

Abstract
Cytomegalovirus (CMV) infects a majority of the population worldwide. It has been implicated in cardiovascular disease, induces microvascular dysfunction, and synergizes with hypercholesterolemia to promote leukocyte and platelet recruitment in venules. Although platelets and platelet-associated P-selectin contribute to cardiovascular disease inflammation, their role in CMV-induced vascular responses is unknown. We assessed the role of platelets in CMV-induced microvascular dysfunction by depleting platelets and developing bone marrow chimeric mice deficient in platelet P-selectin. Wild-type and chimeric mice received mock or murine (m)CMV intraperitoneally. Five weeks later, some mice were switched to a high-cholesterol diet (HC) to investigate the synergism between mCMV and HC. Arteriolar vasodilation and recruitment of leukocytes and donor platelets in venules were measured at 11wk. mCMV with or without HC caused significant endothelial dysfunction in arterioles. Platelet depletion restored normal vasodilation in mCMV-HC but not mCMV-ND mice, whereas protection was seen in both groups for platelet P-selectin chimeras. Only mCMV + HC elevated leukocyte and platelet recruitment in venules. Leukocyte adhesion was reduced to mock levels by acute platelet depletion but was only partially decreased in platelet P-selectin chimeras. Platelets from mCMV-HC mice and, to a lesser extent, mCMV-ND but not mock-HC mice showed significant adhesion in mCMV-HC recipients. Our findings implicate a role for platelets, acting through P-selectin, in CMV-induced arteriolar dysfunction and suggest that the addition of HC leads to a platelet-dependent, inflammatory infiltrate that is only partly platelet P-selectin dependent. CMV appeared to have a stronger activating influence than HC on platelets and may represent an additional therapeutic target in vulnerable patients.
Keywords: platelets, arteriolar dilation, leukocyte adhesion, hypercholesterolemia, microvasculature
cardiovascular disease is the leading cause of death in the United States, regardless of sex or race. Endothelial dysfunction is an early response to risk factors such as hypercholesterolemia, diabetes, and hypertension (8, 28). More recently, pathogens that have a high prevalence in the population have also been implicated in cardiovascular disease (5, 9, 15–17, 19). One such pathogen, human cytomegalovirus (CMV), infects 60–100% of the population and establishes a lifelong persistent infection. It can reduce endothelial nitric oxide release, upregulate adhesion molecules on vascular endothelium, and promote leukocyte-endothelium adhesion (4, 7, 12, 14, 20, 34, 36, 45), all key events in the pathogenesis of cardiovascular disease. The microvasculature becomes dysfunctional in response to cardiovascular risk factors long before clinical symptoms appear, which can enhance the vulnerability of an organ to subsequent ischemic events (25, 26, 29, 37). We have previously shown in a murine model that murine (m)CMV impairs endothelium-dependent vasodilation responses in arterioles (22). Furthermore, because a majority of individuals with “traditional” cardiovascular risk factors are CMV positive, we tested whether this virus can synergize with hypercholesterolemia, and we found that, together, these factors promote a proinflammatory and prothrombogenic phenotype in postcapillary venules at timepoints when neither would alone. While it has been demonstrated that platelets participate in hypercholesterolemia-induced arteriolar dysfunction and leukocyte recruitment (38), whether platelets have a role in CMV-induced responses remains unclear and was the focus of the present study.
Platelets can interact with other cell types, including leukocytes and endothelial cells, via cell surface adhesion molecules such as glycoprotein IIb/IIIa, glycoprotein Ib, and P-selectin (24, 35, 41). The cross-linking of these molecules leads to intracellular signaling that can activate both platelets and nearby cells through the release of factors such as superoxide, chemokines, and cytokines. In this way, platelets can actively participate in inflammatory responses and endothelial dysfunction in response to many stimuli (35, 40). Although primary CMV infection has been associated with thrombotic events in patients (3, 13, 27) and can induce platelet adhesion to the infected endothelium in cell culture (31), little is known about the contribution of platelets to vascular events induced during persistent infection. We have previously shown a role for P-selectin in mCMV-induced arteriolar dysfunction as well as the leukocyte adhesion seen in hypercholesterolemic mCMV-infected mice (33). Although we did not determine whether this was platelet-derived P-selectin (vs. endothelial P-selectin), we were able to show that platelets used their own P-selectin to adhere to the postcapillary venule wall in mice exposed to both mCMV and hypercholesterolemia, supporting such a possibility.
In the present study, we used a platelet depletion approach to first test the role of platelets in mCMV-induced arteriolar dysfunction as well as in arteriolar and venular responses to mCMV plus high cholesterol (HC). We then determined whether platelets were acting via their own P-selectin to promote these microvascular events using chimeric mice that did not express P-selectin on platelets but retained endothelial P-selectin. To determine whether mCMV or HC was the primary driver of the thrombogenic phenotype, we also tested the recruitment of platelets isolated from mice exposed to a single risk factor and infused into recipients exposed to both mCMV and HC and vice versa.
MATERIALS AND METHODS
Animals.
Wild-type (WT) C57BL/6J mice, B6.SJL-Ptprca Pepcb/BoyJ mice (CD45 congenic WT mice), and B6.129S7-Selptm1Bay/J (P-sel−/−) mice (on a C57BL/6J background) were obtained from Jackson Laboratories (Bar Harbor, ME). Mice (3–5 wk old) were injected with mock inoculum or 3 × 104 plaque-forming units of mCMV [Smith strain, prepared as previously described (22)]. After 5 wk, mice were either maintained on normal diet (ND) or placed on a cholesterol-enriched diet (HC; Teklad 94059 containing 1.25% cholesterol and 15.8% fat, Harlan Teklad, Madison, WI). Intravital microscopy was performed at 11 wk postinfection (with or without 6-wk HC). Separate groups of mCMV-infected mice received 125 μl/kg rabbit anti-mouse thrombocyte antiserum to deplete platelets [anti-platelet serum (APS); Accurate Chemicals, Westbury, NY] or rabbit serum (RS) as a control in saline vehicle intraperitoneally 24 h before observation of the microvasculature. The dose was based on previous work and has been shown to not significantly alter leukocyte counts (38). n = 9–14 for all groups. Animal handling procedures were approved by the Louisana State University Health Institutional Animal Care and Use Committee and were in accordance with guidelines of the American Physiological Society.
Bone marrow chimera generation.
Two combinations of chimeras were made as previously described (42). WT → WT chimeras were irradiated CD45 congenic WT mice that received bone marrow cells from WT animals, maintaining P-selectin expression. P-sel−/− → WT chimeras, with P-selectin-deficient hematopoietic cells (or more specifically, platelets) but normal vascular wall P-selectin expression, were generated by transplanting bone marrow from a P-sel−/− mouse into an irradiated CD45 congenic mouse. In both cases, this resulted in a significant increase of circulating leukocytes expressing CD45.2 (of donor origin), from <5% in congenic mice to >90% in chimeric mice, allowing verification of proper chimera reconstitution by flow cytometry, as previously described. Once confirmed, chimeric mice were exposed to the infection and diet protocol described above.
Intravital microscopy.
At 11 wk postinfection, mice were anesthetized with ketamine hydrochloride (150 mg/kg body wt ip) and xylazine (7.5 mg/kg body wt ip). Core body temperature was maintained at 35 ± 0.5°C. Intravital microscopy on the cremaster muscle was used to measure leukocyte rolling, adhesion, and emigration and, where indicated, adhesion of exogenous platelets in postcapillary venules. In some groups, venule observation of leukocytes was followed by the assessment of arteriolar vasodilation responses to ACh (endothelium dependent) and papaverine (endothelium independent), as previously described (22, 38). Rolling leukocytes were identified as leukocytes moving slower than red blood cells, and both the flux (numbers of leukocytes rolling past the section midpoint per minute) and average velocity (in μm/s) were calculated. A leukocyte was defined as adherent if it remained stationary for ≥30 s (expressed as numbers of leukocytes/mm2 vessel wall) and was measured throughout the 1-min observation period. Leukocyte emigration was measured online at the end of each observation period and was expressed as numbers of leukocytes per millimeter squared of the interstitium based on the count per field of view adjacent to the segment under observation.
Once the venule data had been collected, arterioles were chosen for the assessment of vasodilation responses. Arteriolar vasodilation responses to 10−5 M ACh were recorded. These were expressed as percent diameter changes versus baseline. Arteriolar diameters were then allowed to return to baseline with bicarbonate-buffered saline superfusion before papaverine was applied to test for maximal endothelium-independent vasodilation. Arterioles that were not responsive to 10−4 M papaverine were excluded from the study. Data were collected from 1–3 arterioles/mouse (second to fourth order) with baseline diameters of 15–40 μm. In separate groups of mice, arteriolar responses to a dose range of ACh (10−7–10−3 M) were collected, and vasodilation responses to 10−4 M sodium nitroprusside were also determined. Venular data were not collected in these mice.
In some groups, platelets were isolated from donors that were matched or mismatched to the recipients in terms of infection and diet. They were isolated by a series of centrifugation steps and fluorescently labeled using carboxyfluorescein succinimidyl ester, and 108 platelets were infused via the jugular vein into recipients as previously described (11). Venules were chosen for 1-min recordings as described above for leukocytes. Platelets were considered adherent if they stopped for ≥2 s, and platelet adhesion was expressed as numbers of platelets per millimeter squared of the vessel wall. No arteriole measurements were obtained from mice that received mismatched platelets.
Serum cholesterol levels.
Serum was frozen for subsequent measurements of cholesterol levels using a spectrophotometric assay (Sigma Chemicals, St. Louis, MO).
Circulating platelet counts.
A sample of blood was obtained from the tail to determine the circulating platelet count before APS or rabbit serum was injected as well as 24 h later before the experiment was performed. Blood was diluted in ammonium oxalate, and platelets were counted with the aid of a hemocytometer.
Virus interaction with platelets.
To test whether mCMV may exert its effect on platelets through a direct interaction, platelets were isolated from WT mice as described above, and 30 × 106 platelets were incubated at a 1:1 ratio with mCMV in PBS for 1 h at 4°C. Platelets were washed twice to remove free virus, and half of these samples were incubated with 1 mg/ml proteinase K to control for nonspecific binding. Two other controls [mCMV only (to control for virus adsorbing to the tube wall) or platelets only] were incubated and washed as per the platelet + mCMV samples. PCR was performed to test for viral DNA as previously described (22), and results were calculated as the percent input virus remaining in the tube, where a tube containing the equivalent amount of virus but not washed was designated as 100%. Dilutions of this were used to demonstrate detection sensitivity. Results shown represent one of two experiments.
Role of platelets in primary infection.
To test whether platelets contributed to, or perhaps controlled, viral dissemination, we used PCR to measure viral DNA in the liver, spleen, and salivary glands of infected mice at 4 days postinfection as previously described (22). Some mice received APS or rabbit serum on the day of infection and 2 days postinfection.
Statistical analysis.
All values are reported as means ± SE. ANOVA with Bonferroni's post hoc test was used for statistical comparison of experimental groups.
RESULTS
Platelet depletion.
Rabbit serum led to small reductions in blood platelet counts before versus 24 h after injection (14.2% and 8.0% in mCMV-ND and mCMV-HC mice, respectively). In contrast, APS depleted circulating platelets counts by 95%, from 1.597 × 106 ± 0.302 × 106 to 0.083 × 106 ± 0.065 × 106 platelets/ml in mCMV-ND mice and from 1.594 × 106 ± 0.212 × 106 to 0.084 × 106 ± 0.074 × 106 platelets/ml in mCMV-HC mice.
Cholesterol levels.
As has been previously shown, maintenance of mock CMV-treated mice on HC for 6 wk led to a two- to threefold increase in plasma cholesterol levels (Table 1). This was similar for mice that received mCMV, chimeric mice, or mice that received rabbit serum or platelet-depleting serum.
Table 1.
Plasma cholesterol levels in WT and chimeric mice maintained on ND or placed on HC for 6 wk
| Groups | Plasma Cholesterol Levels, mg/dl |
|---|---|
| Mock-ND | 56.6 ± 2.25 |
| Mock-HC | 127.0 ± 10.49* |
| mCMV-ND | 59.0 ± 5.50 |
| mCMV-ND rabbit serum | 63.3 ± 6.79 |
| mCMV-ND anti-platelet serum | 63.3 ± 4.58 |
| mCMV-HC | 122.5 ± 4.59* |
| mCMV-HC rabbit serum | 140.0 ± 24.67* |
| mCMV-HC anti-platelet serum | 147.4 ± 28.96* |
| WT → WT Chim Mock-ND | 43.2 ± 5.72 |
| WT → WT Chim mCMV-ND | 44.8 ± 11.62 |
| WT → P-sel−/− Chim mCMV-ND | 39.1 ± 4.13 |
| WT → WT Chim mCMV-HC | 92.4 ± 1.95† |
| WT → P-sel−/− Chim mCMV-HC | 78.8 ± 2.93† |
Data are expressed as means ± SE. Mock, mock inoculum, ND, normal diet; HC, high-cholesterol diet; mCMV, murine cytomegalovirus; WT, wild type; Chim, chimeric; P-sel−/−, P-selectin deficient.
P < 0.0001 versus all wild-type ND groups;
P ≤ 0.0005 versus ND chimeric groups.
Contribution of platelets to arteriolar vasodilation responses.
Arteriolar dilation to ACh was impaired in mCMV-ND mice compared with mock-ND control mice (Fig. 1, A and B). This was characterized by reduced maximum responsiveness of the arterioles to ACh, whereas EC50 remained similar in both groups (mock-ND mice: 2.099 × 10−6 M and mCMV-ND mice: 1.088 × 10−6 M). A similar profile of arteriolar dysfunction was also observed in mCMV-HC mice (EC50: 2.344 × 10−6 M). In contrast, HC alone did not alter arteriolar responses to ACh (Fig. 1B). Responsiveness to endothelium-independent vasodilators, papaverine (Fig. 1C) and sodium nitroprusside (data not shown), were comparable between groups, suggesting that vascular smooth muscle cell function was not impaired and the vessels were not significantly constricted. For experiments in Fig. 1B and subsequent figures, 10−5 M ACh was used, because this dose was the lowest dose at which we observed both strong vasodilation in control mice and differences between arteriolar responses in uninfected and infected mice. Rabbit serum did not alter the vasodilation responses to ACh in either normocholesterolemic or hypercholesterolemic mCMV groups. Acute depletion of platelets for 24 h before the experiment failed to offer any protection against the impaired arteriolar dilation responses in mCMV-ND mice. To determine whether some protection was present in the mCMV-ND APS group but not detected because the vessels were less sensitive to ACh, dose-response curves were performed for mCMV-ND RS and APS groups, and it was found that APS did not reverse the dysfunction at any ACh dose tested (Fig. 1A). In contrast, thrombocytopenia was associated with complete restoration of ACh-induced vasodilation in mCMV-HC mice (Fig. 1B). When endothelium-independent arteriolar responses were tested with papaverine, there were no significant differences found between groups (Fig. 1C). Although some groups tended to be lower than the mock-ND group, the vessels were clearly capable of better dilation than was observed with ACh in these instances in that the ratio of papaverine to ACh dilation was >2.2 in all five groups exhibiting impaired endothelium-dependent vasodilation, whereas it was <1.4 in the others. Furthermore, there were no significant differences between baseline diameters for any of the groups (Table 2).
Fig. 1.
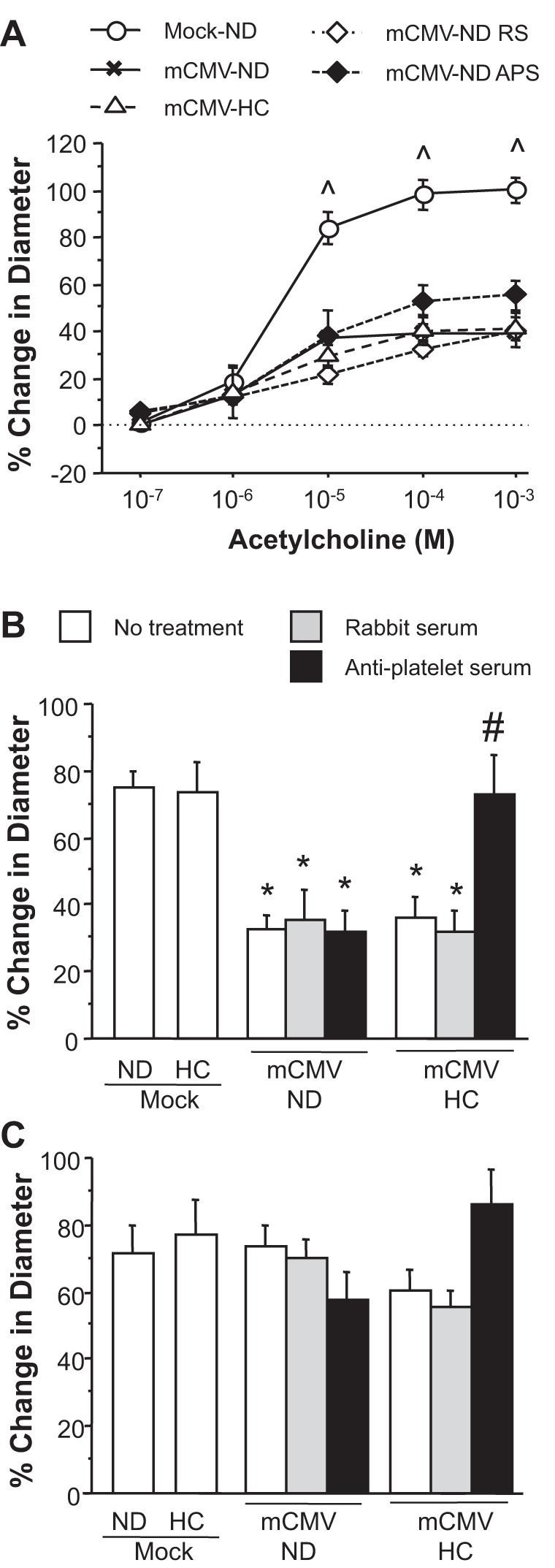
Changes in arteriolar diameter in response to ACh (A and B) or papaverine (C) at 11 wk postinfection in mice injected with mock inoculum (mock) or murine cytomegalovirus (mCMV) and maintained on a normal diet (ND) or placed on a high-cholesterol diet (HC) at 5 wk postinfection. Some mice received rabbit serum (RS) or anti-platelet serum (APS) 24 h before observations. ^P ≤ 0.0001, mock-ND vs. all other mCMV groups; *P < 0.001 vs. mock-ND and mock-HC groups; #P < 0.0005 vs. other mCMV-HC groups.
Table 2.
Baseline diameters of arterioles in the different groups shown in Figs. 1 and 2 as well as wall shear rate in postcapillary venules shown in Figs. 3 and 4
| Groups | Baseline Diameter, μm | Wall Shear Rate, s−1 |
|---|---|---|
| Data shown in Figs. 1 and 3 | ||
| Mock-ND | 22.0 ± 1.61 | 819 ± 97.9 |
| Mock-HC | 19.9 ± 1.08 | 679 ± 48.0 |
| mCMV-ND | 23.0 ± 1.05 | 718 ± 55.2 |
| mCMV-ND rabbit serum | 24.0 ± 1.08 | |
| mCMV-ND anti-platelet serum | 21.8 ± 1.06 | |
| mCMV-HC | 22.6 ± 1.20 | 808 ± 65.6 |
| mCMV-HC rabbit serum | 24.1 ± 0.86 | 728 ± 59.0 |
| mCMV-HC anti-platelet serum | 21.5 ± 1.46 | 649 ± 50.6 |
| Data shown in Figs. 2 and 4 | ||
| WT Mock-ND | 21.0 ± 0.71 | 920 ± 113.1 |
| WT → WT Chim Mock-ND | 19.8 ± 1.16 | 816 ± 198.3 |
| WT mCMV-ND | 24.7 ± 2.16 | |
| WT → WT Chim mCMV-ND | 22.9 ± 1.30 | |
| WT → P-sel−/− Chim mCMV-ND | 19.9 ± 1.57 | |
| WT mCMV-HC | 22.6 ± 2.25 | 732 ± 58.1 |
| WT → WT Chim mCMV-HC | 20.2 ± 0.72 | 648 ± 93.2 |
| WT → P-sel−/− Chim mCMV-HC | 19.6 ± 0.63 | 597 ± 32.4 |
Data are expressed as means ± SE.
Control bone marrow chimeras (WT mice that received WT bone marrow) exposed to mock-ND exhibited the expected strong arteriolar dilation responses to ACh (76.0 ± 4.5% increase in diameter; Fig. 2A). Similar to nonchimeric WT mice, mCMV infection of WT → WT chimeras was associated with impaired vasodilation in mice maintained on ND or placed on HC. In contrast, when chimeras were generated such that platelets were deficient in P-selectin, significant protection was conferred against the mCMV-induced arteriolar dysfunction in normocholesterolemic mice. P-sel−/− → WT chimeras were afforded similar protection against mCMV-HC-induced arteriolar dysfunction, with vasodilation responses comparable to those achieved by acute platelet depletion. P-sel−/− → WT chimera responses remained slightly lower than those in their WT→WT mock-ND counterparts. Although it was not determined if the incomplete protection was due to reduced sensitivity to ACh or a reduced maximal response in P-sel−/− → WT chimeras, it can be concluded that platelet P-selectin plays a major role but is not entirely responsible for the impaired dilation responses. There were no significant differences in endothelium-independent (papaverine-induced) responses (Fig. 2B) or baseline diameters (Table 2) between the chimera groups.
Fig. 2.
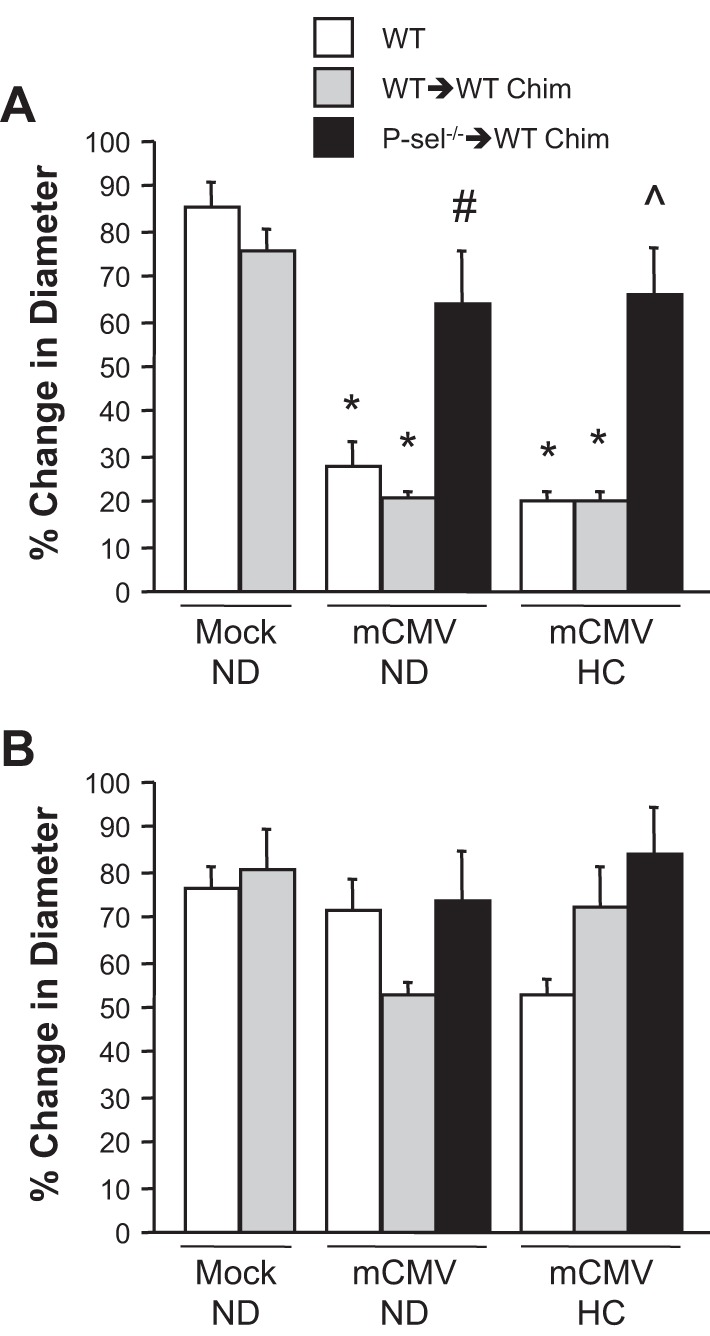
Arteriolar vasodilation in response to ACh (A) or papaverine (B) in wild-type (WT), control chimeric (WT → WT Chim), or P-selectin chimeric (P-sel−/− → WT Chim) mice 11 wk after injection with mock inoculum or mCMV. Some mice were placed on HC at 5 wk postinfection. *P < 0.0001 vs. WT and WT-WT mock-ND groups; #P < 0.001 vs. other mCMV-ND groups; ^P < 0.0001 vs. other mCMV-HC groups.
Role of platelets in leukocyte recruitment in postcapillary venules.
None of the observed alterations in venular responses described below could be explained by differences in wall shear rate (Table 2). mCMV or HC individually did not enhance leukocyte recruitment (Fig. 3, A and B). However, mCMV and hypercholesterolemia synergized to enhance leukocyte adhesion in postcapillary venules compared with mock-ND, mock-HC, or mCMV-ND groups (Fig. 3A). Leukocyte emigration was also increased in mCMV-HC mice over mock-ND mice or mice exposed to either risk factor alone (Fig. 3B). This is in line with what we have previously reported (33). Rabbit serum did not affect leukocyte recruitment in mCMV-HC venules; however, depletion of platelets using APS significantly reduced both leukocyte adhesion and emigration toward levels observed in mock-ND controls. To determine whether platelets were mediating leukocyte adhesion by influencing leukocyte rolling, we measured leukocyte rolling flux and velocity. While not significant, there was a greater than twofold increase in leukocyte flux in WT mCMV-HC mice, and platelet depletion reduced this to levels observed in mock-ND control mice (Fig. 3C). Rolling velocity was not altered from corresponding controls by mCMV-HC or platelet depletion (data not shown).
Fig. 3.
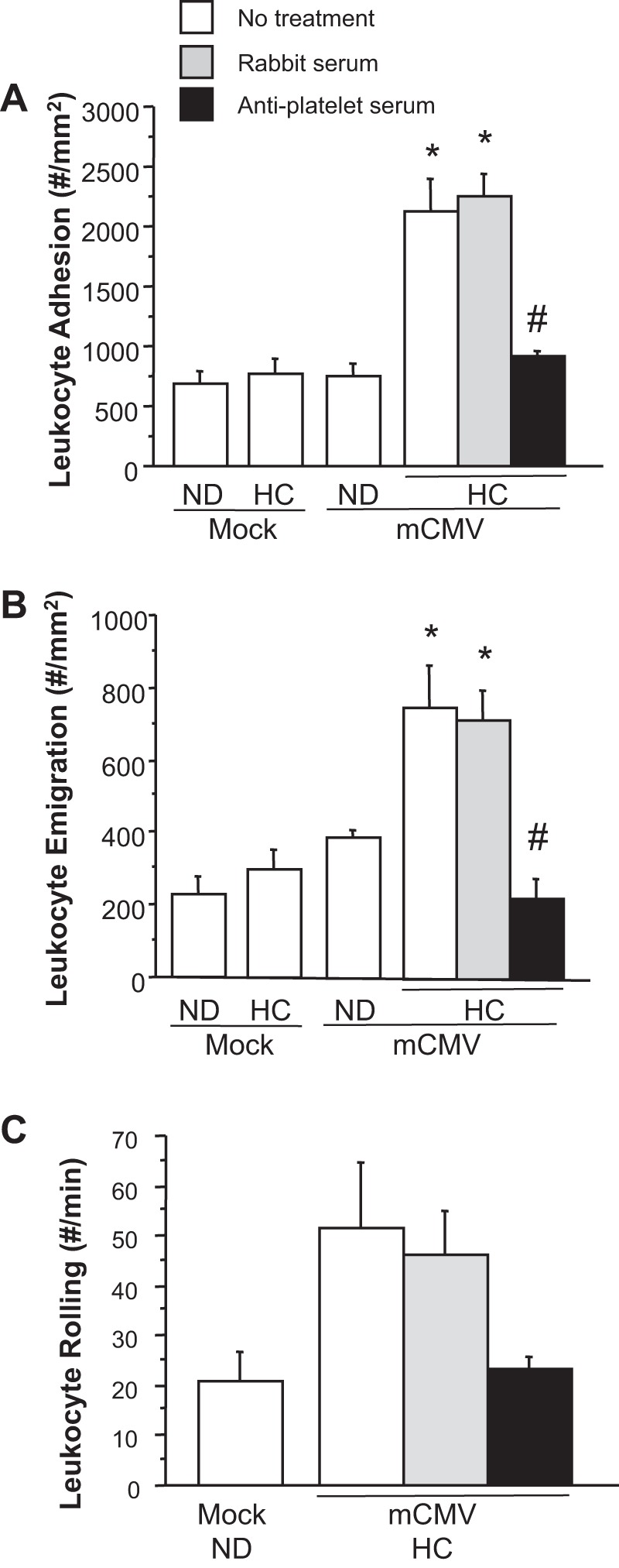
Adherent leukocytes in postcapillary venules (A), emigrated leukocytes in the surrounding tissue (B), and rolling leukocytes in venules (C) of mice 11 wk after they received mock inoculum or mCMV and maintained on ND or placed on HC at 5 wk postinfection. Some mice were treated with RS or APS at 24 h before observations. *P < 0.005 vs. mock-ND, mock-HC, and mCMV-ND groups; #P < 0.0001 vs. other mCMV-HC groups.
The elevated leukocyte adhesion and emigration induced by mCMV-HC that was seen in WT mice was similarly observed in WT → WT chimeras, indicating that bone marrow transplant per se did not alter normal inflammatory responses to mCMV-HC (Fig. 4). The absence of platelet P-selectin in P-sel−/− → WT chimeras afforded only partial protection against mCMV-HC-induced leukocyte adhesion and emigration. Although rolling flux was lower in chimeric mice versus WT mice, there was a greater than twofold increase in mCMV-HC versus mock-ND WT → WT chimeras (Fig. 4C). This elevation of rolling flux was not reduced by the absence of platelet-P-selectin, which, together with the APS data, suggests that platelets may be mediating the reduction in leukocyte adhesion by reducing rolling flux through a platelet P-selectin-independent mechanism.
Fig. 4.
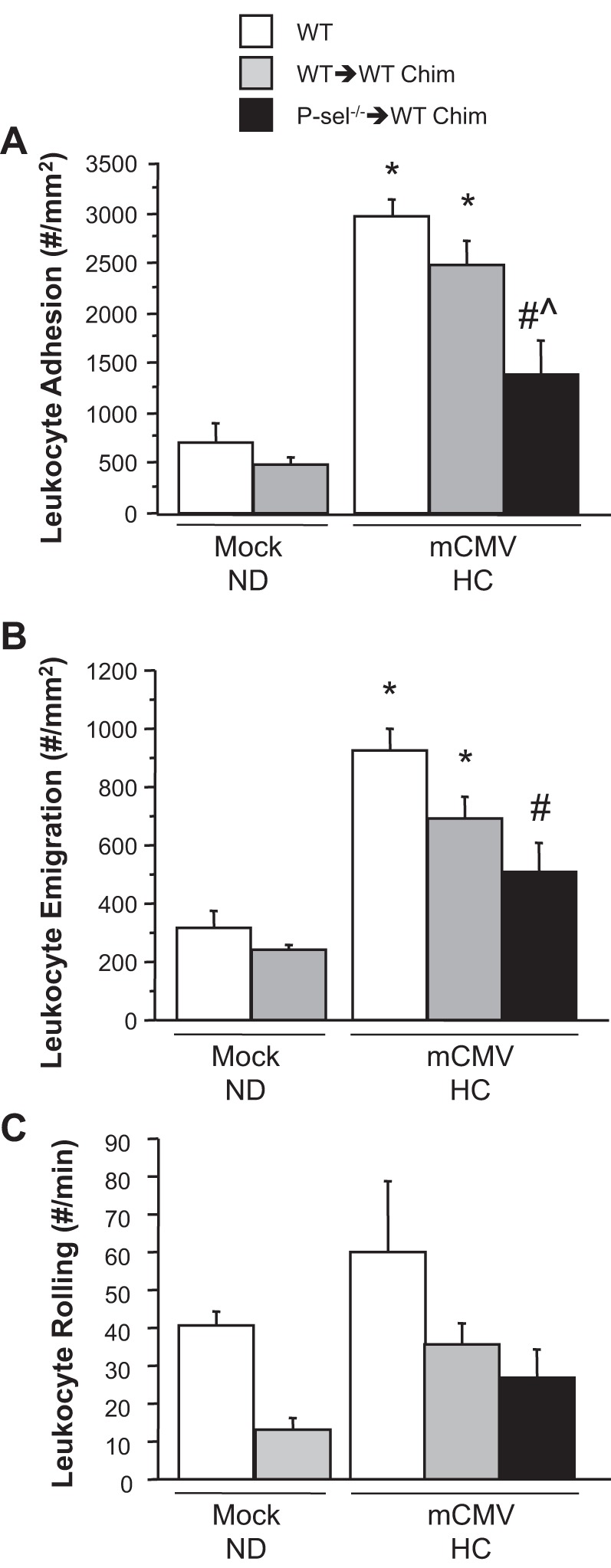
Leukocyte adhesion in postcapillary venules (A), emigration into the interstitium (B), and rolling in venules (C) of WT, WT → WT Chim, and P-sel−/− → WT Chim mice 11 wk after injection with mock inoculum or mCMV. Some mCMV mice were placed on HC at 5 wk postinfection. *P < 0.005 vs. both mock-ND groups; #P < 0.0005 vs. the WT mCMV-HC group; ^P < 0.005 vs. the WT→WT Chim mCMV-HC group.
Impact of mCMV versus HC on platelet recruitment in postcapillary venules.
We have previously shown that platelet adhesion in postcapillary venules is significantly elevated in mCMV-HC mice compared with either risk factor alone or with mock-ND mice (33). To further define how each factor contributes to this synergistic thrombogenic effect of mCMV-HC, we performed experiments on platelets isolated from donor mice, which were fluorescently labeled and infused into and observed in matched or mismatched recipient mice. As expected, platelet adhesion in matched mock-HC or mCMV-ND mice was comparable to that seen in mock-ND mice that received matched platelets (matched donor-recipient experiments denoted by arrows; Fig. 5). However, mCMV-HC platelets infused into matched recipients exhibited significantly increased platelet adhesion. When mCMV-HC platelets were infused into recipients that had been exposed to no risk factor (mock-ND) or only one risk factor (mock-HC or mCMV-ND), no such thrombogenic phenotype was observed. Furthermore, when mock-HC platelets were infused in mCMV-HC recipient mice, platelet adhesion remained at control levels. In contrast, when mCMV-ND platelets were infused into mCMV-HC recipients, their adhesion was significantly elevated, to ∼50% of levels seen for mCMV-HC platelets in the same recipients.
Fig. 5.
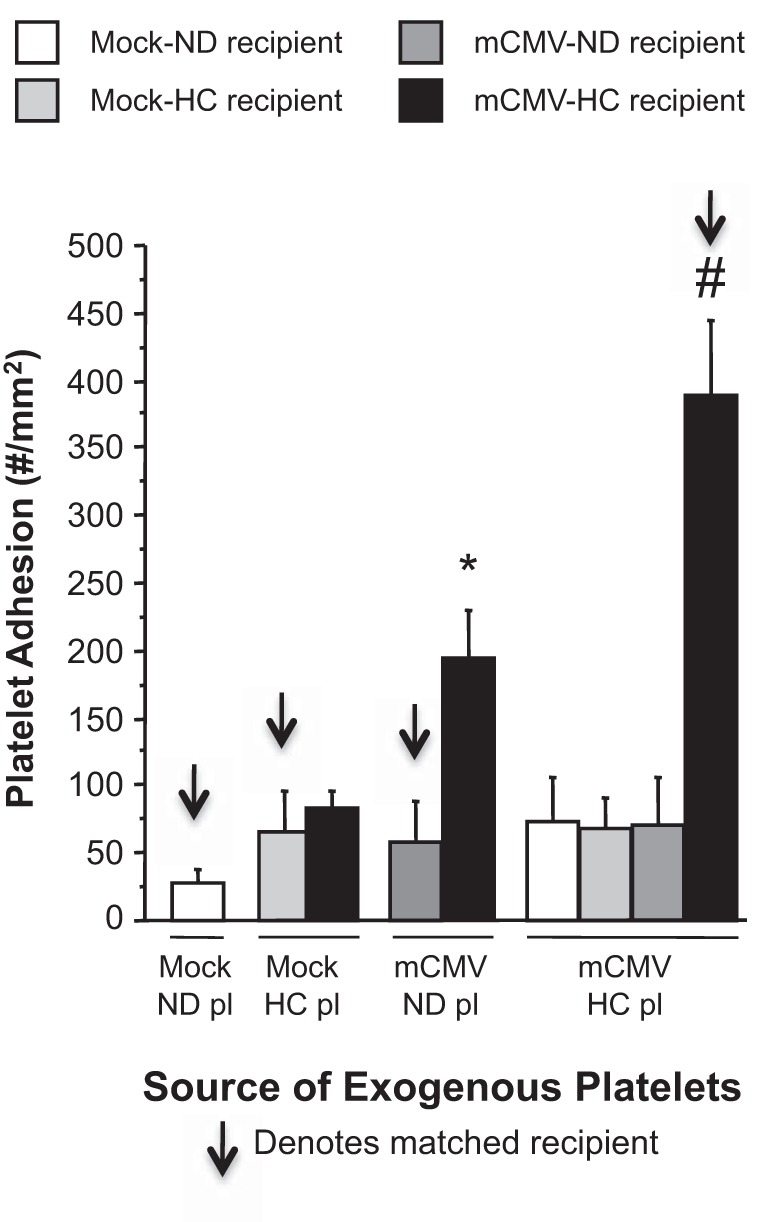
Adhesion of exogenous platelets (Pl) in postcapillary venules of matched (arrows) or mismatched recipients. Mice were injected with mock inoculum or mCMV, and, 5 wk later, some mice were placed on HC, whereas others were kept on ND. Observations were made at 11 wk postinfection. *P < 0.0005 vs. the mock-ND + mock-ND Pl group; #P < 0.0001 vs. all other groups.
Testing of platelet-virus interactions.
Although platelets express receptors, such as Toll-like receptor (TLR)2, that the virus could potentially bind, we were unable to detect viral DNA in platelet samples incubated with mCMV (Fig. 6). In fact, levels of virus detected were similar to those in tubes not containing platelets, which represented a control for viral adsorption to the tube wall. These data suggest that platelets may not directly bind mCMV.
Fig. 6.
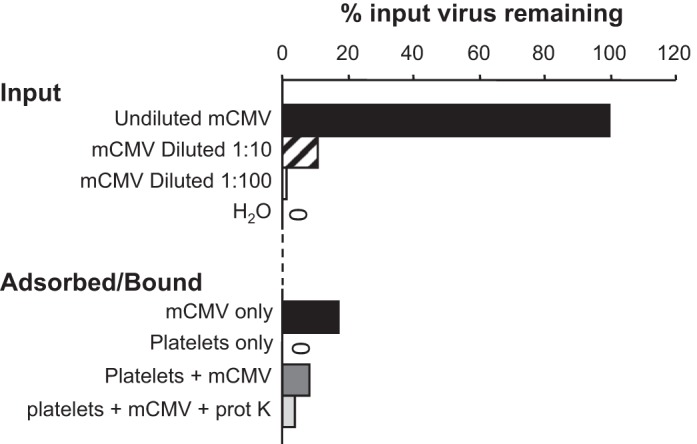
Measurements of mCMV remaining in tubes containing virus or water as a control (input) or tubes containing virus only, platelets only, platelets + virus, or platelets + virus followed by treatment with proteinase K (adsorbed/bound). All of the adsorbed/bound tubes were washed to remove unbound or nonadsorbed virus before detection of viral DNA was performed. Data are expressed as percentages of input virus remaining based on the tube containing undiluted virus and not washed before detection. “0” denotes no signal detected.
Platelets in the early dissemination of mCMV.
Because acute depletion of platelets did not confer protection but long-term absence of platelet P-selectin was protective against arteriolar dysfunction in mCMV-ND mice, we determined whether platelets may contribute to the dissemination of the virus during primary infection, by comparing viral titers in organs from WT mice treated with rabbit serum or APS. We observed no differences between groups (data not shown).
DISCUSSION
Hypercholesterolemia is associated with a heightened activation state of platelets in humans (30, 32). These cells, in turn, play a major role in the microvascular responses to hypercholesterolemia (38). CMV has been implicated in atherosclerotic lesions (6, 21, 43, 44, 47) and also leads to microvascular dysfunction in a murine model (22). This virus has recently been linked to thrombosis in humans (3, 13, 27) and can activate platelets (2). Furthermore, the human CMV-infected endothelium becomes more adhesive to platelets (31). However, these findings are in the setting of primary infection. In the present study, we demonstrate a role for platelets, acting through their surface receptor P-selectin, in the generation of arteriolar dysfunction during persistent mCMV infection. Our findings implicate platelets in the inflammatory phenotype observed in hypercholesterolemic animals that have been exposed to mCMV. However, unlike in hypercholesterolemic mice with no infection (38), there remained a platelet-dependent, but platelet P-selectin-independent, component to the inflammatory response.
Arteriolar dysfunction occurs early in the response to HC [3-wk HC (39)] and represents a shift in sensitivity of the vessels to ACh rather than a blunted maximal response (38). However, this is transient and resolves by 6 wk in uninfected mice (Fig. 1) (33). CMV leads to a sustained endothelium-dependent vasodilation response (18, 22), and the addition of hypercholesterolemia slightly worsens this response in a time-dependent manner (22) [albeit not at the timepoint used in the present study (33)]. We show here that, regardless of diet, there is reduced maximal endothelium-dependent dilation but not altered sensitivity (i.e., EC50) to ACh in mCMV groups. This suggests that mCMV may be acting through a different pathway than HC to induce endothelium-dependent impairment.
Based on our findings with APS, platelets do not contribute to mCMV-induced arteriolar dysfunction in normocholesterolemic mice. In contrast, when these mice were fed HC, platelets acquired an acute role in this dysfunction. A previous study (38) has shown that platelets participate in hypercholesterolemia-induced arteriolar dysfunction, and our findings suggest that mCMV may be prolonging this. Furthermore, the role for platelets shifted from decreasing EC50 for ACh in uninfected HC mice to reducing the maximal response in mCMV-HC mice. Although human CMV has been associated with thrombocytopenia (1, 46), platelet counts were comparable between our groups; therefore, this is unlikely to account for the disparity in the acute role for platelets between mCMV-ND and mCMV-HC mice. Instead, the difference in platelet adhesion in venules of these groups may be responsible, as there is evidence that platelets adherent in venules impair nearby arterioles (23). However, we did not specifically address this.
We have previously demonstrated that 20-h inhibition of P-selectin restores normal arteriolar function in mCMV-ND mice (33). Taken together with the absence of protection observed here in thrombocytopenic mice, it appears that endothelial, rather than platelet, P-selectin mediates arteriolar dysfunction induced by mCMV. Nevertheless, before dismissing a role for platelets in the mCMV-ND group, and because the platelet depletion was limited to the 24 h immediately before observation, we used P-selectin bone marrow chimeras and showed that platelets had a more chronic role in the mCMV-induced arteriolar dysfunction. Such conflicting findings between APS and P-sel−/− → WT chimera groups have been previously found for other parameters and are likely due to the difference in the duration of platelet versus P-selectin deficiency (38). We excluded a role for platelets in the dissemination of mCMV during primary infection, suggesting that this disparity was not due to an attenuation of early infection in chimeric mice. It is also possible that a switch in the underlying mechanisms of dilation responses to ACh may occur in infected mice with long-term deficiency of platelet P-selectin. However, since platelets do not adhere to the arteriolar wall in response to mCMV alone between time of infection and 11 wk postinfection, it is likely that they are acting indirectly to induce arteriolar dysfunction. For example, platelet P-selectin interactions with P-selectin glyocoprotein ligand-1 on circulating blood cells and/or leukocytes recruited to vessels in remote organs may be leading to the release of soluble factors, such as the vasoconstrictor thromboxane A2, that mediate dysfunction. Exactly how and when, over the 11 wk postinfection, platelets exert their effect on the vasculature remain to be elucidated.
Previously, APS or deficiency of P-selectin on platelets have been shown to attenuate the early phase of inflammation in postcapillary venules of noninfected hypercholesterolemic mice (38). In the present study, we found that platelet depletion remained effective in protecting against the prolongation of this inflammatory response in mCMV-HC mice. However, P-selectin chimeric mice were only partially protected against the leukocyte recruitment observed in response to mCMV-HC. In light of our previous findings that treatment of mCMV-HC mice with anti-P-selectin antibody completely abrogated leukocyte adhesion and emigration responses to mCMV-HC (38), this suggests that endothelial P-selectin is in part responsible for this and that the role of platelets in inflammatory responses in HC mice is somehow altered by the addition of mCMV, such that a platelet-dependent, but platelet P-selectin-independent, component is introduced by the virus. In a previous study (33), we showed that P-selectin mediates a majority of leukocyte rolling in this model. Although not conclusive, our present data for leukocyte rolling in APS-treated mice and P-selectin chimeras suggest that platelets are acting at least in part by reducing leukocyte rolling flux through a pathway not involving platelet P-selectin. This indirectly supports a key role for endothelial P-selectin in leukocyte rolling in mCMV-HC mice.
Primary infection of endothelial cells by human CMV leads to enhanced platelet adhesion to these cells. In contrast, during persistent infection, platelet recruitment in postcapillary venules of mCMV-ND mice is only marginally higher than that observed in mock-ND control mice. However, in mCMV-HC mice, platelet adhesion is significantly upregulated, and this is dependent on platelet P-selectin (33). We found a role for platelets, acting via P-selectin, in arteriolar dysfunction in both mCMV-ND and mCMV-HC mice in the present study. This suggests some level of activation, but not necessarily local recruitment, of these cells. Thus, we used mismatched platelet-to-donor transfer to test whether it is likely to be HC or mCMV that exerts more influence on the platelet recruitment observed in mCMV-HC mice. Our findings suggest that mCMV causes a subtle activation or priming of platelets that is not manifested as adhesion in matched cremasteric vessels but is evident when cells are placed in the thrombogenic environment of mCMV-HC mice. Although this is in line with a role for platelet P-selectin in arteriolar dysfunction in mCMV-ND mice, the apparent low level of platelet activation in mCMV-ND mice was not sufficient to cause arteriolar dysfunction acutely (based on APS data). In contrast, based on their adhesion levels, mCMV-HC platelets appear to be more activated, and this may explain why APS was effective at reversing the arteriolar dysfunction in mCMV-HC mice. There is little known about the direct actions of CMV on platelets, although platelets express receptors, such as TLR2, that could potentially bind CMV (10). A recent study (2) has shown that human CMV will bind directly to a small subpopulation (<2%) of platelets and that both human CMV and mCMV can upregulate platelet P-selectin expression through a TLR2-dependent mechanism. In contrast, we were unable to detect binding of the virus to isolated platelets. Whether the disparity with the human CMV study is related to virus strain, dose, or species specificity or to the sensitivity of the detection method (flow cytometry vs. PCR) is unclear. However, taken together with our inability to locate the virus in organs of these mice at 11 wk postinfection, we suggest that mCMV is acting indirectly to activate platelets during persistent infection.
Our study showed that platelets, acting via P-selectin, contribute to the arteriolar dysfunction in mCMV-ND mice. Together with our mismatched platelet experiments, this suggests that platelets in mCMV-ND mice are activated sufficiently to release substances and/or to cause the release of soluble factors from other cells that impair vasodilation. However, when HC is added to these mice, this platelet signal reaches the threshold required to cause inflammation. Although this inflammation may be an extension of what is normally a transient platelet-P-selectin-dependent process in mock-HC mice, exposure to both factors leads to the development of a platelet P-selectin-independent component also. These data add mechanistic insights into how this virus may be mediating microvascular dysfunction through a platelet-dependent mechanism. They also suggest that mCMV alters how other risk factors act via platelets to induce these responses. This could have important implications in the design of therapeutic interventions targeting platelet responses in human CMV-positive cardiovascular disease patients or perhaps a subpopulation that are particularly vulnerable to the impact of human CMV on platelets and the vasculature.
GRANTS
This work was supported by American Heart Association Scientist Development Grant 0730294N and National Institute of General Medical Sciences Grant GM-103433 (to K. Y. Stokes) as well as fellowships from the Malcolm Feist Cardiovascular Research Endowment, Louisiana State University Health Sciences Center (to M. V. Khoretonenko and J. L. Brunson).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.V.K. and K.Y.S. conception and design of research; M.V.K., J.L.B., E.S., I.L.L., C.R.M., and K.Y.S. performed experiments; M.V.K., J.L.B., E.S., I.L.L., C.R.M., and K.Y.S. analyzed data; M.V.K., J.L.B., E.S., I.L.L., C.R.M., and K.Y.S. interpreted results of experiments; M.V.K., J.L.B., E.S., I.L.L., and K.Y.S. prepared figures; M.V.K., J.L.B., and K.Y.S. drafted manuscript; M.V.K. and K.Y.S. edited and revised manuscript; M.V.K., J.L.B., E.S., I.L.L., C.R.M., and K.Y.S. approved final version of manuscript.
REFERENCES
- 1.Aslam M, Anderson JL, Guglietti D, Cardwell D. CMV-induced neonatal thrombocytopenia: a case report and review of the literature. Am J Perinatol 24: 429–434, 2007. [DOI] [PubMed] [Google Scholar]
- 2.Assinger A, Kral J, Yaiw KC, Schrottmaier W, Kurzejamska E, Wang Y, Mohammad AA, Religa P, Rahbar A, Schabbauer G, Butler LM, Soderberg-Naucler C. Human cytomegalovirus-platelet interaction triggers Toll-like receptor 2-dependent proinflammatory and proangiogenic responses. Arterioscler Thromb Vasc Biol 34: 801–809, 2014. [DOI] [PubMed] [Google Scholar]
- 3.Atzmony L, Halutz O, Avidor B, Finn T, Zimmerman O, Steinvil A, Zeltser D, Giladi M, Justo D. Incidence of cytomegalovirus-associated thrombosis and its risk factors: a case-control study. Thromb Res 126: e439–e443, 2010. [DOI] [PubMed] [Google Scholar]
- 4.Bentz GL, Jarquin-Pardo M, Chan G, Smith MS, Sinzger C, Yurochko AD. Human cytomegalovirus (HCMV) infection of endothelial cells promotes naive monocyte extravasation and transfer of productive virus to enhance hematogenous dissemination of HCMV. J Virol 80: 11539–11555, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blum A, Giladi M, Weinberg M, Kaplan G, Pasternack H, Laniado S, Miller H. High anti-cytomegalovirus (CMV) IgG antibody titer is associated with coronary artery disease and may predict post-coronary balloon angioplasty restenosis. Am J Cardiol 81: 866–868, 1998. [DOI] [PubMed] [Google Scholar]
- 6.Burnett MS, Gaydos CA, Madico GE, Glad SM, Paigen B, Quinn TC, Epstein SE. Atherosclerosis in apoE knockout mice infected with multiple pathogens. J Infect Dis 183: 226–231, 2001. [DOI] [PubMed] [Google Scholar]
- 7.Burns LJ, Pooley JC, Walsh DJ, Vercellotti GM, Weber ML, Kovacs A. Intercellular adhesion molecule-1 expression in endothelial cells is activated by cytomegalovirus immediate early proteins. Transplantation 67: 137–144, 1999. [DOI] [PubMed] [Google Scholar]
- 8.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 87: 840–844, 2000. [DOI] [PubMed] [Google Scholar]
- 9.Chiu B, Viira E, Tucker W, Fong IW. Chlamydia pneumoniae, cytomegalovirus, and herpes simplex virus in atherosclerosis of the carotid artery. Circulation 96: 2144–2148, 1997. [DOI] [PubMed] [Google Scholar]
- 10.Compton T, Kurt-Jones EA, Boehme KW, Belko J, Latz E, Golenbock DT, Finberg RW. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J Virol 77: 4588–4596, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cooper D, Chitman KD, Williams MC, Granger DN. Time-dependent platelet-vessel wall interactions induced by intestinal ischemia-reperfusion. Am J Physiol Gastrointest Liver Physiol 284: G1027–G1033, 2003. [DOI] [PubMed] [Google Scholar]
- 12.Craigen JL, Yong KL, Jordan NJ, MacCormac LP, Westwick J, Akbar AN, Grundy JE. Human cytomegalovirus infection up-regulates interleukin-8 gene expression and stimulates neutrophil transendothelial migration. Immunology 92: 138–145, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delbos V, Abgueguen P, Chennebault JM, Fanello S, Pichard E. Acute cytomegalovirus infection and venous thrombosis: role of antiphospholipid antibodies. J Infect 54: e47–50, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Dengler TJ, Raftery MJ, Werle M, Zimmermann R, Schonrich G. Cytomegalovirus infection of vascular cells induces expression of pro-inflammatory adhesion molecules by paracrine action of secreted interleukin-1β. Transplantation 69: 1160–1168, 2000. [DOI] [PubMed] [Google Scholar]
- 15.Epstein SE, Zhu J, Najafi AH, Burnett MS. Insights into the role of infection in atherogenesis and in plaque rupture. Circulation 119: 3133–3141, 2009. [DOI] [PubMed] [Google Scholar]
- 16.Espinola-Klein C, Rupprecht HJ, Blankenberg S, Bickel C, Kopp H, Victor A, Hafner G, Prellwitz W, Schlumberger W, Meyer J. Impact of infectious burden on progression of carotid atherosclerosis. Stroke 33: 2581–2586, 2002. [DOI] [PubMed] [Google Scholar]
- 17.Freeman RB., Jr The “indirect” effects of cytomegalovirus infection. Am J Transplant 9: 2453–2458, 2009. [DOI] [PubMed] [Google Scholar]
- 18.Grahame-Clarke C, Chan NN, Andrew D, Ridgway GL, Betteridge DJ, Emery V, Colhoun HM, Vallance P. Human cytomegalovirus seropositivity is associated with impaired vascular function. Circulation 108: 678–683, 2003. [DOI] [PubMed] [Google Scholar]
- 19.Grattan MT, Moreno-Cabral CE, Starnes VA, Oyer PE, Stinson EB, Shumway NE. Cytomegalovirus infection is associated with cardiac allograft rejection and atherosclerosis. JAMA 261: 3561–3566, 1989. [PubMed] [Google Scholar]
- 20.Grundy JE, Lawson KM, MacCormac LP, Fletcher JM, Yong KL. Cytomegalovirus-infected endothelial cells recruit neutrophils by the secretion of C-X-C chemokines and transmit virus by direct neutrophil-endothelial cell contact and during neutrophil transendothelial migration. J Infect Dis 177: 1465–1474, 1998. [DOI] [PubMed] [Google Scholar]
- 21.Hsich E, Zhou YF, Paigen B, Johnson TM, Burnett MS, Epstein SE. Cytomegalovirus infection increases development of atherosclerosis in apolipoprotein-E knockout mice. Atherosclerosis 156: 23–28, 2001. [DOI] [PubMed] [Google Scholar]
- 22.Khoretonenko MV, Leskov IL, Jennings SR, Yurochko AD, Stokes KY. Cytomegalovirus infection leads to microvascular dysfunction and exacerbates hypercholesterolemia-induced responses. Am J Pathol 177: 2134–2144, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim MH, Granger DN, Harris NR. Mediators of CD18/P-selectin-dependent constriction of venule-paired arterioles in hypercholesterolemia. Microvasc Res 73: 150–155, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kupatt C, Wichels R, Horstkotte J, Krombach F, Habazettl H, Boekstegers P. Molecular mechanisms of platelet-mediated leukocyte recruitment during myocardial reperfusion. J Leukoc Biol 72: 455–461, 2002. [PubMed] [Google Scholar]
- 25.Kurose I, Argenbright LW, Anderson DC, Tolley J, Miyasaka M, Harris N, Granger DN. Reperfusion-induced leukocyte adhesion and vascular protein leakage in normal and hypercholesterolemic rats. Am J Physiol Heart Circ Physiol 273: H854–H860, 1997. [DOI] [PubMed] [Google Scholar]
- 26.Kurose I, Wolf R, Cerwinka W, Granger DN. Microvascular responses to ischemia/reperfusion in normotensive and hypertensive rats. Hypertension 34: 212–216, 1999. [DOI] [PubMed] [Google Scholar]
- 27.Ladd AM, Goyal R, Rosainz L, Baiocco P, DiFabrizio L. Pulmonary embolism and portal vein thrombosis in an immunocompetent adolescent with acute cytomegalovirus hepatitis. J Thromb Thrombolysis 28: 496–499, 2009. [DOI] [PubMed] [Google Scholar]
- 28.Landmesser U, Hornig B, Drexler H. Endothelial function: a critical determinant in atherosclerosis? Circulation 109: II27–II33, 2004. [DOI] [PubMed] [Google Scholar]
- 29.Panes J, Kurose I, Rodriguez-Vaca D, Anderson DC, Miyasaka M, Tso P, Granger DN. Diabetes exacerbates inflammatory responses to ischemia-reperfusion. Circulation 93: 161–167, 1996. [DOI] [PubMed] [Google Scholar]
- 30.Puccetti L, Sawamura T, Pasqui AL, Pastorelli M, Auteri A, Bruni F. Atorvastatin reduces platelet-oxidized-LDL receptor expression in hypercholesterolaemic patients. Eur J Clin Invest 35: 47–51, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Rahbar A, Soderberg-Naucler C. Human cytomegalovirus infection of endothelial cells triggers platelet adhesion and aggregation. J Virol 79: 2211–2220, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanguigni V, Pignatelli P, Lenti L, Ferro D, Bellia A, Carnevale R, Tesauro M, Sorge R, Lauro R, Violi F. Short-term treatment with atorvastatin reduces platelet CD40 ligand and thrombin generation in hypercholesterolemic patients. Circulation 111: 412–419, 2005. [DOI] [PubMed] [Google Scholar]
- 33.Senchenkov E, Khoretonenko MV, Leskov IL, Ostanin DV, Stokes KY. P-selectin mediates the microvascular dysfunction associated with persistent cytomegalovirus infection in normocholesterolemic and hypercholesterolemic mice. Microcirculation 18: 452–462, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shahgasempour S, Woodroffe SB, Garnett HM. Alterations in the expression of ELAM-1, ICAM-1 and VCAM-1 after in vitro infection of endothelial cells with a clinical isolate of human cytomegalovirus. Microbiol Immunol 41: 121–129, 1997. [DOI] [PubMed] [Google Scholar]
- 35.Smyth SS, McEver RP, Weyrich AS, Morrell CN, Hoffman MR, Arepally GM, French PA, Dauerman HL, Becker RC. Platelet functions beyond hemostasis. J Thromb Haemost 7: 1759–1766, 2009. [DOI] [PubMed] [Google Scholar]
- 36.Span AH, Mullers W, Miltenburg AM, Bruggeman CA. Cytomegalovirus induced PMN adherence in relation to an ELAM-1 antigen present on infected endothelial cell monolayers. Immunology 72: 355–360, 1991. [PMC free article] [PubMed] [Google Scholar]
- 37.Stokes K, Granger DN. The microcirculation: a motor for the systemic inflammatory response and large vessel disease induced by hypercholesterolemia? J Physiol 562: 647–653, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stokes KY, Calahan L, Russell JM, Gurwara S, Granger DN. Role of platelets in hypercholesterolemia-induced leukocyte recruitment and arteriolar dysfunction. Microcirculation 13: 377–388, 2006. [DOI] [PubMed] [Google Scholar]
- 39.Stokes KY, Dugas TR, Tang Y, Garg H, Guidry E, Bryan NS. Dietary nitrite prevents hypercholesterolemic microvascular inflammation and reverses endothelial dysfunction. Am J Physiol Heart Circ Physiol 296: H1281–H1288, 2009. [DOI] [PubMed] [Google Scholar]
- 40.Stokes KY, Granger DN. Platelets: a critical link between inflammation and microvascular dysfunction. J Physiol 590: 1023–1034, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tailor A, Granger DN. Hypercholesterolemia promotes P-selectin-dependent platelet-endothelial cell adhesion in postcapillary venules. Arterioscler Thromb Vasc Biol 23: 675–680, 2003. [DOI] [PubMed] [Google Scholar]
- 42.Tomita Y, Sachs DH, Sykes M. Myelosuppressive conditioning is required to achieve engraftment of pluripotent stem cells contained in moderate doses of syngeneic bone marrow. Blood 83: 939–948, 1994. [PubMed] [Google Scholar]
- 43.Vliegen I, Duijvestijn A, Grauls G, Herngreen S, Bruggeman C, Stassen F. Cytomegalovirus infection aggravates atherogenesis in apoE knockout mice by both local and systemic immune activation. Microbes Infect 6: 17–24, 2004. [DOI] [PubMed] [Google Scholar]
- 44.Vliegen I, Stassen F, Grauls G, Blok R, Bruggeman C. MCMV infection increases early T-lymphocyte influx in atherosclerotic lesions in apoE knockout mice. J Clin Virol 25, Suppl 2: S159–S171, 2002. [DOI] [PubMed] [Google Scholar]
- 45.Weis M, Kledal TN, Lin KY, Panchal SN, Gao SZ, Valantine HA, Mocarski ES, Cooke JP. Cytomegalovirus infection impairs the nitric oxide synthase pathway: role of asymmetric dimethylarginine in transplant arteriosclerosis. Circulation 109: 500–505, 2004. [DOI] [PubMed] [Google Scholar]
- 46.Yaari S, Koslowsky B, Wolf D, Chajek-Shaul T, Hershcovici T. CMV-related thrombocytopenia treated with foscarnet: a case series and review of the literature. Platelets 21: 490–495, 2010. [DOI] [PubMed] [Google Scholar]
- 47.Yi L, Wang DX, Feng ZJ. Detection of human cytomegalovirus in atherosclerotic carotid arteries in humans. J Formos Med Assoc 107: 774–781, 2008. [DOI] [PubMed] [Google Scholar]