

Abstract
Evolution of metabolic syndrome is associated with a progressive reduction in skeletal muscle microvessel density, known as rarefaction. Although contributing to impairments to mass transport and exchange, the temporal development of rarefaction and the contributing mechanisms that lead to microvessel loss are both unclear and critical areas for investigation. Although previous work suggests that rarefaction severity in obese Zucker rats (OZR) is predicted by the chronic loss of vascular nitric oxide (NO) bioavailability, we have determined that this hides a biphasic development of rarefaction, with both early and late components. Although the total extent of rarefaction was well predicted by the loss in NO bioavailability, the early pulse of rarefaction developed before a loss of NO bioavailability and was associated with altered venular function (increased leukocyte adhesion/rolling), and early elevation in oxidant stress, TNF-α levels, and the vascular production of thromboxane A2 (TxA2). Chronic inhibition of TNF-α blunted the severity of rarefaction and also reduced vascular oxidant stress and TxA2 production. Chronic blockade of the actions of TxA2 also blunted rarefaction, but did not impact oxidant stress or inflammation, suggesting that TxA2 is a downstream outcome of elevated reactive oxygen species and inflammation. If chronic blockade of TxA2 is terminated, microvascular rarefaction in OZR skeletal muscle resumes, but at a reduced rate despite low NO bioavailability. These results suggest that therapeutic interventions against inflammation and TxA2 under conditions where metabolic syndrome severity is moderate or mild may prevent the development of a condition of accelerated microvessel loss with metabolic syndrome.
Keywords: skeletal muscle perfusion, vascular remodeling, vascular reactivity, rodent models of obesity, nitric oxide bioavailability, chronic inflammation
epidemiological studies have clearly and consistently demonstrated that both the incidence and prevalence of the metabolic syndrome is continuing to increase in Western society and in most developed economies worldwide (4, 9, 24). Although each of the constituent systemic pathologies (e.g., impaired glycemic control, atherogenic dyslipidemia, hypertension, obesity) have been well documented to increase the risk for development of impaired vascular structure/function and peripheral vascular disease (PVD) (10, 11, 13, 14, 16, 25, 29, 45), the significance of this multi-pathology state is that it increases the risk for individuals to develop PVD well beyond that for any single contributing element. Given the impact of PVD on life expectancy, quality of life, depression, and the direct (health care) and indirect (lost productivity) economic costs to society (3, 41, 44), considerable emphasis has been placed on not only the study of PVD and its contributing elements in human subjects but also for the detailed investigation of appropriate animal models of vasculopathy in the metabolic syndrome (1, 49, 51). However, although the existing literature is replete with descriptions of correlations between the systemic pathologies of the metabolic syndrome and specific indexes of vasculopathy, our understanding of these relationships is hampered since insufficient knowledge has been presented, which details the developments of vascular impairments that accompany this constellation of pathologies.
The obese Zucker rat is an animal model of the metabolic syndrome that has become important for the study of vascular dysfunction in this setting (7, 20, 31). Due to its characteristic dysfunctional leptin receptor gene, the obese Zucker rat (OZR) experiences chronic hyperphagia and rapidly becomes obese compared with its control strain, the lean Zucker rat (LZR). Stemming from this, the OZR becomes progressively insulin-resistant, dyslipidemic (moderate hypercholesterolemia and severe hypertriglyceridemia), and moderately hypertensive (1, 15, 20, 49). Although considerable evidence has been presented by our laboratory (5, 27) and by numerous others (18, 30, 32, 35, 36, 47, 53) regarding the correlation between indexes of vascular dysfunction with characteristics of the metabolic syndrome in these animals at specific ages, minimal evidence has been produced regarding the temporal development of the vascular impairments in these animals, and how these correlate with the evolution of the metabolic syndrome, and other putative markers of dysfunction (e.g., markers of inflammation).
A consistent observation that has been made in the metabolic syndrome in both OZR and in human subjects (33, 38, 43) is a loss in peripheral microvessel density (rarefaction) that is associated with this disease state. As defined by Goligorsky (26), microvascular rarefaction is a form of microvascular adaptation in response to a challenged or disease state that results in a reduction in arteriolar or capillary density within an affected tissue. In OZR, this process plateaus at an ∼25% reduction in microvessel density compared with control levels in LZR (21, 42, 48), with clear implications for mass transport/exchange and muscle fatigue resistance (46). Furthermore, previous results have demonstrated that this response is not dependent on the development of hypertension in OZR (22) and that it is associated with the development of insulin resistance and low bioavailability of nitric oxide (NO) within the microvasculature (23). However, at present we do not have a detailed understanding of either how or why microvessels begin to degrade in skeletal muscle of OZR with the metabolic syndrome, and our understanding of the time course of this evolving rarefaction is superficial at best. The primary purpose of the present study was to address this gap in our understanding of skeletal muscle microvasculopathy in the metabolic syndrome and to determine the initiating factors for and temporal development of the microvascular rarefaction that evolves in this tissue in OZR. Our initial hypothesis was that skeletal muscle microvessel density declines in OZR in parallel to the evolving reduction to vascular NO bioavailability.
MATERIALS AND METHODS
Animals.
Male LZR and OZR (Harlan) were fed standard chow and drinking water ad libitum, unless otherwise indicated, and were housed in the animal care facility at the West Virginia University Health Sciences Center. All protocols received prior Institutional Animal Care and Use Committee approval. Animals were used for terminal experiments at 7, 10, 13, 17, or 20 wk of age (n = 8–10 animals in each age group for OZR; n = 6–8 animals in each age group for LZR).
At each age, after an overnight fast, rats were anesthetized with injections of pentobarbital sodium (50 mg/kg ip) and received tracheal intubation to facilitate maintenance of a patent airway. In all rats, a carotid artery and an external jugular vein were cannulated for determination of arterial pressure and for infusion of supplemental anesthetic or pharmacological agents, as necessary. Blood samples were drawn from the venous cannula within ∼20 min of implantation for determination of glucose (Freestyle; Abbott Diabetes Care, Alameda, CA) and insulin concentrations (Cayman Chemical, Ann Arbor, MI). Plasma nitrotyrosine levels and markers of inflammation were determined using commercially available ELISA systems (Luminex 100 PS; EMD Millipore, Billerica, MA). Glucose levels were determined at the time of the blood draw. All other samples were spun to remove the plasma and snap frozen in liquid N2 until they could be analyzed as groups.
Preparation of isolated skeletal muscle resistance arterioles.
In all rats, the intramuscular continuation of the gracilis artery was removed and cannulated (19). Within each arteriole, vessel reactivity was evaluated in response to application of ACh (10−9 M - 10−6 M), arachidonic acid (10−9 M - 10−6 M), hypoxia (reduction in PO2 from ∼135 mmHg to ∼50 mmHg), and sodium nitroprusside (10−9 - 10−6 M). Subsequently, vessels were treated with TEMPOL (10−4 M) or Nω-nitro-l-arginine methyl ester (l-NAME; 10−4 M) to assess the contribution of vascular oxidant stress and endothelium-dependent NO production to these mechanical responses.
Measurement of vascular NO bioavailability.
From each rat, the abdominal aorta was removed and vascular NO production was assessed using amperometric sensors (World Precision Instruments, Sarasota, FL). Briefly, aortae were isolated, sectioned longitudinally, pinned in a silastic-coated dish, and superfused with warmed (37°C) physiological salt solution equilibrated with 95% O2 and 5% CO2. An NO sensor (ISO-NOPF 100) was placed in close apposition to the endothelial surface, and a baseline level of current was obtained. Subsequently, increasing concentrations of methacholine (10−10-10−6 M) were added to the bath and the changes in current were determined. To verify that responses represented NO release, these procedures were repeated following pretreatment of the aortic strip with l-NAME (10−4 M).
Determination of vascular metabolites of arachidonic acid.
Vascular production of 6-keto-prostaglandin F1α(6-keto-PGF1α; the stable breakdown product of PGI2) (34, 39), and 11-dehydro-thromboxane B2 (11-dehydro-TxB2; the stable plasma breakdown product of TxA2) (6) in response to challenge with reduced PO2 was assessed using pooled conduit arteries (femoral, saphenous, iliac) from LZR and OZR. Pooled arteries from each animal were incubated in microcentrifuge tubes in 1 ml of physiological salt solution for 30 min under control conditions (21% O2). After this time, the superfusate was removed, stored in a new microcentrifuge tube, and frozen in liquid N2, while a new aliquot of PSS was added to the vessels and the equilibration gas was switched to 0% O2 for the subsequent 30 min. After the second 30-min period, this new PSS was transferred to a fresh tube, frozen in liquid N2, and stored at −80°C. Metabolite release by the vessels was determined using commercially available EIA kits for 6-keto-PGF1α and 11-dehydro-TxB2 (Cayman).
Histological determination of microvessel density.
From each rat, the gastrocnemius muscle from the left leg was removed, rinsed in PSS, and fixed in 0.25% formalin. Muscles were embedded in paraffin and cut into 5 μm cross sections. Sections were incubated with Griffonia simplicifolia I lectin (GS-1), alkaline phosphatase (AlkPhos), and dipeptidylpeptidase IV (DPEP IV), for subsequent determination of microvessel density. GS-1 is a general stain that labels all microvessels <20 μm in diameter (28), AlkPhos preferentially stains arteriolar ends of capillaries (2), and DPEP IV preferentially stains venular ends of capillaries (37). Labeled microvessel density was determined using fluorescent (for GS-1) or light microscopy (for AlkPhos and DPEP IV) as described previously (22).
Leukocyte-endothelial cell interactions.
In a separate cohort of LZR and OZR at 7 and 10 wk of age, an in situ extensor digitorum longus muscle was prepared as described previously (50). After preparation of the muscle for imaging, 10 random high magnification fields of view (Plan-Neo 20×/0.5 and 40×/0.75; Zeiss) were recorded using a high-speed cooled digital imaging system (Axiocam HSm; Zeiss) for later analyses. Animals received a bolus injection of rhodamine 6G (0.3 mg/kg; Sigma) to permit visualization of leukocytes. Rhodamine 6G-labeled vascular cells were visualized by epi-illumination at 510–560 nm, using a 590-nm emission filter. The number of rolling and adherent leukocytes in venules was expressed as cells per squared millimeter of endothelial surface, calculated from the diameter and length of the venular segment. Leukocytes were considered adherent if they remained stationary for a 30-s observation period. Leukocytes were considered rolling if their velocity was less than 250 μm/s (54).
Contribution of TNF-α and TxA2.
In a separate cohort of rats, OZR were treated with daily injections of either pentoxifylline (TNF-α production inhibitor; 30 mg·kg−1·day−1) or SQ-29548 (PGH2/TxA2 receptor antagonist; 2 mg·kg−1·day−1) from 7 wk to 13 wk of age to assess the contributions of TNF-α production or the actions of TxA2 on the progression of microvascular rarefaction. Animals were euthanized at 7, 10, or 13 wk of age, and microvessel density was determined as described above, with biochemical determinations of plasma TNF-α, TxA2, and nitrotyrosine. In a final group of animals, OZR that had received chronic injections of SQ-29548 had treatments terminated at 13 wk of age and were left untreated out to 20 wk of age. These OZR were used at 17 and 20 wk of age to determine the impact of cessation of treatment on the progression of microvascular rarefaction.
Data and statistical analyses.
The mechanical responses of isolated arterioles following pharmacological challenge were fit with the three-parameter logistic equation: y = min + [(max − min)/(1 + 10logED50−x)], where y represents the change in arteriolar diameter; min and max represent the lower and upper bounds, respectively, of the change in arteriolar diameter with increasing agonist concentration; x is the logarithm of the agonist concentration; and logED50 represents the logarithm of the agonist concentration (x) at which the response (y) is halfway between the lower and upper bounds.
All data are presented as means ± SE. Statistically significant differences in measured and calculated parameters in the present study were determined using ANOVA. In all cases, Student-Newman-Keuls post hoc test was used when appropriate, and P < 0.05 was taken to reflect statistical significance. For the analyses of inflammatory and other putative biomarkers between groups, we used discriminant techniques to eliminate the univariate nature of ANOVA and issues of independent variable co-linearity, which minimizes the utility of regression techniques. Discriminant analyses represent a classification technique for evaluating the contribution of multiple variables to the establishment of differences between experimental groups. These analyses result in a rank ordering of ceteris paribus correlation coefficients in terms of their significance for the establishment of differences between groups.
RESULTS
Data describing the baseline characteristics of LZR and OZR at the different age ranges in the present study are summarized in Table 1. Although both strains of animals demonstrated a steady growth in their size with maturation, OZR were consistently much heavier than LZR at the same age and gradually demonstrated an increase in the presentation of the metabolic syndrome with age. By ∼13 wk of age, OZR were exhibiting the full manifestation of the metabolic syndrome, with profound obesity, impaired glycemic control, and dyslipidemia and the establishment of a moderate hypertension.
Table 1.
Characteristics of animal groups within the present study
| Age, weeks | LZR | OZR | OZR-Pentox | OZR-SQ-29548 | |
|---|---|---|---|---|---|
| Mass, g | 7 | 153 ± 5 | 229 ± 7* | 234 ± 7* | 250 ± 9* |
| 10 | 249 ± 4 | 420 ± 10* | 404 ± 8* | 392 ± 10* | |
| 13 | 311 ± 8 | 534 ± 14* | 512 ± 11* | 501 ± 11* | |
| 17 | 361 ± 5 | 664 ± 11* | — | — | |
| 20 | 375 ± 6 | 706 ± 14* | — | — | |
| Mean arterial pressure, mmHg | 7 | 102 ± 3 | 99 ± 4 | 97 ± 5 | 98 ± 4 |
| 10 | 104 ± 4 | 105 ± 6 | 96 ± 6 | 101 ± 6 | |
| 13 | 101 ± 5 | 116 ± 4 | 105 ± 5 | 101 ± 5 | |
| 17 | 104 ± 6 | 128 ± 5* | — | — | |
| 20 | 105 ± 6 | 134 ± 7* | — | — | |
| Glucose, mg/dl | 7 | 90 ± 6 | 91 ± 4 | 94 ± 6 | 96 ± 8 |
| 10 | 92 ± 5 | 94 ± 9 | 95 ± 7 | 99 ± 7 | |
| 13 | 92 ± 7 | 114 ± 6* | 108 ± 9* | 116 ± 8* | |
| 17 | 98 ± 8 | 154 ± 9* | — | — | |
| 20 | 102 ± 7 | 360 ± 15* | — | — | |
| Insulin, ng/ml | 7 | 1.0 ± 0.2 | 3.0 ± 0.4* | 3.1 ± 0.5* | 3.0 ± 0.5* |
| 10 | 1.0 ± 0.3 | 4.6 ± 0.6* | 4.4 ± 0.5* | 4.8 ± 0.5* | |
| 13 | 1.2 ± 0.3 | 6.9 ± 0.5* | 7.1 ± 0.6* | 6.8 ± 0.6* | |
| 17 | 1.4 ± 0.3 | 7.6 ± 0.6* | — | — | |
| 20 | 1.5 ± 0.4 | 8.2 ± 0.6* | — | — |
Values are means ± SE. OZR, obese Zucker rat.
P < 0.05 vs. lean Zucker rat (LZR).
Figure 1 summarizes the progressive rarefaction of the skeletal muscle microcirculation in LZR and OZR across the age ranges in the present study (Fig. 1A). Although microvessel density was similar between LZR and OZR at ∼7 wk of age, OZR demonstrated an initial phase of microvascular rarefaction, such that microvessel density was significantly decreased by 10 wk of age. Microvessel density was stable in OZR from that point until ∼13 wk of age wherein a second phase of rarefaction occurred that resulted in another significant reduction to microvessel density. Microvessel density was only modestly reduced from that point to 20 wk of age in OZR. Figure 1B presents the magnitude of microvessel loss over the four age ranges of the present study. These results further clarify that there are two periods of time in which the rate of rarefaction in skeletal muscle of OZR is substantially elevated, between 7 and 10 and 13 and 17 wk of age, with more quiescent periods with regard to microvessel density changes between 10 and 13 and 17 and 20 wk of age.
Fig. 1.
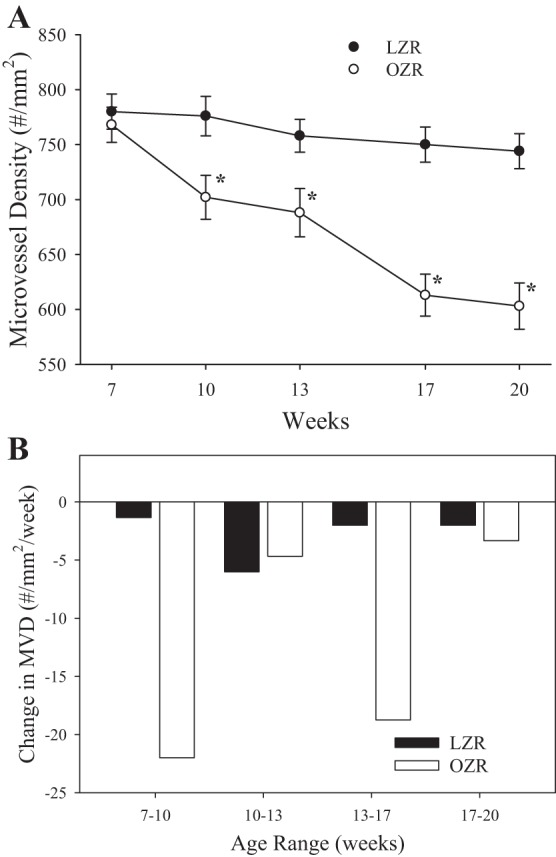
The change in skeletal muscle microvessel density (MVD) in lean Zucker rat (LZR) and obese Zucker rat (OZR) between 7 and 20 wk of age. Data are presented as means ± SE; n = 8–10 animals in each age group for OZR and n = 6–8 animals in each age group for LZR. A: raw changes in skeletal muscle microvessel density. B: slope of the changes in microvessels density over the 4 age ranges of the present study (7–10 wk, 10–13 wk, 13–17 wk, and 17–20 wk) for the 2 groups. *P < 0.05 vs. LZR at that age.
Data describing arteriolar reactivity to dilator stimuli from LZR and OZR at 7 wk of age are summarized in Fig. 2. In response to increasing concentrations of ACh (Fig. 2A), the dilator response in arterioles from OZR was not different from that in LZR. Pretreatment of vessels with the antioxidant TEMPOL had no impact on dilator responses to ACh in either strain, although pretreatment with the NO synthase inhibitor l-NAME virtually abolished all mechanical reactivity to ACh in OZR. For increasing concentrations of arachidonic acid (Fig. 2B), arterioles from OZR exhibited a similar dilator response compared with LZR, and this was not impacted by TEMPOL treatment. However, application of l-NAME reduced arachidonic acid-induced dilation in arterioles from OZR. Dilator responses to sodium nitroprusside (Fig. 2C) did not differ between arterioles of LZR and OZR at 7 wk of age and were unaffected by treatment with either TEMPOL or l-NAME. Hypoxic dilation of resistance arterioles from OZR was not different from that in LZR and was unaffected following pretreatment with TEMPOL. Pretreatment with l-NAME resulted in a consistent reduction to hypoxic dilation in OZR. In all cases, responses of LZR following treatment with TEMPOL were not significantly impacted, whereas l-NAME treatment significantly attenuated responses to ACh, arachidonic acid, and hypoxia (data not shown).
Fig. 2.

Data describing the dilator reactivity of ex vivo first order skeletal muscle (gracilis) resistance arterioles from LZR and OZR at 7 wk of age. Data are presented for the dilator responses of arterioles in response to increasing concentrations of ACh (A), arachidonic acid (B), sodium nitroprusside (C), and reduced oxygen tension (D). Data are presented as means ± SE; n = vessels from 8–10 animals in for OZR and n = vessels from 6–8 animals for LZR. *P < 0.05 in the upper bound (A–C) or magnitude of the response (D) vs. LZR; †P < 0.05 in the upper bound (A–C) or magnitude of the response (D) vs. OZR. l-NAME, Nω-nitro-l-arginine methyl ester.
Figure 3 describes dilator responses of arterioles from LZR and OZR at 10 wk of age in response to imposed stimuli. ACh caused a concentration-dependent dilation of arterioles from OZR that was very similar to that in LZR and was not significantly impacted by pretreatment with TEMPOL (Fig. 3A). However, pretreatment with l-NAME abolished reactivity to ACh in arterioles from OZR. Dilator responses to arachidonic acid (Fig. 3B) were reduced in vessels from OZR versus LZR, and pretreatment with TEMPOL restored much of this lost reactivity. Pretreatment with l-NAME reduced dilator responses of arterioles from OZR with increasing concentrations of arachidonic acid. Dilator responses to increasing concentrations of sodium nitroprusside were not different between LZR and OZR. Hypoxic dilation of resistance arterioles from OZR was reduced compared with that for vessels from LZR, but was nearly restored following pretreatment with TEMPOL (Fig. 3D). Incubation of vessels with l-NAME resulted in a significant reduction to hypoxic dilation in vessels from OZR. In all cases, responses of vessel from LZR following treatment with TEMPOL were not impacted, whereas l-NAME treatment significantly attenuated responses to ACh, arachidonic acid, and hypoxia (data not shown).
Fig. 3.

Data describing the dilator reactivity of ex vivo first order skeletal muscle (gracilis) resistance arterioles from LZR and OZR at 10 wk of age. Data are presented for the dilator responses of arterioles in response to increasing concentrations of ACh (A), arachidonic acid (B), sodium nitroprusside (C), and reduced oxygen tension (D). Data are presented as means ± SE; n = vessels from 8–10 animals in for OZR and n = vessels from 6–8 animals for LZR. *P < 0.05 in the upper bound (A–C) or magnitude of the response (D) vs. LZR; †P < 0.05 in the upper bound (A–C) or magnitude of the response (D) vs. OZR.
Data describing the dilator responses of arterioles from LZR and OZR at 13 wk of age in response to imposed challenges are summarized in Fig. 4. In response to increasing concentrations of ACh, dilator responses from arterioles of OZR were reduced from that in LZR and were restored by pretreatment with TEMPOL (Fig. 4A). As above, pretreatment with l-NAME abolished arteriolar reactivity to ACh in vessels from OZR. Dilator responses to arachidonic acid were reduced in OZR at 13 wk of age, and pretreatment with TEMPOL was able to improve this response (Fig. 4B). Pretreatment of vessels with l-NAME resulted in a significant reduction to arachidonic acid-induced dilation in OZR. Dilator responses of resistance arterioles from LZR and OZR in response to sodium nitroprusside were not impacted at this age (Fig. 4C). Hypoxic dilation (Fig. 4D) in arterioles from OZR was reduced compared with that in LZR, and pretreatment with TEMPOL resulted in a significant improvement to dilator responses to reduced PO2. As above, pretreatment with l-NAME significantly reduced dilator responses to low oxygen tension in OZR at this age range. In all cases, responses of vessel from LZR following treatment with TEMPOL were not impacted, whereas l-NAME treatment significantly attenuated responses to ACh, arachidonic acid, and hypoxia (data not shown).
Fig. 4.

Data describing the dilator reactivity of ex vivo first order skeletal muscle (gracilis) resistance arterioles from LZR and OZR at 13 wk of age. Data are presented for the dilator responses of arterioles in response to increasing concentrations of ACh (A), arachidonic acid (B), sodium nitroprusside (C), and reduced oxygen tension (D). Data are presented as means ± SE; n = vessels from 8–10 animals in for OZR and n = vessels from 6–8 animals for LZR. *P < 0.05 in the upper bound (A–C) or magnitude of the response (D) vs. LZR; †P < 0.05 in the upper bound (A–C) or magnitude of the response (D) vs. OZR.
Figures 5 and 6 present the data describing arteriolar dilator reactivity from LZR and OZR at 17 and 20 wk of age, respectively. Although the magnitude of the individual impairments and the specific effects of acute pharmacological intervention were somewhat different between the two ages, they were directionally consistent. In OZR, dilator responses to increasing concentrations of ACh were impaired versus responses in LZR (Figs. 5A and 6A). In both cases, treatment with TEMPOL resulted in significant improvement to the response, whereas treatment with l-NAME abolished all reactivity to ACh. Similarly, responses of arterioles from OZR to arachidonic acid were significantly blunted compared with that in LZR (Figs. 5B and 6B). Pretreatment with TEMPOL improved reactivity, although this appeared to have a diminished effectiveness at the 20 wk age, and treatment with l-NAME was virtually without effect. Responses to nitroprusside were largely intact, although the maximum dilator response was increasingly blunted in vessels from OZR with increasing age, and this was not altered by treatment with TEMPOL or l-NAME, suggesting that this may reflect a structural effect (Figs. 5C and 6C). Dilator responses to hypoxia were reduced in OZR at both age groups. Although TEMPOL improved the response at 17 and 20 wk of age, pretreatment with l-NAME did not have a significant impact.
Fig. 5.

Data describing the dilator reactivity of ex vivo first order skeletal muscle (gracilis) resistance arterioles from LZR and OZR at 17 wk of age. Data are presented for the dilator responses of arterioles in response to increasing concentrations of ACh (A), arachidonic acid (B), sodium nitroprusside (C), and reduced oxygen tension (D). Data are presented as means ± SE; n = vessels from 8–10 animals in for OZR and n = vessels from 6–8 animals for LZR. *P < 0.05 in the upper bound (A–C) or magnitude of the response (D) vs. LZR; †P < 0.05 in the upper bound (A–C) or magnitude of the response (D) vs. OZR.
Fig. 6.

Data describing the dilator reactivity of ex vivo first order skeletal muscle (gracilis) resistance arterioles from LZR and OZR at 20 wk of age. Data are presented for the dilator responses of arterioles in response to increasing concentrations of ACh (A), arachidonic acid (B), sodium nitroprusside (C), and reduced oxygen tension (D). Data are presented as means ± SE; n = vessels from 8–10 animals in for OZR and n = vessels from 6–8 animals for LZR. *P < 0.05 in the upper bound (A–C) or magnitude of the response (D) vs. LZR; †P < 0.05 in the upper bound (A–C) or magnitude of the response (D) vs. OZR.
Figure 7 summarizes data describing the agonist-induced NO bioavailability in arteries of LZR and OZR at increasing age. In arteries of rats at 7 wk (Fig. 7A) and 10 wk (Fig. 7B) of age, vascular NO bioavailability was not different between LZR and OZR and was unaffected as a result of pretreatment with the antioxidant TEMPOL (Fig. 7A). However, by 13 wk (Fig. 7C), 17 wk (Fig. 7D), and 20 wk (Fig. 7E) of age, the vascular NO bioavailability from OZR was significantly reduced compared with that in LZR and demonstrated a worsening with increasing age. Treatment with TEMPOL was effective at restoring NO bioavailability in OZR with increasing age, although the magnitude of this effect appeared to be diminished with increasing age. At all ages, pretreatment of vessels with l-NAME nearly abolished any methacholine-induced NO bioavailability in LZR or OZR.
Fig. 7.

Data describing the bioavailability of vascular-produced nitric oxide (NO) from ex vivo skeletal muscle conduit arteries from LZR and OZR at 7 (A), 10 (B), 13 (C), 17 (D), and 20 (E) wk of age. Data are presented as the slope of the NO level with increasing concentrations of methacholine-pooled arteries under control conditions and following pretreatment with either TEMPOL or Nω-nitro-l-arginine methyl ester (l-NAME). Data are presented as means ± SE; n = arteries from 8–10 animals in for OZR and n = arteries from 6–8 animals for LZR. *P < 0.05 vs. responses in LZR under control conditions; †P < 0.05 vs. responses in OZR under control conditions.
Figure 8 presents the correlation of vascular NO bioavailability with skeletal muscle microvessel density in LZR and OZR at two age ranges, 7–10 wk (Fig. 8A) and 17–20 wk (Fig. 8B). In younger OZR, vascular NO bioavailability is an extremely poor predictor of the early loss in microvessel density, suggesting that NO-independent processes may be responsible for the early pulse of rarefaction in these animals. However, in older OZR, the severity of the reduction in NO bioavailability is a very strong predictor of microvessel density (Fig. 8B).
Fig. 8.
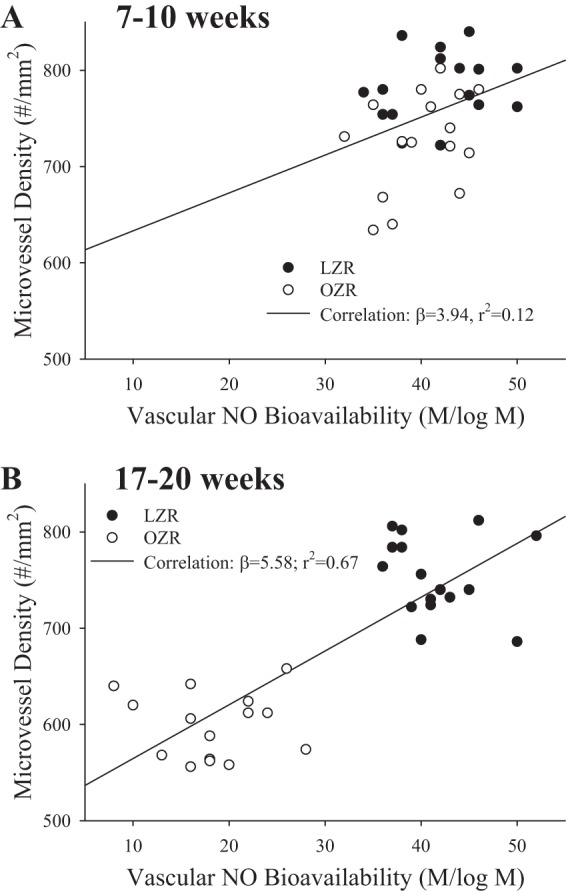
The correlations between vascular nitric oxide bioavailability (from Fig. 7) and skeletal muscle microvessel density (from Fig. 1) for LZR and OZR at 7–10 (A) and 17–20 (B) wk of age. On each panel are the lines of best linear fit with the slope (β) coefficient and the r2 for each.
The vascular production of 6-keto-PGF1α (Fig. 9A; the stable breakdown product of PGI2) and 11-dehyro-TxB2 (Fig. 9B; the stable breakdown product of TxA2) in arteries from LZR and OZR in response to hypoxia over the age ranges of the present study is summarized in Fig. 9. As shown in Fig. 9A, hypoxia-stimulated production of PGI2 was comparable between LZR and OZR at younger ages. However, as OZR continued to age, and with increasing severity of the metabolic syndrome, these responses began to diverge and the production of PGI2 in arteries of OZR was significantly reduced compared with responses in LZR. In contrast, vascular production of TxA2 was consistently elevated in OZR compared with LZR under resting conditions at all ages (Fig. 9B). This disparity was exacerbated with increasing age, such that the vascular production of TxA2 in OZR was consistently much higher than in LZR, although it did appear to plateau between 13 and 17 wk of age.
Fig. 9.
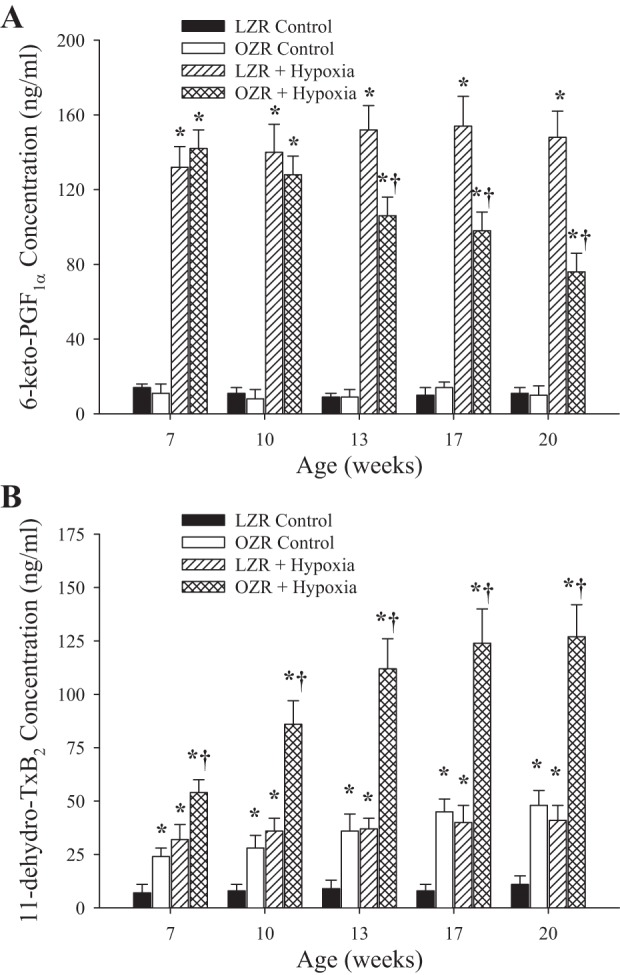
Arterial production of PGI2 (A; estimated from 6-keto-PGF1α) and thromboxane A2 (TxA2; B; estimated from 11-dehydro-TxB2) in LZR and OZR between 7 and 20 wk of age. Data are presented under control conditions and following challenge with hypoxia. Data are presented as means ± SE; n = arteries from 8–10 animals in for OZR and n = arteries from 6–8 animals for LZR. *P < 0.05 vs. responses in LZR under control conditions; †P < 0.05 vs. responses in OZR under control conditions.
With the use of differential immunohistochemistry, the changes in microvessel density in the skeletal muscle of LZR and OZR at increasing ages are summarized in Fig. 10. Although staining with GS-1 presented an aggregate change in microvessel density and clearly demonstrated the progressive rarefaction in OZR with increasing age, staining with AlkPhos (which preferentially stains terminal arterioles and the arteriolar ends of capillaries) (2, 34) demonstrated an extremely consistent response to that determined with GS-1 at 7 wk (Fig. 10A), 10 wk (Fig. 10B), 13 wk (Fig. 10C), 17 wk (Fig. 10D), and 20 wk (Fig. 10E) of age. However, staining with DPEP IV (which preferentially stains small venules and venular ends of capillaries) demonstrated reduced venular capillary/venular staining that preceded any reduction in either GS-1 or AlkPhos staining at 7 wk of age and demonstrated a more severe reduction at each age in OZR compared with GS-1 and AlkPhos.
Fig. 10.

The change in skeletal muscle microvessel density in LZR and OZR at 7 (A), 10 (B), 13 (C), 17 (D), and 20 (E) wk of age. Results are presented following tissue staining with Griffonia simplicifolia 1 lectin (GS-1 lectin), alkaline phosphatase (AlkPhos), and dipeptidylpeptidase IV (DPEP IV). Data are presented as means ± SE; n = 8–10 animals in each age group for OZR and n = 6–8 animals in each age group for LZR. *P < 0.05 vs. LZR at that age.
Figure 11 presents data describing leukocyte adhesion and rolling in post-capillary skeletal muscle venules of LZR and OZR at 7–10 wk of age (data from both ages were combined to produce 1 panel for each). The number of adherent (Fig. 11A) and rolling (Fig. 11B) leukocytes in these vessel segments in the skeletal muscle of OZR was significantly elevated compared with that in LZR at both 7 and 10 wk of age.
Fig. 11.
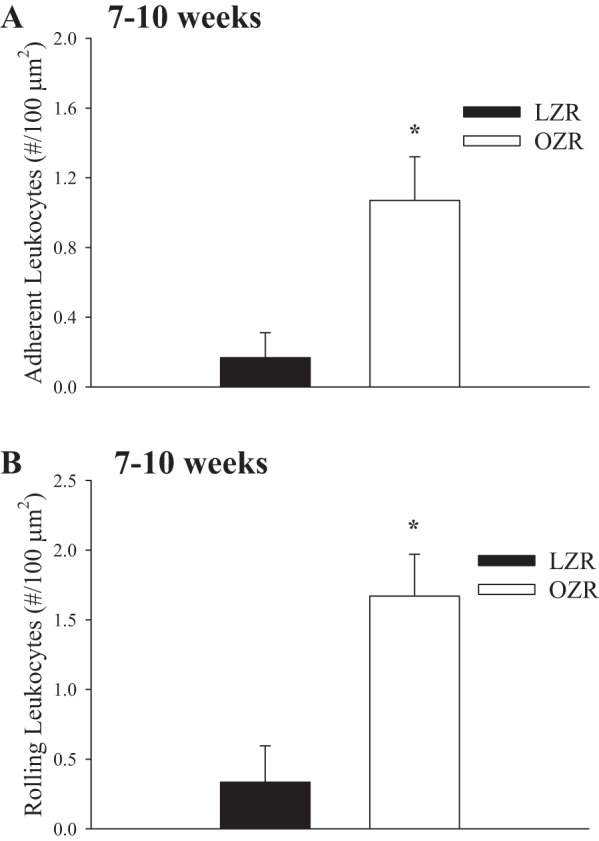
Data describing the adhesion (A) and rolling (B) of leukocytes in the postcapillaries of venules of in situ extensor digitorum longus muscle of LZR and OZR at 7–10 wk of age. Rhodamine 6G-labeled vascular cells were visualized by epi-illumination. The number of rolling and adherent leukocytes in venules was expressed as number of cells/are of endothelial surface with the denominator calculated from the length and diameter of the venular segment. *P < 0.05 vs. LZR.
Based on these results, we engaged in a discovery process to determine biomarkers that correlated well with the early pulse of microvascular rarefaction in OZR, with the results summarized in Table 2. Although plasma levels of the inflammatory biomarkers TNF-α, monocyte chemoattractant protein (MCP-1), sICAM, and sVCAM all demonstrated the appropriate temporal characteristics, TNF-α and both oxidant stress (plasma nitrotyrosine; Table 1) and vascular production of TxA2 (Fig. 9B) were the three strongest predictors of early reduction in microvessel density in skeletal muscle of OZR based on discriminant analysis (Table 3). MCP-1, sICAM, and sVCAM represented the grouping of the next three strongest predictors of the early phase of rarefaction with all other measured biomarkers assuming an inconsistent or not statistically significant role.
Table 2.
Plasma markers of oxidant stress and inflammation in the present study
| Age, weeks | LZR | OZR | |
|---|---|---|---|
| N-tyrosine, ng/ml | 7 | 8 ± 2 | 12 ± 3 |
| 10 | 10 ± 2 | 22 ± 3* | |
| 13 | 14 ± 3 | 41 ± 5* | |
| 17 | 16 ± 4 | 54 ± 6* | |
| 20 | 20 ± 3 | 56 ± 7* | |
| sVCAM, pg/ml | 7 | 8 ± 1 | 9 ± 1 |
| 10 | 10 ± 1 | 14 ± 2 | |
| 13 | 11 ± 1 | 21 ± 3* | |
| 17 | 12 ± 2 | 27 ± 4* | |
| 20 | 13 ± 3 | 29 ± 5* | |
| TNF-α, pg/ml | 7 | 1.6 ± 0.4 | 4.0 ± 1.0* |
| 10 | 2.4 ± 0.4 | 9.9 ± 0.9* | |
| 13 | 2.8 ± 0.3 | 11.4 ± 1.8* | |
| 17 | 3.2 ± 0.4 | 13.4 ± 1.6* | |
| 20 | 3.4 ± 0.5 | 13.2 ± 1.7* | |
| Monocyte chemoattractant protein-1, pg/ml | 7 | 29 ± 5 | 35 ± 6 |
| 10 | 32 ± 6 | 65 ± 8* | |
| 13 | 35 ± 6 | 91 ± 11* | |
| 17 | 38 ± 5 | 102 ± 12* | |
| 20 | 40 ± 6 | 114 ± 11* | |
| sICAM, pg/ml | 7 | 28 ± 5 | 40 ± 6 |
| 10 | 29 ± 7 | 63 ± 5* | |
| 13 | 34 ± 6 | 64 ± 6* | |
| 17 | 38 ± 5 | 72 ± 7* | |
| 20 | 42 ± 5 | 80 ± 7* |
P < 0.05 vs. LZR.
Table 3.
Structure matrix of results from discriminant analyses for markers of inflammation between animal groups in the present study
| Age | 7 Weeks |
10 Weeks |
13 Weeks |
17 Weeks |
17 Weeks |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Functions | 1 | 2 | 1 | 2 | 1 | 2 | 1 | 2 | 1 | 2 |
| Percent variance | 86.4 | 11.8 | 80.8 | 10.9 | 64.2 | 8.4 | 38.4 | 12.8 | 21.4 | 6.4 |
| Canonical correlation | 0.841 | 0.720 | 0.812 | 0.684 | 0.622 | 0.564 | 0.412 | 0.480 | 0.167 | 0.212 |
| TNF-α | 0.584* | 0.633* | 0.588* | 0.663* | 0.424 | 0.468 | — | — | — | — |
| Thromboxane A2 | 0.622* | 0.708* | 0.637* | 0.589* | 0.324 | 0.408 | 0.118 | 0.204 | — | — |
| Reactive oxygen species | 0.588* | 0.628* | 0.484* | 0.824* | 0.624* | 0.588* | 0.601* | 0.384 | 0.542* | 0.486* |
Represents largest absolute correlation between each variable and any discriminant function.
Data describing the effects of chronic treatment of OZR with either pentoxifylline (inhibitor of TNF-α production) or SQ-29548 (PGH2/TxA2 receptor blocker) from 7 to 13 wk of age on microvessel density in OZR and LZR are presented in Fig. 12. In control OZR, microvascular rarefaction proceeded as described above. However, in response to chronic treatment with either pentoxifylline or SQ-29548, the rarefaction that normally develops in OZR up to 13 wk of age was severely attenuated, and there was almost no reduction in microvessel density compared with responses in LZR (Fig. 12A). As a result of the chronic treatment with pentoxifylline, levels of both vascular TxA2 production (Fig. 12C) and systemic oxidant stress (Fig. 12D) were reduced, although still elevated compared with levels in LZR. However, chronic inhibition of the effects of TxA2 did not alter circulating levels of TNF-α (Fig. 12B) or oxidant stress (Fig. 12D) compared with untreated OZR.
Fig. 12.

The changes in skeletal muscle microvessel density in LZR and OZR between 7 and 13 wk of age under control (CON) conditions and in response to chronic treatment with either pentoxifylline or SQ-29548 (SQ; A). Also presented are the changes in plasma concentrations of TNF-α (B), TxA2 (C), and nitrotyrosine (D) from these same 4 groups of animals. Data are presented as means ± SE; n = 5 animals for each group of OZR and n = 5 animals for LZR. *P < 0.05 vs. responses in LZR under control conditions; †P < 0.05 vs. responses in OZR under control conditions.
The effects of chronically treating OZR with SQ-29548 from 7 wk to 13 wk of age, followed by termination of treatment and the animal being allowed to age out to 20 wk are summarized in Fig. 13. As above, chronic treatment of OZR with the antagonist of the PGH2/TxA2 receptor nearly abrogated the early pulse of rarefaction in OZR to 13 wk of age. Subsequent withdrawal of treatment resulted in gradual rarefaction of the microvascular network, although the rate of this reduction in microvessel density in OZR was sufficiently slow that by 20 wk of age, the microvessel density in OZR was still not different from that determined in untreated age-matched LZR.
Fig. 13.
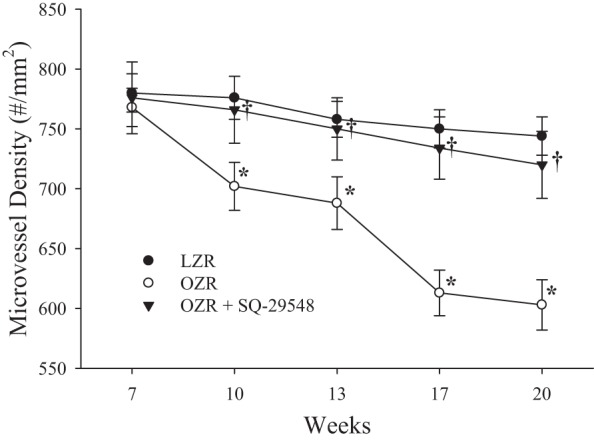
The changes in skeletal muscle microvessel density in LZR and OZR between 7 and 20 wk of age under control conditions and in response to chronic treatment with SQ-29548 out to 13 wk of age and removal of treatment at that time point. Data are presented as means ± SE. *P < 0.05 vs. responses in LZR; †P < 0.05 vs. responses in OZR.
DISCUSSION
One of the established microvascular consequences of the progressive development of the metabolic syndrome in both rodent models and human subjects is an evolving rarefaction of the microvessel networks. This progressive reduction in microvessel density has multiple implications with regard to the integrated processes of mass transport and exchange within tissue/organs and represents a significant contributor to the clinical manifestation of peripheral vascular disease in afflicted subjects and the inherent reduction in patient quality of life and resistance to treatment and healing, as well as accelerated mortality. However, although there is a general understanding of the extent of microvessel rarefaction in the OZR model of the metabolic syndrome, and preliminary evidence that the magnitude of the reduction to microvessel density is associated with the reduction in vascular NO bioavailability, there is a very limited understanding of the initiating factors underlying microvascular rarefaction and how the condition develops. This study was designed to address this current gap in our understanding.
The primary observation from the current study is that the evolution of microvascular rarefaction in the skeletal muscle of OZR occurs via two general phases, and early phase of rarefaction between 7 and 10 wk of age, followed by a slower rate of decay between 13 and 17 wk of age, culminating with a final later phase that occurs between 13 and 17 wk of age (Fig. 1). There is an additional lower rate of microvessel loss after 17 wk of age, but our unpublished work suggests that net microvessel loss develops asymptotic behavior after 17 wk of age. What was most interesting about this observation is that our previous results suggested that the change in skeletal muscle microvessel density would parallel the changes to vascular NO bioavailability (21, 23). However, as demonstrated in Figs. 2–6 and especially in Fig. 7, this assumption is not supported by the present data. Although vascular NO bioavailability does decay with progression of the metabolic syndrome in OZR, this is delayed by several weeks compared with the decay in skeletal muscle microvessel density, such that the initial reduction in vascular density cannot be predicted by a change in vascular NO levels (Fig. 8A). Interestingly, the ultimate extent of rarefaction is well correlated by the loss in NO bioavailability, as suggested by the results from previous studies (8, 12, 17, 26, 40), where physiological and pharmacological interventions to maintain NO bioavailability also blunt the severity of the reductions to microvessel density. Taken together, the results of the present study and the prior ones suggest that although the ultimate loss in vascular NO bioavailability is a strong predictor and potentially a causal contributor to the extent of microvascular rarefaction in OZR, it cannot and does not explain the initial phase of rarefaction that occurs between 7 and 10 weeks of age where NO bioavailability remains intact.
This study represents one of a limited number that not only details the progression of specific characteristics of the metabolic syndrome in OZR but also ties this risk factor progression to specific indexes of vascular/microvascular dysfunction. With its severe leptin resistance and impaired satiety reflex, OZR are significantly heavier than LZR by 7 wk of age, and this difference continues to grow out to 20 wk of age. This is paralleled by a steadily increasing severity of insulin resistance out to about 17 wk of age, although after this time, our OZR began exhibiting type II diabetes mellitus. The development of hypertension also was delayed somewhat, becoming clearly evident by 17 wk of age. The development of the more traditional characteristics of the metabolic syndrome can also be tracked with the progression of chronic inflammation, since levels of TNF-α were elevated very quickly with the presence of obesity and impaired glycemic control, followed by MCP-1 and the adhesion molecules (Table 2). This allows for a very interesting observation - that microvascular network structure (i.e., microvessel density) may be altered in the OZR before there is a significant deterioration to arteriolar endothelial function or vascular reactivity. This is a particularly intriguing possibility, since it is frequently assumed that the changes to endothelial function are the initiating factor for the genesis of microvasculopathy. Clearly, this may not be fully accurate under conditions of the metabolic syndrome, and the contributions of venular dysfunction (rather than arteriolar) and other indexes of endothelial function under these conditions of elevated CVD risk may warrant future investigation.
With the use of differential immunohistochemistry, the present results suggest that microvessel rarefaction does not reflect a general global degradation of microvessels or microvessel segments. Rather, based on the results presented in Fig. 10, it is apparent that microvessel degradation is initiated at the venular ends of capillaries or at the level of the smallest venules and proceeds in a retrograde fashion toward the arteriolar ends of capillaries/terminal arterioles, as the fall in DPEP IV staining preceded the fall in either GS-1 lectin or alkaline phosphatase staining at all ages. This observation suggests that changes in venular endothelial function may be the initiating factor for the development of rarefaction in the skeletal muscle of OZR. This hypothesis received further support since indexes of leukocyte adhesion (Fig. 11A) and rolling (Fig. 11B) for in situ skeletal muscle of OZR were significantly increased in animals between 7 and 10 wk of age, during the initial phase of rarefaction.
Tightly intertwined with the general processes of leukocyte adhesion and rolling are elevations in circulating markers of inflammation, oxidant stress, and their associated sequelae. From this, we engaged in a biomarker discovery process to determine which (if any) markers of inflammation were associated with the development of the metabolic syndrome in OZR before 10 wk of age and, through the use of multivariate discriminant analyses, were strongly predictive of the change in microvessel density over that time period. As shown in Table 2, the inflammatory biomarkers MCP-1, TNF-α, sICAM, and sVCAM and the oxidant stress marker nitrotyrosine were all associated with the development of the metabolic syndrome at that age range. However, the markers TNF-α, nitrotyrosine, and TxA2 production were the most powerfully predictive of the early phase of rarefaction (Table 3), with MCP-1, sICAM, and sVCAM being somewhat less powerfully predictive.
To investigate the potential contributions of TxA2 and TNF-α on the early pulse of microvessel rarefaction in skeletal muscle of OZR, animals were given daily treatments to block the action of TxA2 (SQ-29548) or the production of TNF-α (pentoxifylline) from 7 wk of age out to 13 wk. As shown in Fig. 12A, treatment with either SQ-29548 or pentoxifylline was effective at blunting the initial phase of microvascular rarefaction in OZR compared with the reductions in microvessel density determined in the untreated control animals. Interestingly, treatment with pentoxifylline reduced plasma levels of TNF-α (Fig. 12B), nitrotyrosine (Fig. 12D), and vascular production of TxA2 (Fig. 12C) in OZR. However, treatment of OZR with SQ-29548 had virtually no effect on these three biomarkers, but had a very similar beneficial outcome for microvessel density. Although not definitive, these results suggest that increased oxidant stress and TNF-α that are present in the vasculature of OZR may reside upstream of the elevations in TxA2 and that alterations in arachidonic acid metabolism are an outcome of the changes to oxidant stress and inflammation rather than a contributing factor to them.
To provide further insight into the effects of blunting the early phase of rarefaction on the final extent of the microvessel degradation in OZR, a separate cohort of rats was treated with daily injections of SQ-29548 out to 13 wk of age, after which time treatment was discontinued and the animal was allowed to age further without subsequent intervention. As shown in Fig. 13, treatment with the PGH2/TxA2 receptor antagonist provided the expecting blunting in the development of skeletal muscle microvascular rarefaction in OZR to 13 wk of age, after which the reduction in microvessel density began. However, whether this is a re-initiation of the TxA2-dependent early pulse of rarefaction or the NO-dependent later pulse of rarefaction (since this is the temporal window where NO bioavailability was becoming significantly compromised) is unknown at this time and represents a potential area for future investigation. Although an initial observation, this has the potential for excellent translational importance, since it suggests that if the early pulse of rarefaction can be prevented in subjects where the severity of the metabolic syndrome is still moderate, the rate at which microvessels degrade with further progression/duration of the metabolic syndrome can be blunted, with potential reductions to the severity of the negative outcomes associated with chronic metabolic syndrome. This may be an especially compelling observation that warrants further validation and targeted investment into the specific vascular, metabolic, and proteomic environments that may have been altered to impact the severity of microvascular rarefaction development.
No experimental study is without its limitations and the present one is no exception. In this study, we examined microvascular structure/function and made comparisons between three distinct muscles, gracilis muscle (resistance arteriolar reactivity), gastrocnemius muscle (microvessel density), and EDL (leukocyte adhesion/rolling). Although this was a function of the infrastructure and expertise present in the laboratories of the research team, this does allow for the introduction of potential bias owing to disparities between the microvascular and skeletal muscle cell environment between the three muscles. It is unclear at this time if either this bias or its severity was actually present, and this does need to be kept in mind. Additionally, the agent used for inhibition of TNF-α (pentoxifylline) does have other nonspecific effects beyond simply blocking the production of this inflammatory biomarker, including its actions as a mild phosphodiesterase inhibitor and as a weak antagonist of adenosine type 2 receptors (52). Finally, although the results from the present study provide compelling evidence for the pursuit of higher resolution mechanistic contributors to rarefaction, the loss of microvessel density in OZR clearly demonstrates an asymptotic relationship as the animal approaches 20 wk of age. Whether this represents a decline in the stimuli for rarefaction (unlikely) or an increase in the intensity of the stimuli for angiogenesis as a compensation is unknown, and the mechanistic integration of these stimuli represents areas for further investigation.
Summary.
Although it has been well established that one of the microvasculopathies that accompanies the chronic presentation of the metabolic syndrome is a rarefaction of the skeletal muscle microvascular networks, how this develops from a temporal perspective and the mechanistic contributors have not been well elucidated. The results from the present study suggest that the reduction to microvessel density develops along a distinct time line, with an early phase of rarefaction that is dependent on the generation and actions of reactive oxygen species, specific markers of inflammation and dysfunction to the venular endothelium and the actions of TxA2. Subsequently, the networks enter into a moderately quiescent period, and then into a second phase of rarefaction that is associated with the evolving reductions to vascular NO bioavailability. Furthermore, the importance of the venular dysfunction is highlighted by the apparent retrograde behavior of microvessel degradation, implicating the venular endothelium as the initiating site of rarefaction. Although specific signaling pathways and cellular environmental influences underlying this loss in microvessel density in the metabolic syndrome remain to be elucidated, the temporally distributed nature of the mechanisms and the potential for an early intervention that can prevent a larger long-term negative outcome have clear implications for the possible reduction in the negative health outcomes associated with chronic presentation of the metabolic syndrome.
GRANTS
This study was supported by the American Heart Association (IRG 14330015, PRE 16850005, EIA 0740129N) and the National Institutes of Health (RR-2865AR; P20-RR-016477).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.C.F., A.G.G., J.T.B., R.W.B., and P.D.C. conception and design of research; J.C.F., A.G.G., J.T.B., R.W.B., and I.M.O. performed experiments; J.C.F., A.G.G., S.J.F., J.T.B., R.W.B., I.M.O., E.R.D., and P.D.C. analyzed data; J.C.F., A.G.G., S.J.F., R.W.B., I.M.O., E.R.D., and P.D.C. interpreted results of experiments; J.C.F. prepared figures; J.C.F. drafted manuscript; J.C.F., A.G.G., S.J.F., J.T.B., R.W.B., I.M.O., E.R.D., and P.D.C. edited and revised manuscript; J.C.F., A.G.G., S.J.F., J.T.B., R.W.B., I.M.O., E.R.D., and P.D.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the Office of Laboratory Animal Research at West Virginia University for expertise and assistance during the performance of procedures. We acknowledge the support provided through Center for Cardiovascular and Respiratory Sciences at the West Virginia University Health Sciences Center.
REFERENCES
- 1.Aleixandre de Artiñano A, Miguel Castro M. Experimental rat models to study the metabolic syndrome. Br J Nutr 102: 1246–1253, 2009. [DOI] [PubMed] [Google Scholar]
- 2.Bell MA, Scarrow WG. Staining for microvascular alkaline phosphatase in thick celloidin sections of nervous tissue: morphometric and pathological applications. Microvasc Res 27: 189–203, 1984. [DOI] [PubMed] [Google Scholar]
- 3.Boudreau DM, Malone DC, Raebel MA, Fishman PA, Nichols GA, Feldstein AC, Boscoe AN, Ben-Joseph RH, Magid DJ, Okamoto LJ. Health care utilization and costs by metabolic syndrome risk factors. Metab Syndr Relat Disord 7: 305–314, 2009. [DOI] [PubMed] [Google Scholar]
- 4.Boutayeb A, Boutayeb S, Boutayeb W. Multi-morbidity of non-communicable diseases and equity in WHO Eastern Mediterranean countries. Int J Equity Health 12: 60, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butcher JT, Goodwill AG, Stanley SC, Frisbee JC. Differential impact of dilator stimuli on increased myogenic activation of cerebral and skeletal muscle resistance arterioles in obese zucker rats. Microcirculation 20: 579–589, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Catella F, Healy D, Lawson JA, FitzGerald GA. 11-Dehydrothromboxane B2: a quantitative index of thromboxane A2 formation in the human circulation. Proc Natl Acad Sci USA 83: 5861–5865, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen D, Wang MW. Development and application of rodent models for type 2 diabetes. Diabetes Obes Metab 7: 307–317, 2005. [DOI] [PubMed] [Google Scholar]
- 8.Dai X, Faber JE. Endothelial nitric oxide synthase deficiency causes collateral vessel rarefaction and impairs activation of a cell cycle gene network during arteriogenesis. Circ Res 106: 1870–1881, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Carvalho Vidigal F, Bressan J, Babio N, Salas-Salvadó J. Prevalence of metabolic syndrome in Brazilian adults: a systematic review. BMC Public Health 13: 1198, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeVallance E, Fournier SB, Donley DA, Bonner DE, Lee K, Frisbee JC, Chantler PD. Is obesity predictive of cardiovascular dysfunction independent of cardiovascular risk factors? Int J Obes (Lond). In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Donley DA, Fournier SB, Reger BL, DeVallance E, Bonner DE, Olfert IM, Frisbee JC, Chantler PD. Aerobic exercise training reduces arterial stiffness in metabolic syndrome. J Appl Physiol 116: 1396–1404, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faber JE, Zhang H, Lassance-Soares RM, Prabhakar P, Najafi AH, Burnett MS, Epstein SE. Aging causes collateral rarefaction and increased severity of ischemic injury in multiple tissues. Arterioscler Thromb Vasc Biol 31: 1748–1756, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Falkner B, Cossrow ND. Prevalence of metabolic syndrome and obesity-associated hypertension in the racial ethnic minorities of the United States. Curr Hypertens Rep 16: 449, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan J, Song Y, Chen Y, Hui R, Zhang W. Combined effect of obesity and cardio-metabolic abnormality on the risk of cardiovascular disease: a meta-analysis of prospective cohort studies. Int J Cardiol 168: 4761–4768, 2013. [DOI] [PubMed] [Google Scholar]
- 15.Fellmann L, Nascimento AR, Tibiriça E, Bousquet P. Murine models for pharmacological studies of the metabolic syndrome. Pharmacol Ther 137: 331–340, 2013. [DOI] [PubMed] [Google Scholar]
- 16.Ferdinand KC, Rodriguez F, Nasser SA, Caballero AE, Puckrein GA, Zangeneh F, Mansour M, Foody JM, Pemu PE, Ofili EO. Cardiorenal metabolic syndrome and cardiometabolic risks in minority populations. Cardiorenal Med 4: 1–11, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandes T, Nakamuta JS, Magalhães FC, Roque FR, Lavini-Ramos C, Schettert IT, Coelho V, Krieger JE, Oliveira EM. Exercise training restores the endothelial progenitor cells number and function in hypertension: implications for angiogenesis. J Hypertens 30: 2133–2143, 2012. [DOI] [PubMed] [Google Scholar]
- 18.Focardi M, Picchi A, Donnini S, Cameli M, Ziche M, Marzilli M, Mondillo S. Hydrogen peroxide mediates endothelium-dependent dilation of coronary arterioles in obese rats on a low-carbohydrate diet. Microcirculation 20: 599–608, 2013. [DOI] [PubMed] [Google Scholar]
- 19.Fredricks KT, Liu Y, Lombard JH. Response of extraparenchymal resistance arteries of rat skeletal muscle to reduced PO2. Am J Physiol Heart Circ Physiol 267: H706–H715, 1994. [DOI] [PubMed] [Google Scholar]
- 20.Frisbee JC, Delp MD. Vascular function in the metabolic syndrome and the effects on skeletal muscle perfusion: lessons from the obese Zucker rat. Essays Biochem 42: 145–161, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Frisbee JC, Samora JB, Peterson J, Bryner R. Exercise training blunts microvascular rarefaction in the metabolic syndrome. Am J Physiol Heart Circ Physiol 291: H2483–H2492, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Frisbee JC. Hypertension-independent microvascular rarefaction in the obese Zucker rat model of the metabolic syndrome. Microcirculation 12: 383–392, 2005. [DOI] [PubMed] [Google Scholar]
- 23.Frisbee JC. Reduced nitric oxide bioavailability contributes to skeletal muscle microvessel rarefaction in the metabolic syndrome. Am J Physiol Regul Integr Comp Physiol 289: R307–R316, 2005. [DOI] [PubMed] [Google Scholar]
- 24.Fu J, Prasad HC. Changing epidemiology of metabolic syndrome and type 2 diabetes in Chinese youth. Curr Diab Rep 14: 447, 2014. [DOI] [PubMed] [Google Scholar]
- 25.García-Cruz E, Leibar-Tamayo A, Romero J, Piqueras M, Luque P, Cardeñosa O, Alcaraz A. Metabolic syndrome in men with low testosterone levels: relationship with cardiovascular risk factors and comorbidities and with erectile dysfunction. J Sex Med 10: 2529–2538, 2013. [DOI] [PubMed] [Google Scholar]
- 26.Goligorsky MS. Microvascular rarefaction: the decline and fall of blood vessels. Organogenesis 6: 1–10, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goodwill AG, Frisbee SJ, Stapleton PA, James ME, Frisbee JC. Impact of chronic anticholesterol therapy on development of microvascular rarefaction in the metabolic syndrome. Microcirculation 16: 667–684, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greene AS, Lombard JH, Cowley AW, Jr, Hansen-Smith FM. Microvessel changes in hypertension measured by Griffonia simplicifolia I lectin. Hypertension 15: 779–783, 1990. [DOI] [PubMed] [Google Scholar]
- 29.Huang RC, Beilin LJ, Ayonrinde O, Mori TA, Olynyk JK, Burrows S, Hands B, Adams LA. Importance of cardiometabolic risk factors in the association between nonalcoholic fatty liver disease and arterial stiffness in adolescents. Hepatology 58: 1306–1314, 2013. [DOI] [PubMed] [Google Scholar]
- 30.Iliescu R, Chade AR. Progressive renal vascular proliferation and injury in obese Zucker rats. Microcirculation 17: 250–258, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kasiske BL, O′Donnell MP, Keane WF. The Zucker rat model of obesity, insulin resistance, hyperlipidemia, and renal injury. Hypertension 19: I110–I115, 1992. [DOI] [PubMed] [Google Scholar]
- 32.Katakam PV, Snipes JA, Steed MM, Busija DW. Insulin-induced generation of reactive oxygen species and uncoupling of nitric oxide synthase underlie the cerebrovascular insulin resistance in obese rats. J Cereb Blood Flow Metab 32: 792–804, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lind L, Lithell H. Decreased peripheral blood flow in the pathogenesis of the metabolic syndrome comprising hypertension, hyperlipidemia, and hyperinsulinemia. Am Heart J 125: 1494–1497, 1993. [DOI] [PubMed] [Google Scholar]
- 34.Liu Y, Fredricks KT, Roman RJ, Lombard JH. Response of resistance arteries to reduced PO2 and vasodilators during hypertension and elevated salt intake. Am J Physiol Heart Circ Physiol 273: H869–H877, 1997. [DOI] [PubMed] [Google Scholar]
- 35.Lobato NS, Filgueira FP, Prakash R, Giachini FR, Ergul A, Carvalho MH, Webb RC, Tostes RC, Fortes ZB. Reduced endothelium-dependent relaxation to anandamide in mesenteric arteries from young obese Zucker rats. PLoS One 8: e63449, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu S, Xiang L, Clemmer JS, Gowdey AR, Mittwede PN, Hester RL. Impaired vascular KATP function attenuates exercise capacity in obese zucker rats. Microcirculation 20: 662–669, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mrázková O, Grim M, Carlson BM. Enzymatic heterogeneity of the capillary bed of rat skeletal muscles. Am J Anat 177: 141–148, 1986. [DOI] [PubMed] [Google Scholar]
- 38.Nazzaro P, Schirosi G, Mezzapesa D, Petruzzellis M, Pascazio L, Serio G, De Benedittis L, Federico F. Effect of clustering of metabolic syndrome factors on capillary and cerebrovascular impairment. Eur J Intern Med 24: 183–188, 2013. [DOI] [PubMed] [Google Scholar]
- 39.Nies AS. Prostaglandins and the control of the circulation. Clin Pharmacol Ther 39: 481–488, 1986. [DOI] [PubMed] [Google Scholar]
- 40.Okutsu M, Call JA, Lira VA, Zhang M, Donet JA, French BA, Martin KS, Peirce-Cottler SM, Rembold CM, Annex BH, Yan Z. Extracellular superoxide dismutase ameliorates skeletal muscle abnormalities, cachexia, and exercise intolerance in mice with congestive heart failure. Circ Heart Fail 7: 519–530, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oliveros E, Somers VK, Sochor O, Goel K, Lopez-Jimenez F. The concept of normal weight obesity. Prog Cardiovasc Dis 56: 426–433, 2014. [DOI] [PubMed] [Google Scholar]
- 42.Roudier E, Chapados N, Decary S, Gineste C, Le Bel C, Lavoie JM, Bergeron R, Birot O. Angiomotin p80/p130 ratio: a new indicator of exercise-induced angiogenic activity in skeletal muscles from obese and non-obese rats? J Physiol 587: 4105–4119, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmid-Schönbein GW. An emerging role of degrading proteinases in hypertension and the metabolic syndrome: autodigestion and receptor cleavage. Curr Hypertens Rep 14: 88–96, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shamseddeen H, Getty JZ, Hamdallah IN, Ali MR. Epidemiology and economic impact of obesity and type 2 diabetes. Surg Clin North Am 91: 1163–1172, 2011. [DOI] [PubMed] [Google Scholar]
- 45.Shin JA, Lee JH, Lim SY, Ha HS, Kwon HS, Park YM, Lee WC, Kang MI, Yim HW, Yoon KH, Son HY. Metabolic syndrome as a predictor of type 2 diabetes, and its clinical interpretations and usefulness. J Diabetes Investig 4: 334–343, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stainsby WN, Snyder B, Welch HG. A pictographic essay on blood and tissue oxygen transport. Med Sci Sports Exerc 20: 213–221, 1988. [DOI] [PubMed] [Google Scholar]
- 47.Tajbakhsh N, Sokoya EM. Compromised endothelium-dependent hyperpolarization-mediated dilations can be rescued by NS309 in obese Zucker rats. Microcirculation. In press. [DOI] [PubMed] [Google Scholar]
- 48.Toblli JE, Cao G, DeRosa G, Di Gennaro F, Forcada P. Angiotensin-converting enzyme inhibition and angiogenesis in myocardium of obese Zucker rats. Am J Hypertens 17: 172–180, 2004. [DOI] [PubMed] [Google Scholar]
- 49.Tofovic SP, Jackson EK. Rat models of the metabolic syndrome. Methods Mol Med 86: 29–46, 2003. [DOI] [PubMed] [Google Scholar]
- 50.Tyml K, Budreau CH. A new preparation of rat extensor digitorum longus muscle for intravital investigation of the microcirculation. Int J Microcirc Clin Exp 10: 335–343, 1991. [PubMed] [Google Scholar]
- 51.Wang Y, Yu Q, Chen Y, Cao F. Pathophysiology and therapeutics of cardiovascular disease in metabolic syndrome. Curr Pharm Des 19: 4799–4805, 2013. [DOI] [PubMed] [Google Scholar]
- 52.Ward A, Clissold Pentoxifylline SP. A review of its pharmacodynamic and pharmacokinetic properties, and its therapeutic efficacy. Drugs 34: 50–97, 1987. [DOI] [PubMed] [Google Scholar]
- 53.Xiang L, Hester RL, Fuller WL, Sebai ME, Mittwede PN, Jones EK, Aneja A, Russell GV. Orthopedic trauma-induced pulmonary injury in the obese Zucker rat. Microcirculation 17: 650–659, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zeintl H, Sack FU, Intaglietta M, Messmer K. Computer assisted leukocyte adhesion measurement in intravital microscopy. Int J Microcirc Clin Exp 8: 293–302, 1989. [PubMed] [Google Scholar]