

Summary
Genetic studies have established anaplastic lymphoma kinase (ALK), a cell surface receptor tyrosine kinase, as a tractable molecular target in neuroblastoma. We describe comprehensive genomic, biochemical, and computational analyses of ALK mutations across 1596 diagnostic neuroblastoma samples. ALK tyrosine kinase domain mutations occurred in 8% of samples; at three hotspots plus 13 minor sites – and correlated significantly with poorer survival in high- and intermediate-risk neuroblastoma. Biochemical and computational studies distinguished oncogenic (constitutively activating) from non-oncogenic mutations and allowed robust computational prediction of their effects. We also established differential in vitro crizotinib sensitivity of mutated variants. Our studies identify ALK genomic status as a clinically important therapeutic stratification tool in neuroblastoma, and will allow tailoring of ALK-targeted therapy to specific mutations.
Introduction
Neuroblastomas are embryonal tumors that arise from the sympathetic nervous system, and represent the most frequently diagnosed malignancy in the first year of life (Cheung and Dyer, 2013; Maris, 2010). Despite improvements in treatment over recent decades, cure rates for patients with high-risk neuroblastoma (Maris, 2010) lag significantly behind those of other common childhood cancers (Smith et al., 2010). Current treatments rely on dose-intensive chemotherapy, radiation therapy, and immunotherapeutic targeting of the disialoganglioside GD2 (Maris, 2010; Yu et al., 2010). Most recent clinical studies in neuroblastoma have focused on escalating dose intensity in both induction and consolidation therapy, with evidence that this improves outcome (Pearson et al., 2008). The potential long-term adverse effects of increasing treatment intensity on survivors of this childhood cancer are a major concern (Hobbie et al., 2008; Smith et al., 2010), however – making it imperative that more effective treatment strategies are developed.
One promising avenue for targeted therapy in neuroblastoma focuses on anaplastic lymphoma kinase (ALK), a cell-surface neural receptor tyrosine kinase (RTK) expressed at significant levels only in the developing embryonic and neonatal brain (Iwahara et al., 1997; Morris et al., 1997). Germline mutations in intact ALK were recently identified as the major cause of hereditary neuroblastoma (Mossé et al., 2008). These mutations cause single amino acid missense substitutions in the ALK tyrosine kinase domain (TKD) that promote constitutive, ligand-independent, activation of this RTK. Somatically acquired ALK-activating mutations are also found as oncogenic drivers in neuroblastoma (Chen et al., 2008; George et al., 2008; Hallberg and Palmer, 2013; Janoueix-Lerosey et al., 2008; Mossé et al., 2008). In addition, ALK gene amplification imparts an oncogenic dependency in some cases (Janoueix-Lerosey et al., 2008; Mossé et al., 2008). ALK has thus emerged as a tractable oncogene for targeted therapy in neuroblastoma. The same tyrosine kinase is also found in oncogenic ALK fusion proteins that arise from chromosomal translocations in non-small-cell lung cancers (NSCLC) (Soda et al., 2007) and anaplastic large cell lymphomas (Morris et al., 1994), for example, motivating development of small molecule ALK kinase inhibitors. Dramatic response rates to crizotinib (an ALK/Met/Ros1 inhibitor) were seen in pretreated patients with advanced relapsed/refractory NSCLC harboring ALK rearrangements (Kwak et al., 2010; Shaw and Engelman, 2013). These studies validated ALK as a therapeutic target, and led to the expedited FDA approval of crizotinib for ALK-translocated NSCLC.
Rapid clinical translation of findings with ALK in neuroblastoma prompted a phase 1 trial of crizotinib (NCT00939770) in patients with recurrent or refractory cancer. Results from this trial highlighted the differential sensitivity to ALK kinase inhibition of ALK-translocated versus ALK-mutated disease (Mossé et al., 2013). The study also underlined the need for further detailed investigation of ALK mutations in order to optimize clinical application of ALK inhibitors in neuroblastoma. To achieve this goal a detailed analysis of the spectrum of ALK mutations, their clinical significance in neuroblastoma, and their biochemical properties is essential. The resulting data will underpin future approaches for identifying patients likely to benefit from ALK inhibition in neuroblastoma, and for predicting which newly emerging mutations are clinically relevant.
Results
To examine the spectrum of ALK mutations in neuroblastoma, we analyzed germline and somatic ALK DNA alterations – at diagnosis – in samples from a cohort of 1596 neuroblastoma patients assembled in collaboration with the Children's Oncology Group (COG; Table 1).
Table 1. Clinical, Genomic, and Survival Characteristics of Overall Patient Cohorta.
| Patient cohort | n (%) | 5-year EFSb ± std error (%) | EFSb p value | 5-year OSc ± std error (%) | OSc p value |
|---|---|---|---|---|---|
|
| |||||
| Overall | 1596 | 67 ± 1.6 | N/A | 75 ± 1.4 | N/A |
|
| |||||
| Age | |||||
| < 18 mo | 756 (47%) | 81 ± 1.9 | < 0.0001 | 91 ± 1.4 | < 0.0001 |
| ≥ 18 mo | 840 (53%) | 55 ± 2.3 | 62 ± 2.2 | ||
|
| |||||
| Risk groupd | |||||
| Low | 626 (40%) | 87 ± 1.8 | < 0.0001g | 97 ± 0.9 | < 0.0001g |
| Intermediate | 292 (18%) | 85 ± 2.8 | 94 ± 1.9 | ||
| High | 664 (42%) | 40 ± 2.5 | 46 ± 2.6 | ||
| Unknown | 14 | ||||
|
| |||||
| INSSe Stage | |||||
| Stage 1,2,3,4s | 927 (58%) | 83 ± 1.6 | < 0.0001 | 93 ± 1.1 | < 0.0001 |
| Stage 4 | 661 (42%) | 44 ± 2.6 | 51 ± 2.6 | ||
| Unknown | 8 | ||||
|
| |||||
| INPCf histology | |||||
| Favorable | 791 (54%) | 86 ± 1.6 | < 0.0001 | 96 ± 0.9 | < 0.0001 |
| Unfavorable | 683 (46%) | 49 ± 2.5 | 57 ± 2.5 | ||
| Unknown | 122 | ||||
|
| |||||
| Ploidy | |||||
| Hyperdiploid | 998 (64%) | 73 ± 1.8 | < 0.0001 | 82 ± 1.6 | < 0.0001 |
| Diploid | 555 (36%) | 58 ± 2.9 | 65 ± 2.8 | ||
| Unknown | 43 | ||||
|
| |||||
| MYCN status | |||||
| Not Amplified | 1239 (78%) | 74 ± 1.6 | < 0.0001 | 83 ± 1.4 | < 0.0001 |
| Amplified | 340 (22%) | 39 ± 4.1 | 46 ± 4.2 | ||
| Unknown | 17 | ||||
|
| |||||
| 11q | |||||
| No loss | 636 (74%) | 77 ± 2.1 | < 0.0001 | 84 ± 1.8 | < 0.0001 |
| LOH | 219 (26%) | 59 ± 3.9 | 69 ± 3.6 | ||
| Unknown | 741 | ||||
|
| |||||
| 1p | |||||
| No loss | 696 (78%) | 77 ± 1.9 | < 0.0001 | 84 ± 1.7 | < 0.0001 |
| LOH | 198 (22%) | 52 ±4.4 | 61 ±4.3 | ||
| Unknown | 702 | ||||
|
| |||||
| ALK mutation | |||||
| Present | 126 (8%) | 53 ± 6.0 | 0.001 | 67 ± 5.9 | 0.02 |
| Absent | 1458 (92%) | 68 ± 1.6 | 6 ± 1.5 | ||
| Unknown | 12 | ||||
|
| |||||
| Site of ALK mutation | |||||
| F1174 | 38 (30%) | 51 ± 11.9 | 0.76 | 60 ± 12.7 | 0.32 |
| F1245 | 15 (12%) | 46 ± 15.0 | 53 ± 14.8 | ||
| R1275 | 54 (43%) | 54 ± 9.1 | 72 ± 8.5 | ||
| Other mutation | 19 (15%) | 63 ± 14.5 | 73 ± 13.4 | ||
|
| |||||
| ALK Copy Number | |||||
| Amplified | 24 (2%) | 24 ± 12.2 | < 0.0001 | 23 ± 11.7 | < 0.0001 |
| Gain | 195 (15%) | 47 ±4.6 | 57 ±4.6 | ||
| No gain/ not amp | 1109 (83%) | 68 ± 1.9 | 77 ± 1.7 | ||
| Loss | 6 (<1%) | 40 ± 31.0 | 60 ± 26.8 | ||
| Unknown status | 262 | ||||
|
| |||||
| ALK aberration | |||||
| Mutation/amplification/gain/loss | 335 (25%) | 47 ± 36 | < 0.0001 | 59 ± 3.6 | < 0.0001 |
| None of the above | 1015 (75%) | 70 ± 2.0 | 78 ± 1.8 | ||
| Unknown status | 246 | ||||
All tumor samples were derived from the initial diagnostic procedure.
Event-free survival.
Overall survival.
As defined (Maris, 2010).
International Neuroblastoma Staging System.
International Neuroblastoma Pathology Classification.
Low/intermediate versus high risk.
ALK mutations
Sequencing of ALK exons 21-28, encompassing the TKD-encoding region, identified 126 diagnostic samples with at least one mutation, corresponding to 8% of subjects (Table 1). Putative disease-associated mutations were distributed throughout the ALK TKD (Figure 1; Table S1), with an additional mutation at R1060 (between the TKD and transmembrane domain). Three additional ALK TKD sequence variations (R1231Q, I1250T, and D1349H) were observed that had previously been listed in the NCBI database of single nucleotide polymorphisms (dbSNP), but with no known clinical significance or annotation in COSMIC (Forbes et al., 2011).
Figure 1. Distribution of ALK mutations in tumor samples from neuroblastoma patients.
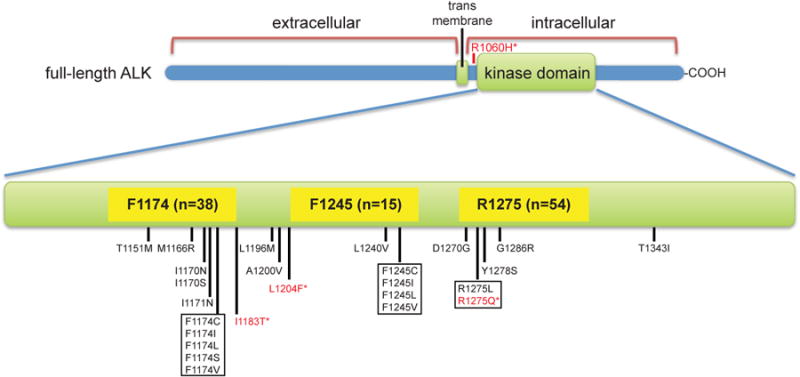
The 126 potentially disease-related mutations observed were distributed over the 16 positions marked in the ALK TKD plus R1060 (between the TKD and the transmembrane domain, upper part of figure). Variants with red asterisks (in red text) were also found in germline DNA. Mutations not previously reported in neuroblastoma include R1060H, I1170N, I1183T, L1204F, D1270G, G1286R, and T1343I. See also Table S1 and Figure S1.
Three ‘hotspot’ residues accounted for 85% of mutations (Figure 1, Table 1): R1275 (43%), F1174 (30%), and F1245 (12%), consistent with previous studies (Chen et al., 2008; George et al., 2008; Hallberg and Palmer, 2013; Janoueix-Lerosey et al., 2008; Mossé et al., 2008). R1275 was substituted with glutamine or leucine in 3.4% of tumors (95% CI: 2.5, 4.3%), F1174 was altered (to L, I, V, C, or S) in 2.4% of tumors (95% CI: 1.6, 3.0%), and F1245 (to L, I, V, or C) in 0.9% (95% CI: 0.5, 1.4%). Two tumor samples harbored mutations at I1170 (to N or S), and another two at I1171 (to N). Single incidences of substitution were seen at a further 15 positions, of which 7 represent mutations not found in current databases (Table S1). Matched constitutional DNA was available for 88 of the 126 ALK-mutated samples, and contained the observed ALK substitution (indicating its presence in the germline) in just 7 cases. Although no information about family history is available, this is an expected rate based on previous analyses (Knudson and Strong, 1972; Mossé et al., 2008). Two were germline R1275Q mutations, and the remaining 5 examples were R1060H, I1183T, L1204F, R1231Q (also in dbSNP), and I1250T (also in dbSNP) mutations (Table S1).
ALK mutations were found in 10.9% of MYCN-amplified tumors, versus 7.2% of those without MYCN amplification. Mutations seen alongside MYCN amplification were biased towards F1174 substitutions (41% in MYCN-amplified cases compared with 30% overall). Further, MYCN amplification occurred in 39% of F1174-mutated tumors, compared with an expected overall frequency of 21% (p<0.01). These data support previous suggestions that F1174 mutations are over-represented in MYCN-amplified tumors, but indicate a less skewed distribution than reported earlier (De Brouwer et al., 2010). Consistent with earlier results, however, patients with both amplified MYCN and F1174-mutated ALK had a significantly worse event-free survival (EFS, p<0.0001) than patients with neither.
ALK copy number
Copy number variation at the ALK locus was observed in 17% of cases for which it was measured (Table 1). Gain of the ALK locus (3-10 copies) was seen in 195 cases (15%), with high-level amplification more rare (24 samples), and whole gene deletion rarer still (6 samples). The ALK gene is located 13.2 megabases centromeric of MYCN. ALK amplification occurred concurrently with MYCN amplification in all but one case – consistent with a previous report (Bagci et al., 2012). Accordingly, since MYCN amplification identifies a patient as a member of the high-risk group (Maris, 2010), ALK amplification occurred exclusively in this group (Table S2) and was associated with inferior outcome (5-year overall survival/OS of 23%, versus 48% in high-risk patients without ALK amplification, p=0.03; Table S3). In the 108/126 ALK-mutated tumors for which we also had copy number data, ALK mutation and high-level gene amplification were mutually exclusive (except for one G1286 variant). Further, 13% of ALK-mutated cases had low-level gain of the ALK locus, similar to the percentage seen in the overall cohort. None demonstrated allelic deletion.
ALK aberration and ALK mutation are prognostic biomarkers of inferior survival
ALK mutations were observed in tumors from all clinical risk groups, and were more commonly observed in those from older patients (Table S2). Across the whole cohort, presence of an ALK aberration (mutation or amplification) correlated with reduced EFS and OS (Figure 2A,B) – as did ALK mutation (Table 1). Presence of any ALK aberration also correlated with reduced EFS and OS in the high-risk group (Figure 2C,D). In univariable analysis, presence of an ALK mutation was also correlated with reduced EFS in intermediate-risk patients (Table S3), a heterogeneous group consisting mainly of very young patients with metastatic disease, or patients of any age with large, unresectable primary tumors. Patient outcome did not differ significantly according to location of the mutation in any analysis. Whereas robust biomarkers to assign outcome probability have been characterized for patients with low- and high-risk disease, the most appropriate therapy for patients with intermediate-risk disease is less well-defined, and our findings suggest that ALK genetic status can be used to identify cases within this group with the highest risk of treatment failure.
Figure 2. Event-free and overall survival based on the presence or absence of an ALK aberration.

Kaplan-Meier curves comparing patients with and without an ALK aberration (mutation and/or amplification): (A) event-free survival (EFS) for the entire neuroblastoma cohort, aberration (n=149) vs. no aberration (n=1,199), p<0.0001; (B) overall survival (OS) for the entire neuroblastoma cohort, aberration (n=149) vs. no aberration (n=1,199), p=0.0002; (C) EFS for the high-risk cohort, aberration (n=88) vs. no aberration (n=540), p=0.0043; (D) OS for the high-risk cohort, aberration (n=88) vs. no aberration (n=540), p=0.0018. See also Table S2.
In multivariable analysis of the overall cohort, presence of an ALK mutation was found to have an independent influence on survival, with a 1.4-fold greater risk of an event (or hazard ratio, HR) within 5 years (95% CI: 1.1, 1.9) compared to cases without an ALK mutation (Table 2). Similarly, independent statistically significant correlation with outcome was seen in three separate multivariable models (adjusted for other known prognostic factors) for:
Any ALK aberration (p=0.0006; HR=1.4, 95% CI: 1.2, 1.7)
ALK copy number gain (p=0.04; HR=1.3, 95% CI: 1.0, 1.6)
ALK amplification (p=0.003; HR=2.2, 95% CI: 1.3, 3.6).
Table 2. Multivariable Analyses Testing the Independence of Correlations between ALK mutation Status and EFS.
| Factors independently statistically significant | Group with increased risk | p value | Hazard Ratio (95% CI) |
|---|---|---|---|
| OVERALL COHORT (n=1530a) | |||
| INSS Stage | Stage 4 | <0.0001 | 2.9 (2.3, 3.6) |
| MYCN status | Amplified | <0.0001 | 1.7 (1.4, 2.1) |
| 11q status | Aberration | <0.0001 | 1.8 (1.4, 2.4) |
| Age | ≥18 months | <0.0001 | 1.6 (1.3, 1.9) |
| Diploidy | Diploid | 0.04 | 1.2 (1.01, 1.5) |
| ALK mutation status | Positive | 0.02 | 1.4 (1.06, 1.9) |
| LOW RISK (n=620a) | |||
| 11q status | Aberration | 0.009 | 2.4 (1.2, 4.7) |
| Histologic category | Unfavorable | 0.01 | 2.2 (1.2, 4.1) |
| Age | ≥18 months | 0.02 | 1.6 (1.3, 1.9) |
| ALK mutation status | Positive | Not significant | |
| INTERMEDIATE RISK (n=275a) | |||
| Histologic category | Unfavorable | 0.03 | 3.3 (1.1, 9.3) |
| ALK mutation status | Positive | 0.007 | 3.1 (1.4, 7.2) |
| HIGH RISK (n=546a) | |||
| Histologic category | Unfavorable | 0.04 | 1.8 (1.02, 3.2) |
| INSS Stage | Stage 4 | 0.0007 | 1.8 (1.3, 2.5) |
| Diploidy | Diploid | 0.01 | 1.4 (1.1, 1.7) |
| ALK mutation status | Positive | 0.03 | 1.5 (1.04, 2.1) |
Sample size reflects the number of patients with known data for all factors included in the model.
See also Table S3.
In a multivariable model adjusted for histologic category (Table 2), ALK mutation status showed statistically significant correlation with reduced EFS in both the intermediate-risk and high-risk patient groups – although not in the low-risk group, again arguing for the utility of ALK mutation status in clinical evaluation.
Predicting signaling consequences of ALK TKD mutations
Our results establish that the presence of an ALK mutation in neuroblastoma is associated with a more aggressive tumor phenotype. Not all ALK substitutions observed in neuroblastoma patients are likely to be oncogenic drivers, however. It will be important in refining therapeutic ALK mutation-based patient stratification to predict as they are observed which mutations are truly drivers that cause ligand-independent ALK signaling, and which are ‘passengers’ or variants of unknown significance. Current methods for predicting functional consequences of patient-derived mutations fall short – especially for activating mutations in kinases. Algorithms such as PolyPhen-2 (Adzhubei et al., 2010), SIFT (Kumar et al., 2009) and the consensus classifier PredictSNP (Bendl et al., 2014) estimate the likelihood of a given mutation having a deleterious effect based on sequence conservation and other criteria. PolyPhen-2 or SIFT (or both) predict that every single ALK substitution listed in Table S1 adversely affects protein function (or is ‘damaging’), except R1060H and R1231Q (a SNP). It seems highly unlikely given our understanding of kinases that even the ALK TKD C-lobe mutations L1204F, T1343I, and D1349H (also in dbSNP) are oncogenic. Moreover, the logic of conservation-based algorithms is not appropriate for predicting gain-of-function mutations in oncogenic kinase domains (Gnad et al., 2013). Indeed, a recent analysis of >400 activating substitutions (Molina-Vila et al., 2014) revealed that most driver mutations in oncogenic kinases do not occur at conserved residues at all, and that their accurate prediction will require explicit attention to kinase regulation mechanisms. Motivated by these considerations, we assessed the effects of all ALK mutations defined here (Figure 1 and Table S1) on in vitro kinase activity – to guide development of better prediction algorithms.
Biochemical effects of clinically-observed ALK TKD mutations
We first monitored autophosphorylation of purified mutated ALK TKDs using native gel electrophoresis (Figure S1) as described (Bresler et al., 2011). The well-studied F1174L and R1275Q mutations greatly accelerated TKD autophosphorylation as expected. So did substitutions at 5 of the other positions listed in Table S1 (M1166, I1170, I1171, F1245, and Y1278), as did the previously reported G1128A and R1192P germline neuroblastoma mutations (Bourdeaut et al., 2011; Janoueix-Lerosey et al., 2008; Mossé et al., 2008). Mutations at T1151, L1196, and G1286 also promoted modest constitutive activation. By contrast, substitutions at five other sites (I1183, A1200, R1231, T1343, and D1349) and those found in dbSNP (R1231Q and D1349H) failed to activate the isolated TKD (Figure S1), signifying that these are unlikely to be clinically significant. D1270G-mutated ALK TKD failed to become autophosphorylated at all, suggesting that this is an inactivating mutation – as expected since D1270 lies in the conserved DFG motif essential for Mg2+-ATP binding to kinases. We did not analyze ALK TKDs harboring L1204F, L1240V, or I1250T mutations in this assay, since they were poorly expressed as recombinant proteins.
For more quantitative analyses, we assayed the ability of the mutated TKDs to phosphorylate a peptide corresponding to ALK's activation loop, and determined values for kcat, KM, ATP, and kcat/KM in vitro. We analyzed both fully auto-phosphorylated ALK TKDs and non-phosphorylated proteins (phosphatase treated as described in Supplemental Experimental Procedures). Non-phosphorylated ALK TKD represents the ‘basal’ kinase state for each receptor variant, whereas autophosphorylated ALK TKDs represent the corresponding activated states – with kcat increased by ∼45-fold in the case of wild-type ALK (Bresler et al., 2011).
Effects of mutations on basal activity of non-phosphorylated ALK TKD
The effects of mutations on non-phosphorylated ALK TKD activity vary according to their location in the kinase. F1174 and F1245 mutations have the strongest effect, increasing kcat by 36-39 fold (Figure 3A; Table S4) – close to the 45-fold increase caused by autophosphorylation of wild-type ALK TKD (Bresler et al., 2011). F1174 and F1245 contribute to a cluster of phenylalanine side-chains (red in Figure 3: the ‘Phe core’) that normally stabilizes the autoinhibited TKD conformation (Bossi et al., 2010; Lee et al., 2010). Mutating these residues will destabilize ALK's autoinhibitory interactions and promote activation. Almost all of the other significantly activating mutations (increasing kcat by >10-fold) occur either at residues in the αC-helix (M1166, I1170, I1171) or in the short α-helix within the activation loop of the inactive TKD (R1275, Y1278). These residues (blue in Figure 3) all participate in autoinhibitory interactions between helix αC and the activation loop α-helix that normally stabilize the inactive conformation of non-phosphorylated ALK TKD (Bossi et al., 2010; Lee et al., 2010), and are disrupted by the mutations analyzed here. Mutations in the N-lobe (green) or phosphate-binding P-loop (cyan) have much smaller effects on ALK TKD (Figure 3A). The only exception is the germline R1192P mutation (which increases kcat of non-phosphorylated ALK TKD by 15 fold). Mutations in the ALK TKD active site (magenta) or C-lobe (grey) have little or no influence on kcat (<3-fold increase), except for the L1196M ‘gatekeeper’ mutation, which increases kcat by nearly 5 fold. Peptide phosphorylation studies further confirmed that the D1270G variant is inactive (Table S4), and revealed a reduced activity for the I1250T (SNP) variant, consistent with previous work (Schönherr et al., 2011a).
Figure 3. kcat and KM, ATP values for mutated ALK TKD variants.
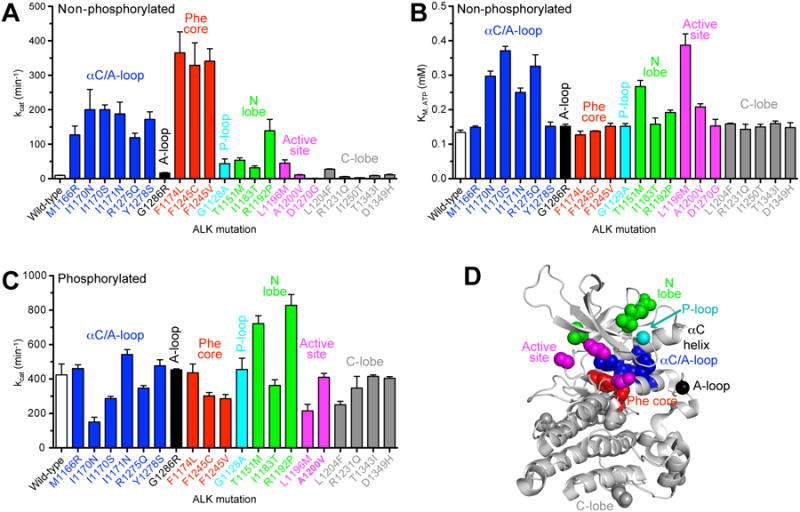
(A) kcat values determined for non-phosphorylated ALK TKD variants at saturating (2 mM) ATP, with 2 mM YYY peptide and 10 mM MgCl2. (B) KM, ATP for non-phosphorylated ALK TKD variants, determined with YYY peptide at 1.0 mM, ATP concentrations from 0.3125 to 2.0 mM, and MgCl2 at 10 mM. Data for wild-type, F1174L, and R1275Q variants were reported previously (Bresler et al., 2011). (C) kcat values for phosphorylated ALK TKD variants, determined as in (A). Data (obtained at 25°C) are all shown as mean ± SEM from at least 3 independent experiments. (D) ALK TKD crystal structure from PDB entry 3LCS (Lee et al., 2010), with important structural regions colored as follows: αC/activation loop interface (blue); Phe core (red); P-loop (cyan); N-lobe (green); active site (magenta); and C-lobe (gray). See also Figure S2 and Tables S4 and S5.
KM, ATP values for non-phosphorylated ALK TKD variants (discussed in more detail below) all fell within a narrow range from 0.13 mM (wild-type) to 0.39 mM (L1196M) – suggesting that all variants are saturated with ATP under physiological conditions (Figure 3B). Accordingly, catalytic efficiencies (kcat/KM, ATP) for non-phosphorylated ALK TKD variants (Figure S2A) follow very similar trends to those seen for kcat. The same is true for kcat/KM, peptide values (Figure S2B).
Effects of neuroblastoma mutations on activity of fully autophosphorylated ALK TKD
The effects of patient-derived ALK mutations on the activity of fully autophosphorylated ALK TKD (prepared as described in Supplemental Experimental Procedures) were much more modest. With the exception of the I1170N variant (for which kcat was just 35% of wild-type), no variant was altered by more than 2-fold in kcat (Figure 3C, Table S5). Overall, therefore, neuroblastoma-derived mutations have their greatest effects on the activity of non-phosphorylated ALK TKD, promoting its constitutive autophosphorylation and thus ligand-independent signaling by the intact receptor.
Transforming ability of mutated ALK variants
To assess how biochemical characteristics relate to transforming ability we measured the ability of intact ALK variants harboring the same TKD mutations to induce focus formation in NIH 3T3 cells (Figure 4A). As shown in Figure 4B and C, quantitation of focus formation assays (Table S6) reveals a remarkably close correspondence between transforming potential and in vitro kcat value for the corresponding non-phosphorylated TKD variants (Figure 3A). A plot of transforming ability against kcat for non-phosphorylated TKD (Figure 4C) yields a straight line with correlation coefficient (r) of 0.95 (p<0.0001). Relative outliers were G1128A (in the P-loop) and L1196M (in the active site), which both appeared relatively more transforming than suggested by in vitro biochemical data, and M1166R, which appeared less transforming than expected by this simple correlation. Transforming potential correlates with kcat of the non-phosphorylated TKD slightly better than it does with either measure of catalytic efficiency: kcat/KM, ATP (r=0.88) or kcat/KM, peptide (r=0.89). By contrast, when transforming ability is plotted against kcat for phosphorylated TKD variants, the slope does not deviate significantly from zero (p=0.68), indicating no correlation (Figure 4C, Figure S2C).
Figure 4. Transformation potential of ALK mutants from NIH 3T3 focus formation assays.

(A) Representative focus formation assays for NIH 3T3 cells transfected with intact ALK variants or empty vector. (B) Quantitation of data from (A). To correct for transfection efficiency, the number of foci for each transfection was divided by the number of G418-resistant colonies, and transforming ability plotted as foci per G418-resistant colony. Each independent experiment was performed in duplicate and data are presented as mean ± SEM of at least three independent experiments (see Table S6). (C) Plots of ALK transforming ability against in vitro kcat for non-phosphorylated ALK TKD (left) or autophosphorylated ALK TKD (right). The lines are linear regressions to the data. Correlation coefficients were 0.95 (non-phosphorylated) and 0.095 (phosphorylated), with significant deviation from zero for non-phosphorylated ALK TKD (p<0.0001) but not phosphorylated ALK TKD (p=0.68).
Taken together, the data in Figures 3 and 4 argue that activity of the non-phosphorylated ALK TKD is an excellent predictor of ALK's transforming ability in NIH 3T3 cells. An increase of just 4.6 – 4.8-fold in the kcat of non-phosphorylated ALK TKD appears sufficient for NIH 3T3 cell transformation, judging from results with the G1128A (cyan) and L1196M (magenta) variants (Figure 4, Table S4). The one exception to this correlation is the N-lobe T1151M variant, for which a relatively reduced kcat/KM, peptide value (Figure S2B, Table S4) may explain failure to transform NIH 3T3 cells (presumably because of elevated KM, peptide). It is important to note that none of the three ALK mutations previously reported in dbSNP (R1231Q, I1250T, and D1349H, all in the C-lobe) were associated with ALK activation in transformation or biochemical assays (Figures 3 and 4); these are silent or passenger mutations. Moreover, analysis of transforming ability in Figure 4B (as a measure of oncogenicity) paints a very different functional picture from that predicted by PolyPhen-2 or SIFT across the spectrum of ALK mutations. These algorithms predict that all mutations (except R1060H and R1231Q) are damaging or affect function – whereas our experimental analysis shows that 11 of the 24 mutations have no significant activating effect. ALK is unlikely to be an important driver in neuroblastoma cases with any of these non-activating mutations, and crizotinib is very unlikely to be therapeutically useful in these contexts. The ‘silent’ mutations account for one of every ∼11 of ALK-mutated patients, and it is important to identify them.
Crizotinib sensitivities of recombinant ALK TKD variants
At least two of the ALK driver mutations observed in neuroblastoma (F1174L and L1196M) have also arisen as acquired crizotinib resistance mutations in tumors driven by oncogenic ALK fusion proteins (Choi et al., 2010; Sasaki et al., 2010). These findings suggest that primary resistance will be a major concern in crizotinib treatment of ALK-mutated neuroblastoma, as we have discussed previously (Bresler et al., 2011). A mere ∼3-fold reduction in KM, ATP appeared sufficient to impair in vivo crizotinib sensitivity of F1174L-mutated ALK when compared with the R1275Q variant, consistent with the appearance of F1174L as an acquired resistance mutation in both a crizotinib-treated inflammatory myofibroblastic tumor (Sasaki et al., 2010) and in vitro screens for crizotinib-resistance mutations in EML4-ALK (Zhang et al., 2011). Table 3 lists KM, ATP values for the other non-phosphorylated ALK TKD variants described here. Based solely on reduced KM, ATP values (as for F1174L), and assuming unchanged crizotinib affinity, the G1128A, M1166R, F1245C, F1245V, or Y1278S variants would be suggested to be relatively crizotinib resistant, whereas the I1170N, I1170S, I1171N, and L1196M (gatekeeper) variants would be argued to resemble R1275Q in being sensitive to crizotinib (with relatively larger KM, ATP values). R1192P is intermediate.
Table 3. Crizotinib Sensitivities of Non-phosphorylated ALK-TKD Mutants at 2.0 mM ATPa.
| ALK TKD variant | KM, ATP (mM)c | Crizotinib IC50 (nM) at 2 mM ATPc | Predicted crizotinib sensitivity | |
|---|---|---|---|---|
|
| ||||
| from KM, ATP | from IC50 | |||
|
| ||||
| G1128A | 0.152 ± 0.008 | 138 ± 21 | - | - |
| M1166R | 0.149 ± 0.004 | 76.1 ± 2.4 | - | + |
| I1170N | 0.297 ± 0.015 | 77.5 ± 3.5 | + | + |
| I1170S | 0.371 ± 0.013 | 76.4 ± 7.7 | + | + |
| I1171N | 0.250 ± 0.013 | 157 ± 11 | + | - |
| F1174Lb | 0.127 ± 0.011 | 130 ± 10 | - | - |
| R1192P | 0.192 ± 0.007 | 90.5 ± 4.7 | -/+ | -/+ |
| L1196M | 0.387 ± 0.033 | 521 ± 4 | + | - |
| F1245C | 0.138 ± 0.001 | 79.2 ± 7.5 | - | + |
| F1245V | 0.152 ± 0.009 | 86.9 ± 2.0 | - | + |
| R1275Qb | 0.326 ± 0.033 | 84.6 ± 8.0 | + | + |
| Y1278S | 0.152 ± 0.012 | 113 ± 5 | - | - |
Crizotinib concentration was varied from 0 to 25,600 nM, with ATP at 2.0 mM, YYY peptide at 0.5 mM, and enzyme held fixed at 50 nM.
Values previously determined and reported (Bresler et al., 2011).
Results are reported as mean ± SEM of at least 3 independent experiments.
See also Figure S3.
Direct measurement of crizotinib IC50 values in vitro (at 2 mM ATP), however, reveals that the correlation with KM, ATP is not simple. The L1196M (gatekeeper) variant is resistant to crizotinib (Figure S3) despite its high KM, ATP – indicating a differential effect of this mutation on ATP and crizotinib binding, as expected from its appearance as an acquired resistance mutation (Katayama et al., 2011). Similarly, I1171N-mutated ALK is less crizotinib sensitive in vitro than a simple view of ATP competition (and KM, ATP) would suggest – further supported by the fact that mutations at I1171 emerged alongside F1174 mutations in screens for crizotinib-resistant variants of EML4-ALK (Zhang et al., 2011). By contrast, the M1166R, F1245V, and F1245C variants are more sensitive to crizotinib in vitro than simple consideration of KM, ATP would suggest – resembling the R1275Q variant in our in vitro crizotinib inhibition studies (Table 3) and NIH 3T3 focus assays (not shown). This finding is particularly important given that F1245 is the third most commonly mutated site in neuroblastoma, accounting for ∼12% of mutations. Our data suggest that cases with F1245 mutations should respond to crizotinib just as well as R1275Q-mutated tumors.
The data in Table 3 make it clear that the individual ALK TKD mutations affect binding of ATP and crizotinib in different ways, and assessment of IC50 values for a selection of mutations in NIH 3T3 focus assays (not shown) concurs. For example, the L1196M mutation impairs crizotinib binding more than ATP binding – thus causing resistance to the drug. It will therefore be important to compare the relative abilities of next generation ALK inhibitors to inhibit the variants studied here. One new inhibitor, called ceritinib, was recently shown to inhibit ALK variants with crizotinib-resistant L1196M, or I1171T mutations (Friboulet et al., 2014), but not to overcome resistance of an F1174 mutation. Other inhibitors are also known to have differential effects on crizotinib-resistance mutations acquired in ALK-rearranged lung cancers (Johnson et al., 2014; Sakamoto et al., 2011). The data in Table 3 suggest that neuroblastoma patients with ALK-activating M1166R, I1170N/S or F1245 mutations should respond to crizotinib just as well as R1275Q-mutated patients. Patients with I1171 or L1196 mutations are likely to respond better to ceritinib, and those with F1174L (and possible other) mutations might be more responsive to the macrocyclic inhibitor PF-06463922 (Johnson et al., 2014). Y1278, R1192 and G1128 mutations appear to be intermediate between these two groups, and comparative studies of ALK inhibitors are needed to determine which inhibitor would be best for these cases.
Computational analysis of mutated ALK TKDs
The poor performance of existing informatics-based approaches in distinguishing activating from non-activating amino acid substitutions prompted us to investigate structure-based computational methods for assessing newly emerging ALK mutations. Importantly, apart from already published data, the computational analysis was done without prior knowledge of biochemical results emerging from this study. As described in Supplemental Experimental Procedures, we simulated molecular dynamics (MD) trajectories for the inactive conformation of all mutated ALK TKD variants, and for wild-type ALK TKD, in both active and inactive conformations. The resulting MD trajectories were analyzed for three key structural properties:
i). Hydrogen bonding network
Distinct sets of key intramolecular hydrogen bonds characterize the active and inactive ALK TKD configurations. Those that maintain the (autoinhibited) positions of the activation loop and αC helix in the inactive TKD (in Figure 3D) are absent in the active structure. A simple scoring function (see Supplemental Experimental Procedures) was used to determine whether each mutation promotes a more ‘active-like’ or ‘inactive-like’ hydrogen-bonding network (Table 4).
Table 4. Computational Prediction of Effects of ALK TKD Mutations.
| Variant | kcat (min-1)a | Activated in vitro ?b | Changes indicative of kinase activation in MD simulations of: | Overall prediction of activation in silico ? | Transforming in NIH 3T3 ?c | PolyPhen-2 Prediction (probability)d | ||
|---|---|---|---|---|---|---|---|---|
| H-bonds | SASA/ FEP | PCA | ||||||
| F1174L | 365 | ● | ● | - | ● | ● | ● | ● (0.70) |
| F1245V | 341 | ● | ● | - | - | ● | ● | ● (1.00) |
| F1245C | 329 | ● | - | - | - | - | ● | ● (1.00) |
| I1170N | 200 | ● | ● | ● | - | ● | ● | ● (1.00) |
| I1170S | 200 | ● | - | ● | - | ● | ● | ● (1.00) |
| I1171N | 188 | ● | - | - | - | - | ● | ● (1.00) |
| Y1278S | 172 | ● | - | - | ● | ● | ● | ● (0.99) |
| R1192P | 139 | ● | ● | - | ● | ● | ● | ● (0.99) |
| M1166R | 127 | ● | ● | - | - | ● | ○ | ● (0.99) |
| R1275Q | 119 | ● | ● | - | - | ● | ● | ● (1.00) |
| L1196M | 45 | ● | - | - | - | - | ○ | ● (1.00) |
| G1128A | 43 | ● | ● | - | - | ● | ○ | ● (1.00) |
| I1183T | 32 | - | - | - | - | - | - | ● (0.96) |
| L1204F | 28 | - | - | - | - | - | - | ● (0.99) |
| G1286R | 16 | - | - | - | - | - | - | ● (0.98) |
| A1200V | 11 | - | - | - | - | - | - | ● (0.67) |
| D1349H | 11 | - | - | - | - | - | - | ● (0.94) |
| Wild-type | 9 | - | NA | NA | NA | NA | - | NA |
| T1343I | 9 | - | - | - | - | - | - | ● (0.84) |
| R1231Q | 5 | - | - | - | - | - | - | - (0.01) |
| I1250T | 3 | - | - | ● | - | ●e | - | ●e(1.00) |
| D1270Ge | 1 | - | ● | - | - | ●e | - | ●e(1.00) |
kcat for non-phosphorylated TKD is listed – from Table S4.
A variant is considered ‘activated’ in vitro if kcat for non-phosphorylated TKD exceeds 4.6 times that of wild-type (see text).
Open circles represent weak transformation (see Figure 4).
Filled black circles in PolyPhen-2 column indicate that this algorithm predicts that the mutation is damaging. Probabilities in parentheses taken from PolyPhen-2 batch run at http://genetics.bwh.harvard.edu/pph2/ (Adzhubei et al., 2010).
D1270G and I1250T mutations are known to be inactivating (this work and Schönherr et al., 2011a). D1270G disrupts the DFG motif, I1250T expresses poorly, suggesting compromised folding.
ii). Hydrophobic interaction network
As mentioned above, key autoinhibitory interactions are stabilized in the inactive conformation by residues with hydrophobic side-chains – notably those in the Phe-core (red in Figure 3D) and contacts between the αC-helix and short activation-loop helix (blue in Figure 3D). Disruption of these autoinhibitory hydrophobic interactions can be assessed readily by monitoring the solvent-accessible surface area (SASA) of relevant residues throughout the MD trajectories. If SASA increases as a result of a neuroblastoma mutation, the mutation is classed as ‘activating’. To further determine whether observed changes in SASA favor the active state, free energy perturbation (FEP) simulations were used to determine whether each mutation significantly destabilizes the inactive state relative to the active state (in which case it is classed as ‘activating’).
iii). Principal component analysis (PCA)
PCA reveals correlated global motions across the MD trajectory. The top 10 dominant modes are considerably different (greater) in the active conformation than in inactive ALK TKD (indicating greater motion), as seen for other kinases (Shih et al., 2011). As outlined in Supplemental Experimental Procedures, each mutant with a top eigenvalue above 200 Å2 is scored as activating (indicating destabilization of key autoinhibitory interactions).
A mutation is predicted to be ‘activating’ overall if it scores as such in one or more of the three criteria outlined above. As shown in Table 4, predictions for each mutated ALK TKD variant studied here agree quite well with our experimental studies. Moreover, the computational analysis suggests a possible mechanism of activation for each mutation, i.e., by perturbing hydrophilic interactions, hydrophobic interactions, or global conformation. All of the mutations that elevate kcat of non-phosphorylated ALK TKD by over 5-fold were predicted correctly except two (I1171N, and F1245C), as listed in Table 4. Perhaps more importantly, our computational analysis correctly predicts the majority of mutations that are not activating – thus showing its value in distinguishing passenger from driver mutations and its potential utility for patient stratification. There are a few exceptions, however. The T1151M mutation was designated as activating in our computational analysis, but did not transform NIH 3T3 cells (Figure 4). Although biochemical analysis indicated an elevated kcat for this variant, it has a reduced kcat/KM, peptide, apparently arising from an elevated KM, peptide that would not be captured computationally. The I1250T and D1270G mutations – both also predicted to be activating using our computational approach – are special cases. D1270 is the conserved DFG aspartate, and loss of its side-chain removes a functional group essential for Mg2+ binding to the TKD. The I1250T mutation impairs protein stability and/or folding (as assessed by poor expression) in a way that the model cannot predict, so no activating effect is seen experimentally. The computational analysis also failed to predict three transforming mutations (Table 4): F1245C, I1171N, and L1196M (the gatekeeper mutation), for reasons that are less clear. Overall, however, our computational approach correctly predicts 75% of the transforming mutations and – importantly – over 75% of silent mutations (excepting special cases). By contrast, PolyPhen-2 (Table 4, right-most column) predicts only one silent mutation correctly and is plagued with false positives. It is important to reiterate that our computational studies were undertaken with no prior knowledge of the results shown in Figures 3 and 4 (with the exception of previously published results).
Discussion
Discovery of activating mutations in the intact ALK gene as drivers in neuroblastoma provided the first example of a pediatric cancer caused by germline mutations in an oncogene, and the only druggable mutation in a pediatric solid tumor. We characterized the spectrum and frequency of germline and somatic alterations in ALK across all neuroblastoma disease subsets in 1596 patients. This dataset is powered to identify ALK mutations in neuroblastoma that, while rare, are still clinically relevant, and to have sufficient power to determine the prognostic capability of ALK alterations within each neuroblastoma risk group (high, intermediate, and low). In addition, cataloguing ALK mutations in these tumors allowed us to correlate sequence variations with oncogenic potency. Some of the observed mutations are unlikely to be oncogenic, and the activated variants differ in their sensitivity to crizotinib – with potentially important therapeutic implications. In multivariable models of the overall cohort, and within each risk group, both the presence of an ALK mutation (except within the low-risk group) and the presence of any ALK aberration were shown to correlate independently with worse EFS. These findings illustrate the value of determining ALK status for prognostic patient stratification, and also support the potential importance of ALK as a therapeutic target.
ALK mutations were observed in 8% of neuroblastoma patients, and span the entire spectrum of disease, including INSS Stage 4 disease, congenital cases, and adolescents/young adults. The fact that ALK mutations occur at the highest frequency (17%) in patients older than 10 suggests differences in the occurrence of genetic mutations based on age, reminiscent of the recently reported age distribution of ATRX mutations in neuroblastoma (Cheung et al., 2012). Within the high-risk subset of neuroblastoma patients the overall frequency of ALK aberration is 14% (10% mutation, 4% amplification). High-risk patients have the poorest outcomes, with approximately only 50% OS despite intensive multi-modal therapy including chemotherapy, surgery, myeloablative conditioning with bone marrow transplant, radiation therapy and immunotherapy plus retinoic acid (Maris, 2010) – making these patients excellent candidates for ALK-targeted therapy. Within the low- and intermediate-risk groups, the frequency of ALK aberration is 6% and 8% respectively. In low-risk cases, therapy usually involves observation, with or without surgical intervention, whereas patients with intermediate-risk disease are treated with conventional cytotoxic chemotherapy and are at risk for the associated late effects. Our results suggest an important opportunity within the intermediate-risk group to identify those with an activating ALK mutation for treatment with ALK inhibitors and de-escalation of traditional cytotoxic therapy.
Of the 24 different patient-derived ALK mutations assessed, only 13 drove transformation of NIH 3T3 cells. Where our studies overlap with other analyses of transformation by ALK variants found in neuroblastoma, they are in complete agreement (Chand et al., 2013; George et al., 2008; Schönherr et al., 2011a; Schönherr et al., 2011b). Importantly, every mutation that promoted transformation also constitutively activated the ALK TKD, with remarkable correlation between transforming activity and in vitro kcat value for the non-phosphorylated TKD. Eleven of the 24 mutations (representing 9% of ALK-mutated patients in this study) appeared silent or even inactivating. Of the activating mutations, in vitro analyses suggest that only 6 (including R1275Q) will be sensitive to inhibition by crizotinib in vivo. Some others are likely to respond better to next generation inhibitors that appear capable of inhibiting L1196M and I1171 variants (ceritinib) or F1174 variants (PF-06463922). Further comparative studies with these inhibitors are required to identify which is optimal for each variant.
Using a molecular dynamics (MD)-based computational approach, we showed that we can predict which mutations are activating – and more importantly which are not activating – with a success rate that greatly exceeds methods such as SIFT, PolyPhen-2, PredictSNP, and others. For example, our method correctly (and blindly) predicted the consequences of all newly described ALK mutations presented in this report, with D1270G as the one exception (where a catalytically crucial residue is mutated). It is important to note that our computational analysis assesses kinase activation, and not transformation itself – but our biochemical data indicate that elevating kcat of non-phosphorylated ALK TKD by 4.6-fold or more causes the receptor to be transforming. Our computational analysis of ALK mutations has significant promise as a clinical tool, and will improve with further training and testing using additional clinically observed (and experimental) mutations. It will also be important to apply computational approaches similar to those that we have employed for EGFR (Park et al., 2012) in efforts to predict inhibitor sensitivity.
Our findings allow us to formulate molecular diagnostic screening recommendations for newly diagnosed neuroblastoma patients, which will be important in clinical evaluation of ALK inhibitors for childhood cancer. We demonstrate that ALK is a predictive therapeutic biomarker of disease status, and also provides a therapeutic target in a select group of patients. With additional molecularly targeted therapeutics and computational models that leverage biochemical understanding to predict the effect of newly emerging ALK mutations, we should now be able to make upfront predictions about which patients are most likely to respond to crizotinib or other ALK inhibitors and (importantly) identify those that will not. Related approaches have been successful with imatinib in chronic myeloid leukemia, gefitinib in NSCLC, and most recently crizotinib in ALK-translocated NSCLC – although functional stratification of individual mutations along the lines described here has not yet been achieved. We are now poised to develop a responder hypothesis as part of a therapeutic strategy that incorporates pharmacologic ALK inhibition into the backbone of contemporary treatment regimens in neuroblastoma. Our phase 1 trial of crizotinib showed some promising responses even in heavily pretreated patients with ALK mutations with recurrent or refractory late-stage disease (Mossé et al., 2013), despite the facts that these heavily pre-treated patients are frequently multi-drug resistant, and also show an increased prevalence of p53 mutations at relapse (Carr-Wilkinson et al., 2010) – confounding factors that were not assessed. In newly diagnosed patients, where these issues are not relevant (and p53 mutations are rare), we expect upfront ALK-targeted treatment guided by the results described here to yield dramatically improved results.
In addition, our findings will help advance the management of individuals with neuroblastoma predisposition. Individuals with a germline ALK variation of unknown significance may have siblings who also harbor these variants, emphasizing the importance of understanding which alleles are indeed risk-alleles so as to determine their risk of developing neuroblastoma, and to offer appropriate clinical screening. No models have yet been established for effective early detection strategies or improving clinical outcomes when germline ALK variations are detected. Implementing clinical surveillance strategies for unaffected children (possibly even adults) with neuroblastoma carrying a germline ALK variant should be guided by data such as those presented here, recognizing the implications of the use of predictive genetic screening and surveillance practices and the absence of evidence of benefit from early detection in these individuals.
Experimental Procedures
Patient population
ALK genomic status was evaluated in 1596 diagnostic tumor DNA samples from patients enrolled on COG neuroblastoma biology protocol ANBL00B1. Matched germline DNA was available in 88 of the 126 cases with identified ALK mutations. Most samples were annotated with clinical and genomic information including age, site of origin, International Neuroblastoma Staging System (INSS) stage, International Neuroblastoma Pathologic Classification (INPC), MYCN copy number, DNA index, registration on clinical trial(s), event-free and overall survival, and tumor DNA copy number/LOH status at chromosome bands 1p36 and 11q23 (Attiyeh et al., 2005; Morowitz et al., 2003). All cases had a confirmed diagnosis of neuroblastoma, and had associated informed consent and enrollment on a COG biology study. This research was approved by the Children's Hospital of Philadelphia Institutional Review Board and the Cancer Therapy Evaluation Program.
Sequencing and copy number determination
Sanger-based DNA re-sequencing of the ALK TKD (exons 21-28) was performed by Beckman Coulter Genomics using BigDye Terminator v3.1 Cycle Sequencing chemistry on an ABI PRISM 3730×l sequencer. Electropherograms were analyzed using Sequencher 4.9 software. For each tumor harboring a somatic mutation, we sequenced the corresponding amplicon in matched germline DNA where available. Variations were crosschecked for polymorphism status in dbSNP and COSMIC databases, and a panel of 218 normal control alleles. Copy number determination employed whole-genome SNP-array analyses (550k) for 577 samples, and realtime quantitative PCR for 1122 (see Supplemental Experimental Procedures).
Statistical considerations
Risk factors tested for prognostic significance were age (<18 months versus ≥18 months), INSS stage (stage 1,2,3,4s versus stage 4), histology (favorable versus unfavorable), ploidy (hyperdiploid versus diploid), MYCN status (not amplified versus amplified), 11q status (no loss versus LOH), and 1p status (no loss versus LOH). For univariable analyses, EFS and OS times were calculated as described in Supplemental Experimental Procedures. A chi-squared test was used to test association between each risk factor and presence of ALK mutation, presence of ALK copy number variation, and presence of any ALK aberration. p values were not adjusted for multiple comparisons, with p<0.05 considered statistically significant. For multivariable analyses, using backwards selection model building, a Cox proportional hazards regression model was used to test for the independent statistical significance of ALK mutations or copy number variations after adjustment for statistically significant risk factors, and to calculate the p value and hazard ratio (HR) for each factor.
Recombinant protein expression, purification, and analysis
Histidine-tagged wild-type and mutated ALK TKD variants were produced from baculovirus-infected Sf9 cells in non-phosphorylated and fully autophosphorylated forms as described (Bresler et al., 2011), and assessed for kinase activity as described in Supplemental Experimental Procedures.
Focus formation assays
Focus formation assays were performed as described (Burke et al., 1997), with details in Supplemental Experimental Procedures.
Supplementary Material
Figure S1: Native Gel Analysis of ALK TKD Mutants.
(related to Figure 1)
Figure S2: Catalytic Efficiencies of ALK TKD Variants.
(related to Figure 3)
Figure S3: The L1196M Mutant is Crizotinib-Resistant Despite its high KM, ATP.
(related to Table 3)
Table S1: Genomic Coordinates of Mutations Identified (and Resulting Amino Acid Substitutions).
(related to Figure 1)
Table S2: ALK Mutation and Amplification Status by NB Risk Group and Age.
(related to Figure 2)
Table S3: ALK Aberrations by Risk Group.
(related to Table 2)
Table S4: Kinetic Properties of Non-phosphorylated ALK TKD Variants.
(related to Figure 3)
Table S5: Kinetic Properties of Phosphorylated ALK TKD Variants.
(related to Figure 3)
Table S6: Quantitation of Focus Formation Assays for Intact ALK Variants in NIH 3T3 Cells.
(related to Figure 4)
Significance.
This work establishes pre-clinical rationale for therapeutic stratification of neuroblastoma patients for ALK inhibition therapy based on ALK genomic status. Beyond previously established risk factors, presence of an ALK aberration (mutation or amplification) is a prognostic biomarker for poorer survival. Presence of an ALK tyrosine kinase domain mutation also correlates with worse prognosis in high- and intermediate risk patients. Biochemical and functional cellular analyses allowed us to determine which mutations are oncogenic drivers, and to develop robust computational approaches for identifying disease-significant ALK mutations as they emerge clinically. Assessing in vitro sensitivity to crizotinib also suggests strategies for its application in the clinic. Stratification based on ALK status thus has significant potential for improving outcomes for a significant patient subset.
Acknowledgments
This work was supported in part by NIH grant R01-CA140198 (Y.P.M.), U.S. Army Peer Reviewed Medical Research Program grants W81XWH-10-1-0212/3 (M.A.L. and Y.P.M), a European Commission Grant FP7-ICT-2011-9-600841 (R.R.), Predoctoral Fellowship 11PRE7670020 from the Great Rivers Affiliate of the American Heart Association (J.H.P.), NIH Training Grant in Structural Biology (T32-GM008275 to S.C.B), an NSF Graduate Research Fellowship (P.J.H.), and by the Institute for Translational Medicine and Therapeutics of the University of Pennsylvania (supported by NCRR Grant UL1RR024134). Computational resources were provided in part by the National Partnership for Advanced Computational Infrastructure under Grant No. MCB060006 from XSEDE (R.R.). We thank members of the Lemmon, Mossé, Ferguson, and Radhakrishnan laboratories for valuable discussions, and Pfizer for their gift of crizotinib. Y.P.M. is an inventor on a patent filed by the Children's Hospital of Philadelphia (WO/2009/103061 or PCT/US2009/034288: Methods and Compositions for Identifying, Diagnosing, and Treating Neuroblastoma).
Footnotes
Author Contributions: M.A.L., Y.P.M., and R.R. conceived and designed the project. S.C.B. designed and performed all biochemical and cellular experiments, with contributions from J.H.P. (supervised by M.A.L.). D.A.W., K.K., and M.L. coordinated sequence analysis and D.A.W., H.R. and E.F.R. designed and performed copy number analyses (supervised by Y.P.M., A.C.W., and M.D.H.). P.J.H. performed all computational studies of ALK TKD (supervised by R.R.). W.B.L. and P.W.M. performed statistical analysis of patient-derived data. S.C.B., D.A.W., Y.P.M., and M.A.L. wrote and edited the manuscript. P.J.H., R.R., and M.D.H. edited the manuscript. S.C.B., D.A.W., and P.J.H. share equal authorship.
Supplemental Information: Supplemental Information includes Supplemental Experimental Procedures, three figures, and six tables, and can be found with this article online.
All other authors declare that they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attiyeh EF, London WB, Mossé YP, Wang Q, Winter C, Khazi D, McGrady PW, Seeger RC, Look AT, Shimada H, et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med. 2005;353:2243–2253. doi: 10.1056/NEJMoa052399. [DOI] [PubMed] [Google Scholar]
- Bagci O, Tumer S, Olgun N, Altungoz O. Copy number status and mutation analyses of anaplastic lymphoma kinase (ALK) gene in 90 sporadic neuroblastoma tumors. Cancer Letts. 2012;317:72–77. doi: 10.1016/j.canlet.2011.11.013. [DOI] [PubMed] [Google Scholar]
- Bendl J, Stourac J, Salanda O, Pavelka A, Wieben ED, Zendulka J, Brezovsky J, Damborsky J. PredictSNP: Robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comp Biol. 2014;10:e1003440. doi: 10.1371/journal.pcbi.1003440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossi RT, Saccardo MB, Ardini E, Menichincheri M, Rusconi L, Magnaghi P, Orsini P, Avanzi N, Borgia AL, Nesi M, et al. Crystal structures of anaplastic lymphoma kinase in complex with ATP competitive inhibitors. Biochemistry. 2010;49:6813–6825. doi: 10.1021/bi1005514. [DOI] [PubMed] [Google Scholar]
- Bourdeaut F, Ferrand S, Brugières L, Hilbert M, Ribeiro A, Lacroix L, Bénard J, Combaret V, Michon J, Valteau-Couanet D, et al. ALK germline mutations in patients with neuroblastoma: a rare and weakly penetrant syndrome. Eur J Hum Genet. 2011;20:291–297. doi: 10.1038/ejhg.2011.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresler SC, Wood AC, Haglund EA, Courtright J, Belcastro LT, Plegaria JS, Cole K, Toporovskaya Y, Zhao H, Carpenter EL, et al. Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma. Sci Transl Med. 2011;3:108ra114. doi: 10.1126/scitranslmed.3002950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke CL, Lemmon MA, Coren BA, Engelman DM, Stern DF. Dimerization of the p185neu transmembrane domain is necessary but not sufficient for transformation. Oncogene. 1997;14:687–696. doi: 10.1038/sj.onc.1200873. [DOI] [PubMed] [Google Scholar]
- Carr-Wilkinson J, O'Toole K, Wood KM, Challen CC, Baker AG, Board JR, Evans L, Cole M, Cheung NK, Boos J, et al. High frequency of p53/MDM2/p14ARF pathway abnormalities in relapsed neuroblastoma. Clin Canc Res. 2010;16:1108–1118. doi: 10.1158/1078-0432.CCR-09-1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chand D, Yamazaki Y, Ruuth K, Schönherr C, Martinsson T, Kogner P, Attiyeh EF, Maris J, Morozova O, Marra MA, et al. Cell culture and Drosophila model systems define three classes of anaplastic lymphoma kinase mutations in neuroblastoma. Dis Model Mech. 2013;6:373–382. doi: 10.1242/dmm.010348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Takita J, Choi Y, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455:971–974. doi: 10.1038/nature07399. [DOI] [PubMed] [Google Scholar]
- Cheung NK, Dyer MA. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer. 2013;13:397–411. doi: 10.1038/nrc3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung NK, Zhang J, Lu C, Parker M, Bahrami A, Tickoo SK, Heguy A, Pappo AS, Federico S, Dalton J, et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA. 2012;307:1062–1071. doi: 10.1001/jama.2012.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, Yatabe Y, Takeuchi K, Hamada T, Haruta H, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363:1734–1739. doi: 10.1056/NEJMoa1007478. [DOI] [PubMed] [Google Scholar]
- De Brouwer S, De Preter K, Kumps C, Zabrocki P, Porcu M, Westerhout EM, Lakeman A, Vandesompele J, Hoebeeck J, Van Maerken T, et al. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin Canc Res. 2010;16:4353–4362. doi: 10.1158/1078-0432.CCR-09-2660. [DOI] [PubMed] [Google Scholar]
- Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39(Database issue):D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, Michellys PY, Awad MM, Yanagitani N, Kim S, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014;4:662–673. doi: 10.1158/2159-8290.CD-13-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George R, Sanda T, Hanna M, Fröhling S, Luther W. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455:975–978. doi: 10.1038/nature07397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnad F, Baucom A, Mukhyala K, Manning G, Zhang Z. Assessment of computational methods for predicting the effects of missense mutations in human cancers. BMC Genomics. 2013;14(Suppl 3):S7. doi: 10.1186/1471-2164-14-S3-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer. 2013;13:685–700. doi: 10.1038/nrc3580. [DOI] [PubMed] [Google Scholar]
- Hobbie WL, Moshang T, Carlson CA, Goldmuntz E, Sacks N, Goldfarb SB, Grupp SA, Ginsberg JP. Late effects in survivors of tandem peripheral blood stem cell transplant for high-risk neuroblastoma. Pediatr Blood Cancer. 2008;51:679–683. doi: 10.1002/pbc.21683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, Mori S, Ratzkin B, Yamamoto T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 1997;14:439–449. doi: 10.1038/sj.onc.1200849. [DOI] [PubMed] [Google Scholar]
- Janoueix-Lerosey I, Lequin D, Brugières L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455:967–970. doi: 10.1038/nature07398. [DOI] [PubMed] [Google Scholar]
- Johnson TW, Richardson PF, Bailey S, Brooun A, Burke BJ, Collins MR, Cui JJ, Deal JG, Deng YL, Dinh D, et al. Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J Med Chem. 2014;57:4720–4744. doi: 10.1021/jm500261q. [DOI] [PubMed] [Google Scholar]
- Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM, Shakespeare WC, Iafrate AJ, Engelman JA, Shaw AT. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci, USA. 2011;108:7535–7540. doi: 10.1073/pnas.1019559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson AG, Jr, Strong LC. Mutation and cancer: neuroblastoma and pheochromocytoma. Am J Hum Genet. 1972;24:514–532. [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SHI, Dezube BJ, Jänne PA, Costa DB, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CC, Jia Y, Li N, Sun X, Ng K, Ambing E, Gao MY, Hua S, Chen C, Kim S, et al. Crystal structure of the ALK (anaplastic lymphoma kinase) catalytic domain. Biochem J. 2010;430:425–437. doi: 10.1042/BJ20100609. [DOI] [PubMed] [Google Scholar]
- Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010;362:2202–2211. doi: 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Vila MA, Nabau-Moretó N, Tornador C, Sabnis AJ, Rosell R, Estivill X, Bivona T, Marino-Buslje C. Activating mutations cluster in the “molecular brake” regions of protein kinases and do not associate with conserved or catalytic residues. Hum Mutat. 2014;35:318–328. doi: 10.1002/humu.22493. [DOI] [PubMed] [Google Scholar]
- Morowitz M, Shusterman S, Mossé Y, Hii G, Winter CL, Khazi D, Wang Q, King R, Maris JM. Detection of single-copy chromosome 17q gain in human neuroblastomas using real-time quantitative polymerase chain reaction. Modern Pathology. 2003;16:1248–1256. doi: 10.1097/01.MP.0000097364.64566.81. [DOI] [PubMed] [Google Scholar]
- Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1994;263:1281–1284. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- Morris SW, Naeve C, Mathew P, James PL, Kirstein MN, Cui X, Witte DP. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin's lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK) Oncogene. 1997;14:2175–2188. doi: 10.1038/sj.onc.1201062. [DOI] [PubMed] [Google Scholar]
- Mossé YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455:930–935. doi: 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossé YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, Rolland D, Balis FM, Maris JM, Weigel BJ, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children's Oncology Group phase 1 consortium study. Lancet Oncol. 2013;14:472–480. doi: 10.1016/S1470-2045(13)70095-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Liu Y, Lemmon MA, Radhakrishnan R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem J. 2012;448:417–423. doi: 10.1042/BJ20121513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson AD, Pinkerton CR, Lewis IJ, Imeson J, Ellershaw C, Machin D European Neuroblastoma Study Group, and Children's Cancer and Leukaemia Group. High-dose rapid and standard induction chemotherapy for patients aged over 1 year with stage 4 neuroblastoma: a randomised trial. Lancet Oncol. 2008;9:247–256. doi: 10.1016/S1470-2045(08)70069-X. [DOI] [PubMed] [Google Scholar]
- Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, Fukami TA, Oikawa N, Tsukuda T, Ishii N, Aoki Y. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011;19:679–690. doi: 10.1016/j.ccr.2011.04.004. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Okuda K, Zheng W, Butrynski J, Capelletti M, Wang L, Gray NS, Wilner K, Christensen JG, Demetri G, et al. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010;70:10038–10043. doi: 10.1158/0008-5472.CAN-10-2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönherr C, Ruuth K, Eriksson T, Yamazaki Y, Ottmann C, Combaret V, Vigny M, Kamaraj S, Palmer RH, Hallberg B. The neuroblastoma ALK(I1250T) mutation is a kinase-dead RTK in vitro and in vivo. Transl Oncol. 2011a;4:258–265. doi: 10.1593/tlo.11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönherr C, Ruuth K, Yamazaki Y, Eriksson T, Christensen J, Palmer RH, Hallberg B. Activating ALK mutations found in neuroblastoma are inhibited by crizotinib and NVP-TAE684. Biochem J. 2011b;440:405–413. doi: 10.1042/BJ20101796. [DOI] [PubMed] [Google Scholar]
- Shaw AT, Engelman JA. ALK in lung cancer: past, present, and future. J Clin Oncol. 2013;31:1105–1111. doi: 10.1200/JCO.2012.44.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AJ, Telesco SE, Choi SH, Lemmon MA, Radhakrishnan R. Molecular dynamics analysis of conserved hydrophobic and hydrophilic bond-interaction networks in ErbB family kinases. Biochem J. 2011;436:241–251. doi: 10.1042/BJ20101791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Seibel NL, Altekruse SF, Ries LA, Melbert DL, O'Leary M, Smith FO, Reaman GH. Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol. 2010;28:2625–2634. doi: 10.1200/JCO.2009.27.0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay KK, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363:1324–1334. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Wang F, Keats J, Zhu X, Ning Y, Wardwell SD, Moran L, Mohemmad QK, Anjum R, Wang Y, et al. Crizotinib-resistant mutants of EML4-ALK identified through an accelerated mutagenesis screen. Chem Biol Drug Des. 2011;78:999–1005. doi: 10.1111/j.1747-0285.2011.01239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Native Gel Analysis of ALK TKD Mutants.
(related to Figure 1)
Figure S2: Catalytic Efficiencies of ALK TKD Variants.
(related to Figure 3)
Figure S3: The L1196M Mutant is Crizotinib-Resistant Despite its high KM, ATP.
(related to Table 3)
Table S1: Genomic Coordinates of Mutations Identified (and Resulting Amino Acid Substitutions).
(related to Figure 1)
Table S2: ALK Mutation and Amplification Status by NB Risk Group and Age.
(related to Figure 2)
Table S3: ALK Aberrations by Risk Group.
(related to Table 2)
Table S4: Kinetic Properties of Non-phosphorylated ALK TKD Variants.
(related to Figure 3)
Table S5: Kinetic Properties of Phosphorylated ALK TKD Variants.
(related to Figure 3)
Table S6: Quantitation of Focus Formation Assays for Intact ALK Variants in NIH 3T3 Cells.
(related to Figure 4)