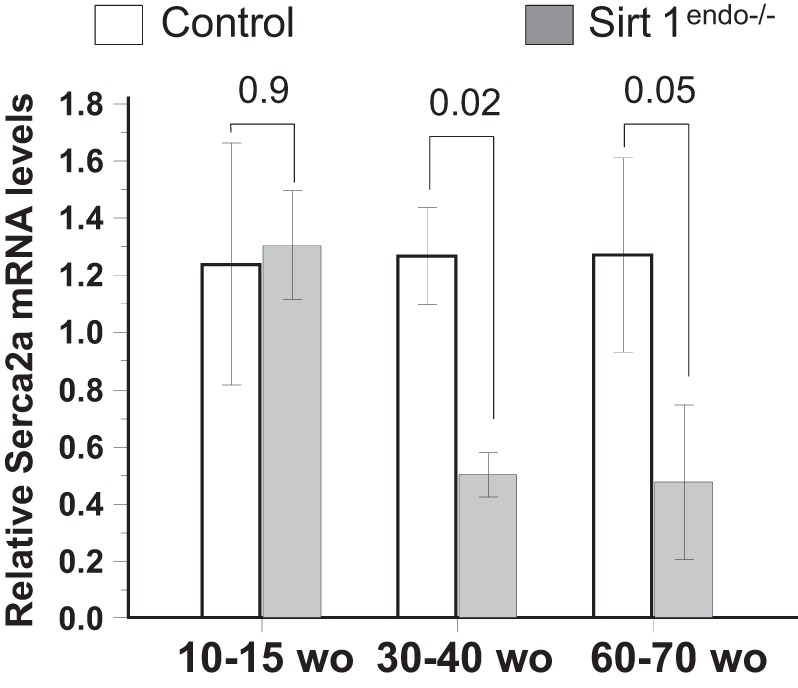
Cardiac expression of Serca2a measured by quantitative PCR in the LV of control and Sirt1endo−/− animals at different ages. Expression of Serca2a mRNA in the LV of Sirt1endo−/− animals decreased after 30–40 wk of age compared with control animals.
Fig. 3.

CD-31 staining (brown stain) in the LV of control and Sirt1endo−/− animals at different ages. Quantification of the CD31 staining is presented in the bar graph as the number of vessels per square millimeter. Note that the capillary density in the LV myocardium of the Sirt1endo−/− mice was decreased after 30–40 wk of age compared with control mice.
Fig. 4.

A: Masson's trichrome staining (blue stain) in the LV of control and Sirt1endo−/− animals at different ages. Quantification of the fibrosis is presented in the bar graph as % of Masson's trichrome-positive area. No evidence of exaggerated fibrosis in Sirt1endo−/− animals is seen. Masson's trichrome staining did not show any significant differences of LV fibrosis between Sirt1endo−/− and control animals at different ages. B: cardiac expression of Collagen 1 measured by quantitative PCR in the LV of control and Sirt1endo−/− animals at different ages. mRNA expression of collagen 1 in the LV of Sirt1endo−/− and control animals was not different.
Sirtuin 6 mRNA expression in the myocardium was decreased in the Sirt1endo−/− animals compared with the control animals after 30–40 wk of age and preceded the decreased Sirtuin 3 mRNA expression (Fig. 5, A and B), but PGC1a mRNA expression remained similar between groups (Fig. 5C). The Sirt1endo−/− animals presented a higher short-to-long endoglin mRNA expression in 30- to 40-wk-old and older animals (Fig. 5D).
Fig. 5.

Cardiac expression of metabolic (Sirtuin 3, Sirtuin 6, and PGC1a) and senescence (short:long endoglin) markers measured by quantitative PCR in the LV of control and Sirt1endo−/− animals at different ages. mRNA expression of Sirtuin 6 (A) and Sirtuin 3 (B) decreased with age in the myocardium of Sirt1endo−/− animals. PGC1a mRNA abundance (C) was not different from control animals. Compared with control mice, the mRNA expression of short:long endoglin (D) increased in the Sirt1endo−/− mice with aging.
To explain the decreased angiogenesis in our Sirt1endo−/− animals we explored the protein expression of HIF1α, an angiogenic factor activated by a Sirtuin 1 deacetylation. We did not detect any difference in the protein expression or in the proportion of acetylated HIF1α in the myocardium of the different groups of animals (Fig. 6A). VEGFa mRNA expression was also similar in 30- to 40-wk-old and younger mice but decreased in the 60- to 70-wk-old Sirt1endo−/− animals compared with control animals (Fig. 6B). Thus, the above studies showed that the cardiac phenotype of Sirt1endo−/− animals is mild, indicating that Sirtuin 1 is dispensable under basal conditions. This finding necessitated further investigation of the cardiac phenotype of these mice under applied stress.
Fig. 6.

A: expression of total protein HIF1α and immunoprecipitated acetylated HIF1α measured by immunoblotting in the LV of control and Sirt1endo−/− animals. B: cardiac expression of VEGFa measured by quantitative PCR in the LV of control and Sirt1endo−/− animals at different ages.
Consequences of left ventricular pressure overload for cardiac functions of Sirt1endo−/− mice.
LV mass increased after surgery in a similar manner in both strains of mice. Also, systolic function decreased with surgery but did not differ between control and Sirt1endo−/− animals (Fig. 7). In contrast, a diastolic dysfunction (IVRT and Tei index) appeared 7 wk after the TAC in the Sirt1endo−/− animals. At that time the mRNA expression of Serca2a in the LV was decreased in the TAC animals, but no difference was found between the Sirt1endo−/− TAC and control TAC animals (Fig. 8). Similarly, Sirtuin 3 mRNA expression decreased in the myocardium of both TAC groups, but we did not find any differences of Sirtuin 6 mRNA expression (Fig. 9, A and B). PGC1α mRNA expression decreased similarly in both TAC groups (Fig. 9C).
Fig. 7.

Comparisons of body weight, LV weight/body weight, shortening fraction, IVRT, and Tei index between control animals without surgery, control animals with transverse aortic constriction (TAC), and Sirt1endo−/− TAC animals measured by echocardiography. Measurements were performed before the surgery (baseline) and 1 and 7 wk after the surgery. Inactivation of Sirtuin 1 in the coronary endothelial cells worsens the cardiac consequences due to pressure overload provoked by TAC. The diastolic dysfunction (IVRT and Tei index) of the Sirt1endo−/− animals subjected to TAC was more profound on echocardiography than in control TAC animals. LV weight/body weight increased and shortening fraction decreased similarly after surgery between the 2 groups. *P < 0.05, Sirt1endo−/− TAC vs. controls TAC; **P < 0.05, controls vs. Sirt1endo−/− TAC and controls TAC.
Fig. 8.
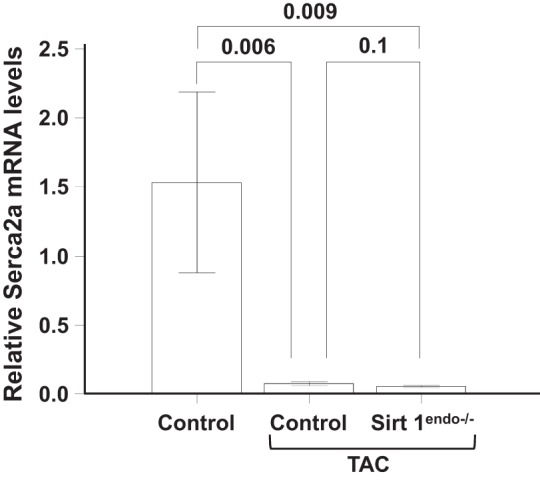
Cardiac expression of Serca2a measured by quantitative PCR in the LV of control animals without surgery, control animals with TAC, and Sirt1endo−/− TAC animals 7 wk after surgery. Expression of Serca2a mRNA in the LV decreased in both constricted TAC groups but was not different between Sirt1endo−/− and control animals.
Fig. 9.
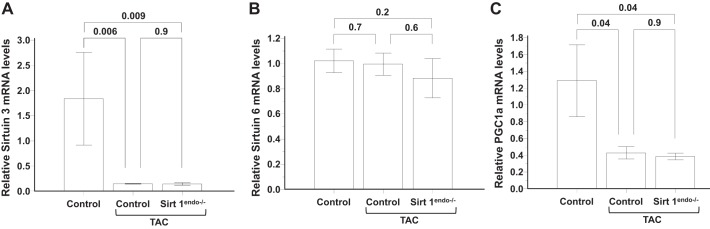
Cardiac expression of metabolic (Sirtuin 3, Sirtuin 6, and PGC1a) markers measured by quantitative PCR in the LV of control animals without surgery, control animals with TAC, and Sirt1endo−/− TAC animals 7 wk after surgery. A: mRNA expression of Sirtuin 3 in the LV decreased in both TAC groups. B: Sirtuin 6 mRNA expression levels in the LV remained similar between the different groups. C: mRNA expression of PGC1a in the LV decreased in both constricted groups.
The capillary density in the TAC Sirt1endo−/− animals remained lower than in the control TAC mice (Fig. 10). The development of fibrosis and the mRNA expression of Collagen 1 were significantly higher in the Sirt1endo−/− TAC group compared with the two other groups (Fig. 11).
Fig. 10.

CD31 staining (brown stain) in the LV of control animals without surgery, control animals with TAC, and Sirt1endo−/− TAC animals 7 wk after surgery. Quantification of the CD31 staining is presented in the bar graph as the number of vessels per square millimeter. Note that the capillary density in the control TAC animals increased, but the opposite occurred in the Sirt1endo−/− TAC group of mice.
Fig. 11.

A: Masson's trichrome staining (blue stain) in the LV of control animals without surgery, control animals with TAC, and Sirt1endo−/− TAC animals 7 wk after surgery. Quantification of the fibrosis is presented in the bar graph as % of Masson's trichrome-positive area. LV fibrosis increased in both groups of TAC animals, but the extent of fibrosis was significantly higher in the Sirt1endo−/− animals. B: cardiac expression of Collagen 1 measured by quantitative PCR in the LV of control animals without surgery, control animals with TAC, and Sirt1endo−/− TAC animals 7 wk after surgery. Collagen 1 mRNA expression in the myocardium of the Sirt1endo−/− constricted animals significantly increased, but there was no significant difference with the control group.
The aortic constriction increased the expression of the HIF1α protein in the LV and was more prominent in the Sirt1endo−/− TAC animals. However, the proportion of acetylated to total HIF1α remained constant among mice with and without TAC (Fig. 12A). This indicated that the abundance of acetylated HIF1α was the highest in the Sirt1endo−/− mice subjected to TAC. VEGFa mRNA expression increased similarly in both TAC groups (Fig. 12B).
Fig. 12.

A: expression of total protein HIF1α and immunoprecipitated acetylated HIF1α measured by immunoblotting in the LV of control animals without surgery, control animals with TAC, and Sirt1endo−/− TAC animals 7 wk after surgery. Immunoblotting of HIF1α in the LV shows the increased expression of total HIF1α in the LV of Sirt1endo−/− of TAC animals. Expression of lysine-acetylated HIF1α was similar between the TAC groups; therefore the proportion of deacetylated HIF1α was higher in the Sirt1endo−/− animals. B: cardiac expression of VEGFa measured by quantitative PCR in the LV of control animals without surgery, control animals with TAC, and Sirt1endo−/− TAC animals 7 wk after surgery. mRNA expression of VEGFa increased in the LV of both constricted groups.
Consequences of Adriamycin injections.
Similar studies were conducted in mice with Adriamycin cardiotoxicity, a model of dystrophic myocardial injury. No differences between Sirt1endo−/− and control Adriamycin-treated animals were detectable on echocardiography (Fig. 13). All animals that received Adriamycin injections developed systolic and diastolic dysfunctions associated with an increased LV mass. This cardiomyopathy was nonetheless similar between Sirt1endo−/− and control animals. On morphological examination, Adriamycin treatment was associated with a similar increase in fibrosis and decrease in capillary density in the LV of both Sirt1endo−/− and control animals (Fig. 14).
Fig. 13.

Comparisons of LV weight/body weight, shortening fraction, IVRT, and Tei index measured by echocardiography between Sirt1endo−/− and control animals subjected to Adriamycin injections. Echocardiography was performed at baseline and 5 wk and 9 wk after the beginning of treatment.
Fig. 14.

A: Masson's trichrome staining (blue stain) in the LV of control animals without Adriamycin, control animals with Adriamycin, and Sirt1endo−/− Adriamycin animals 9 wk after the beginning of treatment. Quantification of fibrosis is presented in the bar graph as % of Masson's trichrome-positive area. B: CD31 staining (brown stain) in the LV of control animals without Adriamycin, control animals with Adriamycin, and Sirt1endo−/− Adriamycin animals 9 wk after the beginning of treatment. Quantification of CD31 staining is presented in the bar graph as the number of vessels per square millimeter.
Cell culture and ex vivo aortic angiogenesis assay.
To understand why capillary density remained lower in the Sirt1endo−/− TAC mice despite the higher VEGFa mRNA expression in this group, we analyzed the mRNA expression of VEGF receptors in HUVECs exposed to Sirt1 inhibitor. The administration of Sirt1 inhibitor over 5 days to HUVECs was associated with a significant decrease of VEGF receptors FLT1 and FLK1 mRNA expression (Fig. 15). This can explain why the angiogenesis remained altered in the Sirt1endo−/− animals despite the higher expression of VEGF in the aortic constriction groups.
Fig. 15.
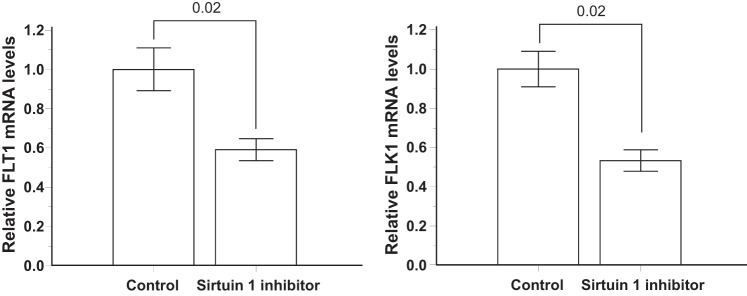
Expression of the VEGF receptors FLT1 and FLK1 measured by quantitative PCR in cultured immortalized human umbilical vein endothelial cells (HUVECs) exposed to 5 days of Sirtuin 1 inhibitor or placebo (Control).
In the aortic sprouting assay, the angiogenesis of the control aortic rings showed a trend (P = 0.08) toward increase of the number of capillary sprouts with the addition of VEGF in the culture medium, but this increase was absent in the Sirt1endo−/− aortic rings (Fig. 16). Moreover, in the VEGF-enriched medium the number of capillary sprouts was significantly higher in control than Sirt1endo−/− rings. This confirms the impaired angiogenic response to VEGF in the Sirt1endo−/− mice.
Fig. 16.

Ex vivo capillary sprouting from aortic explants of control and Sirt1endo−/− mice after 6 days of culture in medium with or without VEGF. The numbers of capillary sprouts per aortic explant are presented in the bar graph.
DISCUSSION
Decline of angiogenic capacity is a hallmark of the aging process and has also been described in many cardiac diseases (11, 18). Here we show for the first time the complex consequences for the myocardium of angiogenic failure due to decreased expression of the Sirtuin 1 protein in coronary endothelial cells. In our model the decreased expression of the VEGF receptors observed in vitro in endothelial cells exposed to Sirtuin 1 inhibitor can explain the impaired capillary density observed in our Sirt1endo−/− animals. A decreased number of capillaries has been previously described as a mechanism of cardiac failure (18, 27). Especially in pathological situations increasing LV work (e.g., hypertension), the development of LVH requires a matching level of angiogenesis (13, 25). The disruption of coordination between cardiomyocyte growth and angiogenesis leads to the development of heart failure (27). Angiogenesis is controlled by several pathways; among them VEGF and its receptors (FLT1 and FLK1) play important roles. Knockout of Sirtuin 1 in coronary endothelial cells is associated with a decreased expression of FLT1 and FLK1, compromising the angiogenesis (21). In the absence of aortic constriction, Sirt1endo−/− animals develop diastolic dysfunction without any significant myocardial fibrosis or increased expression of the HIF1α protein in the myocardium. Once aortic banding is created, the increased oxygen demands required by the development of LVH unmask the inability of those animals to adapt normally to LV pressure overload. Those animals showed worsened diastolic dysfunction, cardiac fibrosis, and overexpression of HIF1α protein. Despite the overexpression of HIF1α and VEGF, the capillary density remained insufficient, perhaps because of the impaired expression of the VEGF receptors FLT1 and FLK1. Interestingly, these observations in Sirt1endo−/− animals with hypertrophic cardiomyopathy (aortic banding) were not observed in the dystrophic model of Adriamycin injections in the Sirt1endo−/− animals.
Although Sirtuin 1 is genetically deleted in endothelial cells, young 10- to 15-wk-old animals do not exhibit any cardiac abnormalities, as judged by echocardiography, histology, and molecular biology studies. The cardiac alterations appear later, after 30 wk of age. This could mean several things. First, this could be related to the insufficient sensitivity of our technological platform (echocardiography, molecular biology, and histology), which, however, is a gold standard for such investigations. Second, this could mean that the absence of Sirtuin 1 expression in endothelial cells is not sufficient to provoke cardiac alterations. In accord with this view, we show here that aging and pressure overload are required to unmask cardiac abnormalities. Therefore, sirtuin expression in endothelial cells is mandatory for the adaptation of cardiac functions to aging and pressure overload. In addition, the aortic ring experiments performed in our study were conducted with rings obtained from 12-wk-old animals. At this age the animals do not show any differences in aortic sprouting without VEGF application (Fig. 16). In contrast, exposing rings to VEGF provokes the discrepant response of aortic sprouting between control and Sirt1endo−/− animals. This finding emphasizes the role played by endothelial Sirtuin 1 in the angiogenesis induced by VEGF.
The only age-dependent cardiac modification observed in our control animals is the mild increase of cardiac fibrosis on the Masson's trichrome staining. However, this increase is similar in control and Sirt1endo−/− animals, and the area of fibrosis is rather small even in the older group. It is therefore likely that this mild increase of fibrosis cannot be responsible for the observed changes in the cardiac function of our animals. It is also possible that 60–70 wk of age is not sufficiently advanced to reveal any significant modifications. The absence of alterations in echocardiography and in molecular biology parameters in the hearts of the 60- to 70-wk-old control animals does not preclude later abnormalities.
The mRNA expression of Serca2a is decreased in the myocardium of our Sirt1endo−/− animals but not in the control animals. This is in accordance with the diastolic dysfunction observed on echocardiography. The protein Serca2a is known to be related to the relaxation of the cardiomyocyte by sequestering Ca2+ from the cytosol into the sarcoplasmic reticulum (20). Serca2a is also involved in angiogenesis. The protein Serca2a is a key step in redox-sensitive Ca2+ store regulation via S-glutathionylation. This glutathionylation of Serca2a is induced by the accumulation of reactive oxygen species and enhances ATP-dependent Ca2+ uptake, decreases cytoplasmic Ca2+ levels, and promotes endothelial angiogenic responses in vitro (8). Therefore the decreased expression of Serca2a in the myocardium of our aging or pressure-overloaded Sirt1endo−/− animals can be also responsible for the decreased angiogenesis (30).
Sirtuin 3 and 6 expressions do not decrease with aging in our control animals, but they are decreased with aging in the Sirt1endo−/− myocardium. Sirtuin 3 is expressed in the mitochondria and plays a role in the response to low energy input in mitochondria (23). Sirtuin 6 is localized to the nucleus, and its expression has been related to life span and protection against the development of cardiac hypertrophy (28). Little is known about the regulation of Sirtuin 3 and 6 expression in heart. Along with Sirtuin 1, Sirtuin 3 and 6 levels increase in response to calorie restriction. With aging (59- to 70-yr-old subjects) the Sirtuin 3 expression in the vastus lateralis muscle of humans was shown to decrease (15), but in the myocardium it has never been described. Our results show that Sirtuin 3 and 6 mRNA expression is decreased with aging (mostly in 60- to 70 wk-old animals) in the Sirt1endo−/− animals but not in control animals. This finding suggests that in the myocardium Sirtuin 3 and 6 expression during aging are decreased because of the absence of Sirtuin 1 in the endothelial cells.
The mRNA expression of PGC1a was not different between the experimental groups, although PGC1a is a known target of Sirt1. PGC1a regulates the angiogenesis in the cardiac tissue by driving the expression of VEGF (1). The knocked-down expression of PGC1a in cardiac tissue leads to an impaired angiogenesis and the development of a peripartum cardiomyopathy (19). The administration of VEGF in those animals partly rescued the peripartum cardiomyopathy. The Sirtuin 1 gene in our model is knocked out only in the endothelial cells. Therefore, it is possible that the expressions of Sirtuin 1 and PGC1a in the myocardium undergo compensatory changes, which are reflected in the results obtained from the LV.
In conclusion, a model of hypertrophic cardiomyopathy, as opposed to dystrophic cardiomyopathy, demonstrates the inability of endothelial cells deficient in Sirtuin 1 to create new vessels and adapt to a challenging condition, like pressure overload, leading to the development of diastolic dysfunction.
GRANTS
Studies were supported in part by National Institutes of Health grants and the Westchester Artificial Kidney Foundation. This work was supported by the Societe Francaise de Nephrologie, la Societe de Reanimation de Langue Francaise, le Conseil Regional de Picardie, and the University Hospital of Amiens (J. Maizel).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.M., S.X., J.C., C.H.S.L., R.V., and M.S.G. conception and design of research; J.M., S.X., J.C., C.H.S.L., and R.V. performed experiments; J.M., J.C., and M.S.G. analyzed data; J.M., J.C., and M.S.G. interpreted results of experiments; J.M. prepared figures; J.M. and M.S.G. drafted manuscript; J.M., S.X., and M.S.G. edited and revised manuscript; J.M., S.X., J.C., C.H.S.L., R.V., and M.S.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We are indebted to Drs. Leonard and Carol Eisenberg for help in using echocardiography equipment.
REFERENCES
- 1.Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, Baek KH, Rosenzweig A, Spiegelman BM. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature 451: 1008–1012, 2008. [DOI] [PubMed] [Google Scholar]
- 2.Bertazzoli C, Bellini O, Magrini U, Tosana MG. Quantitative experimental evaluation of adriamycin cardiotoxicity in the mouse. Cancer Treat Rep 63: 1877–1883, 1979. [PubMed] [Google Scholar]
- 3.Brodsky S, Chen J, Lee A, Akassoglou K, Norman J, Goligorsky MS. Plasmin-dependent and -independent effects of plasminogen activators and inhibitor-1 on ex vivo angiogenesis. Am J Physiol Heart Circ Physiol 281: H1784–H1792, 2001. [DOI] [PubMed] [Google Scholar]
- 4.Chen J, Xavier S, Moskowitz-Kassai E, Chen R, Lu CY, Sanduski K, Špes A, Turk B, Goligorsky MS. Cathepsin cleavage of sirtuin 1 in endothelial progenitor cells mediates stress-induced premature senescence. Am J Pathol 180: 973–983, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci USA 100: 10794–10799, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dahab GM, Kheriza MM, El-Beltagi HM, Fouda AM, El-Din OA. Digital quantification of fibrosis in liver biopsy sections: description of a new method by Photoshop software. J Gastroenterol Hepatol 19: 78–85, 2004. [DOI] [PubMed] [Google Scholar]
- 7.deAlmeida AC, van Oort RJ, Wehrens XH. Transverse aortic constriction in mice. J Vis Exp 38: 1729, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evangelista AM, Thompson MD, Weisbrod RM, Pimental DR, Tong X, Bolotina VM, Cohen RA. Redox regulation of SERCA2 is required for vascular endothelial growth factor-induced signaling and endothelial cell migration. Antioxid Redox Signal 17: 1099–1108, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Facchetti F, Monzani E, Cavallini G, Bergamini E, La Porta CA. Effect of a caloric restriction regimen on the angiogenic capacity of aorta and on the expression of endothelin-1 during ageing. Exp Gerontol 42: 662–667, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature 460: 587–591, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giordano FJ, Gerber HP, Williams SP, VanBruggen N, Bunting S, Ruiz-Lozano P, Gu Y, Nath AK, Huang Y, Hickey R, Dalton N, Peterson KL, Ross J, Chien KR, Ferrara N. A cardiac myocyte vascular endothelial growth factor paracrine pathway is required to maintain cardiac function. Proc Natl Acad Sci USA 98: 5780–5785, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goligorsky MS. SIRTing out the link between autophagy and ageing. Nephrol Dial Transplant 25: 2434–2436, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Izumiya Y, Shiojima I, Sato K, Sawyer DB, Colucci WS, Walsh K. Vascular endothelial growth factor blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload. Hypertension 47: 887–893, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol 230: 230–242, 2001. [DOI] [PubMed] [Google Scholar]
- 15.Lanza IR, Short DK, Short KR, Raghavakaimal S, Basu R, Joyner MJ, McConnell JP, Nair KS. Endurance exercise as a countermeasure for aging. Diabetes 57: 2933–2942, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 322: 1561–1566, 1990. [DOI] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−DeltaDeltaCT method. Methods 25: 402–408, 2001. [DOI] [PubMed] [Google Scholar]
- 18.Oka T, Akazawa H, Naito AT, Komuro I. Angiogenesis and cardiac hypertrophy: maintenance of cardiac function and causative roles in heart failure. Circ Res 114: 565–571, 2014. [DOI] [PubMed] [Google Scholar]
- 19.Patten IS, Rana S, Shahul S, Rowe GC, Jang C, Liu L, Hacker MR, Rhee JS, Mitchell J, Mahmood F, Hess P, Farrell C, Koulisis N, Khankin EV, Burke SD, Tudorache I, Bauersachs J, del Monte F, Hilfiker-Kleiner D, Karumanchi SA, Arany Z. Cardiac angiogenic imbalance leads to peripartum cardiomyopathy. Nature 485: 333–338, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Periasamy M, Bhupathy P, Babu GJ. Regulation of sarcoplasmic reticulum Ca2+ ATPase pump expression and its relevance to cardiac muscle physiology and pathology. Cardiovasc Res 77: 265–273, 2008. [DOI] [PubMed] [Google Scholar]
- 21.Potente M, Ghaeni L, Baldessari D, Mostoslavsky R, Rossig L, Dequiedt F, Haendeler J, Mione M, Dejana E, Alt FW, Zeiher AM, Dimmeler S. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev 21: 2644–2658, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reed MJ, Karres N, Eyman D, Vernon RB. Culture of murine aortic explants in 3-dimensional extracellular matrix: a novel, miniaturized assay of angiogenesis in vitro. Microvasc Res 73: 248–252, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sack MN, Finkel T. Mitochondrial metabolism, sirtuins, and aging. Cold Spring Harb Perspect Biol 4: a013102, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sahn DJ, DeMaria A, Kisslo J, Weyman A. Recommendations regarding quantitation in M-mode echocardiography: results of a survey of echocardiographic measurements. Circulation 58: 1072–1083, 1978. [DOI] [PubMed] [Google Scholar]
- 25.Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, Shimizu I, Asahara T, Hamada H, Tomita S, Molkentin JD, Zou Y, Komuro I. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 446: 444–448, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Shimada T, Takeshita Y, Murohara T, Sasaki K, Egami K, Shintani S, Katsuda Y, Ikeda H, Nabeshima Y, Imaizumi T. Angiogenesis and vasculogenesis are impaired in the precocious-aging Klotho mouse. Circulation 110: 1148–1155, 2004. [DOI] [PubMed] [Google Scholar]
- 27.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest 115: 2108–2118, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sundaresan NR, Vasudevan P, Zhong L, Kim G, Samant S, Parekh V, Pillai VB, Ravindra PV, Gupta M, Jeevanandam V, Cunningham JM, Deng CX, Lombard DB, Mostoslavsky R, Gupta MP. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat Med 18: 1643–1650, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tei C, Nishimura RA, Seward JB, Tajik AJ. Noninvasive Doppler-derived myocardial performance index: correlation with simultaneous measurements of cardiac catheterization measurements. J Am Soc Echocardiogr 10: 169–178, 1997. [DOI] [PubMed] [Google Scholar]
- 30.Thompson MD, Mei Y, Weisbrod RM, Silver M, Shukla PC, Bolotina VM, Cohen RA, Tong X. Glutathione adducts on sarcoplasmic/endoplasmic reticulum Ca2+ ATPase Cys-674 regulate endothelial cell calcium stores and angiogenic function as well as promote ischemic blood flow recovery. J Biol Chem 289: 19907–19916, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Kerckhoven R, van Veghel R, Saxena PR, Schoemaker RG. Pharmacological therapy can increase capillary density in post-infarction remodeled rat hearts. Cardiovasc Res 61: 620–629, 2004. [DOI] [PubMed] [Google Scholar]
- 32.Vasko R, Xavier S, Chen J, Lin CH, Ratliff B, Rabadi M, Maizel J, Tanokuchi R, Zhang F, Cao J, Goligorsky MS. Endothelial sirtuin 1 deficiency perpetrates nephrosclerosis through downregulation of matrix metalloproteinase-14: relevance to fibrosis of vascular senescence. J Am Soc Nephrol 25: 276–291, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]