

Abstract
11C-LY2795050 is a novel kappa opioid receptor (KOR) antagonist tracer for positron emission tomography (PET) imaging. The purpose of this first-in-human study was to determine the optimal kinetic model for analysis of 11C-LY2795050 imaging data. Sixteen subjects underwent baseline scans and blocking scans after oral naltrexone. Compartmental modeling and multilinear analysis-1 (MA1) were applied using the arterial input functions. Two-tissue compartment model and MA1 were found to be the best models to provide reliable measures of binding parameters. The rank order of 11C-LY2795050 distribution volume (VT) matched the known regional KOR densities in the human brain. Blocking scans with naltrexone indicated no ideal reference region for 11C-LY2795050. Three methods for calculation of the nondisplaceable distribution volume (VND) were assessed: (1) individual VND estimated from naltrexone occupancy plots, (2) mean VND across subjects, and (3) a fixed fraction of cerebellum VT. Approach (3) produced the lowest intersubject variability in the calculation of binding potentials (BPND, BPF, and BPP). Therefore, binding potentials of 11C-LY2795050 can be determined if the specific binding fraction in the cerebellum is presumed to be unchanged by diseases and experimental conditions. In conclusion, results from the present study show the suitability of 11C-LY2795050 to image and quantify KOR in humans.
Keywords: brain imaging, kinetic modeling, positron emission tomography, receptors, receptor imaging
Introduction
The kappa opioid receptor (KOR) is one of the three major subtypes of opioid receptors. The distribution of KOR in human brain has been investigated in vitro with autoradiography, or radioligand binding assays using homogenized brain tissue.1, 2, 3, 4 Kappa opioid receptor exists abundantly in amygdala, anterior cingulate cortex and insula, with moderate levels in the neocortical regions and putamen, followed by caudate, thalamus, globus pallidus, hippocampus, pons, and substantia nigra. Using reverse transcription-PCR detection of human KOR transcripts in human brain,5 a signal was detected in most regions including the cerebellum. A low density of KOR was observed in cortex white matter by autoradiography using 3H-U69593.4
Multiple lines of evidence from preclinical and clinical studies have implicated KOR in a variety of neuropsychiatric disorders, including substance abuse,6,7 epilepsy,8,9 Alzheimer's disease10,11 and major depression.12, 13, 14 As a result, considerable efforts have been made to develop radiotracers to image KOR in humans and probe its involvement in the pathophysiology of these disorders. A number of ligands have been developed, including 11C-GR103545,15 11C-MeJDTic16 and 11C-LY2795050.17 11C-GR103545 is an agonist tracer extensively evaluated in nonhuman primates,18, 19, 20 and recently in humans.21 However, KOR agonists at relatively low mass doses elicit dysphoric22 and psychomimetic14 effects. Therefore, the use of agonist radiotracers in human positron emission tomography (PET) imaging requires careful control of the injected mass. On the other hand, KOR antagonists have been targeted for development as potential pharmacological agents for the treatment of a wide range of conditions such as drug addiction, depression, and feeding behavior,23 and the application of antagonist radiotracers will make it possible to more easily perform KOR imaging in human. For antagonist tracers, 11C-MeJDTic had high KOR affinity (Ki=1.01±0.17 nmol/L; human cloned KOR)24 and was evaluated in mice,16 but no reports of its use in nonhuman primates or humans have been published. We have recently developed 11C-LY2795050 (Ki=0.72 nmol/L; human cloned KOR)17 as a novel, KOR-selective antagonist radiotracer and showed its suitability to image KOR in rhesus monkey.17,25 The in vitro selectivity for KOR over mu or delta opioid receptor was estimated to be 35.8 and 212.5 times, respectively.17 The affinity of LY2795050 for the opioid receptors was measured by radioligand displacement experiments with cloned human opioid receptors and the opioid antagonist radioligand 3H-diprenorphine, and naltrexone was used to define nonspecific binding.26
In this paper, we present the results from our first-in-human study with the selective KOR antagonist 11C-LY2795050. Our goals are (1) to determine the appropriate model to describe its in vivo kinetics and (2) to choose a suitable method to define the nondisplaceable distribution volume (VND) for derivation of binding potentials.
Materials and methods
Human Subjects
Sixteen healthy subjects (24 to 56 years of age; 8 men and 8 women, body weight 75±10 kg) were included. Studies were performed under protocols approved by the Yale University School of Medicine Human Investigation Committee and the Yale-New Haven Hospital Radiation Safety Committee, and in accordance with the United States federal guidelines and regulations for the protection of human research subjects contained in Title 45 Part 46 of the Code of Federal Regulations (45 CFR 46). Written informed consent was obtained from all subjects. As part of the subject evaluation, magnetic resonance (MR) images were acquired on all subjects to eliminate those with structural brain abnormalities and for PET image registration. The MR imaging was performed on a 3-T whole-body scanner (Trio, Siemens Medical Systems, Erlangen, Germany) with a circularly polarized head coil. The dimension and pixel size of MR images were 256 × 256 × 176 and 0.98 × 0.98 × 1.0 mm3, respectively.
Radiotracer Synthesis
11C-LY2795050 was synthesized as previously described.17 Radiochemical purity of 11C-LY2795050 in the final product solution was >99%.
Positron Emission Tomography Imaging Experiments
Subjects underwent two PET scans on the same day: a baseline 11C-LY2795050 PET scan followed by a second scan at ~75 minutes after an oral administration of 150 mg naltrexone, a nonselective opioid receptor antagonist. The time between tracer injections was 4.8±0.9 hours. For one subject with 11C-LY2795050, baseline and blocking scans were performed 1 month apart.
Positron emission tomography scans were conducted on the High Resolution Research Tomograph (HRRT) (Siemens Medical Solutions, Knoxville, TN, USA), which acquires 207 slices (1.2 mm slice separation) with a reconstructed image resolution (full width at half maximum) of ~3 mm. Before tracer administration, a 6-minute transmission scan was conducted for attenuation correction. Each scan was acquired in list mode for 90 minutes after intravenous administration of tracer over 1 minute by an automatic pump (Harvard PHD 22/2000; Harvard Apparatus, Holliston, MA, USA). The injected mass limit was 10 μg. Dynamic scan data were reconstructed in 27 frames (6 × 0.5 minutes, 3 × 1 minutes, 2 × 2 minutes, 16 × 5 minutes) with corrections for attenuation, normalization, scatter, randoms, and deadtime using the MOLAR algorithm.27 Event-by-event motion correction28 was included in the reconstruction based on measurements with the Polaris Vicra sensor (NDI Systems, Waterloo, Canada) with reflectors mounted on a swim cap worn by the subject.
Input Function Measurement
For each subject, the radial artery was catheterized for blood sampling. An automated blood counting system (PBS-101; Veenstra Instruments, Joure, The Netherlands) was used to measure the radioactivity in whole blood during the first 7 minutes. Thirteen samples (2 to 10 mL) were collected manually at selected time points after tracer administration starting at 3 minutes. For each sample, plasma was obtained by centrifugation at 4°C (2,930 g for 5 minutes). Whole blood and plasma were counted in crosscalibrated gamma counters (1480 & 2480 WIZARD; Perkin-Elmer, Waltham, MA, USA).
To determine radioactivity in plasma for the first 7 minutes, the whole blood-to-plasma ratios were calculated from the hand-drawn samples. The ratio from 3 to 90 minutes was fitted to the following equation: at+b, and the plasma time-activity curve (TAC) in the first 7 minutes was calculated from the measured whole blood TAC and the extrapolated ratio. These data were combined with those from the plasma samples to produce the final curve of total radioactivity in plasma. To reduce noise in these data, the total plasma curve from ~5 minutes onward was fitted to a sum of exponentials.
Plasma Metabolite Analysis
Analysis of the metabolite profile in the arterial plasma was performed using a modified automatic column-switching HPLC method.29 Plasma samples collected at 5, 15, 30, 60, and 90 minutes after injection were mixed with urea (8 mol/L) and then filtered through 1.0 μm Whatman 13 mm GD/X syringe filters (GE, Florham Park, NJ, USA). Up to 5 mL of plasma filtrate was injected to the automatic HPLC system equipped with a Gemini-NX analytical column (4.6 × 250 mm, 5 μm; Phenomenex, Torrance, CA, USA) and eluted with a mobile phase consisting of 45% acetonitrile and 55% 0.1 mol/L ammonium formate (v/v) at a flow rate of 1.5 mL/min. The HPLC eluate was fraction-collected and counted in the gamma counters. The fraction counts were corrected for volume and decay. The unmetabolized parent fraction was calculated as the ratio of the sum of radioactivity in fractions containing the parent compound to the total amount of radioactivity collected, and fitted to an integrated gamma function (four fitted parameters: a, b, c, and d):
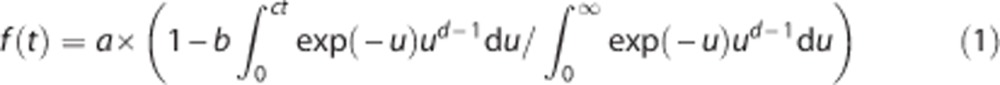 |
In addition, the time-varying extraction efficiency of radioactivity in filtered plasma samples was determined, and normalized to that of reference plasma sample. The plasma input function was calculated as the product of the total plasma activity, the parent HPLC fraction, and the normalized extraction efficiency.
Measurement of Tracer-Free Fraction in Plasma
Arterial blood samples were taken immediately before tracer injection for analysis of plasma-free fraction (fP). An ultrafiltration (Millipore Centrifree micropartition device, 4104, Billerica, MA, USA) method was used for measuring fP of tracer in plasma in triplicate. The free fraction fP was determined from the count ratio of ultrafiltrate to plasma.
Image Registration and Definition of Regions of Interest
Regions of interest (ROIs) were taken from the AAL (Automated Anatomical Labeling) for SPM230 in MNI (Montreal Neurological Institute) space.31 For each subject, the dynamic PET images after hardware motion correction were coregistered to the early summed PET images (0 to 10 minutes after injection) using a 6-parameter mutual information algorithm32 (FLIRT of FSL) to eliminate any residual motion. The summed PET image was then coregistered to the subject's T1-weighted 3 T MR image (6-parameter rigid registration), which was subsequently coregistered to the AAL template in MNI space using a nonlinear transformation (Bioimage suite).33 Using the combined transformations from template to PET space, regional TACs were generated for 14 ROIs: amygdala, caudate, centrum semiovale, cerebellum, anterior cingulate cortex, posterior cingulate cortex, frontal lobe, globus pallidus, hippocampus, insula, occipital lobe, putamen, temporal lobe, and thalamus.
Quantitative Analysis
Outcome measures were derived with kinetic analysis of the regional TACs using the arterial plasma TAC as an input function. The distribution volume (VT)34 was calculated using one- and two-tissue compartment models (1TC and 2TC), as well as the multilinear analysis-1 (MA1) method.35 The time stability of VT estimates was evaluated by fitting the model to regional TACs with shortened scan durations, ranging from 90 to 30 minutes for 2TC, and from 90 to 50 minutes for MA1 (t*=30 minutes) model. The ratio of VT value from the shortened scan to that from the 120-minute scan was computed for each ROI and duration. The following two criteria were used to determine a minimum scan duration36: (a) the average of the ratio was between 0.95 and 1.05; and (b) the interindividual standard deviation of the ratio was <0.1. The nondisplaceable distribution volume (VND) required for computing binding potentials was calculated from the occupancy plots (see below). The simplified reference tissue model (SRTM) with the cerebellum as a reference region was also applied to the regional TACs. Due to the lack of a suitable reference region, the estimated binding potential (BPND) values were corrected (see below). All modeling was performed with in-house programs written with IDL 8.0 (ITT Visual Information Solutions, Boulder, CO, USA). For parameter estimation, data points were weighted based on noise equivalent counts in each frame. Percentage standard error (%s.e.) was estimated from the theoretical parameter covariance matrix.
The KOR occupancy (r) by naltrexone and nondisplaceable distribution volume (VND) were calculated from the following equation:37
For each subject, the percentage of specific binding was calculated as the difference between VT and VND divided by VT. All regions were used for the occupancy plots in the naltrexone blocking study, assuming uniform KOR occupancy. On the basis of the estimated VND, the three binding potentials, BPND, BPP and BPF were calculated using MA1-based VT estimates. The value of VND was calculated in three ways: (1) individual VND from each occupancy plot, (2) the mean VND from all occupancy plots (i.e., a constant value for all subjects), and (3) the fraction of cerebellum VT corresponding to nonspecific binding. Approach (3) was used for both MA1 and SRTM models. Assuming that VT CER=αVND (α is a constant value) in the pseudo reference tissue model,38 the corrected BPND is described as α(BPND+1)−1. The fraction value α was determined as the average ratio of the cerebellum VT to the VND estimated from individual occupancy plots used in method (1).
Results
Injection Parameters
In the baseline and blocking scans, the subjects received radioactivity doses of 334±149 MBq and 334±152 MBq, respectively, with specific activity of 14.8±6.5 GBq/μmol and 16.2±6.8 GBq/μmol at the time of injection. Injections were performed by computer-controlled syringe pump. Injected mass was 9.3±0.9 μg and 8.7±2.1 μg for baseline and blocking scans, respectively. In most cases, the mass limit of 10 μg was the limiting factor, thus there was very little variability in the magnitude of injected mass.
Plasma Analysis
In either the baseline or blocking scan, total plasma activity stabilized at a constant level after 20 minutes after injection (Figure 1A). 11C-LY2795050 metabolized fairly quickly in plasma (Figure 1B), with the parent fraction decreasing to 44±8% and 18±5%, respectively, at 30 and 90 minutes after injection in the baseline scans (n=16). The parent fractions in the naltrexone blocking scans were similar to those from the baseline scans (Figure 1B). The estimated metabolite-corrected radioactivity time course in the arterial plasma is shown in Figure 1C. The plasma-free fraction (fP) of 11C-LY2795050 was 0.77±0.16% for baseline scans (n=16) and 0.75±0.16% for blocking scans (n=16).
Figure 1.

Mean±s.d. of (A) total plasma activity, (B) parent fraction in the plasma, and (C) metabolite-corrected plasma activity over time after injection of 11C-LY2795050 in the baseline (closed circles, n=16) and blocking (open circles, n=16) scans. (A) and (C) Displayed in SUV units (concentration/(injected dose/body weight)). SUV, standard uptake value.
Brain Uptake and Kinetics
Uptake images from the baseline and naltrexone-blocking scans are shown in Figure 2. Regional TACs for representative brain regions are shown in Figure 3. 11C-LY2795050 displayed favorable imaging properties in the brain, with rapid entry, heterogeneous regional accumulation and fast kinetics. Activity peaked at ~4 minutes in all brain regions (Figures 3A and 3B). Pretreatment with naltrexone reduced the uptake of the radiotracer in all brain regions, suggesting specific binding of the radiotracer in the brain (Figures 2C, 3C, and 3D), given the lack of change in the input function (Figure 1C).
Figure 2.

Images from a typical subject (female, 32 years old, 77 kg body weight). (A) Magnetic resonance (MR) images. (B and C) Coregistered positron emission tomography (PET) images summed from 30 to 90 minutes after injection of 11C-LY2795050. (B) Baseline scan and (C) postnaltrexone scan. Activity is expressed as SUV (concentration/(injected dose/body weight)). PET images were spatially smoothed by three-dimensional Gaussian filter with full width at half maximum (FWHM) (3.6 mm). SUV, standard uptake value.
Figure 3.

Regional time-activity curves in five regions of interest (ROIs) after injection of 11C-LY2795050 from the baseline (A and B) and naltrexone blocking (C and D) scans. Panels A and C display the 1TC (dotted line) and 2TC (solid line) fits and panels B and D display the MA1 fits. For each region, the symbols correspond to the measured regional activity.
Kinetic Model Assessment
The baseline scans were used to assess the best model for kinetic analysis. The 1TC and 2TC models reached convergence for every scan in all regions. The mean value of K1 in the 1TC model ranged from 0.04 mL/cm3 per minute in the centrum semiovale to 0.11 mL/cm3 per minute in the occipital cortex. The k4 value ranged from 0.025±0.003/min (centrum semiovale) to 0.054±0.038/min (cerebellum). The thalamus showed a high k4 value (0.085±0.031/min).
The 2TC model was favored over the 1TC model according to the AIC and visual assessment of the quality of fits (Figures 3A and 3C). The F test indicated a significantly better fit for the 2TC model in 223 out of 224 regions. In two cases, the 2TC model provided moderately large VT estimate with large %s.e. (>10%) in the posterior cingulate. Due to the lack of fit with 1TC model and the variability in 2TC VT estimates, the MA1 model was also evaluated. While the VT values derived from the 1TC model were slightly lower than those from the 2TC model (VT (1TC)=0.94 VT (2TC)−0.09, R2=0.96), the VT values from MA1 matched extremely well with those from the 2TC model (VT (MA1, t*=30 min)=0.98 VT (2TC)+0.06, R2=0.98) (Supplementary Figure S1). The setting for t* in MA1 had almost no effect on VT estimates (Supplementary Figure S2) for t*≥30 minutes. Note that these comparisons were conducted for the regions with good identifiability, i.e., %s.e. of VT<10% with the 2TC model.
The VT values derived from 1TC, 2TC, and MA1 model and the minimum scan time for 2TC and MA1 models are shown in Table 1. High VT values were seen in amygdala, insula, and anterior cingulate cortex. Intermediate VT values were found in globus pallidus, putamen, temporal cortex, frontal cortex, and occipital cortex. Lower VT values were in hippocampus, caudate, posterior cingulate cortex, thalamus, and centrum semiovale, with the lowest VT value in the cerebellum. The intersubject VT variability was low in all models (average of %COV=10 to 12%). The minimum scan duration was 70 minutes to satisfy all stability criteria in all regions for both 2TC and MA1 models (Table 1).
Table 1. Regional distribution volumes of 11C-LY2795050 in baseline and blocking scans.
| Regions |
Baseline (n=16) |
Blocking with naltrexone (n=16) |
Minimum scan duration (minutes) |
|||
|---|---|---|---|---|---|---|
| 1TC (%COV) | 2TC (%COV) | MA1 (%COV) | MA1 (%COV) | 2TC | MA1 | |
| Amygdala | 3.76 (13%) | 3.95 (13%) | 3.95 (14%) | 1.62 (15%) | 60 | 70 |
| Insula | 3.09 (9%) | 3.41 (10%) | 3.41 (10%) | 1.68 (14%) | 70 | 50 |
| Ant. cingulate cortex | 2.98 (10%) | 3.23 (11%) | 3.25 (10%) | 1.71 (13%) | 60 | 50 |
| Globus pallidus | 2.86 (11%) | 3.11 (12%) | 3.11 (12%) | 1.90 (14%) | 50 | 60 |
| Putamen | 2.65 (10%) | 3.00 (11%) | 2.94 (10%) | 1.81 (14%) | 70 | 50 |
| Temporal cortex | 2.44 (9%) | 2.74 (11%) | 2.71 (10%) | 1.70 (13%) | 70 | 60 |
| Frontal cortex | 2.34 (9%) | 2.66 (11%) | 2.63 (10%) | 1.64 (14%) | 70 | 50 |
| Occipital cortex | 2.24 (8%) | 2.58 (10%) | 2.54 (9%) | 1.77 (12%) | 70 | 60 |
| Hippocampus | 2.07 (11%) | 2.31 (12%) | 2.35 (12%) | 1.58 (14%) | 60 | 50 |
| Caudate | 1.99 (16%) | 2.19 (17%) | 2.16 (16%) | 1.40 (19%) | 60 | 50 |
| Post. cingulate cortex | 1.93 (13%) | 2.24 (17%) | 2.24 (12%) | 1.61 (13%) | 70 | 60 |
| Thalamus | 2.03 (10%) | 2.14 (10%) | 2.18 (11%) | 1.78 (14%) | 30 | 50 |
| Centrum semiovale | 1.90 (8%) | 2.30 (10%) | 2.28 (10%) | 1.86 (15%) | 70 | 70 |
| Cerebellum | 1.76 (8%) | 1.96 (9%) | 1.95 (8%) | 1.57 (13%) | 70 | 50 |
MA1, multilinear analysis-1; 1TC, one-tissue compartment model; 2TC, two-tissue compartment model.
%COV is variability across subjects.
Blocking of Specific Binding by Naltrexone
In all regions, the VT values displayed statistically significant reduction in blocking scans after oral naltrexone (P<0.00001) (Table 1), i.e., no region was found that would serve as a reference region. As determined from the occupancy plots (Figure 4), 150 mg of oral naltrexone occupied 93±6% of specific binding. The nondisplaceable distribution volume (VND) for 11C-LY2795050 was estimated as 1.61±0.25 mL/cm3 (range: 1.13 to 2.06 mL/cm3). VND or occupancy values did not have any significant correlation with either gender or subject weight.
Figure 4.

A typical occupancy plot of 11C-LY2795050 using VT in all regions of interest (ROIs) from the baseline and blocking scans (150 mg naltrexone). Occupancy is measured as the slope of the regression line, and 11C-LY2795050 VND is the x-axis intercept.
The specific binding percentage in the cerebellum (=(VT CER(baseline)−VND)/VT CER(baseline)), the region with the lowest VT, was estimated at 17±10% (range: 4 to 38%), suggesting that cerebellar VT might be a useful estimator of VND, if corrected. The slope and intercept of the regression line of VND estimates against cerebellum VT were 1.00±0.29 (95% confidence interval: 0.37 to 1.6) and −0.34±0.57 (−2.4 to 0.97), respectively. Thus, the intercept was not significantly different from 0 (P=0.57), and the slope was not significantly different from 1/1.17 (P=0.63). Thus, we evaluated three approaches to define VND to calculate binding potentials: (1) individual VND from the occupancy plot, (2) mean VND across subjects from the occupancy plots (1.61 mL/cm3), and (3) a fixed fraction of each individual's cerebellum VT (VT CER (baseline)/1.17). Among these three ways to estimate VND, method (3) provided the lowest intersubject variability in all binding potentials (BPND, BPP, and BPF). The binding potential values from MA1 and SRTM with method (3) are summarized in Table 2. In method (3), the intersubject variability was smallest in BPND (mean across regions: 22%), followed by BPP (23%), and BPF (30%). For method (2), using the average VND, variability was substantially higher: BPND (32%), BPP (32%), and BPF (36%). Using individual VND values in method (1) produced the highest intersubject variability: BPND (39%), BPP (28%), and BPF (35%). The corrected BPND values derived from SRTM were lower than those from the MA1 model (corrected BPND (SRTM)=0.89 × corrected BPND (MA1)+0.06, R2=0.98).
Table 2. Regional binding potential values of 11C-LY2795050 from SRTM and MA1.
| Regions |
Baseline (n=16) |
|||
|---|---|---|---|---|
| SRTM |
MA1 |
|||
| BPND (%COV) | BPND (%COV) | BPP (%COV) | BPF (%COV) | |
| Amygdala | 1.28 (18%) | 1.37 (19%) | 2.29 (21%) | 303.09 (23%) |
| Insula | 1.00 (11%) | 1.05 (11%) | 1.75 (14%) | 234.97 (22%) |
| Ant. cingulate cortex | 0.91 (18%) | 0.96 (18%) | 1.59 (17%) | 212.65 (24%) |
| Globus pallidus | 0.84 (16%) | 0.87 (17%) | 1.45 (19%) | 194.38 (25%) |
| Putamen | 0.75 (15%) | 0.77 (15%) | 1.28 (16%) | 170.54 (21%) |
| Temporal cortex | 0.61 (15%) | 0.63 (15%) | 1.05 (17%) | 140.97 (23%) |
| Frontal cortex | 0.57 (17%) | 0.59 (18%) | 0.97 (18%) | 129.71 (24%) |
| Occipital cortex | 0.57 (19%) | 0.53 (14%) | 0.88 (16%) | 117.22 (22%) |
| Hippocampus | 0.45 (21%) | 0.42 (28%) | 0.69 (29%) | 91.33 (32%) |
| Caudate | 0.29 (58%) | 0.30 (57%) | 0.50 (57%) | 65.37 (61%) |
| Post. cingulate cortex | 0.45 (30%)a | 0.36 (43%) | 0.58 (42%) | 78.61 (51%) |
| Thalamus | 0.40 (23%) | 0.37 (21%) | 0.52 (31%) | 68.87 (34%) |
| Centrum semiovale | 0.33 (29%) | 0.32 (32%) | 0.62 (22%) | 82.65 (28%) |
| Cerebellum | 0.17 (0%) | 0.29 (8%) | 39.24 (23%) | |
BP, binding potential; MA1, multilinear analysis-1; SRTM, simplified reference tissue model.
%COV is variability across subjects.
VND was assumed to be 85% of the distribution volume in the cerebellum.
The values with %s.e.>100% were excluded (n=10).
In Vivo Affinity of 11C-LY2795050
The in vivo BPF values from the present study were correlated with unweighted averages of previously reported regional KOR concentrations (Bmax) obtained in vitro from ligand competition binding assays in brain tissue homogenates or autoradiography studies using radioligand 3H-diprenorphine,1 3H-etrophine,2,3 or 3H-ethylketocyclazocine3 in the presence of different displacing agents or 3H-U69593.4 In the in vitro literature, the unit of specific binding is fmol/mg protein. This unit can be converted to fmol/mg of wet tissue by assuming that there is ~0.1 mg protein per mg of wet tissue.39 A correlation plot of the regional binding potential BPF(=Bavail/KD) and the in vitro Bmax is shown in Figure 5. A statistically significant correlation was found with the regression equation of Bmax=0.028 × BPF (R2=0.20, P<0.0001, n=10). Since BPF=Bmax/KD, the slope of this regression line represents the in vivo KD of 11C-LY2795050. We also correlated the in vivo BPF values with in vitro Bmax values obtained from the individual studies that contributed to the average Bmax values used above. The KD estimates ranged from 0.019 nmol/L (3H-diprenorphine, R2=0.43, P<0.0001, n=9) to 0.056 nmol/L (3H-etrophine by Cross et al, R2=0.61, P<0.05, n=3) (Supplementary Figure S3). In the analysis, the determination coefficient was very low (R2<0.1) with in vitro Bmax if amygdala, putamen, and globus pallidus measured by 3H-etrophine are included. Thus, these three regions were excluded from the comparison when using 3H-etrophine data.
Figure 5.

Correlations between regional BPF estimates of 11C-LY2795050 and kappa receptor Bmax values measured in vitro. In vitro Bmax values were the unweighted averages as measured in autoradiography studies with the radioligand 3H-diprenorphine (Pfeiffer et al1), 3H-etrophine (Delay-Goet et al3 and Cross et al2), 3H-U69593 (Barg et al4), or 3H-ethylketocyclazocine with various displacing agents (Delay-Goet et al3). Ten regions of interest (ROIs) were used to compare in vivo data with in vitro measurements of kappa opioid receptor (KOR) Bmax: amygdala (AMY), insula (INS), temporal cortex (TMP), frontal cortex (FRO), globus pallidus (GP), putamen (PUT), caudate (CAU), cingulate cortex (CIN), hippocampus (HIP), and thalamus (THA). For the cingulate cortex, BPF values were computed as the unweighted average between anterior and posterior cingulates. The regression equation was derived as Bmax=0.028 × BPF (R2=0.20). BP, binding potential.
Receptor occupancy by the carrier mass of LY2795050 was calculated by 100 × F/(F+KD), where F is the mean value (from 60 to 90 minutes after injection) of the metabolite-corrected and protein-unbound plasma concentration (expressed in nmol/L), and in vivo KD is assumed to be 0.028 nmol/L (estimated in this study). In all scans, the occupancy by LY2795050 was <5%. Receptor occupancy estimated using the in vitro Ki (0.72 nmol/L) of LY2795050 was even lower.
Discussion
In this study, we conducted the first-in-human evaluation with the selective KOR antagonist 11C-LY2795050. Our goals were (1) to determine the optimal model to describe its kinetics, (2) to assess the specific binding component from blocking study with 150 mg of oral naltrexone, and (3) to determine an appropriate method to estimate the nondisplaceable volume of distribution from the blocking data.
The metabolism profile of 11C-LY2795050 in human (44% parent fraction at 30 minutes) was similar to that of rhesus monkey (40% parent fraction at 30 minutes).17 Similar to the monkey study, 11C-LY2795050 readily entered into the human brain, and was washed out rapidly (Figure 3). The radioactivity in the brain reached peak levels at ~4 minutes after injection. This suggests that 11C-LY2795050 has favorable properties as a radiotracer. The rank order of uptake was also similar to that in the rhesus monkey and consistent with regional KOR densities measured in human postmortem studies in vitro (Figure 5).
The 2TC model17 and MA1 model with t*=40 minutes25 were used as the models of choice for analysis of 11C-LY2795050 imaging data in rhesus monkeys, with 2TC providing better fits than 1TC. This was also true in humans. The MA1 VT values matched extremely well with those from 2TC. Thus, the 2TC model and MA1 model were selected as suitable models to describe 11C-LY2795050 kinetics in the human brain. In addition, the t* setting did not have a strong effect on VT estimates (Supplementary Figure S2). While a late t* value (e.g., 60 minutes) would produce unstable VT estimates due to a smaller number of data points for fitting, MA1 analysis with t*=30, 40, or 50 minutes all produced reliable VT estimates. One caveat is that the 2TC model in the present human study produced VT estimates with a large standard error in a few cases. Higher levels of noise are usually found in human imaging data compared with those in nonhuman primates, so larger errors in the model parameters are expected. The MA1 model would be more suitable for parametric imaging due to a reasonable computation time. Additional data from more subjects would help with the evaluation of an optimal model for analysis.
Given the low in vivo KD estimate (0.028 nmol/L), small k4 values with the 2TC model are possible. For 11C-LY2795050, the average k4 value across all regions was 0.046±0.015/min. The value was similar to the estimates from 11C-FLB 457 (k4: 0.02~0.05/min),40 which also has a high affinity to D2/D3 receptors (0.02 nmol/L). Note that the instability of k4 estimation should be considered when discussing a relationship between KD and k4 values.
The centrum semiovale was included in the occupancy plot measurements since its VT was decreased by naltrexone blocking. This blockade was consistent with in vitro data; in the autoradiography study using 3H-U69593, low density of KOR was detected in the white matter. The occupancy plot relies on the assumption that VND is the same in all included regions, which could be different in white matter, but in retrospect, the centrum semiovale was not an outlier on occupancy plots. Occupancy and VND values from the occupancy plots without the centrum semiovale were only 2±1% lower than those with the centrum semiovale included.
In the rhesus monkey study with 11C-LY2795050, the cerebellum was found to be a suitable reference region, since there was no difference between VT values at baseline and following varying blocking doses of LY2795050. On the other hand, VT values in the present study with naltrexone blocking were significantly reduced in all regions, indicating the lack of an ideal reference region for 11C-LY2795050 in humans. Therefore, we assessed three methods for the determination of VND, using intersubject variability of the resulting BPND, BPP, and BPF as an evaluation and selection criterion.
For all binding potentials, the lowest variability was seen when VND was estimated as a fraction of the cerebellar VT (method (3)). This suggests that the estimated cerebellum VT correlates with VT estimates for other regions, and therefore the use of the corrected cerebellar VT value as VND reduces intersubject variability by cancellation of common error or variability. When binding potentials were estimated using individual VND values derived from occupancy plot of individual subjects (method (1)), high variability in the estimates was found. This is because individual VND had higher intersubject variability (%COV=15%) than the fractional cerebellum VT (%COV=8%). Individual VND was determined as the x-axis intercept of the occupancy plot (Figure 4), which is often associated with larger estimation error compared with slope estimation. Interestingly, using the mean VND (constant value for all subjects, method (2)) yielded binding potential variability that was intermediate between methods (1) and (3), which either suggests that there is true intersubject variability in VND, or that the cancellation of inherent method-related variability is important. On the basis of these assessments, we chose method (3), i.e., using the fractional cerebellum VT as VND to estimate binding potentials. In method (3) the binding potential BPND in the cerebellum is assumed to have a constant value (i.e., α−1). Thus, the intersubject variability of BPND in the cerebellum is not taken into account. This approach will be valuable if the specific binding fraction (i.e., BPND) in the cerebellum is unchanged by diseases and experimental conditions. We also applied the fractional correction to the BPND estimates from SRTM. The corrected BPND values correlated very well with those from the MA1 model, but with an underestimation of ~10%.
By correlating BPF values measured here with KOR Bmax values measured in vitro, we determined an in vivo KD value of 0.028 nmol/L for 11C-LY2795050 in the human brain. We first compared this value with that obtained in rhesus monkeys. An in vivo ED50 value of 15.6 μg/kg was derived from a 11C-LY2795050 PET study with coinjection of unlabeled LY2795050 in rhesus monkey.25 We calculated the relationship between the injected LY2795050 dose and plasma concentration from the rhesus monkey data used in the paper by Kim et al.25 To obtain plasma concentration in nmol/L, the measured arterial input functions (Bq/mL) were corrected for protein binding (fP, 0.018±0.002 in rhesus monkeys) and parent fraction, divided by the specific activity of injected 11C-LY2795050, and then averaged over 40 to 90 minutes after injection of 11C-LY2795050. Using the regression line (plasma concentration (nmol/L)=0.0065 × injected dose (μg/kg), R2=0.997, Supplementary Figure S4), the in vivo ED50 of LY2795050 was converted to KD (0.10 nmol/L).
The in vivo KD estimates of 0.10 nmol/L in rhesus monkeys and of 0.028 nmol/L (0.019 to 0.056 nmol/L using regional Bmax values from individual in vitro studies) in humans for 11C-LY2795050 were smaller than the inhibition coefficient (Ki) of 0.72 nmol/L measured in vitro using cloned human KOR. The discrepancy between in vivo KD values and in vitro Ki could be attributed to a number of factors. First, the in vitro Ki values are usually determined from radioligand competition assays performed at room temperature (22°C), such as in this case for LY2795050, while in vivo KD values are derived from imaging experiments conducted at body temperature (37°C). Temperature sometimes exerts a significant effect on the binding affinities of radioligands, although the direction of changes is not readily predictable (see Elfving et al41). For example, the in vitro Ki of fallypride for the dopamine D2 receptor in the rat striatum was 0.04 nmol/L at 22°C and 2.03 nmol/L at 37°C, while the in vivo KD derived from imaging experiments with 18F-fallypride in baboons was 0.2 nmol/L.42 For another D2 ligand IBF, the in vivo KD (0.081 nmol/L for 123I-IBF) was very similar to the Ki measured in vitro at 22°C (0.06 nmol/L) and 37°C (0.10 nmol/L).43 The benzodiazepine receptor ligand iomazenil is another case in which the in vivo KD (0.54 nmol/L for 123I-iomazenil) derived from imaging experiments was quite similar to the in vitro Ki measures either at 22°C (0.35 nmol/L) or at 37°C (0.66 nmol/L).44
The uncertainty, or measurement errors in ligand-free fraction in the plasma (fP), is the second factor that might contribute to the discrepancy between in vivo KD and in vitro Ki. fP values are required in the determination of in vivo KD, as they are used to estimate the free ligand concentrations in the brain. In both rhesus monkeys and humans, 11C-LY2795050 fP was very small (<2%), and any small errors in its measurement accuracy will contribute to the uncertainty in the KD estimate.
In conclusion, we conducted successfully the first in vivo evaluation of 11C-LY2795050 in humans. The uptake pattern of 11C-LY2795050 was in good accordance with the known KOR distribution. 11C-LY2795050 displayed favorable kinetic properties and can be used for quantitative PET measurement of KOR in human brain. The 2TC and MA1 models were selected as the best model to describe its kinetics and derive binding parameters. Blocking experiments showed that 150 mg of oral naltrexone provided >90% KOR occupancy, and that there was no ideal reference region for 11C-LY2795050 in the human brain. The use of the cerebellum VT corrected for its small specific binding fraction as VND was proposed as a method to calculate binding potentials.
Acknowledgments
The authors appreciate the excellent technical assistance of the staff at the Yale University PET Center.
Johannes Tauscher was employed by Eli Lilly and Company at the time of study.
Footnotes
Supplementary Information accompanies the paper on the Journal of Cerebral Blood Flow & Metabolism website (http://www.nature.com/jcbfm)
This study was supported by Eli Lilly and Company, and by a research grant from NIMH (1 R01 MH091537 to YH). This publication was also made possible by CTSA Grant UL1 RR024139 from the National Center for Research Resources (NCRR) and the National Center for Advancing Translational Sciences (NCATS), components of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NIH.
Supplementary Material
References
- Pfeiffer A, Pasi A, Mehraein P, Herz A. Opiate receptor binding sites in human brain. Brain Res. 1982;248:87–96. doi: 10.1016/0006-8993(82)91150-7. [DOI] [PubMed] [Google Scholar]
- Cross AJ, Hille C, Slater P. Subtraction autoradiography of opiate receptor subtypes in human brain. Brain Res. 1987;418:343–348. doi: 10.1016/0006-8993(87)90101-6. [DOI] [PubMed] [Google Scholar]
- Delay-Goyet P, Zajac JM, Javoy-Agid F, Agid Y, Roques BP. Regional distribution of mu, delta and kappa opioid receptors in human brains from controls and parkinsonian subjects. Brain Res. 1987;414:8–14. doi: 10.1016/0006-8993(87)91321-7. [DOI] [PubMed] [Google Scholar]
- Barg J, Belcheva M, Rowinski J, Ho A, Burke WJ, Chung HD, et al. Opioid receptor density changes in Alzheimer amygdala and putamen. Brain Res. 1993;632:209–215. doi: 10.1016/0006-8993(93)91155-l. [DOI] [PubMed] [Google Scholar]
- Simonin F, Gaveriaux-Ruff C, Befort K, Matthes H, Lannes B, Micheletti G, et al. kappa-Opioid receptor in humans: cDNA and genomic cloning, chromosomal assignment, functional expression, pharmacology, and expression pattern in the central nervous system. Proc Natl Acad Sci USA. 1995;92:7006–7010. doi: 10.1073/pnas.92.15.7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mash DC, Staley JK. D3 dopamine and kappa opioid receptor alterations in human brain of cocaine-overdose victims. Ann NY Acad Sci. 1999;877:507–522. doi: 10.1111/j.1749-6632.1999.tb09286.x. [DOI] [PubMed] [Google Scholar]
- Shippenberg TS. The dynorphin/kappa opioid receptor system: a new target for the treatment of addiction and affective disorders. Neuropsychopharmacology. 2009;34:247. doi: 10.1038/npp.2008.165. [DOI] [PubMed] [Google Scholar]
- de Lanerolle NC, Williamson A, Meredith C, Kim JH, Tabuteau H, Spencer DD, et al. Dynorphin and the kappa 1 ligand [3H]U69,593 binding in the human epileptogenic hippocampus. Epilepsy Res. 1997;28:189–205. doi: 10.1016/s0920-1211(97)00044-2. [DOI] [PubMed] [Google Scholar]
- Loacker S, Sayyah M, Wittmann W, Herzog H, Schwarzer C. Endogenous dynorphin in epileptogenesis and epilepsy: anticonvulsant net effect via kappa opioid receptors. Brain. 2007;130:1017–1028. doi: 10.1093/brain/awl384. [DOI] [PubMed] [Google Scholar]
- Cohen RM, Andreason PJ, Doudet DJ, Carson RE, Sunderland T. Opiate receptor avidity and cerebral blood flow in Alzheimer's disease. J Neurol Sci. 1997;148:171–180. doi: 10.1016/s0022-510x(96)05315-4. [DOI] [PubMed] [Google Scholar]
- Mathieu-Kia AM, Fan LQ, Kreek MJ, Simon EJ, Hiller JM. Mu-, delta- and kappa-opioid receptor populations are differentially altered in distinct areas of postmortem brains of Alzheimer's disease patients. Brain Res. 2001;893:121–134. doi: 10.1016/s0006-8993(00)03302-3. [DOI] [PubMed] [Google Scholar]
- Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC, Jr., et al. Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther. 2003;305:323–330. doi: 10.1124/jpet.102.046433. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Jr., Beguin C, DiNieri JA, Baumann MH, Richards MR, Todtenkopf MS, et al. Depressive-like effects of the kappa-opioid receptor agonist salvinorin A on behavior and neurochemistry in rats. J Pharmacol Exp Ther. 2006;316:440–447. doi: 10.1124/jpet.105.092304. [DOI] [PubMed] [Google Scholar]
- Ranganathan M, Schnakenberg A, Skosnik PD, Cohen BM, Pittman B, Sewell RA, et al. Dose-related behavioral, subjective, endocrine, and psychophysiological effects of the kappa opioid agonist Salvinorin A in humans. Biol Psychiatry. 2012;72:871–879. doi: 10.1016/j.biopsych.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravert HT, Mathews WB, Musachio JL, Scheffel U, Finley P, Dannals RF. [11C]-methyl 4-[(3,4-dichlorophenyl)acetyl]-3-[(1-pyrrolidinyl)-methyl]-1- piperazinecarboxylate ([11C]GR89696): synthesis and in vivo binding to kappa opiate receptors. Nucl Med Biol. 1999;26:737–741. doi: 10.1016/s0969-8051(99)00043-8. [DOI] [PubMed] [Google Scholar]
- Poisnel G, Oueslati F, Dhilly M, Delamare J, Perrio C, Debruyne D, et al. [11C]-MeJDTic: a novel radioligand for kappa-opioid receptor positron emission tomography imaging. Nucl Med Biol. 2008;35:561–569. doi: 10.1016/j.nucmedbio.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Zheng MQ, Nabulsi N, Kim SJ, Tomasi G, Lin SF, Mitch C, et al. Synthesis and evaluation of 11C-LY2795050 as a kappa-opioid receptor antagonist radiotracer for PET imaging. J Nucl Med. 2013;54:455–463. doi: 10.2967/jnumed.112.109512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot PS, Narendran R, Butelman ER, Huang Y, Ngo K, Slifstein M, et al. 11C-GR103545, a radiotracer for imaging kappa-opioid receptors in vivo with PET: synthesis and evaluation in baboons. J Nucl Med. 2005;46:484–494. [PubMed] [Google Scholar]
- Schoultz BW, Hjornevik T, Willoch F, Marton J, Noda A, Murakami Y, et al. Evaluation of the kappa-opioid receptor-selective tracer [11C]GR103545 in awake rhesus macaques. Eur J Nucl Med Mol Imaging. 2010;37:1174–1180. doi: 10.1007/s00259-010-1384-6. [DOI] [PubMed] [Google Scholar]
- Tomasi G, Nabulsi N, Zheng MQ, Weinzimmer D, Ropchan J, Blumberg L, et al. Determination of in vivo Bmax and Kd for 11C-GR103545, an agonist PET tracer for kappa-opioid receptors: a study in nonhuman primates. J Nucl Med. 2013;54:600–608. doi: 10.2967/jnumed.112.112672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganawa M, Jacobsen LK, Zheng MQ, Lin SF, Banerjee A, Byon W, et al. Evaluation of the agonist PET radioligand [11C]GR103545 to image kappa opioid receptor in humans: Kinetic model selection, test-retest reproducibility and receptor occupancy by the antagonist PF-04455242. Neuroimage. 2014;99:69–79. doi: 10.1016/j.neuroimage.2014.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J Neurosci. 2008;28:407–414. doi: 10.1523/JNEUROSCI.4458-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf MD, Coop A. Kappa opioid antagonists: past successes and future prospects. AAPS J. 2005;7:E704–E722. doi: 10.1208/aapsj070371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JB, Atkinson RN, Vinson NA, Catanzaro JL, Perretta CL, Fix SE, et al. Identification of (3R)-7-hydroxy-N-((1S)-1-[[(3R,4R)-4-(3-hydroxyphenyl)- 3,4-dimethyl-1-piperidinyl]methyl]-2-methylpropyl)-1,2,3,4-tetrahydro- 3-isoquinolinecarboxamide as a novel potent and selective opioid kappa receptor antagonist. J Med Chem. 2003;46:3127–3137. doi: 10.1021/jm030094y. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Zheng MQ, Nabulsi N, Labaree D, Ropchan J, Najafzadeh S, et al. Determination of the in vivo selectivity of a new kappa-opioid receptor antagonist PET tracer C-11-LY2795050 in the rhesus monkey. J Nucl Med. 2013;54:1668–1674. doi: 10.2967/jnumed.112.118877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitch CH, Quimby SJ, Diaz N, Pedregal C, de la Torre MG, Jimenez A, et al. Discovery of aminobenzyloxyarylamides as kappa opioid receptor selective antagonists: application to preclinical development of a kappa opioid receptor antagonist receptor occupancy tracer. J Med Chem. 2011;54:8000–8012. doi: 10.1021/jm200789r. [DOI] [PubMed] [Google Scholar]
- Carson RE, Barker WC, Liow JS, Johnson CA. Design of a motion-compensation OSEM list-mode algorithm for resolution-recovery reconstruction for the HRRT. IEEE Nucl Sci Symp Conf Rec (2003) 2003;5:3281–3285. [Google Scholar]
- Jin X, Chan C, Mulnix T, Panin V, Casey ME, Liu C, et al. List-mode reconstruction for the Biograph mCT with physics modeling and event-by-event motion correction. Phys Med Biol. 2013;58:5567–5591. doi: 10.1088/0031-9155/58/16/5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton J, Yokoi F, Dannals RF, Ravert HT, Szabo Z, Wong DF. Column-switching HPLC for the analysis of plasma in PET imaging studies. Nucl Med Biol. 2000;27:627–630. doi: 10.1016/s0969-8051(00)00125-6. [DOI] [PubMed] [Google Scholar]
- Tzourio-Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage. 2002;15:273–289. doi: 10.1006/nimg.2001.0978. [DOI] [PubMed] [Google Scholar]
- Holmes CJ, Hoge R, Collins L, Woods R, Toga AW, Evans AC. Enhancement of MR images using registration for signal averaging. J Comput Assist Tomogr. 1998;22:324–333. doi: 10.1097/00004728-199803000-00032. [DOI] [PubMed] [Google Scholar]
- Viola P, Wells WM., III Alignment by maximization of mutual information. Int J Comput Vis. 1997;24:137–154. [Google Scholar]
- Papademetris X, Jackowski M, Rajeevan N, Constable RT, Staib LH. Bioimage suite: an integrated medical image analysis suite. Insight J. 2005. [PMC free article] [PubMed]
- Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN, et al. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab. 2007;27:1533–1539. doi: 10.1038/sj.jcbfm.9600493. [DOI] [PubMed] [Google Scholar]
- Ichise M, Toyama H, Innis RB, Carson RE. Strategies to improve neuroreceptor parameter estimation by linear regression analysis. J Cereb Blood Flow Metab. 2002;22:1271–1281. doi: 10.1097/01.WCB.0000038000.34930.4E. [DOI] [PubMed] [Google Scholar]
- Frankle WG, Huang Y, Hwang DR, Talbot PS, Slifstein M, Van Heertum R, et al. Comparative evaluation of serotonin transporter radioligands 11C-DASB and 11C-McN 5652 in healthy humans. J Nucl Med. 2004;45:682–694. [PubMed] [Google Scholar]
- Cunningham VJ, Rabiner EA, Slifstein M, Laruelle M, Gunn RN. Measuring drug occupancy in the absence of a reference region: the Lassen plot re-visited. J Cereb Blood Flow Metab. 2010;30:46–50. doi: 10.1038/jcbfm.2009.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn RN, Murthy V, Catafau AM, Searle G, Bullich S, Slifstein M, et al. Translational characterization of [11C]GSK931145, a PET ligand for the glycine transporter type 1. Synapse. 2011;65:1319–1332. doi: 10.1002/syn.20966. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Vanisberg MA, Maloteaux JM. Regional and subcellular localization in human brain of [3H]paroxetine binding, a marker of serotonin uptake sites. Biol Psychiatry. 1988;24:299–309. doi: 10.1016/0006-3223(88)90198-9. [DOI] [PubMed] [Google Scholar]
- Olsson H, Halldin C, Swahn CG, Farde L. Quantification of [11C]FLB 457 binding to extrastriatal dopamine receptors in the human brain. J Cereb Blood Flow Metab. 1999;19:1164–1173. doi: 10.1097/00004647-199910000-00013. [DOI] [PubMed] [Google Scholar]
- Elfving B, Bjornholm B, Ebert B, Knudsen GM. Binding characteristics of selective serotonin reuptake inhibitors with relation to emission tomography studies. Synapse. 2001;41:203–211. doi: 10.1002/syn.1076. [DOI] [PubMed] [Google Scholar]
- Slifstein M, Hwang DR, Huang Y, Guo N, Sudo Y, Narendran R, et al. In vivo affinity of [18F]fallypride for striatal and extrastriatal dopamine D2 receptors in nonhuman primates. Psychopharmacology (Berl) 2004;175:274–286. doi: 10.1007/s00213-004-1830-x. [DOI] [PubMed] [Google Scholar]
- Laruelle M, al-Tikriti MS, Zea-Ponce Y, Zoghbi SS, Baldwin RM, Charney DS, et al. In vivo quantification of dopamine D2 receptor parameters in nonhuman primates with [123I]iodobenzofuran and single photon emission computerized tomography. Eur J Pharmacol. 1994;263:39–51. doi: 10.1016/0014-2999(94)90521-5. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Abi-Dargham A, al-Tikriti MS, Baldwin RM, Zea-Ponce Y, Zoghbi SS, et al. SPECT quantification of [123I]iomazenil binding to benzodiazepine receptors in nonhuman primates: II. Equilibrium analysis of constant infusion experiments and correlation with in vitro parameters. J Cereb Blood Flow Metab. 1994;14:453–465. doi: 10.1038/jcbfm.1994.56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.