

Abstract
The genotype-phenotype map is an essential object to understand organismal complexity and adaptability. However, its experimental characterisation is a daunting task. Thus, simple models have been proposed and investigated. They have revealed that genotypes differ in their robustness to mutations; phenotypes are represented by a broadly varying number of genotypes, and simple point mutations suffice to navigate the space of genotypes while maintaining a phenotype. Nonetheless, most current models focus only on one level of the map (folded molecules, gene regulatory networks, or networks of metabolic reactions), so that many relevant questions cannot be addressed. Here we introduce toyLIFE, a multi-level model for the genotype-phenotype map based on simple genomes and interaction rules from which a complex behaviour at upper levels emerges —remarkably plastic gene regulatory networks and metabolism. toyLIFE is a tool that permits the investigation of how different levels are coupled, in particular how and where mutations affect phenotype or how the presence of certain metabolites determines the dynamics of toyLIFE gene regulatory networks. The model can easily incorporate evolution through more complex mutations, recombination, or gene duplication and deletion, thus opening an avenue to explore extended genotype-phenotype maps.
Describing and understanding the intricacies of the genotype-phenotype map counts amongst the most difficult and most essential issues to comprehend organismal complexity and adaptation through natural selection1. High-throughput data obtained from whole genome sequencing and other -omics techniques currently allow a characterisation with unprecedented detail of how genotypic variation affects phenotypes. The analysis of gene networks has demonstrated that phenotypes cannot be understood on the basis of isolated genes2, and that the effects of mutations strongly depend on a genetic background that is expressed at different levels before generating a final phenotype3. While simple point mutations may affect more than one gene4, phenotypes, overall, tend to be extremely robust: populations may sustain a high level of cryptic variation that acts at once as a buffering mechanism5 and as a reservoir of variability to promote rapid adaptation6. An essential part of our improved understanding of the concepts, design principles and general mechanisms underlying the appearance of biological function from organismal genomes arises from the use of in silico tools and models7.
The neutral theory of evolution8,9 posits that most mutations have no, or very little, effect on phenotypes, and are thus ignored by natural selection. This is an amply supported fact, though the level at which mutations cease to have an effect is a matter of research. DNA is translated into proteins and ribozymes which fold into three-dimensional structures. These molecules bind to each other and to the genome itself, enhancing or inhibiting the expression of genes —hence forming highly complex regulatory networks—, and eventually interact with metabolites to produce the metabolic pathways that sustain cellular life10. Redundancy appears at all these levels. Besides the well-known redundancy of the genetic code, many different aminoacid11 —in the case of proteins— or RNA12 —in the case of ribozymes— sequences fold into equivalent three-dimensional structures and exhibit similar interaction sites, thus maintaining their functions. Regulatory and metabolic networks are quite robust to additions, eliminations or substitutions of some of their components as well. For instance, regulatory regions with similar transcriptional output often have little overt sequence similarity, both within and between genomes13. Also, regulatory DNA sequences in different Drosophila species exhibiting the same expression patterns are not conserved14. As of robustness of metabolic networks, one-gene knockout experiments with Saccharomyces cerevisiae show that around 50% of mutants present a selective disadvantage below 1% relative to the wildtype15. Similar results have been obtained with Escherichia coli16.
The huge number of genomic solutions ushering in the same phenotype leads to the concept of genotype networks, that is ensembles of genotypes that yield the same phenotype and can be mutually accessed through mutations17. Genotype networks often traverse the whole space of genotypes, and are highly interwoven: virtually any phenotype is just a few mutations away from any other. These networks reflect the robustness of phenotypes against mutations, and their structure is essential to promote adaptability and evolutionary innovation18,19. Most of our knowledge on the topology of genotype networks relies on information obtained from well-motivated computational models that map genotype onto a simplified phenotype. Classical examples mapping sequence to molecular structure (which acts as a proxy for phenotype) are those of RNA20 or proteins folded through algorithms of variable complexity21,22. Other models have addressed the map between higher expression levels, as those mimicking gene regulatory networks23,24,25 or metabolism26.
Despite the significant conceptual advances provided by those models, there are two crucial elements of the genotype-phenotype map that they disregard: the existence of a hierarchy of expression levels between genotype and phenotype and the bi-directional coupling among the levels. Studies focusing on RNA or proteins assume that the molecular function is mostly determined by their spatial structure. This makes sense for some very specific enzymes27 but, in general, these molecules are pieces of complex regulatory or metabolic networks. Further, molecular interactions are not considered (see Ref. 28 for an exception modelling the quaternary structure of proteins), and there is no representation of the molecular context29. Therefore, cases where a protein may act as an enhancer of the expression of a gene by sticking to its promoter, but may become sequestered and thus inactivated in the presence of another protein are impossible to embody in one-molecule models, among many others.
In turn, models considering higher levels typically disregard the dynamics of underlying sequences. Gene regulatory networks are represented in an effective way through direct interactions among their components, as in Boolean regulatory networks. In this case, gene states are binary variables which interact (enhancing or inhibiting the expression of interacting genes) to determine new states at a subsequent time step30,31. Boolean networks do not consider how mutations at the genome level propagate to upper levels, and only implement straight changes in the Boolean functions. The situation is similar with metabolic models that use the ensemble of metabolic reactions as genotypes, since the kind of mutations considered can thus only be the elimination or addition of reactives or full reactions26.
The current situation is that we lack a model that captures the essentials of the biology at all levels from genome to metabolisms, but which at the same time is sufficiently simple so as to provide useful answers and insights about the genotype-phenotype mapping. In this paper we make one such proposal, that we refer to as toyLIFE. toyLIFE is a model that contains simplified versions of genes, promoters, proteins, and metabolites, which interact with each other under the laws of a simplified chemistry. Besides introducing the model and showing examples of its rich phenomenology, we identify a number of emerging properties that toyLIFE shares with natural systems. Such are the existence of a large number of robust phenotypes, of common metabolic functions (which arise in the absence of any evolutionary fine-tuning), a space-covering map at the sequence-structure level (as observed in RNA and protein folding models) but a small fraction of metabolically functional genomes. Coupling among different levels restricts the diversity of possible Boolean functions, as well as the metabolites that can be broken. In the framework of toyLIFE, mutations can show their effects at different levels before affecting the phenotype, and functional molecules can be co-opted to fulfil different and not previously foreseen functions.
Results
Definition of toyLIFE
The basic building blocks of toyLIFE are toyNucleotides (toyN), toyAminoacids (toyA), and toySugars (toyS). Each block comes in two flavors: hydrophobic (H) or polar (P). Random polymers of basic blocks constitute toyGenes (formed by 20 toyN units), toyProteins (chains of 16 toyA units), and toyMetabolites (sequences of toyS units of arbitrary length). These elements of toyLIFE are defined on the two-dimensional space (Figure 1A).
Figure 1. Building blocks and interactions defining toyLIFE.

(A): The three basic building blocks of toyLIFE are toyNucleotides, toyAminoacids, and toySugars. They can be hydrophobic (H, white) or polar (P, red), and their random polymers constitute toyGenes, toyProteins, and toyMetabolites. (B): Folding of a toyProtein. When a toyGene is expressed, its coding region is translated into a sequence of toyAminoacids, which folds on a 4 × 4 lattice following a self-avoiding walk. As a result, the toyProtein acquires a folding energy, which is the sum of the interaction energies between non-contiguous toyAminoacids of the chain (one, two or three, with energies ranging from 0 to −2). Interaction energy is pairwise additive. A toyProtein folds into the structure that minimises this folding energy. If two structures have the same minimal folding energy, the one with the minimum number of H toyAminoacids on its perimeter is chosen; if this number also coincides, the toyProtein does not fold. toyProteins are therefore characterised by two traits: their perimeter and their folding energy. (C): Possible interactions between pairs of toyLIFE elements. toyGenes interact through their promoter region with toyProteins (including the toyPolymerase and toyDimers); toyProteins can bind to form toyDimers, and interact with the toyPolymerase when bound to a promoter; both toyProteins and toyDimers can bind a toyMetabolite at arbitrary regions along its sequence.
toyGenes
toyGenes are composed of a 4-toyN promoter region followed by a 16-toyN coding region. There are 24 different promoters and 216 coding regions, leading to 220 ≈ 106 toyGenes. An ensemble of toyGenes forms a genotype. If the toyGene is expressed, it will produce a chain of 16 toyA that represents a toyProtein. Translation follows a straightforward rule: H (P) toyN translate into H (P) toyA.
toyProteins
toyProteins correspond to the minimum energy, maximally compact folded structure of the 16 toyA chain arising from a translated gene. Their folded configuration is calculated through the hydrophobic-polar (HP) protein lattice model21 (see Figure 1B). The possible folds are limited to compact 4 × 4 structures on a lattice. There are 38 such structures ignoring symmetries. The energy of a fold is the sum of all pairwise interaction energies between toyA that are not contiguous along the sequence. Pairwise interaction energies stand for the decrease in free energy when HH and HP bonds are formed: EHH = −2, EHP = −0.3, respectively (with EPP = 0), as in Ref. 32. The structure of a toyProtein is its lowest energy fold. If there is more than one fold with the same minimum energy, we select the one with fewer H toyA in the perimeter. If there is still more than one fold fulfilling both conditions, we discard that toyProtein by assuming that it is intrinsically disordered and thus non-functional33. Out of 216 = 65,536 possible toyProteins, 36,642 do not yield unique folds. Among the rest, we find 2,181 different toyProteins —a toyProtein is fully characterised by its folding energy and its perimeter (see below)— with 322 different perimeters.
Molecular interactions in toyLIFE
toyProteins interact through any of their sides with other toyProteins, with promoters of toyGenes, and with toyMetabolites (see Figure 1C). When toyProteins bind to each other, they form a toyDimer, which is the only protein aggregate considered in toyLIFE. The two toyProteins disappear, leaving only the toyDimer. Once formed, toyDimers can also bind to promoters or toyMetabolites through any of their sides —binding to other toyProteins or toyDimers, however, is not permitted. In all cases, the interaction energy (Eint) is the sum of pairwise interactions for all HH, HP and PP pairs formed in the contact —these interactions follow the rules of the HP model as well. Bonds can be created only if the interaction energy between the two molecules Eint is lower than a threshold energy Ethr = −2.6. Note that a minimum binding energy threshold is necessary to avoid the systematic interaction of any two molecules. Other alternatives might be the addition of terms that represent an energetic cost (in other models, as in RNA folding, the threshold is set to 0 because structural elements such as loops or dangling ends yield positive contributions to the total folding energy) or the consideration of stochastic interactions, such that those with higher energy would be less probable. If below threshold, the total energy of the resulting complex is the sum of Eint plus the folding energy of all toyProteins involved. The lower the total energy, the more stable the complex. When several toyProteins or toyDimers can bind to the same molecule, only the most stable complex is formed. Consistently with the assumptions for protein folding, when this rule does not determine univocally the result, no binding is produced (some exceptions apply though; see Methods for details on disambiguation rules).
In the toyLIFE universe, only the folding energy and perimeter of a toyProtein matter to characterise its interactions, so folded chains sharing these two features are indistinguishable. This is a difference with respect to the original HP model, where different inner cores defined different proteins and the composition of the perimeter was not considered as a phenotypic feature. However, subsequent versions of HP had already included additional traits34.
As the length of toyMetabolites is usually longer than 4 toyS (the length of interacting toyProteins sites), there might be several positions where the interaction with a toyProtein has the same energy. In those cases we select the sites that yield the most centered interaction. If ambiguity persists between different sides of the same toyProtein, no bond is formed. Also, no more than one toyProtein/toyDimer is allowed to bind to the same toyMetabolite, even if its length would permit it. toyProteins/toyDimers bound to toyMetabolites cannot bind to promoters.
Dynamics in toyLIFE
Expression of toyGenes occurs through the interaction with the toyPolymerase, which is a special kind of toyProtein (see Figure 1A). The toyPolymerase only has one interacting side (with sequence PHPH) and its folding energy is fixed to value −11. It is always present in the system. The toyPolymerase binds to promoters or to the right side of a toyProtein/toyDimer already bound to a promoter. When the toyPolymerase binds to a promoter, translation is directly activated and the corresponding toyGene is expressed. However, a more stable (lower energy) binding of a toyProtein or toyDimer to a promoter precludes the binding of the toyPolymerase. This inhibits the expression of the toyGene, except if the toyPolymerase binds to the right side of the toyProtein/toyDimer, in which case the toyGene can be expressed (Figure 2).
Figure 2. Regulatory and metabolic functions in toyProteins.

(A): A toyGene is expressed (translated) when the toyPolymerase binds to its promoter region. The sequence of Ps and Hs of the toyProtein will be exactly the same as that of the toyGene coding region. (B): If a toyProtein binds to the promoter region of a toyGene with a lower energy than the toyPolymerase does, it will displace the latter, and the toyGene will not be expressed. This toyProtein acts as an inhibitor. (C): The toyPolymerase does not bind to every promoter region. Thus, not all toyGenes are expressed constitutively. However, some toyProteins will be able to bind to these promoter regions. If, once bound to the promoter, they bind to the toyPolymerase with their rightmost side, the toyGene will be expressed, and these toyProteins act as activators. (D): More complex interactions —involving more elements— appear. For example, a toyProtein that forms a toyDimer with an inhibitor —preventing it from binding to the promoter— will effectively activate the expression of the toyGene. However, it does neither interact with the promoter region nor with the toyPolymerase, and its function is carried out only when the inhibitor is present. We call this kind of toyProteins conditional activators. (E): Two toyProteins can bind together to form a toyDimer that inhibits the expression of a certain toyGene. As they need each other to perform this function, we call them conditional inhibitors. As the number of genes increases, this kind of complex relationships can become very intricate. (F): Catabolism in toyLIFE. A toyDimer is bound to a toyMetabolite when a new toyProtein comes in. If the new toyProtein binds to one of the two units of the toyDimer, forming a new toyDimer energetically more stable than the old one, the two toyProteins will unbind and break the toyMetabolite up into two pieces. We say that the toyMetabolite has been catabolised.
The dynamics of the model proceeds in discrete time steps and variable molecular concentrations are not taken into account. A step-by-step description of toyLIFE dynamics is summarised in Figure 3. There is an initial set of molecules which results from the previous time step: toyProteins (including toyDimers and the toyPolymerase) and toyMetabolites, either endogenous or provided by the environment. These molecules first interact between them to form possible complexes (see previous section) and are then presented to a collection of toyGenes that is kept constant along subsequent iterations. Regulation takes place, mediated by a competition for binding the promoters of toyGenes, possibly causing their activation and leading to the formation of new toyProteins. Binding to promoters is decided in sequence. Starting with any of them (the order is irrelevant), it is checked whether any of the toyProteins/toyDimers available bind to the promoter —remember that complexes bound to toyMetabolites are not available for regulation—, and then whether the toyPolymerase can subsequently bind to the complex and express the accompanying coding region. If it does, the toyGene is marked as active and the toyProtein/toyDimer is released. Then a second promoter is chosen and the process repeated, until all promoters have been evaluated. toyGenes are only expressed after all of them have been marked as either active or inactive. Each expressed toyGene produces one single toyProtein molecule. There can be more units of the same toyProtein, but only if multiple copies of the same toyGene are present.
Figure 3. Dynamics of toyLIFE.

Input molecules at time step t are toyProteins (Ps) (including toyDimers (Ds)) and toyMetabolites, either produced as output at time step t − 1 or environmentally supplied (all toyMetabolites denoted Ms). Ps and Ds interact with Ms to produce complexes P-M and D-M. Next, these complexes, the remaining Ps and Ds, and the toyPolymerase (Pol) interact with toyGenes (G) at the regulation phase. The most stable complexes with promoters are formed (Pol-G, P-G and D-G), activating or inhibiting toyGenes. P-Ms and D-Ms do not participate in regulation. Ps and Ds not in complexes are eliminated and new Ps (dark grey) are formed. These Ps interact with all molecules present and form Ds, new P-M and D-M complexes, and catabolise old D-M complexes. At the end of this phase, all Ms not bound to Ps or Ds are returned to the environment, and all Ps and Ds in P-M and D-M complexes unbind and are degraded. The remaining molecules (Ms just released from complexes, as well as all free Ps and Ds) go to the input set of time step t + 1.
toyProteins/toyDimers not bound to any toyMetabolite are eliminated in this phase. Thus, only the newly expressed toyProteins and the complexes involving toyMetabolites in the input set remain. All these molecules interact yet again, and here is where catabolism can occur. Catabolism happens when, once a toyMetabolite-toyDimer complex is formed, an additional toyProtein binds to one of the units of the toyDimer with an energy that is lower than that of the initial toyDimer. In this case, the latter disassembles in favor of the new toyDimer, and in the process the toyMetabolite is broken (see Figure 2F for an illustration of the catabolism process). The two pieces of the broken toyMetabolites will contribute to the input set at the next time step, as will free toyProteins/toyDimers. However, toyProteins/toyDimers bound to toyMetabolites disappear in this phase —they are degraded—, and only the toyMetabolites are kept as input to the next time step. Unbound toyMetabolites are returned to the environment. This way, the interaction with the environment happens twice in each time step: at the beginning and at the end of the cycle.
toyProteins behave as toyGene switches
The minimal interaction rules that define toyLIFE dynamics endow toyProteins with a set of possible activities not included a priori in the rules of the model (see Figure 2). For example, since the 4-toyN interacting site of the toyPolymerase cannot bind to all promoter regions —because some of these interactions have Eint > Ethr—, translation mediated by a toyProtein or toyDimer binding might allow the expression of genes that would otherwise never be translated. These toyProteins thus act as activators. This process finds a counterpart in toyProteins that bind to promoter regions more stably than the toyPolymerase does, and therefore prevent gene expression. They are acting as inhibitors. There are two additional functions that could not be foreseen and involve a larger number of molecules. A toyProtein that forms a toyDimer with an inhibitor —preventing its binding to the promoter— effectively behaves as an activator for the expression of the toyGene. However, it interacts neither with the promoter region nor with the toyPolymerase, and its activating function only shows up when the inhibitor is present. This toyProtein thus acts as a conditional activator. On the other hand, two toyProteins can bind together to form a toyDimer that inhibits the expression of a particular toyGene. As the presence of both toyProteins is needed to perform this function, they behave as conditional inhibitors. This flexible, context-dependent behaviour of toyProteins, permits the construction of toy Gene Regulatory Networks (toyGRNs).
Gene regulatory networks in toyLIFE are deterministic Boolean networks
Molecular interactions and dynamical rules in toyLIFE can be translated into toyGRN that behave as deterministic Boolean networks30,31. The corresponding Boolean variables are the states (expressed or not expressed) of toyGenes. These variables are transformed through Boolean functions that represent the dynamical rules described, having as input current toyGene states and as output their states at the next time step. Boolean functions depend on the toyProteins present in the system and on the functions they perform. Through iteration of the Boolean map one can characterise the set of attractors of the dynamics and the corresponding basins of attraction.
If the initial set is formed by k genes, we should consider 2k different possible vectors of dimension k that correspond to the initial states (i.e. all combinations of genes being expressed (1) or not expressed (0)). First, the presence of possible toyDimers coming from expressed genes is evaluated, and then their interactions with promoter regions (in competition or cooperation with the toyPolymerase and other toyProteins) are evaluated. This yields an updated set of expressed toyGenes (a different state) to which the previous rules are again applied. In this way, one can construct a truth table that can be subsequently represented in the form of a directed graph (indicating which state maps into which other) and is fully analogous to a deterministic Boolean network. An example of a Boolean network derived from a system of three genes is represented in Figure 4.
Figure 4. Example of a Boolean network produced by toyLIFE rules.

The inputs of the truth table (possible initial states) are all combinations of states of three toyGenes. Whenever a toyGene is active, the protein it codes for is present. The main panel schematically represents all relevant interactions between molecules: in this case the toyPolymerase may bind to the promoter regions of toyGenes 1 and 2 (+ signs), and toyProtein 1 inhibits the expression of toyGene 2 (− signs). The simultaneous presence of toyProteins 1 and 3 leads to toyDimer 1–3, and the simultaneous presence of toyProteins 1 and 2 to toyDimer 1–2. Both toyDimers inhibit the expression of toyGene 2 and activate the expression of toyGene 3. The construction of the Boolean functions codified in the truth table is straightforward given the interactions conditional on presence or absence of each toyProtein. The truth table maps every possible initial state (gi) to its corresponding regulatory output (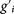 ). When the truth table is represented as a directed graph (summarising the dynamics of the system from all possible initial conditions) it is seen that there are two attractors for the dynamics: (1, 0, 1), whose basin of attraction has size 7, and (1, 0, 0), whose basin of attraction has size 1. (Note that the order of toyGenes in a genome is irrelevant, and only responds to aesthetic reasons.)
). When the truth table is represented as a directed graph (summarising the dynamics of the system from all possible initial conditions) it is seen that there are two attractors for the dynamics: (1, 0, 1), whose basin of attraction has size 7, and (1, 0, 0), whose basin of attraction has size 1. (Note that the order of toyGenes in a genome is irrelevant, and only responds to aesthetic reasons.)
Boolean networks of toyLIFE depend on metabolism
The presence of toyMetabolites may modify toyGRNs by changing the output states of the corresponding Boolean network (Figure 5). According to the dynamical rules of toyLIFE, toyMetabolites may interact with toyProteins or toyDimers. Any molecule bound to a toyMetabolite is no longer available to bind to promoters, and therefore the expression of the toyGRN is modified. An example of how a toyGRN might change can be derived from Figure 4: if a toyMetabolite able to bind to toyDimer 1–3 is added to the input set, state (1, 0, 1) is mapped to (1, 1, 0) (Figure 5).
Figure 5. toyMetabolites change the expression of toyGRNs.

This is the same example illustrated in Figure 4, but with the addition of a toyMetabolite able to bind toyDimers 1–2 and 1–3. When these toyDimers bind to the toyMetabolite, they no longer participate in the regulation phase, and thus states (1, 0, 1), (1, 1, 0) and (1, 1, 1) are all mapped to state (1, 1, 0) in the presence of this toyMetabolite. In other words, the presence of the toyMetabolite changes three entries in the truth table, and therefore the associated Boolean network —whose asymptotic state is now a different one.
Metabolons
The behaviour just described prompts the identification of metabolically functional genotypes that we term metabolons. The term metabolon was first proposed by Paul A. Srere35 in 1985 to refer to a “supramolecular complex of sequential metabolic enzymes and cellular structural elements”, and is here used as a conceptual analogue. A metabolon in toyLIFE is an ensemble of toyGenes able to catabolise at least one toyMetabolite. In the example above, the three toyGenes are a basic metabolon that catabolises in particular the toyMetabolite used as example.
When the toyMetabolite is absent, the dynamics is described in Figure 4 and eventually converges to the steady state (1, 0, 1) —except if the initial state is (1, 0, 0). This state is however disturbed under a constant supply of toyMetabolites able to bind to toyDimer 1–3. In that case, toyGenes 1 and 2 are expressed in the next time step. toyProtein 1 is able to form a toyDimer with itself, binding to unit 1 of the toyDimer-toyMetabolite complex. This latter interaction (which forms toyDimer 1-1) is favored over toyDimer 1–3 and catabolism of the toyMetabolite occurs (see Figures 4 and 5). If at the next time step the two pieces of toyMetabolite are unable to interact with any of the toyProteins in the system, they are eliminated. The toyGRN of this example remains in the new steady state (1, 1, 0) —which is also able to catabolise— as long as toyMetabolites are supplied. The three toyGenes system returns to the former steady state (1, 0, 1) as soon as the external supply stops. A graphical summary of a metabolon in toyLIFE is provided in Supplementary Figure S1.
The genotype-phenotype map in toyLIFE
toyLIFE integrates several levels of complexity: genotypes (sequences of toyGenes) expressing toyProteins (first level) that interact among themselves and with promoters generating toyGRNs (second level), and interactions with the environment (third level) trough catabolism of toyMetabolites. Genotypes are easily identified as the sequences of Hs and Ps making up toyGenes. In toyLIFE, the visible expression of the genotype is best represented through its interaction with the environment, that is with toyMetabolites. Accordingly, the phenotype of a genotype (a collection of toyGenes) is defined as the ensemble of toyMetabolites it can catabolise, once it has reached the attractor of the Boolean dynamics, starting from the initial state (0, 0, 0). If the attractor is a cycle, we consider that a genotype is able to catabolise a toyMetabolite if it can break it in any of the steps of the cycle. For this paper, we will only focus on toyMetabolites of size 8, although different lengths can be considered. There are 28 = 256 different toyMetabolites of size 8, and a genotype can either catabolise (in which case it is a metabolon) or not each one of them. The phenotype is formally defined as a vector of dimension 256 whose components take value 1 at those positions corresponding to toyMetabolites that can be catabolised, and value 0 otherwise. This definition is analogous to others in the literature where metabolic activity is explicitly modelled26.
Point mutations might affect different levels
As it has been defined, there are no mutations of toyGenes explicitly considered in the dynamics of toyLIFE. The initial sequences of toyGenes remain constant as we study properties of the emerging toyGRN and related phenotypes. This nonetheless, those conditions do not prevent an analysis of the effect of mutations in the phenotype. Actually, an interesting product of the multi-level structure of toyLIFE is the possibility of determining at which level is the effect of point mutations observed. Point mutations are changes from a P toyN to an H toyN, or vice versa, in the sequence of a toyGene.
For our present purposes we will focus on the effects of point mutations on the ability of genotypes to catabolise more or less toyMetabolites. Beneficial (accordingly, deleterious) mutations are defined as those mutations enabling the genotype to catabolise more (less) toyMetabolites than before. Lethal mutations transform the metabolon into a genotype that is unable to catabolise any toyMetabolite. The fitness effects of these mutations will depend on the evolutionary dynamics, and we will not consider them here, as they lie outside the aims of this article. A summary of changes caused by point mutations in the metabolon in Figure 4 can be found in Table 1. A mutation causing a change in the perimeter of a toyProtein can leave other functions unchanged, or modify Boolean functions in different ways which might eventually cause —or not— a phenotypic change. Out of the 60 possible mutations (12 affecting the promoters and 48 affecting the coding regions), 8.3% are neutral, 88.3% are deleterious, and only 3.3% are beneficial. Out of 53 deleterious mutations, 50 are lethal (that is, 94.3% of the total). This is a very high percentage of lethal mutations, compared with an average metabolon —the average percentage of lethal mutations, in 104 metabolons chosen at random, is 52.4%. However, note that this metabolon is not special in any way: in particular, since it is not a product of evolution and selection, it needs not have high robustness a priori. The exploration of genotype space through neutral paths can likely lead to metabolons with specific properties, as a higher number of neutral neighbors or a decreased effect of mutations on phenotype.
Table 1. Effect of point mutations in the genotype of the example metabolon shown in Figure 4.
| Effect of mutations | Tot | Neu | Adv | Del | Let |
|---|---|---|---|---|---|
| Different toyProtein folding (same perimeter & Boolean function) | 1 | 1 | 0 | 0 | 0 |
| Different toyProtein folding & perimeter (same Boolean function) | 3 | 1 | 0 | 2 | 0 |
| Different toyProtein folding & perimeter & Boolean function | 44 | 0 | 2 | 42 | 41 |
| Changes in Boolean functions due to the promoter | 12 | 3 | 0 | 9 | 9 |
| All | 60 | 5 | 1 | 53 | 50 |
The table shows the total number (Tot) of mutations causing each effect, and how many of these mutations are neutral (Neu), advantageous (Adv), deleterious (Del) and lethal (Let).
Functional properties of three-toyGenes genotypes
The genotype-phenotype map in toyLIFE is highly redundant and displays ample variations in the number of genotypes representing the same phenotype. Redundancy comes not only from neutral mutations, but also from the existence of compensatory mutations and genomic solutions with mutations in many toyN that yield the same phenotype. The redundancy of the HP model has been discussed in the literature32,36 and is, through the interaction rules of toyLIFE, non-trivially extended to the formation of molecular aggregates and catabolic processes. These are qualitative features that toyLIFE shares with natural systems and that we quantify in the following.
We begin by analysing the navigability of the genotype space. To this end we use metabolons similar to the one represented in Figure 4 and perform random walks on their neutral space. That is, we take three initial gene sequences at random, which form a genotype (or genome) of length 60. After making sure this genome is able to catabolise at least one toyMetabolite, we attempt a point mutation at a randomly chosen genome site. If the phenotype of the mutant is identical to that of the previous genome, the mutation is accepted; otherwise, the mutation is discarded and, in either case, the process is repeated. Mutations do not affect the toyPolymerase. The mutation process is attempted a variable number of times (that is, the random walks are of different lengths: 102, 103 or 104), and repeated for a large number of independent realisations (104 original genotypes). In this way, we obtain the histograms shown in Figure 6. The average number of accumulated substitutions, i.e. the Hamming distance between the original genome and the current one, grows with the number of mutations attempted, yielding genomes that increasingly differ from their ancestors. This behaviour is fully analogous to that observed in RNA secondary structure neutral networks12, in proteins37, and in one-level models of gene regulatory networks24 or metabolism26.
Figure 6. Histograms of Hamming distances from an ancestral genotype obtained through neutral paths.
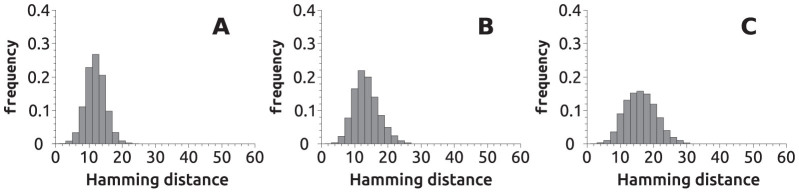
We chose a sample of 104 three-toyGene genotypes at random and, for each of them, computed a neutral random walk (see text). For each random walk, we then measured the Hamming distance between the final genotype and the original one. The histograms show the distribution of Hamming distances. We repeated this experiment with random walks of length 102 (A), 103 (B) and 104 (C). The average distance grows with the length of the random walks: from 12.4 (A) to 14.0 (B) to 16.6 (C). Note that the width of the distributions also grows with the length of the random walk.
Next, we have exhaustively explored the space of genotypes consisting of three toyGenes and evaluated their ability to break toyMetabolites of size 8. In total, there are around 8.1 × 1013 metabolons out of the total of ~ 2 × 1017 three-toyGenes genomes —the number of combinations of all possible toyGenes, 220, in groups of three, with repetitions. That is, only about 0.04% of all possible genomes are able to catabolise toyMetabolites of size 8. In agreement with the definition of phenotype given above, there are up to 2256 ≈ 1077 different phenotypes. However, only 11,981 different phenotypes can be realised by three-toyGene genomes, yielding an average close to 7 × 109 metabolons per phenotype. This average is however not very informative, since the variation in phenotype abundance is enormous (Figure 7A). There is also ample variability in the characteristics of phenotypes. Most toyMetabolites are broken by more than 1013 genomes, but some of them can be broken by far fewer genomes (see Figure 7B), and some toyMetabolites cannot be broken by any genome at all. Specifically, there are 20 toyMetabolites that cannot be broken. They have a particular composition or structure, since they contain 7 consecutive H or P sugars (there are 4 such toyMetabolites) or are palindromes (a total of 16 additional toyMetabolites). In both cases, only symmetrical toyDimers can bind to these toyMetabolites —asymmetrical toyDimers give rise to ambiguous interactions and are discarded. But symmetrical toyDimers bound to a given toyMetabolite cannot be broken by any toyProtein, because both subunits forming the toyDimer have the same perimeter and, again, this gives rise to ambiguous interactions.
Figure 7. Statistical properties of three-toyGene genotypes.

(A): Phenotype abundance. There are 11,981 different phenotypes with abundances that vary in eleven orders of magnitude. The phenotypes, in the x–axis, are rank ordered following the number of genotypes that express them. (B): Number of metabolons able to break a given toyMetabolite. The latter, in the x–axis, are rank-ordered following the number of genotypes that catabolise them. (C): Abundances of Boolean functions in phenotypes. Some Boolean functions are easy to obtain, while others are very rare. Again, Boolean functions are rank ordered according to the number of genotypes that express them.
Finally, many Boolean functions are obtained from different genotypes. For n genes, there are 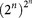 different Boolean functions, because for each of the 2n possible inputs there are 2n possible outputs. For three genes, this is already a very large number, 88 = 16,777,216 Boolean functions. These are reduced to 2,804,480 after discounting permutations of genes. Figure 7C represents the abundances of Boolean functions. As can be seen, there is a highly unequal representation in terms of genotypes, and only about 10% of all possible Boolean functions are actually represented by at least one genotype.
different Boolean functions, because for each of the 2n possible inputs there are 2n possible outputs. For three genes, this is already a very large number, 88 = 16,777,216 Boolean functions. These are reduced to 2,804,480 after discounting permutations of genes. Figure 7C represents the abundances of Boolean functions. As can be seen, there is a highly unequal representation in terms of genotypes, and only about 10% of all possible Boolean functions are actually represented by at least one genotype.
Discussion
Despite their simplicity, models of the genotype-phenotype map provide important conceptual insights. Not only that, some of them have been able to capture qualitative and quantitative features of the natural systems they aimed at representing. However important details might be, these are occasionally offset by universal rules that determine the emerging phenomenology and statistical behaviour both of biological systems and their in silico cartoons. For instance, the HP model of protein folding, which disregards the fine chemical structure of aminoacids and constrains HP polymers to fold on regular lattices, is able to predict the existence of unique folding states for sufficiently large polymers and the formation of hydrophobic cores, among others, in agreement with empirical knowledge38. Computational studies of RNA sequences folding into their minimal energy secondary structure have enlightened a large number of dynamical and structural properties with a clear empirical counterpart, such as punctuated equilibria at the molecular level39 or increases in robustness with phenotype size40, a feature that is quantitatively shared by all genotype-phenotype maps studied to date28,32,41,42. Boolean networks, despite working with a sharp threshold for gene expression, have witnessed notable success, including faithful reproduction of living cell cycles43. toyLIFE constructs a multi-level genotype-phenotype map from simple interactions inspired by the HP model from which the logical architecture of Boolean networks emerges. The addition of metabolic abilities arises as a natural extension of the basic model.
In devising the model here analysed, we had to make some choices regarding energy parameters, number of molecules or genes allowed to interact, or disambiguation rules to define functional molecules. We do not claim that toyLIFE matches biological reality, and it was not our intention to do so. The interaction and dynamical rules in toyLIFE were chosen so as to make the model as simple as possible, while retaining the essentials of molecular genetics. We aimed to explore universal features of complex molecular systems, regardless of the details. In that sense, although similar models with different rules might be devised, we would expect that many of them (if not most) would display a phenomenology comparable to the one here presented. The main principles behind the complex interactions between molecules, regulation and metabolism must be largely independent on these kind of details.
The possibilities of toyLIFE are not exhausted by the cases presented in this work, which constitute a minimal —hopefully illustrative— sample of the kind of complexity toyLIFE might encode for. Still, a deeper exploration of certain emergent behaviours seems worth pursuing. First, toyLIFE gives clues on the level —between genotype and phenotype— where the effect of mutations can be seen. Distance between phenotypes is simple to define in toyLIFE, and a more systematic analysis might allow as well a quantitative comparison with empirical studies measuring the distribution of fitness effects44. This function is an important object in developing models of phenotypic change that effectively incorporate the molecular details of evolution. Second, even the three-toyGenes genomes here studied reveal the emergence of functional abilities not implemented in the basic rules of the model, such as toyProteins behaving as conditional activators. This observation indicates that a protein can be recruited in appropriate molecular contexts to perform additional functions, that is, it can be co-opted to develop a second useful, but non-adaptive, role29. The consideration of larger genomes and larger molecular aggregates should certainly usher in new collective abilities, and very often lead to multi-functional toyProteins. In this scenario, the effect of single mutations might then arise at multiple levels, likely revealing a pleiotropic structure4 in the toyLIFE genotype-phenotype map. The effect of point mutations at different levels and the fraction of neutral or lethal mutations, among others, would be relevant issues to explore. It will also be interesting to study how different metabolons are fit together in a larger genome, developing more complex metabolic networks than the ones shown in this paper. Third, it would be worth comparing the statistical properties of random Boolean networks and other gene regulatory networks with those obtained from toyLIFE. An open and challenging question is how an explicit consideration of genome dynamics modifies or constrains the statistical properties of genotype-phenotype models that discard them. Fourth, for three-toyGenes genomes, we have observed a very high dilution of metabolons in comparison to genomes that cannot break any of the toyMetabolites considered. This result is in qualitative agreement with models of gene regulatory networks24 and metabolism26 that ignore lower levels. An open question is how this dilution changes as we increase the number of participating toyGenes and diversify the set of toyMetabolites that should be catabolised. At present, this study is severely limited by the computational time it requires. Finally, it is easy to implement additional mutational mechanisms in toyLIFE, such as gene duplication or deletion. The implications of such a change on the phenotype cannot be foreseen without an explicit analysis. However, we have found two-toyGenes metabolons whose function is maintained when a third toyGene is added. In this respect, toyLIFE might provide complementary insight on the evolutionary effects of gene duplication, including their lethality and their ability to develop new functions45,46.
Methods
Disambiguation rules in toyLIFE
Interaction rules in toyLIFE have been devised to remove any ambiguity. When more than one rule could be chosen, we opted for computational simplicity, having made sure that the general properties of the model remained unchanged. A detailed list of the specific disambiguation rules implemented in the model follows:
Folding rule: if a toyProtein can fold into two (or more) different configurations with the same energy and the same number of H in the perimeter, it is considered degenerate and does not fold.
One-side rule: any interaction in which a toyProtein can bind any ligand with two (or more) different sides and the same energy is discarded. As a result, for example, a toyProtein having four equal sides is not reactive.
Annihilation rule: if two (or more) toyProteins can bind a ligand with the same energy, the binding does not occur. However, if a third toyProtein can bind the ligand with greater (less stable) energy than the other two, and does so uniquely, it will bind it.
Identity rule: an exception to the Annihilation rule occurs if the competing toyProteins are the same. In this case, one of them binds the ligand and the other(s) remains free.
Stoichiometric rule: an extension of the Identity rule. If two (or more) copies of the same toyProtein/toyDimer/toyMetabolite are competing for two (or more) different ligands, there will be binding if the number of copies of the toyProtein/toyDimer/toyMetabolite equals the number of ligands.
For example, say that P1 binds to P2, P3 and P4 with the same energy. Then, (a) if P1, P2 and P3 are present, no complex will form; (b) if there are two copies of P1, dimers P1–P2 and P1–P3 will both form; but (c) if P4 is added, no complex will form. Conversely, if all ligands are copies as well, the Stoichiometry rule does not apply. For example, three copies of P1 and two copies of P2 will form two copies of dimer P1–P2, and one copy of P1 will remain free.
Author Contributions
All authors developed the model and discussed the results. CFA and PC carried out the numerical simulations. SM and JAC wrote the manuscript.
Supplementary Material
Supplementary Information
Acknowledgments
This work was supported through projects FIS2011-22449 (CFA, PC and JAC) and FIS2011–27569 (SM) of the Spanish MINECO.
References
- Lewis N. E., Nagarajan H. & Palsson B. O. Constraining the metabolic genotype-phenotype relationship using a phylogeny of in silico methods. Nat. Revs. Microbiol. 10, 291–305 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlebach G. & Shamir R. Modelling and analysis of gene regulatory networks. Nat. Revs. Mol. Cell. Biol. 9, 770–780 (2008). [DOI] [PubMed] [Google Scholar]
- Chandler C. H., Chari S. & Dworkin I. Does your gene need a background check? How genetic background impacts the analysis of mutations, genes, and evolution. Trends Genet. 29, 358–366 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner G. P. & Zhang J. The pleiotropic structure of the genotype-phenotype map: the evolvability of complex organisms. Nat. Revs. Genet. 10, 204–213 (2011). [DOI] [PubMed] [Google Scholar]
- Rutherford S. L. From genotype to phenotype: buffering mechanisms and the storage of genetic information. BioEssays 22, 1095–1105 (2000). [DOI] [PubMed] [Google Scholar]
- Paaby A. B. & Rockman M. V. Cryptic genetic variation: evolution's hidden substrate. Nat. Revs. Genet. 15, 247–258 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura B. D., Lemerle C., Michalodimitrakis K. & Serrano L. From in vivo to in silico biology and back. Nature 443, 527–533 (2006). [DOI] [PubMed] [Google Scholar]
- Kimura M. Evolutionary rate at the molecular level. Nature 217, 624–626 (1968). [DOI] [PubMed] [Google Scholar]
- Kimura M. The Neutral Theory of Molecular Evolution (Cambridge University Press, Cambridge, 1984). [Google Scholar]
- Watson J. D. et al. Molecular biology of the gene 7th ed. (Benjamin Cummings, San Francisco, 2013). [Google Scholar]
- Lipman D. J. & Wilbur W. J. Modelling neutral and selective evolution of protein folding. Proc. Roy. Soc. London B 245, 7–11 (1991). [DOI] [PubMed] [Google Scholar]
- Schuster P., Fontana W., Stadler P. F. & Hofacker I. L. From sequences to shapes and back: A case study in RNA secondary structures. Proc. Roy. Soc. London B 255, 279–284 (1994). [DOI] [PubMed] [Google Scholar]
- Weirauch M. T. & Hughes T. R. Conserved expression without conserved regulatory sequence: the more things change, the more they stay the same. Trends Genet. 26, 66–74 (2010). [DOI] [PubMed] [Google Scholar]
- Hare E. E., Peterson B., Iyer V. N., Meier R. & Eisen M. B. Sepsid even-skipped enhancers are functionally conserved in Drosophila despite lack of sequence conservation. PLoS Genet. 4, e1000106 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thatcher J., Shaw J. M. & Dickinson W. Marginal fitness contributions of nonessential genes in yeast. Proc. Natl. Acad. Sci. USA 95, 253–257 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T. et al. Construction of Escherichia coli k-12 in-frame, single-gene knockout mutants: the keio collection. Mol. Sys. Biol. 20, 2006.0008 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A. Genotype networks shed light on evolutionary constraints. Trends Ecol. Evol. 26, 577–584 (2011). [DOI] [PubMed] [Google Scholar]
- Draghi J. A., Parsons T. L., Wagner G. P. & Plotkin J. B. Mutational robustness can facilitate adaptation. Nature 463, 353–355 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A. The origins of evolutionary innovations (Oxford University Press, New York, 2011). [Google Scholar]
- Schuster P. Prediction of RNA secondary structures: From theory to models and real molecules. Rep. Prog. Phys. 69, 1419–1477 (2006). [Google Scholar]
- Dill K. A. Theory for the folding and stability of globular proteins. Biochemistry 24, 1501–1509 (1985). [DOI] [PubMed] [Google Scholar]
- Bastolla U., Vendruscolo M. & Knapp E.-W. A statistical mechanical method to optimize energy functions for protein folding. Proc. Natl. Acad. Sci. USA 97, 3977–3981 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman S. A. The origins of order: self-organization and selection in evolution (Oxford University Press, New York, 1993). [Google Scholar]
- Ciliberti S., Martin O. C. & Wagner A. Innovation and robustness in complex regulatory gene networks. Proc. Natl. Acad. Sci. USA 104, 13591–13596 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne J. L., Moore J. H. & Wagner A. Robustness, evolvability, and the logic of genetic regulation. Artificial Life 20, 111–126 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues J. F. M. & Wagner A. Evolutionary plasticity and innovations in complex metabolic reaction networks. PLoS Comp. Biol. 5(12), e1000613 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultes E. A. & Bartel D. P. One sequence, two ribozymes: implications for the emergence of new ribozyme folds. Science 289, 448–452 (2000). [DOI] [PubMed] [Google Scholar]
- Greenbury S. F., Johnston I. G., Louis A. A. & Ahnert S. E. A tractable genotype-phenotype map modelling the self-assembly of protein quaternary structure. J. Roy. Soc. Interface 6, 20140249 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatigorsky J. Gene sharing and evolution: the diversity of protein functions (Harvard University Press, Cambridge MA, 2007). [Google Scholar]
- Kauffman S. A. Metabolic stability and epigenesis in randomly constructed genetic nets. J. Theor. Biol. 22, 437–467 (1969). [DOI] [PubMed] [Google Scholar]
- Cheng D., Qi H. & Li Z. Analysis and control of boolean networks (Springer, New York, 2011). [Google Scholar]
- Li H., Helling R., Tang C. & Wingreen N. Emergence of preferred structures in a simple model of protein folding. Science 273, 666–669 (1996). [DOI] [PubMed] [Google Scholar]
- Radivojac P. et al. Intrinsic disorder and functional proteomics. Biophys. J. 92, 1439–1456 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque T., Chetty M. & Sattar A. Extended HP model for protein structure prediction. J. Comp. Biol. 16, 85–103 (2009). [DOI] [PubMed] [Google Scholar]
- Srere P. A. The metabolon. Trends Biochem. Sci. 10, 109–110 (1985). [Google Scholar]
- Holzgräfe C., Irbäck A. & Troein C. Mutation-induced fold switching among lattice proteins. J. Chem. Phys. 135, 195101 (2011). [DOI] [PubMed] [Google Scholar]
- Babajide A., Hofacker I. L., Sippl M. J. & Stadler P. F. Neutral networks in protein space: a computational study based on knowledge-based potentials of mean force. Fold. Des. 2, 261–269 (1997). [DOI] [PubMed] [Google Scholar]
- Lau K. F. & Dill K. A. A lattice statistical mechanics model of the conformational and sequence spaces of proteins. Macromolecules 22, 3986–3997 (1989). [Google Scholar]
- Huynen M. A., Stadler P. F. & Fontana W. Smoothness within ruggedness: The role of neutrality in adaptation. Proc. Natl. Acad. Sci. USA 93, 397–401 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre J., Buldú J. M., Stich M. & Manrubia S. C. Topological structure of the space of phenotypes: the case of RNA neutral networks. PLoS ONE 6, e26324 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom J. D., Raval A. & Wilke C. O. Thermodynamics of neutral protein evolution. Genetics 175, 255–266 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dall'Olio G. M., Bertranpetit J., Wagner A. & Laayouni H. Human genome variation and the concept of genotype networks. PLoS ONE 9, e99424 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidich M. I. & Bornholdt S. Boolean network model predicts cell cycle sequence of fission yeast. PLoS ONE 3, e1672 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker A. & Keightley P. D. The distribution of fitness effects of new mutations. Nat. Rev. Genet. 8, 610–618 (2007). [DOI] [PubMed] [Google Scholar]
- Lynch M. & Conery J. S. The evolutionary fate and consequences of duplicate genes. Science 290, 1151–1155 (2000). [DOI] [PubMed] [Google Scholar]
- Kondrashov F. A. Gene duplication as a mechanism of genomic adaptation to a changing environment. Proc. R. Soc. Lond. B 279, 5048–5057 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information