

Abstract
Mice that carry a mutation in a calcium binding domain of Otoferlin, the putative calcium sensor at hair cell synapses, have normal distortion product otoacoustic emissions (DPOAEs), but auditory brain stem responses (ABRs) are absent. In mutant mice mechanotransduction is normal but transmission of acoustic information to the auditory pathway is blocked even before the onset of hearing. The cross-sectional area of the auditory nerve of mutant mice is reduced by 54%, and the volume of ventral cochlear nuclei is reduced by 46% relative to hearing control mice. While the tonotopic organization was not detectably changed in mutant mice, the axons to end bulbs of Held and the end bulbs themselves were smaller. In mutant mice bushy cells in the anteroventral cochlear nucleus (aVCN) have the electrophysiological hallmarks of control cells. Spontaneous miniature excitatory postsynaptic currents (EPSCs) occur with similar frequencies and have similar shapes in deaf as in hearing animals, but they are 24% larger in deaf mice. Bushy cells in deaf mutant mice are contacted by ∼2.6 auditory nerve fibers compared with ∼2.0 in hearing control mice. Furthermore, each fiber delivers more synaptic current, on average 4.8 nA compared with 3.4 nA, in deaf versus hearing control mice. The quantal content of evoked EPSCs is not different between mutant and control mice; the increase in synaptic current delivered in mutant mice is accounted for by the increased response to the size of the quanta. Although responses to shocks presented at long intervals are larger in mutant mice, they depress more rapidly than in hearing control mice.
Keywords: cochlear nucleus, electrophysiology, excitatory postsynaptic currents, mouse, Otoferlin
depriving the cochlear nuclei of acoustic input after the onset of hearing has long been known to affect neuronal circuits in the cochlear nuclei (Pasic and Rubel 1989; Redd et al. 2000; Ryugo et al. 1998; Stakhovskaya et al. 2008; Trune and Kiessling 1988; Trune and Morgan 1988). However, electrical activity occurs even before the onset of hearing in auditory circuits; that activity, especially in the second postnatal week, regulates synaptic transmission from hair cells (Beutner and Moser 2001; Johnson et al. 2011, 2013; Kros et al. 1998). Before hearing begins, supporting cells in the cochlea induce action potentials in small groups of adjacent inner hair cells that in turn evoke periodic bursts of suprathreshold responses in the auditory nerve that are propagated to the cortex (Tritsch et al. 2007, 2010; Tritsch and Bergles 2010). Synaptotagmin IV contributes to shaping synaptic responses in adult inner hair cells and immature outer hair cells (Johnson et al. 2010), but after the third postnatal day (P3) synaptic transmission between inner hair cells and spiral ganglion neurons, the cells whose axons are auditory nerve fibers (ANFs), depends largely on the detection of calcium by Otoferlin (Beurg et al. 2010; Roux et al. 2006). The output from the cochlea is significantly reduced and altered in mice that lack normal Otoferlin (Beurg et al. 2010; Pangrsic et al. 2010). Downstream synapses do not depend on Otoferlin but likely depend on synaptotagmin I as a calcium sensor, as do other central synapses (Wang et al. 2011). Mice that lack Otoferlin are thus naive of most normal, patterned spontaneous activity that originates in the cochlea and propagates throughout the auditory system before the onset of hearing in wild-type animals. Here we examine how the reduction of electrical activity from inner hair cells affects downstream neuronal circuits.
ANFs innervate the ipsilateral ventral (VCN) and dorsal cochlear (DCN) nuclei tonotopically. Fibers that encode low frequencies innervate ventral regions, and fibers that encode high frequencies innervate dorsal regions. ANFs terminate on bushy cells in the anterior VCN (aVCN) with large synapses, end bulbs of Held (Brawer and Morest 1975; Held 1893; Lauer et al. 2013; Ryugo and Sento 1991; Sento and Ryugo 1989). End bulbs release glutamate, which binds to postsynaptic AMPA receptors in the bushy cell membrane (Gardner et al. 1999, 2001; Wang et al. 1998). To understand how the lack of input from inner hair cells even before the onset of hearing affects the function of the auditory pathway, we examine mice with a mutation that prevents synaptic transmission between inner hair cells and spiral ganglion cells and measure synaptic events in the bushy cell targets of spiral ganglion cells that receive input through end bulbs of Held.
The mice studied in these experiments have a point mutation resulting in a nonconserved substitution of I319N in the second of six calcium binding domains of Otoferlin that results in the absence of its expression in cochlear hair cells (Longo-Guess et al. 2007). Otoferlin is thought to sense calcium and regulate synaptic transmission by inner hair cells (Johnson and Chapman 2010; Roux et al. 2006). Otoferlin is a member of the Ferlin family of proteins, all of which have been implicated in calcium-regulated membrane fusion events and are membrane-anchored, cytosolic proteins that are involved in vesicle fusion and/or membrane trafficking (Lek et al. 2012; Yasunaga et al. 2000). Mutations in Otoferlin were first described from human pathologies in several unrelated, consanguineous Lebanese families (Yasunaga et al. 1999). These deaf individuals have an autosomal recessive, nonsyndromic prelingual form of deafness, DFNB9 (Yasunaga et al. 1999). A mutation associated with a splice site in Otoferlin has been associated with auditory neuropathy spectrum disorder in humans (Runge et al. 2013). In humans Otoferlin has multiple long and short alternatively spliced isoforms, but mice have only the long isoform (Yasunaga et al. 1999, 2000). In mice Otoferlin appears in inner hair cells at P4, after which it is responsible for the calcium-induced exocytosis in inner hair cells; synaptotagmin isoforms 1, 2, and 7 are also present in inner hair cells, but none appears responsible for calcium-induced exocytosis before P3 (Beurg et al. 2010). Otoferlin also mediates synaptic transmission from outer hair cells (Beurg et al. 2008) and is required for the replenishment of synaptic vesicles in hair cells (Pangrsic et al. 2010).
MATERIALS AND METHODS
Mice.
Otoferlin mutant animals, Otof deaf5Jcs/Kjn, were purchased from Jackson Lab (stock no. 006128). An ENU-induced point mutation from thymine to adenine in exon 10 of the otoferlin gene causes a nonconserved amino acid change from isoleucine to asparagine in the second calcium binding domain of the protein. Breeding colonies of mutant mice, on a mixed background of C57BL/6J and C3HeB/FeJ, were maintained by crossing deaf Otoferlin mutant males with hearing, heterozygous females. Their offspring were either homozygous deaf mutants or hearing heterozygotes and could be distinguished before experiments by the presence or absence of a Preyer reflex. Wild-type mice were created by mating heterozygous animals; resulting wild-type mice were bred and maintained separately. Genotypes of all mice were confirmed post hoc (Longo-Guess et al. 2007). Homozygous Otoferlin mutant animals will be referred to as “deaf Otoferlin mutant mice”; heterozygotes and wild-type animals will in some cases be lumped and referred to as “hearing control mice” or “Otoferlin control mice.” Animals of both sexes, aged from P11 to P60, with the majority of animals aged P17–23, were used for anatomical experiments and animals aged P16–22 for the electrophysiological experiments. Otoferlin-knockout mice were kindly given to us by Dr. Isabel Roux with permission from Dr. Christine Petit (Roux et al. 2006). Genotyping of these mice was also confirmed post hoc. All procedures were approved by the Institutional Animal Care and Use Committee at the University of Wisconsin, Madison.
Solutions.
The dissection of the cochlear nuclei from the brain stem was done in a high-sucrose extracellular saline solution that contained reduced Na+ and Ca2+ (345 mosM). The composition of the solution (in mM) was as follows: 99 NaCl, 3 KCl, 1.2 KH2PO4, 1 CaCl2, 1.3 MgSO4, 20 NaHCO3, 6 HEPES, 10 glucose, and 72 sucrose, pH 7.3. The cutting solution was kept around 28°C.
The extracellular physiological saline (osmolarity 308 mosM) used to perfuse the tissue (1.5–2 h) after biocytin injections and also for whole cell recordings contained (in mM) 130 NaCl, 3 KCl, 1.2 KH2PO4, 2.4 CaCl2, 1.3 MgSO4, 20 NaHCO3, 6 HEPES, 10 glucose, and 0.4 ascorbic acid, pH 7.3. Whole cell recordings were made in the presence of 10 μM strychnine to block glycinergic inhibition. All salines were saturated with 95% O2-5% CO2 and maintained at 32–33°C. Chemicals were from Sigma-Aldrich, except that sucrose was purchased from Fisher.
The internal pipette solution for voltage- and current-clamp recordings contained (in mM) 108 potassium gluconate, 9 HEPES, 9 EGTA, 4.5 MgCl2, 14 phosphocreatinine (Tris salt), 4 ATP (Na salt), and 0.3 GTP (Tris salt), final osmolarity ∼303 mosM. The pH was adjusted to 7.4 with KOH. The final holding potentials were corrected for a −12 mV junction potential.
Brain slices.
For electrophysiological studies, parasagittal slices of the left cochlear nucleus were cut from the brain stem with a vibrating microtome (Leica VT 1000S) in sections of 200 μm. Cochlear nuclei were superfused continually at ∼3–6 ml/min. The temperature was measured with a Thermalert thermometer (IT-23, Physitemp) and was controlled with a custom-made feedback-controlled heater to remain at 32 or 33°C.
Biocytin injections.
For the anatomy, cochlear nuclei were removed bilaterally from the brain stem by hand with scissors so that the circuitry of the cochlear nuclei could be assessed in its entirety. In a few cases, parasagittal slices up to 420 μm thick of cochlear nuclei were cut with a vibrating microtome (Leica VT 1000S). Tissue was transferred to a holding chamber of ∼0.6 ml and superfused continually at ∼6–8 ml/min for 1.5–2 h. Temperature was kept at 33°C with a custom-made feedback-controlled heater that was connected to a Thermalert thermometer (IT-23, Physitemp). Injections were made under the control of a Wild (M5) dissecting microscope.
Biocytin (1%) was dissolved in extracellular saline and was applied as the fibers were disrupted with a glass pipette. Extracellular injections were made with a Picospritzer, through a glass pipette with a tip diameter of ∼5 μm, along the dorsal/ventral axis of the posteroventral cochlear nucleus (pVCN). Some injections were made only at the nerve root, while others were made in groups of three, with one made ventrally in the nerve root, one medially in the pVCN, and one dorsally in the pVCN. Single injections into the aVCN were made to visualize the tuberculoventral cell projections, which also labeled end bulbs. Tissue was fixed in 4% paraformaldehyde, stored at 4°C, cryoprotected in 30% sucrose, embedded in a gelatin-albumin mixture, and resectioned at 60 μm in frozen sections. Biocytin was visualized with Vectastain ABC Peroxidase Kits (Standard) purchased from Fisher (Golding et al. 1995). Tissues were mounted on subbed slides, dehydrated with alcohol, and stained with cresyl violet to visualize cellular nuclei. Photomicrographs were taken through a Zeiss Axioskop with a Zeiss Axiocam.
Volume and area measurements.
The volumes of the magnocellular regions in the VCN and DCN were measured from camera lucida reconstructions in serial sections from each nucleus. Images were scanned into a computer, outlined, and analyzed with ImageJ software, from which the final volume measurements were imported into Excel and compared by Student's t-tests for statistical analysis. The area of the auditory portion of the eighth nerve was measured in ImageJ from photomicrographs of cross sections through the nerve. Measurements are presented as means ± SD.
Auditory brain stem responses and distortion product otoacoustic emissions.
Animals aged between P20 and P60 were anesthetized with 150 mg/kg Ketaject and 5 mg/kg xylazine. Once animals were unresponsive to a paw pinch, they were placed on a heating pad to maintain body temperature. Auditory brain stem responses (ABRs) and distortion product otoacoustic emissions (DPOAEs) were recorded with Tucker Davis Technologies Sig Gen Software.
To measure ABRs, animals were placed next to a calibrated freefield speaker (ES1) positioned 10 cm away from their left ear, grounded by a subcutaneous electrode behind the contralateral pinna, a subcutaneous reference electrode at the apex of the skull, and a subcutaneous recording electrode directly behind the left ear, as near as possible to the ear. Animals were presented with clicks, 0.1 ms in duration and of alternating polarity between 90 and 10 dB, or with tones, 5 ms in duration with a 3-ms gating time, at 4 kHz, 8 kHz, 16 kHz, and 32 kHz ranging between 90 and 10 dB SPL in 10-dB steps. Final traces were averages of 500–1,500 individual recordings. Thresholds were determined visually. The lowest intensity at which we detected a response was considered the threshold.
DPOAEs are measurements of sounds made with a small microphone placed in the external ear. DPOAEs near 16 kHz were measured in response to presentation of two simultaneous tones of frequencies f1 and f2 presented at equal levels, each through a separate electrostatic speaker (EC1). The microphone and outputs of the two speakers are connected to the ear through a soft plastic fitting that seals them in the external ear.
Electrophysiological recordings.
Whole cell patch-clamp recordings were made with a Multiclamp 700B amplifier (Axon Instruments) under the control of pCLAMP 9 software. Patch electrodes were made from borosilicate glass and had resistances between 3.5 and 8 MΩ. All recordings were digitized at rates more than twice the low-pass limit of the filter to prevent aliasing. Recordings of evoked excitatory postsynaptic currents (eEPSCs) were made at −65 mV, digitized at 40 kHz, and low-pass filtered at 10 kHz. Compensation for the capacitance and series resistance of electrodes was done with the automatic features of the amplifier. The series resistance was compensated 65–80% with a 10-μs lag. EPSCs were evoked by shocks through a Master-8 stimulator and an Iso-flex isolator (AMPI, Jerusalem, Israel) at voltages from ∼0.1 to -10 V and delivered through an extracellular saline-filled glass pipette (∼10-μm tip). Shocks were increased in strength to find the threshold for a synaptic response. Rather than varying the shock strength by constant intervals, the shock strength was increased in increments of between 0.02 and 1.0 V. If an increase in shock strength resulted in a change in EPSC amplitude, several responses were recorded and the voltage for the next stimulus was increased or decreased by small increments; if there was little change in the EPSC, only a single response was recorded and the next shock was given with a larger increment in voltage. When time permitted, the whole series was repeated. Typically analyses were based on ∼40 responses to 20 increments in voltage. The stimulating pipette was ∼50–100 μm away from the recorded bushy cell. Tissue was visualized with a Zeiss Axioskop 2 microscope with a ×63 water immersion lens using a differential interference contrast microscope and infrared illumination. Analyses were performed with pCLAMP (Clampfit 9.0, Axon Instruments) and with Origin software. Spontaneous, miniature EPSCs (mEPSCs) were identified by their shapes with a template based on events in a deaf mouse and selected individually. All quantifications are presented as means ± SD.
Clustering algorithms.
Two assessments were made of the number of steps in the growth of eEPSCs in bushy cells that are an estimate of the number of inputs and of the amount of current delivered per input. The number of steps was estimated by counting the number of jumps in the growth of the peak synaptic current (EPSC) as the shock strength was gradually increased. An estimate of the current delivered by a single input was made from the jump in amplitude between clusters. One count of inputs was made by eye and the second by a new, objective, statistical clustering method.
A manual assessment was based on multiple rounds of analysis. Once the border between clusters was established, the smallest and largest currents from each cluster were averaged to obtain the mean current of the cluster and then the amounts of current per input were calculated.
The second method of quantifying the number of steps/inputs was a customized statistical clustering method. The data analysis in this report was conducted with R (R Core Team, 2013). The quantification was conducted in the following two steps. First, for a set of n observations of individual sweeps of length T, the T × n matrix was factorized by singular value decomposition to find an efficient empirical orthogonal representation of the observations. By choosing the first principal component in the analysis, the overall pattern observed over n sweeps was characterized by the first principal component of length T, while each sweep was characterized by its weight parameter.
Once each sweep was characterized by its weight parameter, then the n parameters were analyzed by a normal mixture model. A normal mixture model is a probabilistic model that assumes the observations are from a mixture of multiple normal distributions. The assumption behind the normal mixture model is that when a distribution has multiple peaks we assume that the observations are from multiple normal distributions without labeling which they belong to. The parameters related to the distribution were estimated by using the expectation-maximization algorithm (Dempster et al. 1977). The estimation of the number of normal distributions was based on the Bayesian information criterion (BIC) (Schwarz 1978). The BIC is based on maximized log-likelihood with a penalty on the number of model parameters, where the larger the value of the BIC, the stronger the evidence for the model (Fraley and Raftery 2007). Comparing the BIC for different numbers of normal distributions, the method can estimate the number of clusters. The probability of each sweep belonging to different clusters was estimated simultaneously.
RESULTS
Distortion product otoacoustic emissions and auditory brain stem responses.
An objective measure of the health of the cochlea in mice as well as in humans is provided by otoacoustic emissions (Avan et al. 2013). In the healthy cochlea, stimulation with pairs of tones produces distortion products that generate traveling waves that can be detected by a microphone in the ear canal as DPOAEs. Figure 1A shows recordings of DPOAEs produced by two tones, f1 = 14,544 Hz, f2 = 17,440 Hz, from a juvenile homozygous, Otoferlin mutant mouse. When those tones were presented at relatively low levels, the most prominent distortion product was at 2f1 − f2, 11,648 Hz (Fig. 1A, dp1). At higher sound pressure levels a second distortion product was evident at 3f1 − 2f2, 8,873 Hz (Fig. 1A, dp2). Both distortion products disappeared when the animal was killed with an overdose of anesthetic (Fig. 1B), indicating that they resulted from the interaction of the tones with a healthy cochlea. This pattern of responses was observed in all mice tested, including Otoferlin knockout, point mutant, heterozygous mutant, and wild-type control mice. Cochlear amplification thus appears normal in all genotypes, including both deaf Otoferlin knockout and mutant.
Fig. 1.
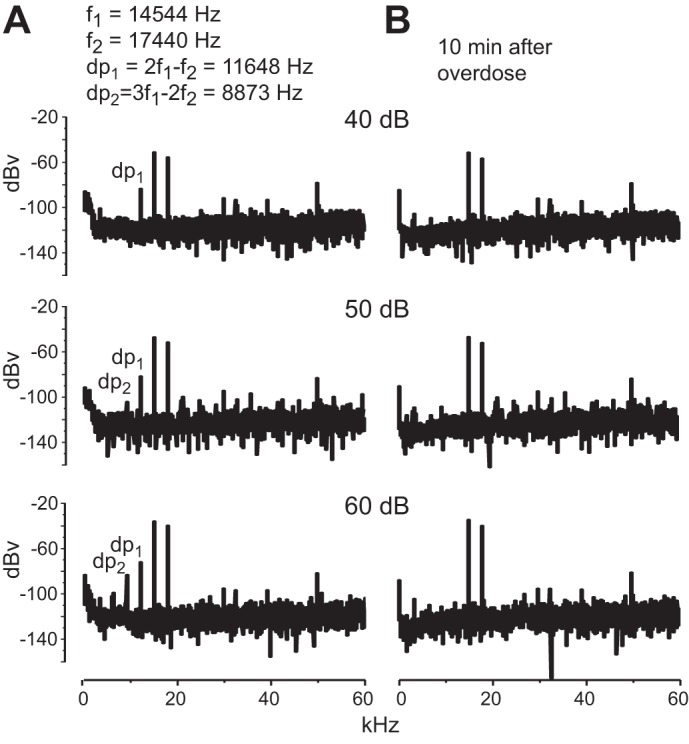
Distortion product otoacoustic emissions. A: 2 tones were delivered to 1 ear, and the distortion products were measured at 3 different sound pressure levels; 2 distortion products were detected, 2f1 − f2 (dp1) and 3f1 − 2f2 (dp2), from a mutant mouse. B: the same animal was then lethally overdosed with anesthetic without moving the tone probes or microphone. Ten minutes after breathing movements ceased the same 2 tones were delivered, but these resulted in no distortion products.
Hearing of mice was further assessed with ABRs, which are measurements of electrical activity associated with the propagation of acoustic information through ANFs to higher auditory centers. Adult (∼P60) and juvenile (∼P19–24) mice of each of the three genotypes associated with the otoferlin point mutation, Mut (n = 13), Het (n = 11), and WT (n = 12), and juvenile mice with the complete knockout of Otoferlin (n = 6) were tested. Figure 2A illustrates typical ABRs from each of the genotypes. All wild-type and heterozygote animals responded to clicks with a small positive wave that was followed by larger positive and negative waves. Every homozygous mutant and knockout animal responded to clicks with only a small positive wave. It seems likely that the first small wave that we observe in all genotypes, including both homozygous mutant and knockout animals and that was also observed by Pangrsic et al. (2010), reflects receptor potentials of hair cells. The lack of electrical activity after the initial small positivity in mice with mutant Otoferlin and without Otoferlin indicates that brain stem circuits are not activated by clicks and that mice that lack a normal copy of the otoferlin gene are deaf. Average thresholds of responses to clicks for hearing and deaf animals are summarized in Fig. 2B. Thresholds in responses to pure tone stimuli are summarized in Fig. 2C.
Fig. 2.
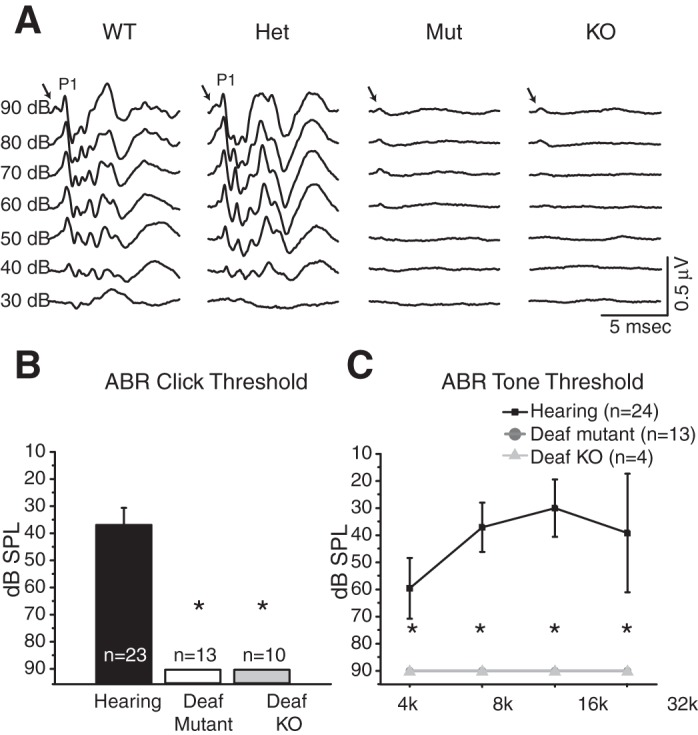
Auditory brain stem responses (ABRs). A: clicks, 0.1 ms in duration and alternating in polarity, evoked a series of positive and negative waves that reflect the activity of neurons in the brain stem in the hearing genotypes, wild-type (WT) and heterozygote (Het) Otoferlin mice. A small positivity that was graded with the intensity of clicks (arrows) was the remnant of a receptor potential that was not removed by averaging responses to clicks of alternating polarity. The first positive wave (P1) reflects the synchronous firing of auditory nerve fibers, while later waves reflect electrical activity of downstream circuits in the cochlear nuclei and superior olivary complex (Melcher and Kiang 1996). The absence of all but the first positivity in the deaf Otoferlin mutant (Mut) and Otoferlin knockout (KO) animals indicates that acoustic signals fail to propagate through the circuits of the brain stem and shows that these animals are deaf. The brief inflection at the beginning of each trace (arrows) likely reflects a receptor potential. B: averaged hearing thresholds for the click stimuli. C: hearing thresholds for the tone stimuli at 4, 8, 16, and 32 kHz show that wild-type and heterozygote hearing control mice hear best at ∼16 kHz and that Otoferlin mutant and Otoferlin knockout animals do not hear at any frequency. *Statistically significant differences relative to hearing controls.
The finding that acoustic emissions are present but ABRs in these same mutant and knockout animals are absent indicates that acoustic information is transduced in the hair cells but not transmitted from the hair cells of the cochlea to the brain stem. These findings are consistent with the conclusion from previous studies that Otoferlin is required for synaptic transmission between hair cells and their spiral ganglion cell targets (Beurg et al. 2010; Johnson and Chapman 2010; Reisinger et al. 2011; Roux et al. 2006).
Reduction in size.
The cochlear nuclei in deaf Otoferlin mutant mice are innervated by smaller auditory nerves than in hearing control mice. The cross-sectional areas of the auditory portion of the eighth nerve in mutants is 0.036 ± 0.008 mm2 (n = 5) compared with 0.078 ± 0.016 mm2 in hearing control mice (n = 10), the difference being statistically significant (P < 0.001). The cross-sectional areas of the auditory nerves are reduced by 54% in deaf Otoferlin mutant mice. Roux and colleagues found that while many ribbon synapses in mutants resemble those of controls, there were only about half the normal number and some ribbons were abnormal and floating in inner hair cells of the Otoferlin mutants (Roux et al. 2006). If every ribbon synapse drives one type I spiral ganglion cell, then it seems likely that the number of type I spiral ganglion cells is reduced.
The cochlear nuclei are also considerably smaller in the deaf Otoferlin mutants than in heterozygotes or wild-type animals. To measure the volumes of the VCN, DCN, and granule cell areas, we outlined them in photomicrographs, measured the areas, and multiplied them by the thickness of sections (60 μm). Figure 3A shows a photomicrograph of a parasagittal section of the cochlear nuclei in which the DCN and VCN are outlined. Figure 3B shows average volumes from juvenile mice of the DCN and VCN (n = 15), five from each of the three otoferlin mutation genotypes. The differences in the average volume of the DCN between hearing and deaf phenotypes were not statistically significant (0.15 ± 0.03 mm3 in deaf Otoferlin mutant relative to 0.18 ± 0.03 mm3 in hearing wild type). However, the volumes of the VCN were significantly (P < 0.001) smaller in the deaf Otoferlin mutant mice (0.19 ± 0.03 mm3) than in hearing mice (0.35 ± 0.04 mm3). The volumes of mutant VCNs were thus reduced by 46% in mutant mice.
Fig. 3.
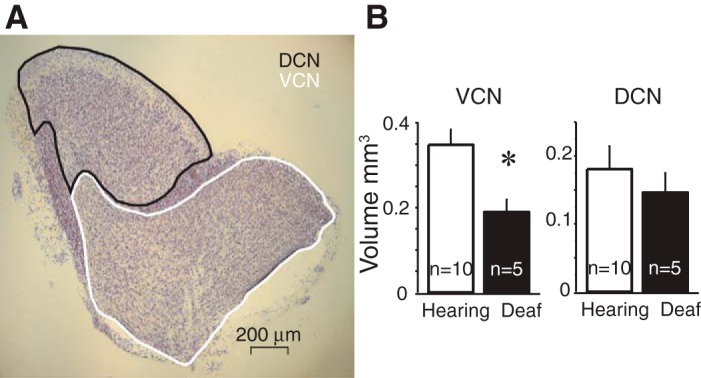
Volumes of cochlear nuclei. A: photomicrograph of a parasagittal section, 60 μm thick, through the cochlear nuclei that had been stained with cresyl violet. The ventral (VCN) and dorsal (DCN) cochlear nuclei are outlined. B: areas were summed across serial sections and multiplied by 60 μm to determine volumes. Average volumes are shown in histograms illustrating the average volume difference of 5 mice from each Otoferlin genotype. The volumes of the VCN were significantly smaller in deaf Otoferlin mutant mice than in hearing control mice (P < 0.001). The difference in the volumes of the DCN was not significant. *Statistically significant difference.
Tonotopy of intrinsic projections.
ANFs innervate the cochlear nuclei systematically as a function of the frequencies to which they are tuned, so that high-frequency-encoding fibers terminate dorsally and low-frequency-encoding fibers terminate ventrally in both the VCN and DCN. This tonotopic organization means that ANFs that encode a narrow range of frequencies innervate narrow bands of neurons in the VCN and DCN. Extracellular injections of 1% biocytin were made into >150 cochlear nuclei. Injections of biocytin into the nerve root and along the dorsal-ventral axis of the pVCN labeled bundles of ANFs that innervated the VCN and DCN topographically as demonstrated previously in other strains of mice. Labeled fibers that bifurcated ventrally in the nerve root innervated the ventral VCN and ventral DCN; fibers that bifurcated dorsally in the nerve root innervated the dorsal aVCN, dorsal pVCN, and dorsal DCN (Cao et al. 2008; Wickesberg and Oertel 1988). In every deaf Otoferlin mutant mouse, as in every hearing control mouse, ANFs ran through the cochlear nuclei in well-organized bundles, suggesting that the tonotopic organization is normal.
Intrinsic connections between the DCN and the VCN follow the tonotopic organization (Wickesberg and Oertel 1988). Biocytin injections into the VCN label bands of tuberculoventral cells in the DCN, where auditory nerve terminals labeled by the same injections also terminate. Figure 4 shows that this organization holds in deaf Otoferlin mutant mice as in hearing heterozygote control mice. When the injection labeling ANFs was made ventrally, the band of labeled tuberculoventral neurons also lay ventrally (Fig. 4, bottom), whereas when the injection labeled fibers more dorsally, labeled tuberculoventral cells also lay more dorsally (Fig. 4, top).
Fig. 4.

Topographic organization of intrinsic cochlear nuclear connections. Photomicrographs of sections from 4 slices show location of injections of biocytin into the anteroventral cochlear nucleus (aVCN, *). The injections labeled a discrete bundle of auditory nerve fibers and a group of tuberculoventral neurons in the deep layer of the DCN; as these labeled neurons are difficult to resolve at low power, they are marked with arrowheads. A: in heterozygotes, injections into the ventral aVCN label tuberculoventral neurons ventrally (bottom) and more dorsal injections label neurons more dorsally (top). B: labeled tuberculoventral neurons in Otoferlin mutant mice follow the same pattern. The more ventral injections label tuberculoventral cells more ventrally than the most dorsal injections. Top left panel is a composite image of photomicrographs of 2 sections; all other panels are composite images of individual sections.
We conclude that within the resolution of these experiments the tonotopic organization of the VCN and DCN is normal in Otoferlin mutant mice. The ANFs are well organized, and the tuberculoventral cell projection follows that organization.
End bulbs of Held.
As in other strains of mice, ANFs in mutant mice terminated in endings of variable size in the multipolar cell area and in uniformly small boutons in the octopus cell area. In the aVCN, where most neurons are bushy cells, many terminals of ANFs end in clusters of boutons that wrap the somas of their targets with the end bulbs of Held (Held 1893). Some synaptic terminals of ANFs in the aVCN are conventional terminal boutons.
The morphology of end bulbs of Held differs in hearing and deaf Otoferlin mutant mice. Figure 5A shows end bulbs from hearing mice. These end bulbs are clusters of boutons connected to fingerlike projections that encompass the target bushy cell soma. We found that the deaf Otoferlin mutant mice had fewer end bulbs compared with their hearing controls, which is not surprising when the volume of the VCN and eighth nerve were reduced. Figure 5B shows end bulbs from deaf Otoferlin mutant mice. These end bulbs are thinner and more wispy than those of hearing control mice. The number of branches per end bulb was counted in 144 end bulbs from hearing mice and 72 in deaf Otoferlin mutant mice. The number of branches per end bulb in hearing mice was significantly greater than in deaf Otoferlin mutant mice; the mean number of branches per end bulb in hearing mice was 13.7 ± 4.1 compared with 11.2 ± 4 in deaf Otoferlin mutant mice (P ≤ 0.001). Our finding that deaf Otoferlin mutant mice have more wispy end bulbs is consistent with the work of others showing that deafness per se can affect the sizes and shapes of end bulbs but it does not eliminate them (Cao et al. 2008; Redd et al. 2000; Ryugo et al. 1997).
Fig. 5.

Auditory nerve fibers and their end bulb terminals. Photomicrographs of end bulbs labeled with extracellular injections of 1% biocytin into the nerve root, aVCN, or posteroventral cochlear nucleus (pVCN). A: end bulbs from hearing mice comprise highly branched clusters of boutons that engulf the cell body of target bushy cells. B: end bulbs from deaf Otoferlin mutant mice are smaller. In some, the axons that lead to the end bulbs are small and thin.
To understand not only the end bulb but also the axon of the ANF leading to the end bulb, we made camera lucida reconstructions of end bulbs together with the segment of axon between the final branch point of ANF and the end bulb. Figure 6 shows five reconstructions of end bulbs and their axons from each phenotype. The reconstructions show that the axons that lead to the end bulbs of hearing mice (Fig. 6A) were thicker than those of deaf Otoferlin mutant mice (Fig. 6B). Measurements of the thickness of axons near the end bulbs in photomicrographs show that the axons in hearing animals (1.36 ± 0.3 μm, n = 10) are significantly thicker than the axons of ANFs from deaf Otoferlin mutant mice (0.93 ± 0.31 μm, n = 5) (P < 0.001).
Fig. 6.
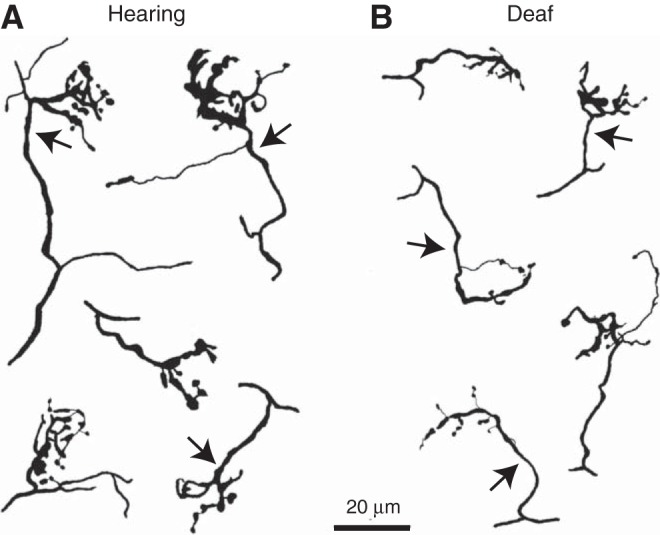
Camera lucida reconstructions of end bulbs. A: end bulbs in hearing animals were reconstructed to the branch of the parent auditory nerve fiber. Arrows mark the axons that lead up to end bulbs. B: similar reconstructions in Otoferlin mutant mice show that end bulbs are smaller and their axons are thinner.
Intrinsic electrical properties of bushy cells.
We recorded from bushy cells in the most anterior regions of the VCN, a region that is occupied mainly by small spherical bushy cells. Large spherical bushy cells are also found most anteriorly in the aVCN, but mice have only a few of these neurons. Globular bushy cells are most frequently found more posteriorly near the nerve root (Tolbert and Morest 1982b; Willard and Ryugo 1983).
Bushy cells from deaf Otoferlin mutant mice have biophysical properties that are largely indistinguishable from those in hearing control mice as assessed by whole cell patch recordings. Figure 7A illustrates responses to depolarizing and hyperpolarizing current pulses in a bushy cell from a deaf Otoferlin mutant mice. As in control hearing mice and in mice of other strains, bushy cells fired one or two action potentials at the onset of the depolarizing current injection and showed rectification (Cao et al. 2007; Oertel 1983). Quantification of the input resistance in bushy cells, based on responses to +10 and −10 pA current pulses, reveals that there is no significant difference between the phenotypes (Fig. 7B); bushy cells in hearing mice had input resistances of 83.7 ± 32.6 MΩ (n = 32), while the average input resistance of bushy cells from deaf Otoferlin mutant mice was 96.1 ± 39.1 MΩ (n = 27). The resting membrane potentials, too, were not different in hearing and deaf mice (Fig. 7C). The bushy cells in hearing animals rested at −66.8 ± 5.0 mV (n = 32), while those in bushy cells from deaf Otoferlin mutant mice rested at −65.9 ± 5.1 mV (n = 27).
Fig. 7.
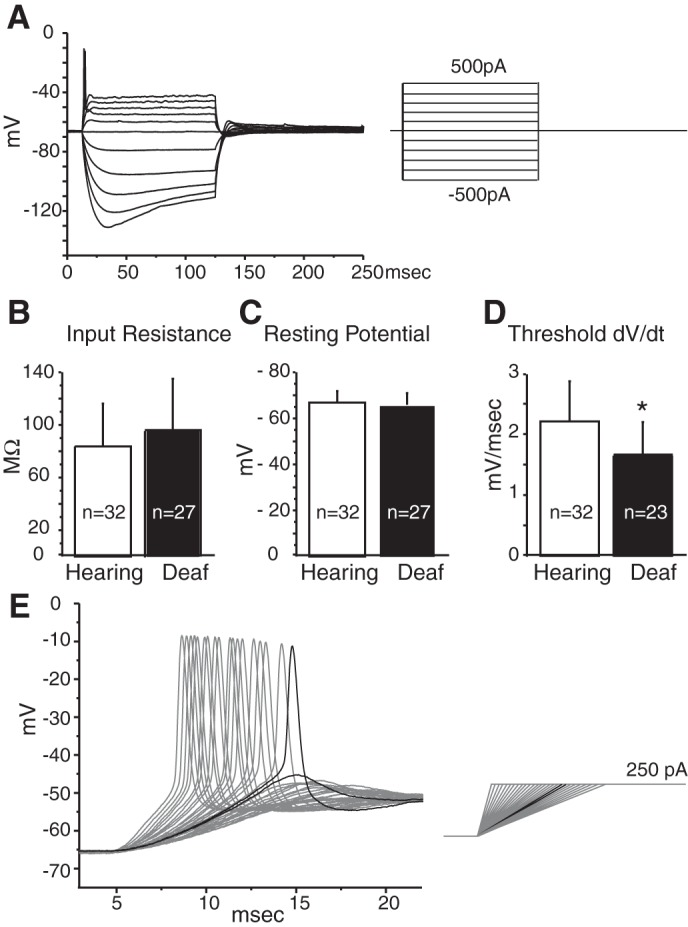
Intrinsic properties of bushy cells. A: superimposed responses to depolarizing and hyperpolarizing current pulses from a bushy cell from a deaf Otoferlin mutant mouse. B: average input resistances from bushy cells in hearing and deaf mice calculated from responses to 10 pA and −10 pA current steps were not significantly different between hearing and deaf Otoferlin mutant mice. C: average resting membrane potentials in bushy cells were not significantly different between hearing and deaf mice. D: rate-of-depolarization (dV/dt) threshold was significantly lower in the bushy cells of deaf than of hearing mice, indicating that bushy cells from hearing mice require a steeper depolarization to fire action potentials than bushy cells from deaf mice. E: representative current-clamp recording from a hearing mouse illustrates how ramped depolarizing current pulses injected into the bushy cell were used to measure the rate-of-depolarization thresholds. Black traces are the responses to the current ramp that were just suprathreshold and just subthreshold. The slope of the black suprathreshold voltage trace was the rate threshold. *Statistically significant difference.
There was, however, a significant difference in the rate of depolarization thresholds between bushy cells of hearing and deaf Otoferlin mutant mice. Bushy cells detect the synchronous firing of ANFs; their sensitivity to rate of depolarization is an intrinsic property that is associated with the time window over which spatial summation occurs (McGinley and Oertel 2006). Bushy cells are more likely to fire action potentials when multiple ANFs are activated synchronously and when excitatory postsynaptic potentials (EPSPs) have a steeper slope of depolarization. To measure the threshold rate of depolarization, current pulses with systematically varied, ramped onsets that rose to a constant value were presented to bushy cells (Fig. 7E). The slope of depolarization that was just steep enough to cause a bushy cell to fire was defined as the threshold rate of depolarization (Fig. 7E). Bushy cells from hearing mice needed to be depolarized on average at least 2.21 ± 0.66 mV/ms to fire action potentials, while those in deaf Otoferlin mutant mice needed to be depolarized on average at least 1.66 ± 0.54 mV/ms to fire action potentials (Fig. 7D). Bushy cells from deaf Otoferlin mutant mice thus fire action potentials in response to less steeply rising depolarizations than bushy cells from hearing mice (P = 0.002), indicating that their requirements for synchronicity are less stringent than those of bushy cells from mice that hear.
Spontaneous EPSCs.
We first compared the spontaneous mEPSCs in bushy cells from mice with differing phenotypes and genotypes. Example recordings of spontaneous postsynaptic currents in 11 overlaid sweeps in bushy cells from hearing and deaf Otoferlin mutant mice are displayed in Fig. 8, A and B, respectively. The averaged miniature events in these same bushy cells from hearing and deaf mice are exhibited in Fig. 8, C and D. To facilitate comparisons, several features of mEPSCs were measured (Fig. 8, E–J; Table 1). The frequency, amplitude, charge, half-width, 10–90% rise time, and 100–37% decay tau were quantified. Of these measures, only the average amplitude and average charge transferred in each mEPSC were significantly greater in bushy cells of deaf Otoferlin mutant than hearing mice (Fig. 8, F and G; Table 1). The average amplitude of mEPSCs in hearing animals was 88 pA, compared with 109 pA in bushy cells of deaf Otoferlin mutant mice (P = 0.02). The average charge transferred by a mEPSC in bushy cells of hearing mice was 27 fC compared with 35 fC in deaf Otoferlin mutant mice (P < 0.001). The shapes of mEPSCs, described by their half-width, 10–90% rise time, and decay time constants, were not significantly different in hearing and deaf mice (Fig. 8, H–J; Table 1). In summary, mEPSCs were larger and carried more charge in deaf mice than in hearing control mice, but their shapes were similar.
Fig. 8.

Miniature excitatory postsynaptic currents (mEPSCs) and characteristics. A: 11 superimposed traces illustrate spontaneous synaptic currents in a bushy cell from a hearing mouse. B: 11 superimposed traces from a bushy cell of a deaf Otoferlin mutant mouse show spontaneous synaptic currents. C: average of all of the individual mEPSCs in A. D: average of all of the individual mEPSCs in B. E: average frequency of mEPSCs varied widely but was not significantly different between bushy cells in hearing and deaf mice. F: average amplitude of mEPSCs was significantly smaller in bushy cells of hearing than of deaf mice (P = 0.02). G: average area of mEPSCs corresponding to charge transferred was significantly smaller in hearing than in deaf mice (P = 0.0003). H: average half-width of the mEPSCs was not significantly different between the phenotypes. I: average 10–90% rise time of the mEPSCs was not significantly different between the hearing and deaf phenotypes. J: average 100–37% decay tau was not significantly different between the hearing and deaf phenotypes. *Statistically significant difference.
Table 1.
Spontaneous mEPSCs
| smEPSCs | Amplitude, pA | Charge, fC | Frequency, s−1 | Half-Width, ms | 10-90% Rise Time, ms | 100-37% Decay tau, ms |
|---|---|---|---|---|---|---|
| Average hearing | 88.2 ± 38.0 | 26.9 ± 8.3 | 58.9 ± 42.5 | 0.27 ± 0.1 | 0.13 ± 0.04 | 0.2 ± 0.1 |
| (n = 35) | (n = 35) | (n = 35) | (n = 35) | (n = 35) | (n = 33) | |
| Average deaf | 108.7 ± 29.7 | 34.9 ± 8.6 | 62.1 ± 90.1 | 0.26 ± 0.1 | 0.15 ± 0.16 | 0.24 ± 0.1 |
| (n = 31) | (n = 31) | (n = 31) | (n = 31) | (n = 31) | (n = 31) | |
| P value | P = 0.02 | P = 0.0003 | P = 0.8 | P = 0.6 | P = 0.6 | P = 0.2 |
Values are means ± SD.
smEPSC, spontaneous miniature excitatory postsynaptic current.
Electrically evoked EPSPs and EPSCs.
Next we compared how bushy cells respond to activation of their auditory nerve inputs. By placing an extracellular stimulating electrode on a bundle of ANFs near a bushy cell that was being monitored in a whole cell patch-clamp recording in current clamp and gradually increasing the strength of shocks delivered through the stimulating pipette, we evoked EPSPs that grew in clustered steps (Fig. 9, A and B). Since ANFs fire all-or-none action potentials, the step increases in slope likely represent the recruitment of fibers that converge on the recorded bushy cell; counts of the numbers of slopes are thus estimates of the numbers of auditory nerve inputs. Both cells have at least two inputs. Most EPSPs in bushy cells from both hearing and deaf Otoferlin mutant mice are suprathreshold. For example, in Fig. 9, A and B, even responses to the weaker shocks showed an inflection indicative of electrically active processes. Responses to strong shocks rise more rapidly and reach a higher peak than responses to smaller shocks.
Fig. 9.
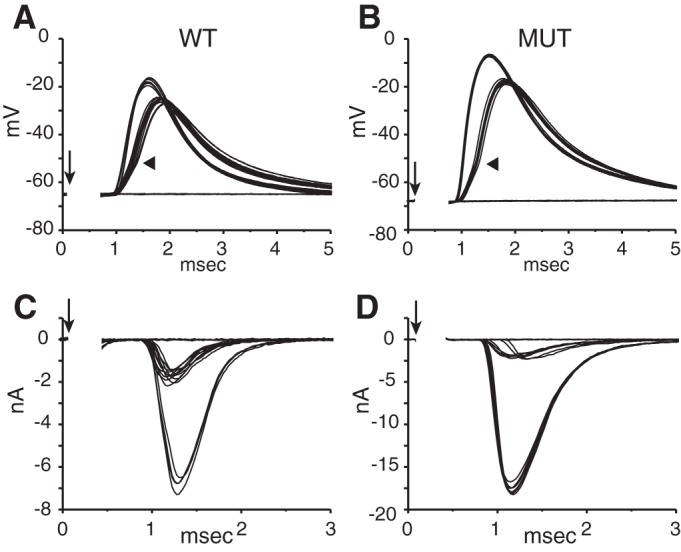
Steps in the growth of shock-evoked excitatory postsynaptic potentials (EPSPs) and EPSCs. As the strength of shocks (↓) to fiber bundles near bushy cells was gradually increased, eEPSPs and eEPSCs grew in steps. Numbers of steps were compared in current clamp and voltage clamp. The first input was suprathreshold because the synaptic response had an inflection (arrowhead) indicative of the generation of an action potential. A: evoked EPSPs (eEPSPs) grew with 2 steps in the same cell from a wild-type hearing mouse as in C. B: eEPSPs grew with 2 steps in the same bushy cell from a mutant deaf mouse as in D. C: eEPSCs in the same wild-type bushy cell as in A grew in 2 steps. D: eEPSCs in the same bushy cell as in B from a deaf Otoferlin mutant mouse grew in 2 steps. EPSCs were recorded at −65 mV; stimulus artifacts have been removed.
Electrically evoked EPSCs were recorded in voltage clamp at a holding potential of −65 mV in the same cells (Fig. 9, C and D). The number of current jumps resolved in the voltage-clamp recordings matched the number of slope clusters measured in current-clamp recordings, generally. In ∼35% of the 45 cells tested, the number of inputs estimated in voltage and current clamp matched. In ∼20% of the recordings one extra step appeared in EPSPs, likely because small current jumps were obscured by variability in EPSCs. However, in ∼45% of cells the number of steps could not be well resolved in current clamp because direct spiking in the bushy cell occluded the synaptic delay. For this reason, the quantification of differences in synaptic transmission between hearing and deaf animals was done only for voltage-clamp recordings.
Electrically evoked EPSCs.
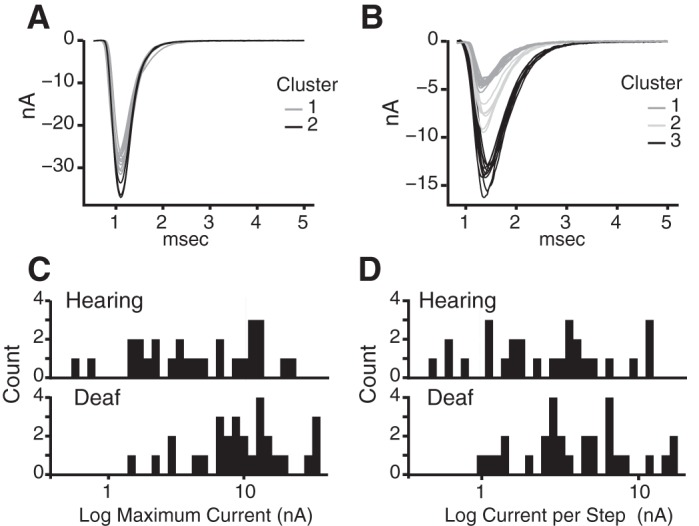
We quantified evoked synaptic transmission from end bulbs to bushy cells in hearing and deaf Otoferlin mutant mice from recordings in voltage clamp made both with and without electrodes that contained QX314 to prevent firing and cesium to make cells more nearly isopotential. Gradual increases in shock strength evoked EPSCs whose peak amplitude grew in steps (Fig. 10, A and B). Shocks were delivered slowly, at 10- to 15-s intervals, to minimize synaptic depression. As ANFs fire all-or-none action potentials, we interpret each jump in current to represent the bringing to threshold of at least one additional ANF that contacted the bushy cell. The total number of steps is thus an estimate of the number of ANFs that converge on the recorded bushy cell (Cao and Oertel 2010). Many of the current steps are large and likely reflect currents delivered through end bulbs of Held. Figure 10, A and B, show four examples of eEPSCs, two cells each from hearing and deaf phenotypes. Figure 10A shows responses from two different bushy cells from hearing, heterozygote mice. Figure 10A, top, shows a bushy cell whose eEPSC jumped in a single step to a peak of ∼11 nA; we thus estimate that this bushy cell had a single ANF input. The second example of an eEPSC in a hearing mouse in Fig. 10A, bottom, grew in two steps, one of 1 nA and the second of 6 nA for a total cumulative current delivered to the cell of ∼7 nA; we conclude that this bushy cell had at least two inputs. Figure 10B illustrates eEPSCs in bushy cells from two different deaf Otoferlin mutant mice. Figure 10B, top, illustrates eEPSCs that grew in two current steps, one of ∼10 nA and another of ∼23 nA, for a peak synaptic current of 33 nA; this cell was likely contacted by two end bulbs. Figure 10B, bottom, shows a second recording from a bushy cell from a deaf mutant mouse whose eEPSCs grew in three jumps, presumably from the recruitment of three ANFs, which delivered 5 nA, 7 nA, and 6 nA, for a total synaptic current of ∼18 nA. The variability in the sizes and shapes of eEPSCs presumably results from the stochastic nature of release of neurotransmitter and from variability in the size of end bulbs. Because it is possible that multiple fibers have a similar threshold, or that some inputs are not stimulated, or that small inputs are not resolvable when synaptic currents are variable, our estimates of numbers of inputs err on the low side.
Fig. 10.

eEPSCs and characteristics. A: eEPSCs in 2 bushy cells from heterozygous, hearing mice grew in 1 step and 2 steps, forming a single cluster and 2 clusters of responses. B: eEPSCs from 2 bushy cells from deaf mice grew in 2 steps or in 3 steps. C: averaged maximal eEPSCs are significantly larger in deaf than in hearing mice. Average maximal current was quantified by averaging the smallest and largest current sweeps from the largest cluster of inputs. D: average number of steps in eEPSCs was also significantly different between bushy cells in hearing and deaf mice, with eEPSCs in the deaf mice growing in more steps. Number of steps was quantified post hoc by eye. E: differences in the current delivered per step approached significance between the phenotypes but was not significant. Amount of current per step was quantified by averaging the smallest and largest current sweeps in a cluster of inputs. F and G: as the variability in amplitudes of eEPSCs was large and the possibility that amplitudes are not normally distributed could not be excluded, we used a t-test to determine whether the logarithms of amplitudes were the same or different. Average logarithms of maximal eEPSCs in hearing and deaf animals were significantly different (P = 0.004). The average current delivered per step also became significant (P = 0.02). Stimulus artifacts were removed from traces shown in A and B. *Statistically significant difference.
Comparisons of eEPSCs in hearing and deaf mice are illustrated in Fig. 10, C–E, and summarized in Table 2. In Fig. 10, C–G, the first assessment of the clustering in synaptic current jumps was determined by eye. The size of a current step was calculated by averaging the amplitudes of the smallest and largest current responses assigned to each cluster. Figure 10C shows that average maximal eEPSCs were significantly larger in bushy cells of deaf than of hearing mice. The average maximal eEPSCs were ∼7 nA in hearing mice, whereas they were ∼13 nA in deaf mice (Table 2). In addition, bushy cells in deaf mice had a significantly greater average number of inputs or current jumps than bushy cells in hearing mice (Fig. 10D; Table 2). Bushy cells of hearing mice had on average ∼2.1 inputs/current step compared with ∼2.7 inputs in deaf Otoferlin mutant mice. The difference between the average currents delivered per step in bushy cells, ∼3.3 nA in bushy cells of hearing mice compared with ∼4.8 nA in bushy cells of deaf Otoferlin mutant mice, approached but did not reach statistical significance (P = 0.06) (Fig. 10E; Table 2).
Table 2.
Evoked EPSCs
| Eye Count |
Statistical Clustering Algorithm |
|||||
|---|---|---|---|---|---|---|
| No. of inputs | Current per input, nA | Maximum current, nA | No. of inputs | Current per input, nA | Maximum current, nA | |
| Statistical test | t-Test | t-Test | t-Test | Permutation test | 2-Sample t-test | 2-Sample t-test |
| Average hearing | 2.11 ± 0.97(n = 27) | 3.33 ± 3.70(n = 57) | 7.03 ± 6.29(n = 27) | 1.96 ± 0.81(n = 27) | 3.49 ± 4.08(n = 53) | 6.81 ± 5.83 (n = 27) |
| Average deaf | 2.68 ± 0.89(n = 29) | 4.77 ± 4.95(n = 78) | 12.81 ± 9.91(n = 29) | 2.55 ± 0.87(n = 29) | 4.92 ± 5.13(n = 74) | 12.55 ± 9.46 (n = 29) |
| P value | P = 0.02 | P = 0.06 | P = 0.01 | P = 0.009 | P = 0.09 | P = 0.009 |
| Statistical test | t-Test Ln of values | t-Test Ln of values | Weighted least square of Ln values | 2-Sample t-test | ||
| Ln average hearing | 7.52 ± 1.2 | 8.42 ± 1.0 | 7.59 ± 1.09 | 8.42 ± 0.99 | ||
| Ln average deaf | 7.98 ± 1.1 | 9.17 ± 0.81 | 7.97 ± 1.16 | 9.16 ± 0.79 | ||
| P value | P = 0.02 | P = 0.004 | P = 0.03 | P = 0.002 | ||
Values are means ± SD.
Because there is considerable physiological variability in the amplitudes of eEPSCs and thus possibly unequal variance in the magnitudes of the eEPSCs, we took the logarithm of each of the amplitudes of the maximum eEPSCs and also of the estimates of current per input based on count of inputs and reran the t-test, using their logarithms (Fig. 10, F and G; Table 2). In the resulting statistical test, the maximal eEPSCs remained significantly different between the hearing and deaf phenotypes, and we also found the difference between average current per input to reach statistical significance between the hearing and deaf mice, with the bushy cells from deaf mice again having larger individual eEPSCs per input (Fig. 10, F and G; Table 2).
For comparing the number of steps with which eEPSCs grew between phenotypes, it was inappropriate to take the logarithm (O'Hara and Kotze 2010). As an alternative, secondary check to the input count made by the experimenter's eye, a rigorous statistical test was conducted based on permutations (Ernst 2004). The statistical significance was calculated as the fraction of permutation values that are at least as extreme as the test statistic derived from the original data. By this test with 10,000 permutations, the mean number of current steps in eEPSCs and as such the number of inputs, was significantly larger in deaf mutant mice than in hearing control mice (P = 0.01).
A potential difficulty with the analysis described above is that it depends on an assessment of clustering by the investigator and cellular responses differed in the clarity of the clustering. We therefore developed an objective clustering algorithm with which the investigator's estimate of the number of steps could be compared. The custom clustering algorithm is based on singular value decomposition to find an efficient yet comprehensive representation of the data. To quantify the current delivered per input, determination of the number of inputs must be made first. Table 2 has all of the results from the clustering algorithm compared with the experimenter's assessment. To determine how many inputs innervate each bushy cell, the clustering algorithm was run on all recordings. A nonparametric permutation was used to quantify inputs between the phenotypes. Throughout, two-sided t-tests were used to assess statistical significance unless otherwise noted (Table 2).
The results of the clustering algorithm parallel the experimenter's assessment; the number of inputs innervating bushy cells in deaf Otoferlin mutant mice was significantly larger than in the hearing mice. The results did vary slightly, such that the clustering algorithm often resulted in slightly fewer input counts than the experimenter quantified (Table 2). Figure 11, A and B, show recordings from two bushy cells in different deaf Otoferlin mutant mice whose clustering is not absolutely clear. The clustering algorithm shows that eEPSCs in the bushy cell illustrated in Fig. 11A grew in two steps. The EPSCs in Fig. 11B grew in three current steps. An additional inference available from this quantification method regarding sample size can be made with simulations. Approximately 25 samples for each phenotype is enough to achieve the 95% statistical power at a significance level of 0.05, which provides a justification of our sample sizes (n = 27 and n = 29 samples per phenotype).
Fig. 11.
Measuring steps with a clustering algorithm. A: a recording from a bushy cell of a deaf mutant mouse was analyzed with an objective clustering algorithm. This cell's eEPSC grew in 2 steps. B: a second recording from a bushy cell of a deaf mutant mouse in which the number of steps was shown by the clustering algorithm to have 3 steps. C: maximal evoked currents in hearing and deaf mice are illustrated in a histogram with current presented logarithmically. D: after quantifying the number of inputs with the clustering algorithm, we can assess how much current is delivered per input. This is illustrated in a logarithmic histogram plot. To deal with the unequal samples resulting from the number of inputs being greater in deaf animals, weights were adjusted to give compared values equal weight. The difference between deaf and hearing animals was statistically significant (P = 0.03). Stimulus artifacts have been removed.
After an input count was assessed, the current per input could be quantified, again by averaging the amplitude of the smallest and largest sweeps from each cluster of inputs. Figure 11, C and D, show the distribution of maximal evoked synaptic currents and current per input per phenotype, respectively, with synaptic currents from deaf mice being larger than in the hearing mice. To take into account the unequal number of inputs between phenotypes (74 in the deaf and 53 in the hearing), a weighted least-squares method was used to assess mean current per input. The averages calculated from more observations have low variability and hence get a higher weight (Faraway 2005). Table 2 illustrates the results based on a one-sided t-test with the alternative of the deaf having greater mean current per input.
Quantal content estimates.
The average charge delivered by mEPSCs from each cell and also the average current delivered in eEPSCs in response to each input having been assessed, estimates of the quantal content can be made (Table 3). The charge delivered per spontaneous mEPSC was averaged for each cell, and then the charge delivered by each evoked input from each cell was divided by the charge for that cell's averaged mEPSC. These resulting estimates of quantal content in steps of eEPSCs were then compared between cells from hearing and deaf animals. There was considerable variation between estimated quantal content of steps, ranging between 3 and 532 quanta. There was not a significant difference between the numbers of quanta released per input in bushy cells from hearing and deaf animals (Table 3). Since the amplitude of current steps, estimates of the current delivered by each auditory nerve input, were larger in deaf than in hearing animals but quantal content was not significantly different, the larger amplitude of EPSCs in deaf animals most likely arose from the upregulation of AMPA receptors at postsynaptic sites in the bushy cells of deaf Otoferlin mutant mice.
Table 3.
Quantal content
| Quantal Content | Statistical Test | ||
|---|---|---|---|
| Sample size | Hearing = 57 inputs from 27 bushy cells | Deaf = 78 inputs from 29 bushy cells | t-Test |
| Mean vesicles per input | Hearing 72.28 ± 78.0 | Deaf 97.87 ± 99.09 | P value = 0.1 |
Values are means ± SD.
Synaptic depression.
To assess synaptic depression, trains of eEPSCs were recorded from bushy cells in deaf Otoferlin mutant mice and their heterozygote and wild-type controls. Five trains, each comprising 10 shocks at 100/s, that produced eEPSCs of maximal amplitude were delivered 5 s apart and in some cases were then averaged for statistical analysis. Figure 12, A and B, show examples of synaptic depression in two hearing mice. Successive eEPSCs generally become smaller than the one before even in hearing animals, but bushy cells from deaf mutant mice show stronger depression and occasional failures (Fig. 12, C–E). The cell shown in Fig. 12D shows an unusual pattern in that the magnitudes of eEPSCs are highly variable, with intermixed large and small responses. The cell illustrated in Fig. 12E shows that some shocks can fail to evoke any response at all. Multiple synaptic failures were seen in 7 of the 19 (37%) bushy cells from deaf Otoferlin mutant mice in which responses to trains of stimuli were recorded. Failures were rare in hearing mice; one synaptic failure was recorded in one heterozygous animal out of 18 bushy cells tested in hearing mice (6%).
Fig. 12.

Trains of eEPSCs. A: in a bushy cell from a wild-type mouse, eEPSCs in response to a single train of 10 shocks at 100 Hz became increasingly smaller, showing the existence of depression. B: in a bushy cell from a heterozygote mouse, depression was similar to that in the wild type. C–E: eEPSCs in bushy cells from deaf mice in responses to stimuli at 100 Hz depress more and sometimes fail altogether. F: the paired-pulse ratio (PPR), the ratio of the amplitude of the eEPSC in response to the second stimulus divided by the amplitude of the eEPSC to the first stimuli, is a measure of synaptic depression. Hearing animals had higher PPRs than deaf mice (P = 0.03). G and H: average values of eEPSCs in bushy cells from hearing and deaf mice show that depression is greater and faster at end bulb-bushy cell synapses in deaf than in hearing mice. Averages were generated by averaging the responses to the first train of stimuli in each cell and then averaging responses across the population of bushy cells. Stimulus artifacts have been removed. *Statistically significant difference.
The paired-pulse ratio (PPR), the amplitude of the second eEPSC divided by the magnitude of the first eEPSC from trains of stimuli, is significantly smaller in deaf mutant cells (P = 0.03) compared with hearing controls. The PPR from bushy cells in hearing mice was 0.65 ± 0.20 (n = 18) compared with 0.48 ± 0.25 (n = 20) in deaf mice. A larger PPR is indicative of less synaptic depression and a lower probability of release, which we found in bushy cells of hearing mice. We also assessed synaptic depression by taking the average of one train of eEPSCs from each bushy cell (Fig. 12, G and H) and averaging across bushy cells to reveal the time course of depression. The average amplitude of EPSCs decays faster in cells from deaf Otoferlin mutant mice than in cells from hearing mice.
DISCUSSION
The absence of synaptic input from early in life is known to affect the development of neuronal circuits in the cochlear nuclei in mammals and birds (Rubel and Fritzsch 2002). We examined the cochlear nuclei of Otoferlin mutant mice in which synaptic transmission between inner hair cells and spiral ganglion cells was blocked both before and after the onset of hearing to learn how downstream neuronal circuits are affected by the near absence of signaling from hair cells throughout life. Comparison of the Otoferlin mutant with other mouse mutants in which signaling is disrupted only after the onset of hearing provides insight into the consequences of signaling before the onset of hearing.
Otoferlin mutant mice have normal mechanotransduction, but calcium-dependent neurotransmitter release from hair cells does not occur, so that electrical activity in hair cells is not transmitted to the brain. Otoferlin serves as a calcium sensor that regulates synaptic transmission between hair cells and their spiral ganglion cell targets both before and after the onset of hearing (Beurg et al. 2010; Roux et al. 2006). Synaptotagmin IV linearizes the synaptic responses to calcium transients in mature hair cells, but it does not itself mediate significant synaptic transmission (Johnson et al. 2010), as mice that express a nonfunctional Otoferlin or that lack Otoferlin are largely unable to activate their auditory nerve targets after P3 (Beurg et al. 2010). Synapses downstream from the hair cells, those of spiral ganglion cells and their targets in the brain stem, depend on synaptotagmin, so these synapses function normally in the absence of Otoferlin. These mutant mice have no detectable ABRs, however, indicating that sensory information from the cochlea fails to be transmitted to the auditory nerve and thence to brain stem auditory circuits. There were no detectable differences in ABRs between mice in which Otoferlin had a point mutation in the second calcium binding domain and those in which the gene for Otoferlin was knocked out in the responses to sound. Mice that lack functional Otoferlin are thus profoundly deaf because acoustic information fails to be transferred from hair cells to spiral ganglion cells.
We find that in deaf Otoferlin mutant mice the VCNs and the auditory nerve are shrunk by nearly half. In ablation studies in mice, as well as in all tested vertebrates (Born and Rubel 1985; Powell and Erulkar 1962), early (P5–6) cochlear ablations dramatically alter both the volume and numbers of neurons in the cochlear nuclei; later ablations (>P11) cause little loss of neurons, leading to the conclusion that the auditory system has an early critical period that occurs before the onset of hearing (Hashisaki and Rubel 1989; Mostafapour et al. 2000; Rubel and Fritzsch 2002; Tierney et al. 1997). Ablations at P6 caused a disproportionately large loss (61–74%) of bushy cells relative to the loss in the total number of VCN cells (62%) (Trune 1982a), and the dendritic profiles of bushy cells were shrunken (Trune 1982b). Deafening with ototoxic drugs or silencing ANFs pharmacologically removes cochlear influence but leaves spiral ganglion cells intact. In kittens neonatal deafening reduced the volume of the VCN more than later deafening, but both caused a shrinkage of bushy cells (Stakhovskaya et al. 2008). The size and appearance of neurons in the VCN of mice are also altered by a conductive hearing loss (Trune and Morgan 1988). Electrical activity in ANFs per se affects bushy cells, as silencing the nerve after the critical period produces a reversible shrinkage of large spherical bushy cells in gerbils (Pasic and Rubel 1989) and electrical stimulation after deafening reverses the shrinkage (Stakhovskaya et al. 2008).
Other forms of nonsyndromic deafness in animals that are produced genetically also target spiral ganglion cells only indirectly. The similarities and differences between two groups of induced deafness and natural deafness mutations provide insights into how spontaneous activity in hair cells before the onset of hearing affects the cochlear nuclei (Johnson et al. 2011, 2013; Kros et al. 1998; Tritsch et al. 2007, 2010; Tritsch and Bergles 2010). Mutations that prevent synaptic transmission from hair cells, including mutations in otoferlin (Pangrsic et al. 2010; Roux et al. 2006), vesicular glutamate transporter3 (Vglut3) (Seal et al. 2008), and Cav1.3 (Platzer et al. 2000), block activation of spiral ganglion cells by electrical activity both before and after the onset of hearing, whereas those that affect mechanotransduction, including mutations in espin in jerker mice (Zheng et al. 2000) and TMC in deafness mice (Kim et al. 2013; Kurima et al. 2002), affect activity after the onset of hearing but not before. One might thus expect mutations that prevent synaptic transmission from hair cells to have more profound consequences than those that affect mechanotransduction but leave prehearing spontaneous activity intact in downstream neuronal circuits. It has been demonstrated that, as in Otoferlin mutants, in Vglut3 mutants a greatly reduced number (∼50%) of ANFs innervates a small (∼50%) VCN (Fig. 3) (Seal et al. 2008). Adult deafness mice have been reported to lose 75% of spiral ganglion neurons (Youssoufian et al. 2005); these mice had shrunken VCNs whose volumes were reduced 37% (Webster 1985).
To a surprising degree the existing fragments of evidence suggest that, while reduced, the cochlear nuclear circuits that are established in these mutants maintain their normal patterns. ANFs in Otoferlin mutant mice and jerker mice innervate the cochlear nuclei topographically as in hearing mice (Fig. 4) (Cao et al. 2008). Even the intrinsic topographic organization of tuberculoventral neurons that project from the DCN to the VCN are well organized in Otoferlin mutant mice (Fig. 4) as in jerker mice (Cao et al. 2008), indicating that the tonotopic organization develops independently of activity before or after the onset of hearing.
End bulbs.
End bulbs terminate on bushy cells and serve as their dominant excitatory input (Bourk 1976; Cant and Morest 1979; Held 1893; Lauer et al. 2013; Ryugo and Sento 1991; Sento and Ryugo 1989; Spirou et al. 2005). Ultrastructurally, it has been shown that synaptic terminals with characteristics of excitatory synapses are of only two or possibly three types (Cant and Morest 1979; Gomez-Nieto and Rubio 2009, 2011; Lauer et al. 2013). In mice all glutamatergic synaptic contacts on somas and dendrites contain large round vesicles and arise from the auditory nerve (Lauer et al. 2013). Terminals with small round vesicles could be cholinergic, serotonergic, or noradrenergic inputs (Klepper and Herbert 1991; Motts et al. 2008; Sherriff and Henderson 1994; Thompson et al. 1995). Excitatory synaptic responses recorded in the present experiments are thus very likely to represent input from the auditory nerve; some of that input arises through end bulbs of Held.
We show that Otoferlin mutant mice have thin axons that terminate in wispy end bulbs compared with the large, almost handlike terminals that encompass the bushy cell somata in the hearing mice (Fig. 5). The diameter of the most distal axon of ANFs that leads up to the end bulbs, assessed in camera lucida reconstructions and measurements of photomicrographs, is significantly thinner in deaf Otoferlin mutant mice than in hearing control mice (Fig. 6). The morphology of end bulbs was also reported to be wispier in jerker mutants (Cao et al. 2008). The differences are reminiscent of the findings in cats by Ryugo and colleagues (Redd et al. 2000; Ryugo et al. 1997). Deaf white cats have a form of nonsyndromic, congenital deafness characterized by degeneration of the organ of Corti early in life with some spiral ganglion atrophy (Heid et al. 1998). End bulbs from deaf white cats appear withered and cover less of the target soma than end bulbs of hearing cats (Redd et al. 2000; Ryugo et al. 1997). Normally hearing cats have approximately double the synaptic area per end bulb as deaf white cats; synapses of hearing cats typically protrude into the end bulb active zone, while those in deaf cats are unusually long and thick and did not form convex bulges into the active zone of the end bulb.
Bushy cells.
Bushy cells of mammals fall into three distinct types, large spherical, small spherical, and globular, that differ not only in histological staining patterns but also, most importantly, in their projection patterns (Brawer and Morest 1975; Cant and Casseday 1986; Osen 1969; Tolbert et al. 1982). Large spherical bushy cells encode low frequencies, occupy the most anterior pole of the aVCN, and project to the medial superior olivary nuclei bilaterally (Smith et al. 1993). Mice have little low-frequency hearing and correspondingly have few large spherical bushy cells and small medial superior olivary nuclei (Webster and Trune 1982; Willard and Ryugo 1983). While the sizes of large and small spherical bushy cells clearly differ in cats (Osen 1969), they are not obviously different in many other mammals such as mice and guinea pigs, for example (Hackney et al. 1990; Willard and Ryugo 1983). Small spherical bushy cells occupy most of the rostral aVCN of mice. They are thought to project to the ipsilateral lateral superior olive (Cant and Casseday 1986). Globular bushy cells are most common in the more posterior aVCN, near the nerve root (Tolbert et al. 1982; Tolbert and Morest 1982a, 1982b). They project across the midline to the contralateral medial nucleus of the trapezoid body (MNTB) (Smith et al. 1991). Recordings in the present study were made from the anterior aVCN; on the basis of their anterior location we conclude that our recordings were largely or exclusively from small spherical bushy cells.
Bushy cells have a threshold rate of depolarization such that depolarizations that are slower than the threshold rate do not evoke firing (McGinley and Oertel 2006). Deaf Otoferlin mutant mice have lower rate thresholds (1.66 ± 0.54 mV/ms) than hearing mice (2.21 ± 0.66 mV/ms). These rate thresholds are in the range thought to be associated with small spherical rather than globular bushy cells (Cao et al. 2008).
Excitation in bushy cells.
We present evidence for both pre- and postsynaptic differences at synapses between ANFs and bushy cells in Otoferlin mutant relative to control mice. Although the end bulbs on bushy cells are smaller in Otoferlin mutant mice than in control mice, the synaptic currents each one evokes are larger, ∼4.8 nA compared with 3.4 nA. Our findings suggest that while the quantal content is similar, the current delivered per quantum is larger in mutant mice. AMPA receptors in bushy cells are composed of GluA3 and 4 isoforms as flop splice variants (Gardner et al. 2001; Wang et al. 1998). In targets of bushy cells it has been demonstrated that GluA4, but not GluA3, is crucial and that GluA2 can substitute for GluA3 but not for GluA4 (Yang et al. 2011). The finding that mEPSCs have similar shapes suggests that the subunit composition of AMPA receptors is similar in Otoferlin mutant and control mice. Their larger size likely reflects the action of spontaneous events on more AMPA receptors in deaf Otoferlin mutant mice than in hearing mice.
Increased synaptic depression seems commonly to accompany deafness. It is evident not only in Otoferlin mutant mice (Fig. 12) but also in the VCN of jerker mice (Cao et al. 2008) and in the MNTB of deafness mice (Oleskevich and Walmsley 2002). Synaptic depression at end bulbs has been attributed to desensitization and depletion of neurotransmitter with high release probabilities (Silver et al. 1998; Wang and Manis 2008; Yang and Xu-Friedman 2008, 2009). A possibility that has not been tested rigorously is that conduction block at branch points somewhere in the auditory nerve terminal arbors also makes a contribution. Our finding that in Otoferlin mutant mice, unlike in control mice, trains of shocks resulted in failures suggests that conduction into end bulbs through smaller axons might lead to conduction failures.
Estimates of convergence of ANFs on bushy cells from the growth of eEPSCs as a function of stimulus strength indicate that deaf Otoferlin mutant mice receive converging inputs from more ANFs than the hearing control mice (Fig. 10). EPSCs in bushy cells of Otoferlin mutant mice grow on average with ∼2.6 steps, while EPSCs in hearing mice grow on average in 2 steps, raising the possibility that the innervation of bushy cells by ANFs undergoes less refinement and synapse elimination in deaf Otoferlin mutant than control mice. Early patterned spontaneous activity determines downstream connectivity in many parts of the brain (Blankenship and Feller 2010; Kirkby et al. 2013). Synapse elimination in the thalamus is known to be decreased by blocking early spontaneous activity in the retina, leaving targets of optic nerve fibers with more inputs than normal (Lee et al. 2014; Penn et al. 1998). Synapse elimination has also been demonstrated in inhibitory connections to the lateral superior olivary nucleus (Clause et al. 2014; Kim and Kandler 2010; Rietzel and Friauf 1998; Sanes and Takacs 1993).
Although biocytin has not been reported to spread between bushy cells (Cao et al. 2007), ultrastructural studies have raised the possibility that bushy cells are electrically coupled to other bushy cells (Gomez-Nieto and Rubio 2009, 2011). Strong electrical coupling could make ANF inputs to one bushy cell be detected in another, increasing the apparent number of auditory nerve inputs, so it is conceivable that the apparent increase in numbers of inputs could arise from stronger coupling. Also, in some way the convergence of ANFs and bushy cells must depend on the relative numbers of surviving spiral ganglion cells and bushy cells, but what determines the viability of these neurons is not understood. It is also possible that convergence is affected by mechanisms that preserve optimal levels of neuronal activation (Shah et al. 2010; Turrigiano 1999).
GRANTS
This work was supported by National Institute on Deafness and Other Communication Disorders Grant R01 DC-00176.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: S.W. and D.O. conception and design of research; S.W. and D.O. performed experiments; S.W., Y.H., and D.O. analyzed data; S.W., Y.H., and D.O. interpreted results of experiments; S.W., Y.H., and D.O. prepared figures; S.W. and D.O. drafted manuscript; S.W., Y.H., and D.O. edited and revised manuscript; S.W., Y.H., and D.O. approved final version of manuscript.
ACKNOWLEDGMENTS
We are grateful for the help we have received from colleagues. Most especially we thank Dr. Xiao-Jie Cao, who contributed in numerous ways to all aspects of this work. Drs. Bret Hanlon, Tom Yin, Robert Fettiplace, Ed Chapman, and Corinna Burger made valuable suggestions for which we are grateful. Dakarai Masunungure made careful measurements of axonal diameters for which we are grateful. This work could not have been done without the help of the staff of the Department of Neuroscience, Ravi Kochhar, Rebecca Welch, Sue Krey, Diane Buechner, and Mary Walker. We also thank Drs. Christine Petit and Isabel Roux for providing us with Otoferlin knockout mice.
REFERENCES
- Avan P, Buki B, Petit C. Auditory distortions: origins and functions. Physiol Rev 93: 1563–1619, 2013. [DOI] [PubMed] [Google Scholar]
- Beurg M, Michalski N, Safieddine S, Bouleau Y, Schneggenburger R, Chapman ER, Petit C, Dulon D. Control of exocytosis by synaptotagmins and otoferlin in auditory hair cells. J Neurosci 30: 13281–13290, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurg M, Safieddine S, Roux I, Bouleau Y, Petit C, Dulon D. Calcium- and otoferlin-dependent exocytosis by immature outer hair cells. J Neurosci 28: 1798–1803, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutner D, Moser T. The presynaptic function of mouse cochlear inner hair cells during development of hearing. J Neurosci 21: 4593–4599, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenship AG, Feller MB. Mechanisms underlying spontaneous patterned activity in developing neural circuits. Nat Rev Neurosci 11: 18–29, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born DE, Rubel EW. Afferent influences on brain stem auditory nuclei of the chicken: neuron number and size following cochlea removal. J Comp Neurol 231: 435–445, 1985. [DOI] [PubMed] [Google Scholar]
- Bourk TR. Electrical Responses of Neural Units in the Anteroventral Cochlear Nucleus of the Cat (PhD thesis) Cambridge, MA: MIT, 1976. [Google Scholar]
- Brawer JR, Morest DK. Relations between auditory nerve endings and cell types in the cat's anteroventral cochlear nucleus seen with the Golgi method and Nomarski optics. J Comp Neurol 160: 491–506, 1975. [DOI] [PubMed] [Google Scholar]
- Cant NB, Casseday JH. Projections from the anteroventral cochlear nucleus to the lateral and medial superior olivary nuclei. J Comp Neurol 247: 457–476, 1986. [DOI] [PubMed] [Google Scholar]
- Cant NB, Morest DK. The bushy cells in the anteroventral cochlear nucleus of the cat. A study with the electron microscope. Neuroscience 4: 1925–1945, 1979. [DOI] [PubMed] [Google Scholar]
- Cao XJ, McGinley MJ, Oertel D. Connections and synaptic function in the posteroventral cochlear nucleus of deaf jerker mice. J Comp Neurol 510: 297–308, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao XJ, Oertel D. Auditory nerve fibers excite targets through synapses that vary in convergence, strength, and short-term plasticity. J Neurophysiol 104: 2308–2320, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao XJ, Shatadal S, Oertel D. Voltage-sensitive conductances of bushy cells of the mammalian ventral cochlear nucleus. J Neurophysiol 97: 3961–3975, 2007. [DOI] [PubMed] [Google Scholar]
- Clause A, Kim G, Sonntag M, Weisz CJ, Vetter DE, Rubsamen R, Kandler K. The precise temporal pattern of prehearing spontaneous activity is necessary for tonotopic map refinement. Neuron 82: 822–835, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster AP, Laird NM, Rubin DB. Maximum likelihood for incomplete data via the EM algorithm. J R Stat Soc 39: 1–38, 1977. [Google Scholar]
- Ernst MD. Permutation methods: a basis for exact inference. Stat Sci 19: 676–685, 2004. [Google Scholar]
- Faraway JJ. Linear Models with R. London: Chapman & Hall/CRC, 2005. [Google Scholar]
- Fraley C, Raftery AE. Bayesian regularization for normal mixture estimation and model-based clustering. J Classif 24: 155–181, 2007. [Google Scholar]
- Gardner SM, Trussell LO, Oertel D. Time course and permeation of synaptic AMPA receptors in cochlear nuclear neurons correlate with input. J Neurosci 19: 8721–8729, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner SM, Trussell LO, Oertel D. Correlation of AMPA receptor subunit composition with synaptic input in the mammalian cochlear nuclei. J Neurosci 21: 7428–7437, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding NL, Robertson D, Oertel D. Recordings from slices indicate that octopus cells of the cochlear nucleus detect coincident firing of auditory nerve fibers with temporal precision. J Neurosci 15: 3138–3153, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Nieto R, Rubio ME. A bushy cell network in the rat ventral cochlear nucleus. J Comp Neurol 516: 241–263, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Nieto R, Rubio ME. Ultrastructure, synaptic organization, and molecular components of bushy cell networks in the anteroventral cochlear nucleus of the rhesus monkey. Neuroscience 179: 188–207, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackney CM, Osen KK, Kolston J. Anatomy of the cochlear nuclear complex of guinea pig. Anat Embryol (Berl) 182: 123–149, 1990. [DOI] [PubMed] [Google Scholar]
- Hashisaki GT, Rubel EW. Effects of unilateral cochlea removal on anteroventral cochlear nucleus neurons in developing gerbils. J Comp Neurol 283: 5–73, 1989. [DOI] [PubMed] [Google Scholar]
- Heid S, Hartmann R, Klinke R. A model for prelingual deafness, the congenitally deaf white cat—population statistics and degenerative changes. Hear Res 115: 101–112, 1998. [DOI] [PubMed] [Google Scholar]
- Held H. Die zentrale Gehoerleitung. Arch Anat Physiol 1893: 201–248, 1893. [Google Scholar]
- Johnson CP, Chapman ER. Otoferlin is a calcium sensor that directly regulates SNARE-mediated membrane fusion. J Cell Biol 191: 187–197, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Eckrich T, Kuhn S, Zampini V, Franz C, Ranatunga KM, Roberts TP, Masetto S, Knipper M, Kros CJ, Marcotti W. Position-dependent patterning of spontaneous action potentials in immature cochlear inner hair cells. Nat Neurosci 14: 711–717, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Franz C, Kuhn S, Furness DN, Ruttiger L, Munkner S, Rivolta MN, Seward EP, Herschman HR, Engel J, Knipper M, Marcotti W. Synaptotagmin IV determines the linear Ca2+ dependence of vesicle fusion at auditory ribbon synapses. Nat Neurosci 13: 45–52, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Kuhn S, Franz C, Ingham N, Furness DN, Knipper M, Steel KP, Adelman JP, Holley MC, Marcotti W. Presynaptic maturation in auditory hair cells requires a critical period of sensory-independent spiking activity. Proc Natl Acad Sci USA 110: 8720–8725, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G, Kandler K. Synaptic changes underlying the strengthening of GABA/glycinergic connections in the developing lateral superior olive. Neuroscience 171: 924–933, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KX, Beurg M, Hackney CM, Furness DN, Mahendrasingam S, Fettiplace R. The role of transmembrane channel-like proteins in the operation of hair cell mechanotransducer channels. J Gen Physiol 142: 493–505, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkby LA, Sack GS, Firl A, Feller MB. A role for correlated spontaneous activity in the assembly of neural circuits. Neuron 80: 1129–1144, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klepper A, Herbert H. Distribution and origin of noradrenergic and serotonergic fibers in the cochlear nucleus and inferior colliculus of the rat. Brain Res 557: 190–201, 1991. [DOI] [PubMed] [Google Scholar]
- Kros CJ, Ruppersberg JP, Rusch A. Expression of a potassium current in inner hair cells during development of hearing in mice. Nature 394: 281–284, 1998. [DOI] [PubMed] [Google Scholar]
- Kurima K, Peters LM, Yang Y, Riazuddin S, Ahmed ZM, Naz S, Arnaud D, Drury S, Mo J, Makishima T, Ghosh M, Menon PS, Deshmukh D, Oddoux C, Ostrer H, Khan S, Riazuddin S, Deininger PL, Hampton LL, Sullivan SL, Battey JF, Jr., Keats BJ, Wilcox ER, Friedman TB, Griffith AJ. Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat Genet 30: 277–284, 2002. [DOI] [PubMed] [Google Scholar]
- Lauer AM, Connelly CJ, Graham H, Ryugo DK. Morphological characterization of bushy cells and their inputs in the laboratory mouse (Mus musculus) anteroventral cochlear nucleus. PLoS One 8: e73308, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Brott BK, Kirkby LA, Adelson JD, Cheng S, Feller MB, Datwani A, Shatz CJ. Synapse elimination and learning rules co-regulated by MHC class I H2-Db. Nature 509: 195–200, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek A, Evesson FJ, Sutton RB, North KN, Cooper ST. Ferlins: regulators of vesicle fusion for auditory neurotransmission, receptor trafficking and membrane repair. Traffic 13: 185–194, 2012. [DOI] [PubMed] [Google Scholar]
- Longo-Guess C, Gagnon LH, Bergstrom DE, Johnson KR. A missense mutation in the conserved C2B domain of otoferlin causes deafness in a new mouse model of DFNB9. Hear Res 234: 21–28, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinley MJ, Oertel D. Rate thresholds determine the precision of temporal integration in principal cells of the ventral cochlear nucleus. Hear Res 216–217: 52–63, 2006. [DOI] [PubMed] [Google Scholar]
- Melcher JR, Kiang NY. Generators of the brainstem auditory evoked potential in cat. III. Identified cell populations. Hear Res 93: 52–71, 1996. [DOI] [PubMed] [Google Scholar]
- Mostafapour SP, Cochran SL, Del Puerto NM, Rubel EW. Patterns of cell death in mouse anteroventral cochlear nucleus neurons after unilateral cochlea removal. J Comp Neurol 426: 561–571, 2000. [DOI] [PubMed] [Google Scholar]
- Motts SD, Slusarczyk AS, Sowick CS, Schofield BR. Distribution of cholinergic cells in guinea pig brainstem. Neuroscience 154: 186–195, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertel D. Synaptic responses and electrical properties of cells in brain slices of the mouse anteroventral cochlear nucleus. J Neurosci 3: 2043–2053, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hara RB, Kotze DJ. Do not log-transform count data. Methods Ecol Evol 1: 118–122, 2010. [Google Scholar]
- Oleskevich S, Walmsley B. Synaptic transmission in the auditory brainstem of normal and congenitally deaf mice. J Physiol 540: 447–455, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osen KK. Cytoarchitecture of the cochlear nuclei in the cat. J Comp Neurol 136: 453–484, 1969. [DOI] [PubMed] [Google Scholar]
- Pangrsic T, Lasarow L, Reuter K, Takago H, Schwander M, Riedel D, Frank T, Tarantino LM, Bailey JS, Strenzke N, Brose N, Muller U, Reisinger E, Moser T. Hearing requires otoferlin-dependent efficient replenishment of synaptic vesicles in hair cells. Nat Neurosci 13: 869–876, 2010. [DOI] [PubMed] [Google Scholar]
- Pasic TR, Rubel EW. Rapid changes in cochlear nucleus cell size following blockade of auditory nerve electrical activity in gerbils. J Comp Neurol 283: 474–480, 1989. [DOI] [PubMed] [Google Scholar]
- Penn AA, Riquelme PA, Feller MB, Shatz CJ. Competition in retinogeniculate patterning driven by spontaneous activity. Science 279: 2108–2112, 1998. [DOI] [PubMed] [Google Scholar]
- Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell 102: 89–97, 2000. [DOI] [PubMed] [Google Scholar]
- Powell TP, Erulkar S. Transneuronal cell degeneration in the auditory relay nuclei of the cat. J Anat 96: 249–268, 1962. [PMC free article] [PubMed] [Google Scholar]
- Redd EE, Pongstaporn T, Ryugo DK. The effects of congenital deafness on auditory nerve synapses and globular bushy cells in cats. Hear Res 147: 160–174, 2000. [DOI] [PubMed] [Google Scholar]
- Reisinger E, Bresee C, Neef J, Nair R, Reuter K, Bulankina A, Nouvian R, Koch M, Buckers J, Kastrup L, Roux I, Petit C, Hell SW, Brose N, Rhee JS, Kugler S, Brigande JV, Moser T. Probing the functional equivalence of otoferlin and synaptotagmin 1 in exocytosis. J Neurosci 31: 4886–4895, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rietzel HJ, Friauf E. Neuron types in the rat lateral superior olive and developmental changes in the complexity of their dendritic arbors. J Comp Neurol 390: 20–40, 1998. [PubMed] [Google Scholar]
- Roux I, Safieddine S, Nouvian R, Grati M, Simmler MC, Bahloul A, Perfettini I, Le Gall M, Rostaing P, Hamard G, Triller A, Avan P, Moser T, Petit C. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell 127: 277–289, 2006. [DOI] [PubMed] [Google Scholar]
- Rubel EW, Fritzsch B. Auditory system development: primary auditory neurons and their targets. Annu Rev Neurosci 25: 51–101, 2002. [DOI] [PubMed] [Google Scholar]
- Runge CL, Erbe CB, McNally MT, Van Dusen C, Friedland DR, Kwitek AE, Kerschner JE. A novel otoferlin splice-site mutation in siblings with auditory neuropathy spectrum disorder. Audiol Neurootol 18: 374–382, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryugo DK, Pongstaporn T, Huchton DM, Niparko JK. Ultrastructural analysis of primary endings in deaf white cats: morphologic alterations in endbulbs of Held. J Comp Neurol 385: 230–244, 1997. [DOI] [PubMed] [Google Scholar]
- Ryugo DK, Rosenbaum BT, Kim PJ, Niparko JK, Saada AA. Single unit recordings in the auditory nerve of congenitally deaf white cats: morphological correlates in the cochlea and cochlear nucleus. J Comp Neurol 397: 532–548, 1998. [DOI] [PubMed] [Google Scholar]
- Ryugo DK, Sento S. Synaptic connections of the auditory nerve in cats: relationship between endbulbs of Held and spherical bushy cells. J Comp Neurol 305: 35–48, 1991. [DOI] [PubMed] [Google Scholar]
- Sanes DH, Takacs C. Activity-dependent refinement of inhibitory connections. Eur J Neurosci 5: 570–574, 1993. [DOI] [PubMed] [Google Scholar]
- Schwarz G. Estimating the dimension of a model. Ann Stat 6: 461–464, 1978. [Google Scholar]
- Seal RP, Akil O, Yi E, Weber CM, Grant L, Yoo J, Clause A, Kandler K, Noebels JL, Glowatzki E, Lustig LR, Edwards RH. Sensorineural deafness and seizures in mice lacking vesicular glutamate transporter 3. Neuron 57: 263–275, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sento S, Ryugo DK. Endbulbs of Held and spherical bushy cells in cats: morphological correlates with physiological properties. J Comp Neurol 280: 553–562, 1989. [DOI] [PubMed] [Google Scholar]
- Shah MM, Hammond RS, Hoffman DA. Dendritic ion channel trafficking and plasticity. Trends Neurosci 33: 307–316, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherriff FE, Henderson Z. Cholinergic neurons in the ventral trapezoid nucleus project to the cochlear nuclei in the rat. Neuroscience 58: 627–633, 1994. [DOI] [PubMed] [Google Scholar]
- Silver RA, Momiyama A, Cull-Candy SG. Locus of frequency-dependent depression identified with multiple-probability fluctuation analysis at rat climbing fibre-Purkinje cell synapses. J Physiol 510: 881–902, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PH, Joris PX, Carney LH, Yin TC. Projections of physiologically characterized globular bushy cell axons from the cochlear nucleus of the cat. J Comp Neurol 304: 387–407, 1991. [DOI] [PubMed] [Google Scholar]
- Smith PH, Joris PX, Yin TC. Projections of physiologically characterized spherical bushy cell axons from the cochlear nucleus of the cat: evidence for delay lines to the medial superior olive. J Comp Neurol 331: 245–260, 1993. [DOI] [PubMed] [Google Scholar]
- Spirou GA, Rager J, Manis PB. Convergence of auditory-nerve fiber projections onto globular bushy cells. Neuroscience 136: 843–863, 2005. [DOI] [PubMed] [Google Scholar]
- Stakhovskaya O, Hradek GT, Snyder RL, Leake PA. Effects of age at onset of deafness and electrical stimulation on the developing cochlear nucleus in cats. Hear Res 243: 69–77, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AM, Moore KR, Thompson GC. Distribution and origin of serotoninergic afferents to guinea pig cochlear nucleus. J Comp Neurol 351: 104–116, 1995. [DOI] [PubMed] [Google Scholar]
- Tierney TS, Russell FA, Moore DR. Susceptibility of developing cochlear nucleus neurons to deafferentation-induced death abruptly ends just before the onset of hearing. J Comp Neurol 378: 295–306, 1997. [DOI] [PubMed] [Google Scholar]
- Tolbert LP, Morest DK. The neuronal architecture of the anteroventral cochlear nucleus of the cat in the region of the cochlear nerve root: electron microscopy. Neuroscience 7: 3053–3067, 1982a. [DOI] [PubMed] [Google Scholar]
- Tolbert LP, Morest DK. The neuronal architecture of the anteroventral cochlear nucleus of the cat in the region of the cochlear nerve root: Golgi and Nissl methods. Neuroscience 7: 3013–3030, 1982b. [DOI] [PubMed] [Google Scholar]
- Tolbert LP, Morest DK, Yurgelun-Todd DA. The neuronal architecture of the anteroventral cochlear nucleus of the cat in the region of the cochlear nerve root: horseradish peroxidase labelling of identified cell types. Neuroscience 7: 3031–3052, 1982. [DOI] [PubMed] [Google Scholar]
- Tritsch NX, Bergles DE. Developmental regulation of spontaneous activity in the mammalian cochlea. J Neurosci 30: 1539–1550, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Yi E, Gale JE, Glowatzki E, Bergles DE. The origin of spontaneous activity in the developing auditory system. Nature 450: 50–55, 2007. [DOI] [PubMed] [Google Scholar]
- Tritsch NX, Zhang YX, Ellis-Davies G, Bergles DE. ATP-induced morphological changes in supporting cells of the developing cochlea. Purinergic Signal 6: 155–166, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trune DR. Influence of neonatal cochlear removal on the development of mouse cochlear nucleus. I. Number, size, and density of its neurons. J Comp Neurol 209: 409–424, 1982a. [DOI] [PubMed] [Google Scholar]
- Trune DR. Influence of neonatal cochlear removal on the development of mouse cochlear nucleus. II. Dendritic morphometry of its neurons. J Comp Neurol 209: 425–434, 1982b. [DOI] [PubMed] [Google Scholar]
- Trune DR, Kiessling AA. Decreased protein synthesis in cochlear nucleus following developmental auditory deprivation. Use of vascular saline perfusion to improve small tissue sample analysis. Hear Res 35: 259–264, 1988. [DOI] [PubMed] [Google Scholar]
- Trune DR, Morgan CR. Stimulation-dependent development of neuronal cytoplasm in mouse cochlear nucleus. Hear Res 33: 141–149, 1988. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. Homeostatic plasticity in neuronal networks: the more things change, the more they stay the same. Trends Neurosci 22: 221–227, 1999. [DOI] [PubMed] [Google Scholar]
- Wang Y, Manis PB. Short-term synaptic depression and recovery at the mature mammalian endbulb of Held synapse in mice. J Neurophysiol 100: 1255–1264, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Wenthold RJ, Ottersen OP, Petralia RS. Endbulb synapses in the anteroventral cochlear nucleus express a specific subset of AMPA-type glutamate receptor subunits. J Neurosci 18: 1148–1160, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Liu H, Gu Y, Chapman ER. Reconstituted synaptotagmin I mediates vesicle docking, priming, and fusion. J Cell Biol 195: 1159–1170, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster DB. The spiral ganglion and cochlear nuclei of deafness mice. Hear Res 18: 19–27, 1985. [DOI] [PubMed] [Google Scholar]
- Webster DB, Trune DR. Cochlear nuclear complex of mice. Am J Anat 163: 103–130, 1982. [DOI] [PubMed] [Google Scholar]
- Wickesberg RE, Oertel D. Tonotopic projection from the dorsal to the anteroventral cochlear nucleus of mice. J Comp Neurol 268: 389–399, 1988. [DOI] [PubMed] [Google Scholar]
- Willard FH, Ryugo DK. Anatomy of the central auditory system. In: The Auditory Psychobiology of the Mouse, edited by Willott JF. Springfield, IL: Thomas, 1983, p. 201–304. [Google Scholar]
- Yang H, Xu-Friedman MA. Relative roles of different mechanisms of depression at the mouse endbulb of Held. J Neurophysiol 99: 2510–2521, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Xu-Friedman MA. Impact of synaptic depression on spike timing at the endbulb of Held. J Neurophysiol 102: 1699–1710, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YM, Aitoubah J, Lauer AM, Nuriya M, Takamiya K, Jia Z, May BJ, Huganir RL, Wang LY. GluA4 is indispensable for driving fast neurotransmission across a high-fidelity central synapse. J Physiol 589: 4209–4227, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasunaga S, Grati M, Chardenoux S, Smith TN, Friedman TB, Lalwani AK, Wilcox ER, Petit C. OTOF encodes multiple long and short isoforms: genetic evidence that the long ones underlie recessive deafness DFNB9. Am J Hum Genet 67: 591–600, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasunaga S, Grati M, Cohen-Salmon M, El-Amraoui A, Mustapha M, Salem N, El-Zir E, Loiselet J, Petit C. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet 21: 363–369, 1999. [DOI] [PubMed] [Google Scholar]
- Youssoufian M, Oleskevich S, Walmsley B. Development of a robust central auditory synapse in congenital deafness. J Neurophysiol 94: 3168–3180, 2005. [DOI] [PubMed] [Google Scholar]
- Zheng L, Sekerkova G, Vranich K, Tilney LG, Mugnaini E, Bartles JR. The deaf jerker mouse has a mutation in the gene encoding the espin actin-bundling proteins of hair cell stereocilia and lacks espins. Cell 102: 377–385, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]