

Abstract
Non-alcoholic fatty liver disease (NAFLD) is a major cause of liver disease around the world. It includes a spectrum of conditions from simple steatosis to non-alcoholic steatohepatitis (NASH) and can lead to fibrosis, cirrhosis, liver failure, and/or hepatocellular carcinoma. NAFLD is also associated with other medical conditions such as obesity, diabetes mellitus (DM), metabolic syndrome, hypertension, insulin resistance, hyperlipidemia, and cardiovascular disease (CVD). In diabetes, chronic hyperglycemia contributes to the development of both macro- and microvascular conditions through a variety of metabolic pathways. Thus, it can cause a variety of metabolic and hemodynamic conditions, including upregulated advanced glycation end-products (AGEs) synthesis. In our previous study, the most abundant type of toxic AGEs (TAGE); i.e., glyceraldehyde-derived AGEs, were found to make a significant contribution to the pathogenesis of DM-induced angiopathy. Furthermore, accumulating evidence suggests that the binding of TAGE with their receptor (RAGE) induces oxidative damage, promotes inflammation, and causes changes in intracellular signaling and the expression levels of certain genes in various cell populations including hepatocytes and hepatic stellate cells. All of these effects could facilitate the pathogenesis of hypertension, cancer, diabetic vascular complications, CVD, dementia, and NASH. Thus, inhibiting TAGE synthesis, preventing TAGE from binding to RAGE, and downregulating RAGE expression and/or the expression of associated effector molecules all have potential as therapeutic strategies against NASH. Here, we examine the contributions of RAGE and TAGE to various conditions and novel treatments that target them in order to prevent the development and/or progression of NASH.
Keywords: Non-alcoholic fatty liver disease, Non-alcoholic steatohepatitis, Advanced glycation end-products, Toxic advanced glycation end-products, Receptor for advanced glycation end-products, Toxic advanced glycation end-products-receptor for advanced glycation end-products system, Diabetes mellitus, Cardiovascular disease, Dietary fructose, Dietary advanced glycation end-products
Core tip: Toxic advanced glycation end-products (TAGE) synthesis is increased by non-alcoholic steatohepatitis (NASH), and patients with NASH exhibit significantly increased serum and hepatic TAGE concentrations. Interactions between TAGE and the receptor for advanced glycation end-products (RAGE) have been suggested to cause oxidative stress and increase the fibrogenic potential of cultured human hepatic stellate cells. Therefore, TAGE signaling via RAGE and the resultant synthesis of reactive oxygen species might play a role in the worsening of hepatic pathology seen in NASH. These observations led us to suggest that extracellular and intracellular TAGE are involved in the pathogenesis of NASH.
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is a major cause of chronic liver disease in developed countries, and hence, is becoming a global public health issue[1]. NAFLD includes a range of conditions, from simple steatosis to non-alcoholic steatohepatitis (NASH)[2-4]. NASH has the potential to progress, which can result in cirrhosis, liver failure, and/or hepatocellular carcinoma[2-4]. NAFLD is regarded as a hepatic symptom of metabolic syndrome (MetS) and is associated with visceral obesity, abnormalities in glucose and lipid metabolism, insulin resistance (IR), and hypertension[5-7]. In NAFLD patients, underlying metabolic conditions such as those described above result in worsening liver dysfunction and a higher incidence of liver fibrosis and are also involved in the development of cardiovascular disease (CVD)[8,9].
Advanced glycation end-products (AGEs) might be involved in the mechanism that links NASH and diabetes mellitus (DM). Accumulating evidence indicates that in diabetic patients chronic hyperglycemia upregulates the production of AGEs (senescent macroprotein derivatives) via non-enzymatic glycation (the Maillard reaction). It has been demonstrated that the binding of AGEs to their receptor (RAGE) induces oxidative stress followed by inflammatory and/or thrombogenic responses in a variety of cell types. Furthermore, in diabetes such binding is considered to be involved in the pathogenesis and worsening of angiopathic conditions[10-16]. In our previous study, the most abundant type of toxic AGEs (TAGE); i.e., glyceraldehyde-derived AGEs (Glycer-AGEs), were found to make a significant contribution to the development of angiopathic conditions in DM[17-20]. In addition, there is a growing consensus that TAGE-RAGE interactions affect gene expression, intracellular signaling, and the secretion of pro-inflammatory factors and induce reactive oxygen species (ROS) production in various cell types including hepatic stellate cells (HSC) and hepatocytes[21,22]. Thus, TAGE-RAGE interactions might play a role in the pathological changes associated with lifestyle-related diseases, particularly NASH. TAGE synthesis is increased in NASH, and NASH patients were found to exhibit significantly higher hepatic and serum TAGE concentrations than individuals with simple steatosis or healthy controls[23]. TAGE-RAGE interactions have also been found to be associated with the induction of oxidative stress and increases in the fibrogenic potential of cultured human HSC[22]. Therefore, it is suggested that TAGE signaling through RAGE and the subsequent ROS production play a role in the worsening of hepatic pathology observed in NASH.
Accordingly, inhibiting the binding of TAGE to RAGE and TAGE synthesis and downregulating RAGE expression and/or the expression of its effectors have potential as treatment strategies for NASH. Here, we examine the contributions of RAGE and TAGE to various conditions and novel treatments that target these molecules in order to prevent the development and/or progression of NASH.
AGEs
The Maillard reaction, in which the N-terminal α-amino or ε-amino regions of protein lysine residues react non-enzymatically with the ketone or aldehyde moieties of reducing sugars, e.g., fructose, glucose, etc., is responsible for synthesizing AGEs. AGEs are known to be involved in protein aging and the pathological complications associated with DM[10-13,17-20,24-27]. In hyperglycemic DM patients, the first step in this process involves the conversion of reversible Schiff base adducts to more stable covalently bound Amadori rearrangement products, which subsequently undergo further rearrangement to produce irreversibly bound moieties (AGEs), and this process can range in duration from days to weeks.
Initially, AGEs were identified based on their fluorescent yellow-brown appearance and their ability to produce cross-links with and between amino groups. However, the term AGEs now refers to numerous products associated with the advanced stages of the glycation process, including N-(carboxyethyl)lysine, N-(carboxymethyl)lysine (CML), and pyrraline, which are colorless and can not form cross-links with proteins[24-29]. In vivo AGE production is affected by the sugar concentration, the rate of turnover of the chemically modified target, and the time available. Increases in the glucose concentration were previously considered to have a major influence on the Maillard reaction; however, glucose is one of the least reactive sugars found in biological organisms[24,30]. As well as extracellular AGE synthesis, the rapid intracellular production of AGEs from intracellular precursors such as trioses, dicarbonyl compounds, and fructose has been gaining attention[31,32]. Due to the great degree of variation in the structures of the AGEs found in vivo and the complex nature of the reactions required for their synthesis, only some AGEs have had their structures identified[33]. Furthermore, even the structures of cytotoxic AGEs are yet to be elucidated.
In a previous study, we found that α-hydroxyaldehydes (glycolaldehyde and glyceraldehyde), fructose, glucose, and dicarbonyl compounds (glyoxal and methylglyoxal, 3-deoxyglucosone) all contribute to protein glycation[27,34-37]. A total of 7 immunochemically distinct AGEs classes [methylglyoxal-derived AGEs; Glycer-AGEs; fructose-derived AGEs; glucose-derived AGEs (Glu-AGEs); 3-deoxyglucosone-derived AGEs; glyoxal-derived AGEs; and glycolaldehyde-derived AGEs] were found in serum samples collected from hemodialysis patients with type 2 DM (T2DM)[27,34-37]. Accordingly, we suggested that the in vivo formation of AGEs occurs via a process involving the Maillard reaction, sugar autoxidation, and sugar metabolism pathways (Figure 1).
Figure 1.
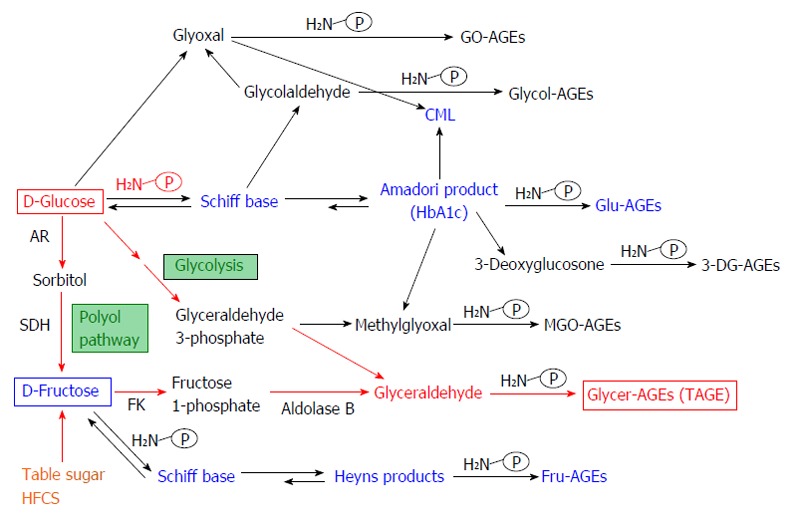
Alternative in vivo advanced glycation end-product synthesis routes. Reducing sugars, such as glucose, fructose and glyceraldehyde, are known to react non-enzymatically with the amino groups of proteins to form reversible Schiff bases and Amadori product/Heyns products. These early glycation products undergo further complex reactions such as rearrangement, dehydration, and condensation to become irreversibly cross-linked, heterogeneous fluorescent derivatives termed advanced glycation end-products (AGEs). Glu-AGEs: Glucose-derived AGEs; Fru-AGEs: Fructose-derived AGEs; Glycer-AGEs: Glyceraldehyde-derived AGEs; Glycol-AGEs: Glycolaldehyde-derived AGEs; MGO-AGEs: Methylglyoxal-derived AGEs; GO-AGEs: Glyoxal-derived AGEs; 3-DG-AGEs: 3-deoxyglucosone-derived AGEs; CML: N-(carboxymethyl)lysine; P-NH2: A free amino residue; HbA1c: Hemoglobin A1c; TAGE: Toxic AGEs; HFCS: High-fructose corn syrup; AR: Aldose reductase; SDH: Sorbitol dehydrogenase; FK: Fructokinase.
PATHWAY FOR THE IN VIVO SYNTHESIS OF GLYCER-AGEs
In vivo, two different pathways are responsible for glyceraldehyde (GLA) production, (1) the fructose metabolic pathway (fructolysis) and (2) the glycolytic pathway (glycolysis)[18-20,38]. In pathway (1) under hyperglycemic conditions a rise in the intracellular glucose concentration stimulates the production of fructose via the polyol pathway in insulin-independent tissues, such as nerve tissue, the kidneys, the lens of the eye, red blood cells, and the brain[39-42]. In addition, fructose is a constituent of sucrose and high-fructose corn syrup (HFCS), and hence, is included in many people’s diets[43,44]. Fructokinase phosphorylates fructose to fructose 1-phosphate, which is then broken down into dihydroxyacetone phosphate and GLA by aldolase B[45,46]. Next, the resultant GLA is transported (or leaks passively) across the cell membrane. GLA induces TAGE synthesis in the both intracellular and extracellular compartments; as for pathway (2) the enzyme glyceraldehyde 3-phosphate (G3P) dehydrogenase (GAPDH) usually breaks down the glycolytic intermediate G3P. However, reductions in GAPDH activity lead to the intracellular accumulation of G3P. As a result, G3P metabolism starts to occur via an alternative pathway, leading to a rise in the concentration of GLA, which promotes the synthesis of Glycer-AGEs, a major form of TAGE. This indicates that a positive feedback mechanism is in operation; namely, that the inhibition of GAPDH activity by TAGE promotes TAGE synthesis (Figure 2).
Figure 2.
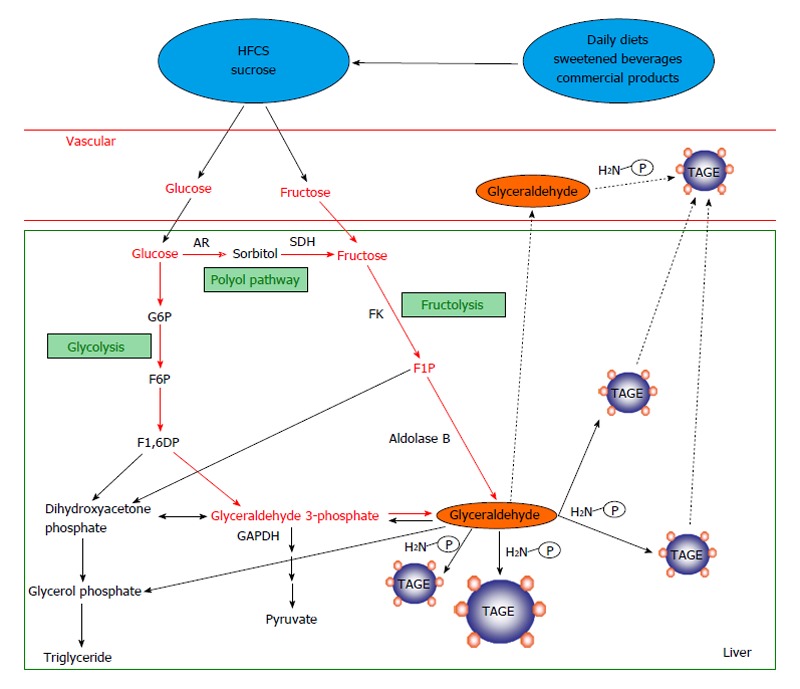
Routes for in vivo glyceraldehyde-derived advanced glycation end-products synthesis. The glycolytic intermediate glyceraldehyde 3-phosphate (G3P) is usually catabolized (glycolysis) by the enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH). However, reductions in GAPDH activity lead to the intracellular accumulation of G3P. As a result, G3P metabolism starts to occur via an alternative pathway, leading to a rise in the concentration of glyceraldehyde, which promotes the synthesis of TAGE. Fructokinase phosphorylates fructose to fructose 1-phosphate, which is then broken down into dihydroxyacetone phosphate and glyceraldehyde by aldolase B (fructolysis). The resultant glyceraldehyde is transported (or leaks passively) across the cell membrane. Glyceraldehyde promotes the formation of TAGE both intracellularly and extracellularly. AGEs: Advanced glycation end-products; TAGE: Toxic AGEs (glyceraldehyde-derived AGEs); HFCS: High-fructose corn syrup; AR: Aldose reductase; SDH: Sorbitol dehydrogenase; FK: Fructokinase; G6P: Glucose 6-phosphate; F6P: Fructose 6-phosphate; F1,6DP: Fructose 1,6-diphosphate; F1P: Fructose 1-phosphate; P-NH2: Free amino residue.
DIETARY FRUCTOSE
It is suspected that fructose is at least partially responsible for the obesity epidemic affecting developed countries. The greater prevalence of fructose in people’s diets results in greater glucose flux and elevated fructose metabolism in hepatocytes. Fructose used to be considered to be a beneficial dietary substance due to the fact it does not stimulate insulin secretion; however, as insulin signaling plays a key role in the development of NAFLD, this property of fructose might be undesirable[47-49]. In adolescents, increased fructose consumption is linked with various CVD risk factors. However, visceral obesity might be responsible for these associations. In the United States, fructose consumption is considered to be associated with the recent rise in the prevalence rates of obesity, fatty liver, and T2DM. The liver is extremely sensitive to variations in dietary content and plays the primary role in the metabolism of simple sugars, such as fructose and glucose[47,48].
The number of calories an individual consumes each day can have a significant influence on their risk of developing NAFLD because excessive energy intake results in obesity, leading to a greater risk of NAFLD. However, the development and progression of NAFLD are also affected by dietary composition. Of all carbohydrates, fructose plays an especially important role in NAFLD progression[50-53]. For example, it has been suggested that fructose consumption is associated with hepatic fat accumulation, fibrosis, and inflammation[54]. The accumulation of visceral adipose tissue and higher plasma triglyceride concentrations have also been linked with fructose consumption[55,56]. Thus, fructose has an important influence on the development of fatty liver disease[57].
Particular dietary sugars (especially fructose) are considered to play a role in the development and progression of NAFLD. The sugar additives (usually HFCS or sucrose) found in beverages and processed foods are widely viewed as the main source of the increased amounts of fructose consumed in developed countries. Dyslipidemia, obesity, and IR have all demonstrated strong associations with greater fructose consumption, and evidence indicating that fructose is involved in the development and progression of NAFLD is accumulating. Human studies have linked fructose consumption to hepatic fat accumulation, fibrosis, and inflammation. At present, it is unclear whether fructose can cause NAFLD on its own or whether it only promotes the condition when consumed in excessive amounts by individuals with a sedentary lifestyle, IR, and/or a positive energy balance. However, there is enough evidence to support a recommendation that the consumption of foods and drinks that are high in added fructose-containing sugars should be limited[54,58].
Although we need to increase our knowledge regarding the influence of fructose on NAFLD, the links between excessive fructose consumption and hypertriglyceridemia, IR, and the accumulation of visceral adipose tissue are sufficiently clear to support a clinical recommendation that NAFLD patients decrease the amount of fructose in their diets.
AGE RECEPTORS
A variety of signaling pathways are activated by AGE synthesis via a series of cell surface receptors. Among AGE receptors, the multi-ligand receptor RAGE has been studied most extensively[59-63]. In addition, various other AGE receptors such as AGE-receptor complexes (AGE-R1/OST-48, AGE-R2/80K-H, and AGE-R3/galectin-3)[64,65] and certain members of the scavenger receptor family (SR-A[66], SR-B:CD36[67,68], SR-BI[69], SR-E:LOX-1[70], FEEL-1, and FEEL-2[71]) have been reported. It was reported that the expression of these AGE receptors varies between different types of cells or tissues and is influenced by metabolic changes, e.g., changes associated with hyperlipidemia, DM, or aging[72]. In vivo and in vitro experiments examining the mechanisms responsible for the effects of AGEs and the factors that regulate their actions, e.g., soluble RAGE (sRAGE), it was suggested that these molecules have significant pathobiological effects[63,73]. A variety of different cell types, such as neurons, hepatocytes, endothelial cells (EC), HSC, microglia, and pericytes, express RAGE[59-61].
In recent in vitro and in vivo studies, we found that protein amino moieties readily react with GLA to produce TAGE[18-20]. Furthermore, TAGE induce vascular inflammation and ROS production, and hence, promote the development of atherosclerosis in DM[74,75]. As TAGE display the greatest affinity for RAGE[74,75] and the binding of TAGE to RAGE adversely affects the vasculature of diabetic patients[18-20], TAGE might contribute to the greater CVD incidence rates seen in DM patients and impaired glucose tolerance (IGT) patients that display postprandial hyperglycemia. Furthermore, we have recently reported that in DM patients TAGE make significant contributions to the pathogenesis of angiopathy[19,20]. Accumulating evidence indicates that TAGE-RAGE interactions induce oxidative stress in various cell types, such as HSC and hepatocytes.
THE TAGE-RAGE SYSTEM IS INVOLVED IN LIVER DISEASE
As for the effects of TAGE on hepatocytes, we demonstrated that in Hep3B cells, a human hepatocellular carcinoma cell line, TAGE-RAGE interactions upregulated the hepatic production of C-reactive protein (CRP) by activating Rac-1[76]. The latter study indicated that at least two CRP expression-inducing signaling pathways are in operation in TAGE-treated Hep3B cells: the nuclear factor kappa B (NF-κB)-Rac-1-induced signal transducer and activator of transcription 3-dependent pathway, which is not directly affected by ROS, and an NADPH oxidase-mediated ROS-dependent pathway involving Rac-1[76]. During the induction of CRP expression by TAGE, the early stages of the process might be ROS-independent, whereas the latter stages might involve a ROS-mediated pathway. In Hep3B cells, the phosphorylation of insulin receptor substrate-1 (IRS-1) at its serine-307 residue and of c-Jun N-terminal kinase (JNK), c-JUN, and IκB kinase were promoted by TAGE. The increased phosphorylation of IκB kinase was associated with reductions in the concentration of IκB[77]. These effects of TAGE on Hep3B cells were abrogated by the overexpression of the dominant negative form of Rac-1. Treatment with curcumin, an inhibitor of NF-κB, or a JNK inhibitor decreased the phosphorylation of IRS-1 at its serine-307 residue in Hep3B cells. In addition, TAGE downregulated the tyrosine phosphorylation of IRS-1, weakened the affinity of the p85 subunit of phosphatidylinositol 3-kinase for IRS-1, and decreased glycogen synthesis in insulin-treated Hep3B cells. All of these effects were abrogated by treatment with NF-κB or JNK inhibitors[77]. Taken together, these results suggest that TAGE activate Rac-1, leading to the induction of the JNK- and IκB kinase-dependent serine phosphorylation of IRS-1, which in turn contributes to hepatic IR.
As the main producers of extracellular matrix molecules in the liver, HSC are important contributors to liver fibrogenesis[78]. In a previous study, we found that TAGE promoted the expression of genes and proteins associated with fibrogenesis or inflammation, e.g., collagen type Iα2, monocyte chemoattractant protein-1 (MCP-1), and transforming growth factor-β1, in cultured HSC via NADPH oxidase-dependent ROS generation[22]. These results increase our knowledge of the role played by TAGE in the pathogenesis of NASH.
INTRACELLULAR TAGE ARE INVOLVED IN LIVER DAMAGE
GLA is a precursor of TAGE. Two GLA-forming pathways are considered to be in operation in the liver: (1) the glycolytic pathway and (2) the fructose metabolic pathway[18-20,38]. As a result, the liver tends to accumulate GLA to a greater extent than other organs.
Abnormalities in fructose and glucose metabolism can result in elevated intracellular GLA levels, which in turn can lead to upregulated intracellular TAGE synthesis, and such processes might play a role in the development of NASH. We found that in Hep3B cells GLA caused the intracellular TAGE concentration to rise and induced apoptosis in a concentration- and time-dependent manner[79]. Conversely, intracellular TAGE production was downregulated and GLA-induced apoptotic cell death was prevented by the addition of aminoguanidine, which inhibits AGE synthesis. Hepatocyte apoptosis was reported to be a characteristic of NASH in previous studies[80,81].
We identified a TAGE-modified protein (approximately 70 kDa) that was initially observed and tended to accumulate in GLA-treated hepatocytes as heat shock cognate 70 (Hsc70)[79]. Hsc70 might be important for GLA-induced cytotoxicity, as TAGE modifications have been demonstrated to have deleterious effects on protein function[82,83]. Furthermore, we found that the mRNA expression level of the acute phase reactant CRP was upregulated by intracellular TAGE[79]. Recently, it was demonstrated that NASH patients have higher plasma high-sensitivity CRP (hs-CRP) concentrations than healthy subjects or patients with simple steatosis[84,85]. Interestingly, in NASH patients a strong correlation was detected between the plasma hs-CRP concentration and the severity of liver damage[84,85]. In addition, intracellular TAGE were reported to induce inflammation, which is a characteristic of NASH. Taken together, these results indicate that intracellular TAGE make a significant contribution to the pathogenesis of NASH and might have potential as targets of treatments for NASH (Figure 3).
Figure 3.
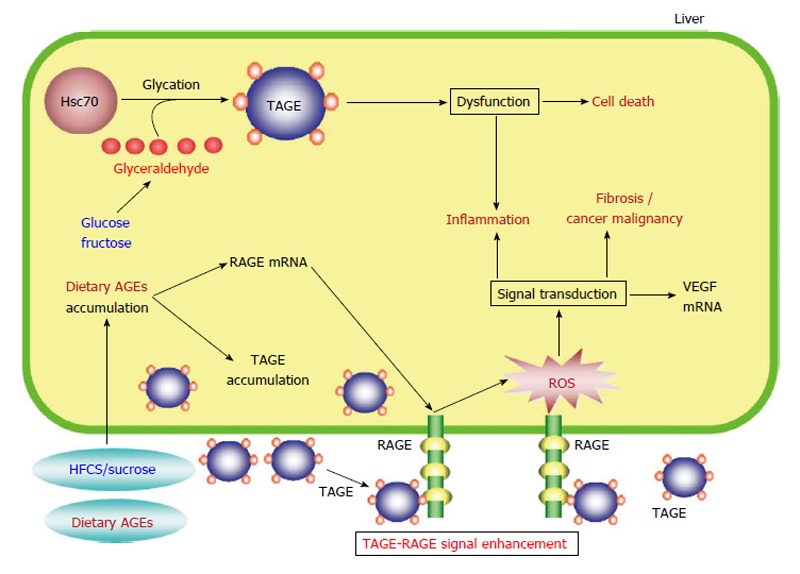
Proposed model for toxic advanced glycation end-products-mediated responses in the liver. HFCS/sucrose and dietary AGEs, which are normally found in sweetened beverages and commercial food products, are taken into the body, where they enhance the production/accumulation of TAGE, upregulate RAGE mRNA expression, and increase serum TAGE concentrations, leading to TAGE-RAGE interactions. The interaction between TAGE and RAGE alters intracellular signaling, gene expression, and the release of pro-inflammatory molecules and also induces oxidative stress in hepatocytes and hepatic stellate cells, which might contribute to the pathological changes observed in NAFLD/NASH. The formation of intracellular TAGE is associated with protein dysfunction followed by inflammation and cell death. Extracellular TAGE induce inflammation and fibrosis/cancer malignancy via RAGE signaling. AGEs: Advanced glycation end-products; TAGE: Toxic AGEs; RAGE: Receptor for AGEs; Hsc70: Heat shock cognate 70; ROS: Reactive oxygen species; VEGF: Vascular endothelial growth factor; HFCS: High-fructose corn syrup; NAFLD: Non-alcoholic fatty liver disease; NASH: Non-alcoholic steatohepatitis.
SERUM TAGE CONCENTRATIONS AND LIVER DISEASE
We measured the serum concentrations of three AGEs (Glu-AGEs, CML, and TAGE) in 66 patients with histologically defined-NASH who were free from liver cirrhosis, 10 patients with simple steatosis, and 30 control subjects to examine whether evaluating circulating AGE concentrations is useful for differentiating between NASH and simple steatosis[23]. The results of the latter study suggested that serum TAGE concentrations are involved in the pathogenesis of NASH and might be useful as biomarkers for differentiating between NASH and simple steatosis as: (1) The NASH patients exhibited significantly increased serum TAGE concentrations compared with the patients with simple steatosis and the healthy controls. According to receiver operating characteristic curves of the subjects’ circulating TAGE concentrations, the optimal cut-off value for predicting NASH was 8.53 units/mL, which resulted in sensitivity and specificity values of 66.7% and 88.9%, respectively; (2) The subjects’ homeostatic model assessment of insulin resistance (HOMA-IR) values and serum adiponectin concentrations (adiponectin is synthesized by adipose tissue and is an anti-inflammatory adipokine that can increase insulin sensitivity) exhibited positive and inverse correlations with their serum TAGE concentrations, respectively; (3) The subjects’ serum TAGE concentrations were not correlated with the severity of their hepatic steatosis or fibrosis, nor were they influenced by the subjects’ glucose tolerance status. The serum TAGE concentrations of the normal and IGT patients did not differ; (4) The NASH patients’ hepatocytes contained TAGE, whereas those belonging to the patients with simple steatosis exhibited negligible TAGE concentrations; and (5) The subjects’ Glu-AGE and CML concentrations did not differ among the groups[23]. The above results indicate that serum TAGE concentrations are useful biomarkers for assessing residual liver function.
PUTATIVE MOLECULAR MECHANISMS RESPONSIBLE FOR THE ASSOCIATION BETWEEN NAFLD AND CARDIOVASCULAR DISEASE
Endothelial progenitor cells (EPC) help to maintain the structure and function of the endothelium, and hence, facilitate angiogenesis and vascular repair. In addition, the number of circulating EPC and their activity levels were found to be inversely correlated with atherosclerotic risk factors. Thus, the number and activity levels of EPC might be useful biomarkers for predicting cardiovascular events. In a recent study, Chiang et al[86] demonstrated that compared with the controls NAFLD patients had significantly fewer circulating EPC and the function of their EPC was impaired. Thus, in NAFLD patients reductions in the number of EPC or their activity might increase the likelihood of cardiovascular events.
In recent studies, we found that: (1) the serum concentration of TAGE, but not CML, was independently associated with HOMA-IR in non-diabetic subjects[87]; (2) in T2DM patients, the serum TAGE concentration, but not those of Glu-AGEs or hemoglobin A1c (HbA1c), can be used as a biomarker of cumulative postprandial hyperglycemia[88]; (3) the serum concentration of TAGE, but not those of HbA1c or CML, was demonstrated to be an independent predictor of vascular inflammation (as evaluated by [18F] fluorodeoxyglucose-positron emission tomography in outpatients who visited Kurume University Hospital)[89]; (4) in healthy subjects, the serum TAGE concentration was found to be independently associated with a reduction in the number of circulating EPC and the impairment of the migratory activity of EPC[90]; and (5) a Japanese trial assessing the utility of pitavastatin and atorvastatin as treatments for acute coronary syndrome reported that high baseline TAGE concentrations were associated with plaque progression[91]. These results suggested that the serum TAGE concentration, but not those of HbA1c, CML, or Glu-AGEs, might be a useful biomarker for predicting atherosclerosis progression and future cardiovascular events. Thus, TAGE-RAGE system activation is considered to lead to a greater risk of cardiovascular events and contribute to the progression of liver damage, which would provide a mechanism link between CVD and NAFLD/NASH.
NOVEL TREATMENT STRATEGIES
The majority of studies about NASH have attempted to assess the relationships between NAFLD/NASH and T2DM, CVD, or chronic kidney disease (CKD)[92]. The results outlined above strongly suggest that the TAGE-RAGE system is involved in the development and progression of NASH. As a result, several therapeutic strategies that target this system, e.g., inhibiting TAGE synthesis, downregulating the expression of RAGE or molecules involved in its downstream pathways, and blocking TAGE-RAGE interactions, have been developed as potential treatments for NASH.
The inhibition of TAGE synthesis: Acarbose
Whilst there are many drugs that are able to improve glycemic control, including patients’ postprandial plasma glucose concentrations, some drugs specifically target postprandial hyperglycemia.
The absorption of carbohydrates from the small intestine can be delayed by treatment with the α-glucosidase inhibitor acarbose, and T2DM patients that were administered acarbose displayed less severe postprandial hyperglycemia[93]. A recent study found that in patients with T2DM or IGT acarbose treatment reduced the rate at which the intimal media of the carotid arteries thickened and led to a lower incidence of CVD[93], indicating that acarbose ameliorates postprandial hyperglycemia, and hence, inhibits the development and progression of CVD. In an in vivo study, we found that protein amino moieties readily react with GLA to produce TAGE, leading to oxidative stress and vascular inflammation. These observations suggested that in DM GLA plays a role in promoting the development of atherosclerosis[18-20]. In a study involving T2DM rats, we demonstrated that the serum concentration of TAGE, but not HbA1c, is a marker of cumulative postprandial hyperglycemia[94]. Based on the abovementioned results, we suggest that acarbose reduces serum TAGE concentrations, which could at least partially explain its cardioprotective effects in vivo. In a previous study, 50 mg acarbose (dosing schedule: thrice a day for a 12-wk period) were administered to 13 Japanese T2DM patients who were free from inflammatory conditions, atherosclerotic heart disease, and microangiopathy and had never taken oral hypoglycemic agents. The patients’ serum TAGE concentrations as well as their serum levels of other biological molecules were assessed before and after the administration of acarbose[88]. The DM patients’ serum free fatty acid and TAGE concentrations had fallen significantly after 12 wk’ acarbose treatment. Acarbose also reduced their postprandial plasma glucose concentrations. These results indicate that HbA1c concentrations might not accurately reflect the ameliorative effects of acarbose on postprandial hyperglycemia. Furthermore, they suggest that serum TAGE concentrations might be useful biomarkers for assessing cumulative postprandial hyperglycemia in T2DM patients. As TAGE have adverse effects on CVD[20], acarbose might be useful for preventing CVD in NASH patients with T2DM or postprandial hyperglycemia.
Inhibiting the binding of TAGE to RAGE using sRAGE
RAGE was found to contribute to acute liver damage in numerous studies, and the blockade of RAGE was demonstrated to reduce cholestatic, toxic, and ischemic liver damage[95-98].
Patients with chronic liver injuries were found to exhibit significantly higher hepatic RAGE expression levels[99], and in NAFLD patients a correlation was detected between the severity of fibrosis and the patients’ serum TAGE concentrations, indicating that RAGE and TAGE make significant contributions to the development of liver disease[23]. In addition, DM, which upregulates AGE synthesis and RAGE expression, has been found to accelerate the progression of fibrosis in a number of human liver conditions, including chronic hepatitis C and NAFLD[100]. Recently, we found that TAGE-RAGE interactions promote inflammation, affect the expression levels of various genes and the activity of intracellular signaling pathways, and induce oxidative stress in various kinds of cells. These effects might be involved in the pathological changes seen in various chronic diseases[17-20].
Endogenous sRAGE has recently been detected in humans[74]. It has been suggested that it is synthesized via the cleavage of a splice variant of RAGE (a type of secretory RAGE exhibiting C-terminal truncation) or full-length cell surface RAGE[74]. Patients with T1DM or T2DM display increased total endogenous sRAGE concentrations[101-104]. In addition, we and others have detected positive correlations between the total serum sRAGE concentration and serum TAGE concentrations in both non-DM and DM subjects[104,105]. Furthermore, body mass index-, sex-, and age-adjusted TAGE concentrations were found to increase significantly in proportion to the rise in the serum sRAGE concentration in non-DM subjects[104,105]. These results indicate that in vivo circulating sRAGE, which functions as a decoy receptor, is unable to bind to and remove the TAGE present in the blood in an efficient manner. As TAGE promote RAGE expression, the blood sRAGE concentration might be a marker of RAGE production within tissues. Furthermore, it might change in response to variations in the serum concentration of TAGE in order to ameliorate TAGE-induced tissue damage including NASH[106-109].
An angiotensin II type 1 receptor blocker: Telmisartan
It has been suggested that the TAGE-RAGE axis interacts with the renin-angiotensin system. In a previous study, we suggested that the angiotensin II type 1 receptor blocker telmisartan reduces RAGE expression via its ability to modulate the peroxisome proliferator-activated receptor-γ (PPAR-γ)[21,110]. We came to this conclusion due to the following observations, which were obtained in experiments involving Hep3B cells: (1) whilst telmisartan downregulated ROS synthesis, TAGE-induced RAGE expression, and CRP expression, candesartan did not induce any of these processes; (2) the PPAR-γ inhibitor GW9662 abrogated the telmisartan-induced inhibition of the expression of RAGE and its associated effector molecules; (3) the effects of ciglitazone and troglitazone, which are full agonists of PPAR-γ, were similar to those of telmisartan; and (4) the administration of curcumin, an inhibitor of NF-κB, or antioxidants abrogated the upregulation of CRP mRNA expression induced by TAGE. Due to its unique ability to modulate PPAR-γ, telmisartan is increasingly considered to be a useful cardiometabolic sartan[21,110,111]. In addition, it has been demonstrated that thiazolidinediones downregulate endothelial RAGE expression via NF-κB suppression[112]. These results suggest that telmisartan has anti-inflammatory effects on TAGE signaling; i.e., it reduces hepatic RAGE expression by activating PPAR-γ, and might also help to protect against NASH.
A hydroxymethyl-glutaryl-CoA reductase inhibitor: Atorvastatin
In a recent study, we found that in Hep3B cells the hydroxymethyl-glutaryl-CoA reductase inhibitor atorvastatin reduced TAGE-induced ROS synthesis in a dose-dependent manner[113]. In addition, atorvastatin and the antioxidant N-acetylcysteine downregulated CRP expression at both the mRNA and protein levels in TAGE-treated Hep3B cells[113]. These results showed that the antioxidative effects of atorvastatin abrogate CRP expression-associated TAGE signaling. Furthermore, they indicate that statins protect blood vessels from damage and abrogate the adverse effects of TAGE by downregulating the activity of their effector molecules.
The consumption of fructose-containing beverages is associated with a greater risk of MetS-related conditions, including NAFLD. Despite the fact that caloric restriction and weight loss is the only effective treatment for NAFLD, it has been demonstrated that atorvastatin is safe for use in NAFLD patients and results in improvements in their hepatic histology. In a previous study, we found that atorvastatin reduced the serum TAGE concentrations of 43 patients with a combination of biopsy-proven NASH and dyslipidemia[114]. After 12 mo atorvastatin treatment (10 mg daily), all of the patients demonstrated significant reductions in their hepatic transaminase (aspartate aminotransferase and alanine aminotransferase (ALT) and γ-glutamyl transpeptidase concentrations. In addition, by end of the treatment their plasma tumor necrosis factor-α (TNF-α) and plasma adiponectin concentrations were reduced by 31% and elevated by 16%, respectively. The patients’ HOMA-IR values were slightly reduced. The patients’ liver/spleen ratios rose significantly from 0.54 ± 0.26 at the baseline to 0.94 ± 0.24 at the end of the treatment; however, their visceral fat area values were unchanged. During the treatment, the patients’ serum TAGE concentrations fell significantly (they were 10.4 ± 3.8, 5.9 ± 3.3, and 2.5 ± 1.1 units/mL before the treatment and after 6 mo and 12 mo treatment, respectively). Correlations were detected between the patients’ serum TAGE concentrations and their serum concentrations of thiobarbituric acid reactive substances (TBARS), TNF-α, procollagen type III propeptide, ALT, and type IV collagen 7S[114].
The administration of atorvastatin to Sprague-Dawley male rats that had consumed a liquid fructose solution (10% w/v) abrogated the inflammatory and metabolic changes induced in the liver by fructose. These beneficial effects were considered to be due to the anti-inflammatory activity of atorvastatin and its downregulation of the hepatic expression of fructokinase, which inhibits fructose metabolism in the liver[115]. Reduced synthesis of GLA (a TAGE precursor and a fructose metabolite) leads to a drop in TAGE synthesis. Atorvastatin is able to reduce the serum TAGE concentration without altering glucose metabolism and does so in a cholesterol-lowering-independent manner. In the abovementioned study, the serum TAGE concentrations of the NASH patients with dyslipidemia fell significantly after the atorvastatin treatment, but their glucose metabolism was unaffected[114]. In conclusion, atorvastatin was demonstrated to be an effective treatment for NASH patients with dyslipidemia who did not respond adequately to diet and exercise therapy. In addition to improving their serum TAGE concentrations, atorvastatin also improved their histological and biochemical data. As atorvastatin decreased the serum TAGE concentrations of NASH patients with dyslipidemia, TAGE might be useful biomarkers for the treatment of NASH[114]. Controlled trials should be performed to further examine the clinical utility of TAGE as biomarkers in NASH.
Dietary AGEs: Kremezin
A study involving mice produced found that AGEs facilitate the progression from simple steatosis to NASH and liver fibrosis[116]. In the methionine choline-deficient rat model of NAFLD, high dietary consumption of AGEs results in elevated hepatic AGE concentrations and increased fibrosis, liver damage, and inflammation. The latter effects are considered to be mediated via the RAGE- and oxidative stress-dependent profibrotic effects of AGEs on activated HSC[117]. The above observations indicate that pharmacological and dietary strategies that target the AGE-RAGE system are able to slow the progression of NAFLD.
In a recent study, we detected increased hepatic expression levels of vascular endothelial growth factor (VEGF) and RAGE in rats that had been administered Glu-AGE-rich beverages. This suggested that dietary AGE consumption is associated with the hepatic expression of liver fibrosis-related genes[118]. Moreover, the abovementioned rats’ livers were found to contain TAGE- and Glu-AGE-positive cells[118]. These results indicate that the consumption of Glu-AGE-rich beverages leads to upregulated hepatic expression of RAGE and VEGF and encourages the build-up of TAGE and Glu-AGEs, resulting in the binding of TAGE to RAGE. Thus, it is important to consider the amounts of Glu-AGEs present in foods to prevent liver disease, especially in people that are at risk of CKD, CVD, NAFLD/NASH, or DM.
It has been demonstrated that Kremezin, an oral adsorbent that consists of porous spherical carbonic particles, is able to attenuate the progression of chronic renal failure (CRF) by removing uremic toxins, e.g., indoxyl sulfate precursors, from the intestine[119]. In CRF patients without DM, 3 mo Kremezin treatment (6 g/d) resulted in markedly reduced serum TAGE and Glu-AGEs concentrations, while the concentrations of these molecules were unaffected in renal function- and age-matched CRF patients that did not receive the drug[120]. The EC in the post-treatment serum samples collected from the Kremezin-treated patients exhibited markedly lower concentrations of MCP-1, vascular cell adhesion molecule-1, and RAGE mRNA than those found in the serum samples collected before treatment[120]. These findings indicate that the pathogenesis of vascular damage is influenced by dietary Glu-AGEs in TAGE-RAGE-related conditions and that reducing the amount of dietary Glu-AGEs taken into the body might represent a useful strategy against NAFLD/NASH.
Further clinical studies might provide insights into whether restricting the consumption of Glu-AGEs would be beneficial for preventing or slowing the progression of NAFLD/NASH and whether Glu-AGEs represent a novel therapeutic target for treatments that aim to reduce the risk of liver disease.
CONCLUSION
TAGE formation and accumulation are known to increase in various tissues during normal aging and to occur at a markedly accelerated rate in DM patients[18-20]. An increasing body of evidence suggests that TAGE are involved in the pathogeneses of various disorders including hypertension, Alzheimer’s disease, diabetic vascular complications, CVD, NAFLD/NASH, and cancer growth and metastasis[7,8,18-23,79,87-91,114,121-126]. We found evidence that TAGE are involved in the pathogenesis of NASH in humans[7,23,114]. TAGE stimulated the proliferation and activation of HSC in vitro via RAGE, which resulted in hepatic inflammation and fibrosis[22]. In addition, NASH patients exhibited significantly higher serum TAGE concentrations than patients with simple steatosis or healthy controls[7,23]. Atorvastatin reduced the serum TAGE concentrations of NASH patients with dyslipidemia, and correlations were detected between the patients’ serum TAGE concentrations and their serum TNF-α, ALT, type IV collagen 7S, procollagen type III propeptide, and TBARS concentrations[114]. In a recent study, we found that non-B or non-C hepatocellular carcinoma (NBNC-HCC) patients had significantly increased circulating TAGE concentrations compared with NASH subjects without HCC and the control subjects[127]. The findings outlined in the present review indicate that TAGE contribute to the pathogenesis of NBNC-HCC and that they might be useful biomarkers for discriminating between NBNC-HCC and NASH.
In conclusion, an increasing amount of evidence indicates that TAGE and RAGE both make important contributions to liver disease. TAGE might play a role in the development and progression of NASH and could be useful biomarkers for differentiating between NASH and NAFLD or between NBNC-HCC and NASH. Further clinical and experimental studies are required to elucidate the mechanisms by which the TAGE-RAGE system affects the development and progression of lifestyle-related conditions including NAFLD/NASH (Figure 4).
Figure 4.
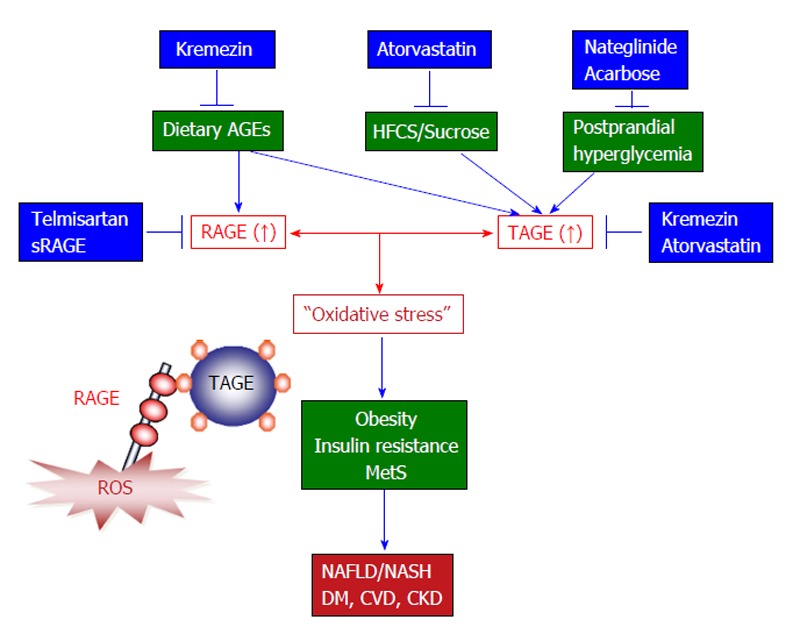
The toxic advanced glycation end-products-receptor for advanced glycation end-products system and novel treatments that target this system to prevent the development and/or progression of non-alcoholic steatohepatitis. Accumulating evidence suggests that TAGE-RAGE interactions affect intracellular signaling, gene expression, and the release of pro-inflammatory molecules and also induce oxidative stress in numerous types of cells, all of which have the potential to contribute to the pathological changes associated with lifestyle-related diseases including NAFLD/NASH. Since TAGE display the strongest binding affinities for RAGE and have adverse effects on diabetic vessels through their interactions with RAGE, TAGE might be partly responsible for the increased risk of cardiovascular disease (CVD) seen in diabetes mellitus (DM) patients and the impaired glucose tolerance observed in patients with postprandial hyperglycemia. NAFLD is considered to be a hepatic symptom of metabolic syndrome (MetS) and is strongly associated with insulin resistance, obesity, and abnormalities in glucose and lipid metabolism. It is important to consider the amounts HFCS/sucrose and AGEs present in foods to prevent liver disease, particularly in individuals that are at high risk of developing NAFLD/NASH, DM, CVD, or chronic kidney disease (CKD). Taken together, the present study suggests that TAGE could be used as novel therapeutic targets for the prevention of lifestyle-related diseases. Therefore, inhibiting the formation of TAGE, blocking TAGE-RAGE interactions, and suppressing the expression of RAGE or its downstream effectors all have potential as therapeutic strategies against lifestyle-related disease including NAFLD/NASH. AGEs: Advanced glycation end-products; TAGE: Toxic AGEs; RAGE: Receptor for AGEs; sRAGE: Soluble form of RAGE; NAFLD: Non-alcoholic fatty liver disease; NASH: Non-alcoholic steatohepatitis; HFCS: High-fructose corn syrup.
Footnotes
Supported by The Japan Society for the Promotion of Science (JSPS) KAKENHI Grant, No. 19300254, 22300264 and 25282029 (Takeuchi M); Kanazawa Medical University, No. SR2012-04 (Tsutsumi M); the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), Regional Innovation Strategy Support Program (Takeuchi M)
P- Reviewer: Basaranoglu M, Czaja MJ, Miyoshi E S- Editor: Ji FF L- Editor: A E- Editor: Liu SQ
References
- 1.Vuppalanchi R, Chalasani N. Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: Selected practical issues in their evaluation and management. Hepatology. 2009;49:306–317. doi: 10.1002/hep.22603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Teli MR, James OF, Burt AD, Bennett MK, Day CP. The natural history of nonalcoholic fatty liver: a follow-up study. Hepatology. 1995;22:1714–1719. [PubMed] [Google Scholar]
- 3.Dam-Larsen S, Franzmann M, Andersen IB, Christoffersen P, Jensen LB, Sørensen TI, Becker U, Bendtsen F. Long term prognosis of fatty liver: risk of chronic liver disease and death. Gut. 2004;53:750–755. doi: 10.1136/gut.2003.019984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–1419. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 5.Chitturi S, Abeygunasekera S, Farrell GC, Holmes-Walker J, Hui JM, Fung C, Karim R, Lin R, Samarasinghe D, Liddle C, et al. NASH and insulin resistance: Insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology. 2002;35:373–379. doi: 10.1053/jhep.2002.30692. [DOI] [PubMed] [Google Scholar]
- 6.Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, McCullough AJ, Natale S, Forlani G, Melchionda N. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001;50:1844–1850. doi: 10.2337/diabetes.50.8.1844. [DOI] [PubMed] [Google Scholar]
- 7.Hyogo H, Yamagishi S. Advanced glycation end products (AGEs) and their involvement in liver disease. Curr Pharm Des. 2008;14:969–972. doi: 10.2174/138161208784139701. [DOI] [PubMed] [Google Scholar]
- 8.Hyogo H, Chayama K, Yamagishi S. Nonalcoholic fatty liver disease and cardiovascular disease. Curr Pharm Des. 2014;20:2403–2411. doi: 10.2174/13816128113199990476. [DOI] [PubMed] [Google Scholar]
- 9.Liu H, Lu HY. Nonalcoholic fatty liver disease and cardiovascular disease. World J Gastroenterol. 2014;20:8407–8415. doi: 10.3748/wjg.v20.i26.8407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamagishi S, Imaizumi T. Diabetic vascular complications: pathophysiology, biochemical basis and potential therapeutic strategy. Curr Pharm Des. 2005;11:2279–2299. doi: 10.2174/1381612054367300. [DOI] [PubMed] [Google Scholar]
- 11.Yamagishi S, Takeuchi M, Inagaki Y, Nakamura K, Imaizumi T. Role of advanced glycation end products (AGEs) and their receptor (RAGE) in the pathogenesis of diabetic microangiopathy. Int J Clin Pharmacol Res. 2003;23:129–134. [PubMed] [Google Scholar]
- 12.Rahbar S, Figarola JL. Novel inhibitors of advanced glycation endproducts. Arch Biochem Biophys. 2003;419:63–79. doi: 10.1016/j.abb.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 13.Vlassara H, Palace MR. Diabetes and advanced glycation endproducts. J Intern Med. 2002;251:87–101. doi: 10.1046/j.1365-2796.2002.00932.x. [DOI] [PubMed] [Google Scholar]
- 14.Wendt T, Bucciarelli L, Qu W, Lu Y, Yan SF, Stern DM, Schmidt AM. Receptor for advanced glycation endproducts (RAGE) and vascular inflammation: insights into the pathogenesis of macrovascular complications in diabetes. Curr Atheroscler Rep. 2002;4:228–237. doi: 10.1007/s11883-002-0024-4. [DOI] [PubMed] [Google Scholar]
- 15.Schmidt AM, Stern D. Atherosclerosis and diabetes: the RAGE connection. Curr Atheroscler Rep. 2000;2:430–436. doi: 10.1007/s11883-000-0082-4. [DOI] [PubMed] [Google Scholar]
- 16.Takenaka K, Yamagishi S, Matsui T, Nakamura K, Imaizumi T. Role of advanced glycation end products (AGEs) in thrombogenic abnormalities in diabetes. Curr Neurovasc Res. 2006;3:73–77. doi: 10.2174/156720206775541804. [DOI] [PubMed] [Google Scholar]
- 17.Takeuchi M, Yamagishi S. TAGE (toxic AGEs) hypothesis in various chronic diseases. Med Hypotheses. 2004;63:449–452. doi: 10.1016/j.mehy.2004.02.042. [DOI] [PubMed] [Google Scholar]
- 18.Sato T, Iwaki M, Shimogaito N, Wu X, Yamagishi S, Takeuchi M. TAGE (toxic AGEs) theory in diabetic complications. Curr Mol Med. 2006;6:351–358. doi: 10.2174/156652406776894536. [DOI] [PubMed] [Google Scholar]
- 19.Takeuchi M, Yamagishi S. Involvement of toxic AGEs (TAGE) in the pathogenesis of diabetic vascular complications and Alzheimer’s disease. J Alzheimers Dis. 2009;16:845–858. doi: 10.3233/JAD-2009-0974. [DOI] [PubMed] [Google Scholar]
- 20.Takeuchi M, Takino J, Yamagishi S. Involvement of the toxic AGEs (TAGE)-RAGE system in the pathogenesis of diabetic vascular complications: a novel therapeutic strategy. Curr Drug Targets. 2010;11:1468–1482. doi: 10.2174/1389450111009011468. [DOI] [PubMed] [Google Scholar]
- 21.Yoshida T, Yamagishi S, Nakamura K, Matsui T, Imaizumi T, Takeuchi M, Koga H, Ueno T, Sata M. Telmisartan inhibits AGE-induced C-reactive protein production through downregulation of the receptor for AGE via peroxisome proliferator-activated receptor-gamma activation. Diabetologia. 2006;49:3094–3099. doi: 10.1007/s00125-006-0437-7. [DOI] [PubMed] [Google Scholar]
- 22.Iwamoto K, Kanno K, Hyogo H, Yamagishi S, Takeuchi M, Tazuma S, Chayama K. Advanced glycation end products enhance the proliferation and activation of hepatic stellate cells. J Gastroenterol. 2008;43:298–304. doi: 10.1007/s00535-007-2152-7. [DOI] [PubMed] [Google Scholar]
- 23.Hyogo H, Yamagishi S, Iwamoto K, Arihiro K, Takeuchi M, Sato T, Ochi H, Nonaka M, Nabeshima Y, Inoue M, et al. Elevated levels of serum advanced glycation end products in patients with non-alcoholic steatohepatitis. J Gastroenterol Hepatol. 2007;22:1112–1119. doi: 10.1111/j.1440-1746.2007.04943.x. [DOI] [PubMed] [Google Scholar]
- 24.Bucala R, Cerami A. Advanced glycosylation: chemistry, biology, and implications for diabetes and aging. Adv Pharmacol. 1992;23:1–34. doi: 10.1016/s1054-3589(08)60961-8. [DOI] [PubMed] [Google Scholar]
- 25.Vlassara H, Bucala R, Striker L. Pathogenic effects of advanced glycosylation: biochemical, biologic, and clinical implications for diabetes and aging. Lab Invest. 1994;70:138–151. [PubMed] [Google Scholar]
- 26.Brownlee M. Advanced protein glycosylation in diabetes and aging. Annu Rev Med. 1995;46:223–234. doi: 10.1146/annurev.med.46.1.223. [DOI] [PubMed] [Google Scholar]
- 27.Takeuchi M, Makita Z. Alternative routes for the formation of immunochemically distinct advanced glycation end-products in vivo. Curr Mol Med. 2001;1:305–315. doi: 10.2174/1566524013363735. [DOI] [PubMed] [Google Scholar]
- 28.Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med. 1988;318:1315–1321. doi: 10.1056/NEJM198805193182007. [DOI] [PubMed] [Google Scholar]
- 29.Monnier VM, Cerami A. Nonenzymatic browning in vivo: possible process for aging of long-lived proteins. Science. 1981;211:491–493. doi: 10.1126/science.6779377. [DOI] [PubMed] [Google Scholar]
- 30.Bunn HF, Higgins PJ. Reaction of monosaccharides with proteins: possible evolutionary significance. Science. 1981;213:222–224. doi: 10.1126/science.12192669. [DOI] [PubMed] [Google Scholar]
- 31.Giardino I, Edelstein D, Brownlee M. BCL-2 expression or antioxidants prevent hyperglycemia-induced formation of intracellular advanced glycation endproducts in bovine endothelial cells. J Clin Invest. 1996;97:1422–1428. doi: 10.1172/JCI118563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giardino I, Edelstein D, Brownlee M. Nonenzymatic glycosylation in vitro and in bovine endothelial cells alters basic fibroblast growth factor activity. A model for intracellular glycosylation in diabetes. J Clin Invest. 1994;94:110–117. doi: 10.1172/JCI117296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poulsen MW, Hedegaard RV, Andersen JM, de Courten B, Bügel S, Nielsen J, Skibsted LH, Dragsted LO. Advanced glycation endproducts in food and their effects on health. Food Chem Toxicol. 2013;60:10–37. doi: 10.1016/j.fct.2013.06.052. [DOI] [PubMed] [Google Scholar]
- 34.Takeuchi M, Makita Z, Yanagisawa K, Kameda Y, Koike T. Detection of noncarboxymethyllysine and carboxymethyllysine advanced glycation end products (AGE) in serum of diabetic patients. Mol Med. 1999;5:393–405. [PMC free article] [PubMed] [Google Scholar]
- 35.Takeuchi M, Makita Z, Bucala R, Suzuki T, Koike T, Kameda Y. Immunological evidence that non-carboxymethyllysine advanced glycation end-products are produced from short chain sugars and dicarbonyl compounds in vivo. Mol Med. 2000;6:114–125. [PMC free article] [PubMed] [Google Scholar]
- 36.Takeuchi M, Yanase Y, Matsuura N, Yamagishi S, Kameda Y, Bucala R, Makita Z. Immunological detection of a novel advanced glycation end-product. Mol Med. 2001;7:783–791. [PMC free article] [PubMed] [Google Scholar]
- 37.Takeuchi M, Iwaki M, Takino J, Shirai H, Kawakami M, Bucala R, Yamagishi S. Immunological detection of fructose-derived advanced glycation end-products. Lab Invest. 2010;90:1117–1127. doi: 10.1038/labinvest.2010.62. [DOI] [PubMed] [Google Scholar]
- 38.Takeuchi M, Yamagishi S. Alternative routes for the formation of glyceraldehyde-derived AGEs (TAGE) in vivo. Med Hypotheses. 2004;63:453–455. doi: 10.1016/j.mehy.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 39.Oates PJ. Polyol pathway and diabetic peripheral neuropathy. Int Rev Neurobiol. 2002;50:325–392. doi: 10.1016/s0074-7742(02)50082-9. [DOI] [PubMed] [Google Scholar]
- 40.Maekawa K, Tanimoto T, Okada S. Gene expression of enzymes comprising the polyol pathway in various rat tissues determined by the competitive RT-PCR method. Jpn J Pharmacol. 2002;88:123–126. doi: 10.1254/jjp.88.123. [DOI] [PubMed] [Google Scholar]
- 41.Iwata T, Popescu NC, Zimonjic DB, Karlsson C, Höög JO, Vaca G, Rodriguez IR, Carper D. Structural organization of the human sorbitol dehydrogenase gene (SORD) Genomics. 1995;26:55–62. doi: 10.1016/0888-7543(95)80082-w. [DOI] [PubMed] [Google Scholar]
- 42.Yabe-Nishimura C. Aldose reductase in glucose toxicity: a potential target for the prevention of diabetic complications. Pharmacol Rev. 1998;50:21–33. [PubMed] [Google Scholar]
- 43.Schalkwijk CG, Stehouwer CD, van Hinsbergh VW. Fructose-mediated non-enzymatic glycation: sweet coupling or bad modification. Diabetes Metab Res Rev. 2004;20:369–382. doi: 10.1002/dmrr.488. [DOI] [PubMed] [Google Scholar]
- 44.Gaby AR. Adverse effects of dietary fructose. Altern Med Rev. 2005;10:294–306. [PubMed] [Google Scholar]
- 45.Hallfrisch J. Metabolic effects of dietary fructose. FASEB J. 1990;4:2652–2660. doi: 10.1096/fasebj.4.9.2189777. [DOI] [PubMed] [Google Scholar]
- 46.Mayes PA. Intermediary metabolism of fructose. Am J Clin Nutr. 1993;58:754S–765S. doi: 10.1093/ajcn/58.5.754S. [DOI] [PubMed] [Google Scholar]
- 47.James J, Thomas P, Cavan D, Kerr D. Preventing childhood obesity by reducing consumption of carbonated drinks: cluster randomised controlled trial. BMJ. 2004;328:1237. doi: 10.1136/bmj.38077.458438.EE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Teff KL, Elliott SS, Tschöp M, Kieffer TJ, Rader D, Heiman M, Townsend RR, Keim NL, D’Alessio D, Havel PJ. Dietary fructose reduces circulating insulin and leptin, attenuates postprandial suppression of ghrelin, and increases triglycerides in women. J Clin Endocrinol Metab. 2004;89:2963–2972. doi: 10.1210/jc.2003-031855. [DOI] [PubMed] [Google Scholar]
- 49.Faeh D, Minehira K, Schwarz JM, Periasamy R, Park S, Tappy L. Effect of fructose overfeeding and fish oil administration on hepatic de novo lipogenesis and insulin sensitivity in healthy men. Diabetes. 2005;54:1907–1913. doi: 10.2337/diabetes.54.7.1907. [DOI] [PubMed] [Google Scholar]
- 50.Nseir W, Nassar F, Assy N. Soft drinks consumption and nonalcoholic fatty liver disease. World J Gastroenterol. 2010;16:2579–2588. doi: 10.3748/wjg.v16.i21.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lim JS, Mietus-Snyder M, Valente A, Schwarz JM, Lustig RH. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol. 2010;7:251–264. doi: 10.1038/nrgastro.2010.41. [DOI] [PubMed] [Google Scholar]
- 52.Nomura K, Yamanouchi T. The role of fructose-enriched diets in mechanisms of nonalcoholic fatty liver disease. J Nutr Biochem. 2012;23:203–208. doi: 10.1016/j.jnutbio.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 53.Yilmaz Y. Review article: fructose in non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2012;35:1135–1144. doi: 10.1111/j.1365-2036.2012.05080.x. [DOI] [PubMed] [Google Scholar]
- 54.Vos MB, Lavine JE. Dietary fructose in nonalcoholic fatty liver disease. Hepatology. 2013;57:2525–2531. doi: 10.1002/hep.26299. [DOI] [PubMed] [Google Scholar]
- 55.Pollock NK, Bundy V, Kanto W, Davis CL, Bernard PJ, Zhu H, Gutin B, Dong Y. Greater fructose consumption is associated with cardiometabolic risk markers and visceral adiposity in adolescents. J Nutr. 2012;142:251–257. doi: 10.3945/jn.111.150219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lê KA, Faeh D, Stettler R, Ith M, Kreis R, Vermathen P, Boesch C, Ravussin E, Tappy L. A 4-wk high-fructose diet alters lipid metabolism without affecting insulin sensitivity or ectopic lipids in healthy humans. Am J Clin Nutr. 2006;84:1374–1379. doi: 10.1093/ajcn/84.6.1374. [DOI] [PubMed] [Google Scholar]
- 57.Basaranoglu M, Basaranoglu G, Sabuncu T, Sentürk H. Fructose as a key player in the development of fatty liver disease. World J Gastroenterol. 2013;19:1166–1172. doi: 10.3748/wjg.v19.i8.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kargulewicz A, Stankowiak-Kulpa H, Grzymisławski M. Dietary recommendations for patients with nonalcoholic fatty liver disease. Prz Gastroenterol. 2014;9:18–23. doi: 10.5114/pg.2014.40845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 60.Schmidt AM, Yan SD, Yan SF, Stern DM. The biology of the receptor for advanced glycation end products and its ligands. Biochim Biophys Acta. 2000;1498:99–111. doi: 10.1016/s0167-4889(00)00087-2. [DOI] [PubMed] [Google Scholar]
- 61.Bucciarelli LG, Wendt T, Rong L, Lalla E, Hofmann MA, Goova MT, Taguchi A, Yan SF, Yan SD, Stern DM, et al. RAGE is a multiligand receptor of the immunoglobulin superfamily: implications for homeostasis and chronic disease. Cell Mol Life Sci. 2002;59:1117–1128. doi: 10.1007/s00018-002-8491-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hudson BI, Bucciarelli LG, Wendt T, Sakaguchi T, Lalla E, Qu W, Lu Y, Lee L, Stern DM, Naka Y, et al. Blockade of receptor for advanced glycation endproducts: a new target for therapeutic intervention in diabetic complications and inflammatory disorders. Arch Biochem Biophys. 2003;419:80–88. doi: 10.1016/j.abb.2003.08.030. [DOI] [PubMed] [Google Scholar]
- 63.Ramasamy R, Vannucci SJ, Yan SS, Herold K, Yan SF, Schmidt AM. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology. 2005;15:16R–28R. doi: 10.1093/glycob/cwi053. [DOI] [PubMed] [Google Scholar]
- 64.Li YM, Mitsuhashi T, Wojciechowicz D, Shimizu N, Li J, Stitt A, He C, Banerjee D, Vlassara H. Molecular identity and cellular distribution of advanced glycation endproduct receptors: relationship of p60 to OST-48 and p90 to 80K-H membrane proteins. Proc Natl Acad Sci USA. 1996;93:11047–11052. doi: 10.1073/pnas.93.20.11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vlassara H, Li YM, Imani F, Wojciechowicz D, Yang Z, Liu FT, Cerami A. Identification of galectin-3 as a high-affinity binding protein for advanced glycation end products (AGE): a new member of the AGE-receptor complex. Mol Med. 1995;1:634–646. [PMC free article] [PubMed] [Google Scholar]
- 66.Araki N, Higashi T, Mori T, Shibayama R, Kawabe Y, Kodama T, Takahashi K, Shichiri M, Horiuchi S. Macrophage scavenger receptor mediates the endocytic uptake and degradation of advanced glycation end products of the Maillard reaction. Eur J Biochem. 1995;230:408–415. doi: 10.1111/j.1432-1033.1995.0408h.x. [DOI] [PubMed] [Google Scholar]
- 67.Ohgami N, Nagai R, Ikemoto M, Arai H, Kuniyasu A, Horiuchi S, Nakayama H. CD36, a member of class B scavenger receptor family, is a receptor for advanced glycation end products. Ann N Y Acad Sci. 2001;947:350–355. doi: 10.1111/j.1749-6632.2001.tb03961.x. [DOI] [PubMed] [Google Scholar]
- 68.Ohgami N, Nagai R, Ikemoto M, Arai H, Miyazaki A, Hakamata H, Horiuchi S, Nakayama H. CD36, serves as a receptor for advanced glycation endproducts (AGE) J Diabetes Complications. 2002;16:56–59. doi: 10.1016/s1056-8727(01)00208-2. [DOI] [PubMed] [Google Scholar]
- 69.Ohgami N, Nagai R, Miyazaki A, Ikemoto M, Arai H, Horiuchi S, Nakayama H. Scavenger receptor class B type I-mediated reverse cholesterol transport is inhibited by advanced glycation end products. J Biol Chem. 2001;276:13348–13355. doi: 10.1074/jbc.M011613200. [DOI] [PubMed] [Google Scholar]
- 70.Jono T, Miyazaki A, Nagai R, Sawamura T, Kitamura T, Horiuchi S. Lectin-like oxidized low density lipoprotein receptor-1 (LOX-1) serves as an endothelial receptor for advanced glycation end products (AGE) FEBS Lett. 2002;511:170–174. doi: 10.1016/s0014-5793(01)03325-7. [DOI] [PubMed] [Google Scholar]
- 71.Tamura Y, Adachi H, Osuga J, Ohashi K, Yahagi N, Sekiya M, Okazaki H, Tomita S, Iizuka Y, Shimano H, et al. FEEL-1 and FEEL-2 are endocytic receptors for advanced glycation end products. J Biol Chem. 2003;278:12613–12617. doi: 10.1074/jbc.M210211200. [DOI] [PubMed] [Google Scholar]
- 72.Vlassara H. The AGE-receptor in the pathogenesis of diabetic complications. Diabetes Metab Res Rev. 2001;17:436–443. doi: 10.1002/dmrr.233. [DOI] [PubMed] [Google Scholar]
- 73.Hudson BI, Harja E, Moser B, Schmidt AM. Soluble levels of receptor for advanced glycation endproducts (sRAGE) and coronary artery disease: the next C-reactive protein? Arterioscler Thromb Vasc Biol. 2005;25:879–882. doi: 10.1161/01.ATV.0000164804.05324.8b. [DOI] [PubMed] [Google Scholar]
- 74.Yonekura H, Yamamoto Y, Sakurai S, Petrova RG, Abedin MJ, Li H, Yasui K, Takeuchi M, Makita Z, Takasawa S, et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem J. 2003;370:1097–1109. doi: 10.1042/BJ20021371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yamamoto Y, Yonekura H, Watanabe T, Sakurai S, Li H, Harashima A, Myint KM, Osawa M, Takeuchi A, Takeuchi M, et al. Short-chain aldehyde-derived ligands for RAGE and their actions on endothelial cells. Diabetes Res Clin Pract. 2007;77 Suppl 1:S30–S40. doi: 10.1016/j.diabres.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 76.Yoshida T, Yamagishi S, Nakamura K, Matsui T, Imaizumi T, Takeuchi M, Ueno T, Sata M. Pigment epithelium-derived factor (PEDF) inhibits advanced glycation end product (AGE)-induced C-reactive protein expression in hepatoma cells by suppressing Rac-1 activation. FEBS Lett. 2006;580:2788–2796. doi: 10.1016/j.febslet.2006.04.050. [DOI] [PubMed] [Google Scholar]
- 77.Yoshida T, Yamagishi S, Nakamura K, Matsui T, Imaizumi T, Takeuchi M, Koga H, Ueno T, Sata M. Pigment epithelium-derived factor (PEDF) ameliorates advanced glycation end product (AGE)-induced hepatic insulin resistance in vitro by suppressing Rac-1 activation. Horm Metab Res. 2008;40:620–625. doi: 10.1055/s-0028-1083785. [DOI] [PubMed] [Google Scholar]
- 78.Moreira RK. Hepatic stellate cells and liver fibrosis. Arch Pathol Lab Med. 2007;131:1728–1734. doi: 10.5858/2007-131-1728-HSCALF. [DOI] [PubMed] [Google Scholar]
- 79.Takino J, Kobayashi Y, Takeuchi M. The formation of intracellular glyceraldehyde-derived advanced glycation end-products and cytotoxicity. J Gastroenterol. 2010;45:646–655. doi: 10.1007/s00535-009-0193-9. [DOI] [PubMed] [Google Scholar]
- 80.Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart LJ, Lindor KD, Gores GJ. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125:437–443. doi: 10.1016/s0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]
- 81.Ribeiro PS, Cortez-Pinto H, Solá S, Castro RE, Ramalho RM, Baptista A, Moura MC, Camilo ME, Rodrigues CM. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am J Gastroenterol. 2004;99:1708–1717. doi: 10.1111/j.1572-0241.2004.40009.x. [DOI] [PubMed] [Google Scholar]
- 82.Hamelin M, Mary J, Vostry M, Friguet B, Bakala H. Glycation damage targets glutamate dehydrogenase in the rat liver mitochondrial matrix during aging. FEBS J. 2007;274:5949–5961. doi: 10.1111/j.1742-4658.2007.06118.x. [DOI] [PubMed] [Google Scholar]
- 83.Kumar PA, Kumar MS, Reddy GB. Effect of glycation on alpha-crystallin structure and chaperone-like function. Biochem J. 2007;408:251–258. doi: 10.1042/BJ20070989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yoneda M, Mawatari H, Fujita K, Iida H, Yonemitsu K, Kato S, Takahashi H, Kirikoshi H, Inamori M, Nozaki Y, et al. High-sensitivity C-reactive protein is an independent clinical feature of nonalcoholic steatohepatitis (NASH) and also of the severity of fibrosis in NASH. J Gastroenterol. 2007;42:573–582. doi: 10.1007/s00535-007-2060-x. [DOI] [PubMed] [Google Scholar]
- 85.Targher G, Bertolini L, Rodella S, Lippi G, Franchini M, Zoppini G, Muggeo M, Day CP. NASH predicts plasma inflammatory biomarkers independently of visceral fat in men. Obesity (Silver Spring) 2008;16:1394–1399. doi: 10.1038/oby.2008.64. [DOI] [PubMed] [Google Scholar]
- 86.Chiang CH, Huang PH, Chung FP, Chen ZY, Leu HB, Huang CC, Wu TC, Chen JW, Lin SJ. Decreased circulating endothelial progenitor cell levels and function in patients with nonalcoholic fatty liver disease. PLoS One. 2012;7:e31799. doi: 10.1371/journal.pone.0031799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tahara N, Yamagishi S, Matsui T, Takeuchi M, Nitta Y, Kodama N, Mizoguchi M, Imaizumi T. Serum levels of advanced glycation end products (AGEs) are independent correlates of insulin resistance in nondiabetic subjects. Cardiovasc Ther. 2012;30:42–48. doi: 10.1111/j.1755-5922.2010.00177.x. [DOI] [PubMed] [Google Scholar]
- 88.Tsunosue M, Mashiko N, Ohta Y, Matsuo Y, Ueda K, Ninomiya M, Tanaka S, Hoshiko M, Yoshiyama Y, Takeuchi M, et al. An alpha-glucosidase inhibitor, acarbose treatment decreases serum levels of glyceraldehyde-derived advanced glycation end products (AGEs) in patients with type 2 diabetes. Clin Exp Med. 2010;10:139–141. doi: 10.1007/s10238-009-0074-9. [DOI] [PubMed] [Google Scholar]
- 89.Tahara N, Yamagishi S, Takeuchi M, Honda A, Tahara A, Nitta Y, Kodama N, Mizoguchi M, Kaida H, Ishibashi M, et al. Positive association between serum level of glyceraldehyde-derived advanced glycation end products and vascular inflammation evaluated by [(18)F]fluorodeoxyglucose positron emission tomography. Diabetes Care. 2012;35:2618–2625. doi: 10.2337/dc12-0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ueda S, Yamagishi S, Matsui T, Noda Y, Ueda S, Jinnouchi Y, Sasaki K, Takeuchi M, Imaizumi T. Serum levels of advanced glycation end products (AGEs) are inversely associated with the number and migratory activity of circulating endothelial progenitor cells in apparently healthy subjects. Cardiovasc Ther. 2012;30:249–254. doi: 10.1111/j.1755-5922.2011.00264.x. [DOI] [PubMed] [Google Scholar]
- 91.Fukushima Y, Daida H, Morimoto T, Kasai T, Miyauchi K, Yamagishi S, Takeuchi M, Hiro T, Kimura T, Nakagawa Y, et al. Relationship between advanced glycation end products and plaque progression in patients with acute coronary syndrome: the JAPAN-ACS sub-study. Cardiovasc Diabetol. 2013;12:5. doi: 10.1186/1475-2840-12-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Armstrong MJ, Adams LA, Canbay A, Syn WK. Extrahepatic complications of nonalcoholic fatty liver disease. Hepatology. 2014;59:1174–1197. doi: 10.1002/hep.26717. [DOI] [PubMed] [Google Scholar]
- 93.Yamagishi S, Matsui T, Ueda S, Fukami K, Okuda S. Clinical utility of acarbose, an alpha-glucosidase inhibitor in cardiometabolic disorders. Curr Drug Metab. 2009;10:159–163. doi: 10.2174/138920009787522133. [DOI] [PubMed] [Google Scholar]
- 94.Kitahara Y, Takeuchi M, Miura K, Mine T, Matsui T, Yamagishi S. Glyceraldehyde-derived advanced glycation end products (AGEs). A novel biomarker of postprandial hyperglycaemia in diabetic rats. Clin Exp Med. 2008;8:175–177. doi: 10.1007/s10238-008-0176-9. [DOI] [PubMed] [Google Scholar]
- 95.Ekong U, Zeng S, Dun H, Feirt N, Guo J, Ippagunta N, Guarrera JV, Lu Y, Weinberg A, Qu W, et al. Blockade of the receptor for advanced glycation end products attenuates acetaminophen-induced hepatotoxicity in mice. J Gastroenterol Hepatol. 2006;21:682–688. doi: 10.1111/j.1440-1746.2006.04225.x. [DOI] [PubMed] [Google Scholar]
- 96.Zeng S, Feirt N, Goldstein M, Guarrera J, Ippagunta N, Ekong U, Dun H, Lu Y, Qu W, Schmidt AM, et al. Blockade of receptor for advanced glycation end product (RAGE) attenuates ischemia and reperfusion injury to the liver in mice. Hepatology. 2004;39:422–432. doi: 10.1002/hep.20045. [DOI] [PubMed] [Google Scholar]
- 97.Cataldegirmen G, Zeng S, Feirt N, Ippagunta N, Dun H, Qu W, Lu Y, Rong LL, Hofmann MA, Kislinger T, et al. RAGE limits regeneration after massive liver injury by coordinated suppression of TNF-alpha and NF-kappaB. J Exp Med. 2005;201:473–484. doi: 10.1084/jem.20040934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Goodwin M, Herath C, Jia Z, Leung C, Coughlan MT, Forbes J, Angus P. Advanced glycation end products augment experimental hepatic fibrosis. J Gastroenterol Hepatol. 2013;28:369–376. doi: 10.1111/jgh.12042. [DOI] [PubMed] [Google Scholar]
- 99.Lohwasser C, Neureiter D, Popov Y, Bauer M, Schuppan D. Role of the receptor for advanced glycation end products in hepatic fibrosis. World J Gastroenterol. 2009;15:5789–5798. doi: 10.3748/wjg.15.5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ratziu V, Munteanu M, Charlotte F, Bonyhay L, Poynard T. Fibrogenic impact of high serum glucose in chronic hepatitis C. J Hepatol. 2003;39:1049–1055. doi: 10.1016/s0168-8278(03)00456-2. [DOI] [PubMed] [Google Scholar]
- 101.Challier M, Jacqueminet S, Benabdesselam O, Grimaldi A, Beaudeux JL. Increased serum concentrations of soluble receptor for advanced glycation endproducts in patients with type 1 diabetes. Clin Chem. 2005;51:1749–1750. doi: 10.1373/clinchem.2005.051961. [DOI] [PubMed] [Google Scholar]
- 102.Nakamura K, Yamagishi S, Adachi H, Kurita-Nakamura Y, Matsui T, Yoshida T, Sato A, Imaizumi T. Elevation of soluble form of receptor for advanced glycation end products (sRAGE) in diabetic subjects with coronary artery disease. Diabetes Metab Res Rev. 2007;23:368–371. doi: 10.1002/dmrr.690. [DOI] [PubMed] [Google Scholar]
- 103.Tan KC, Shiu SW, Chow WS, Leng L, Bucala R, Betteridge DJ. Association between serum levels of soluble receptor for advanced glycation end products and circulating advanced glycation end products in type 2 diabetes. Diabetologia. 2006;49:2756–2762. doi: 10.1007/s00125-006-0394-1. [DOI] [PubMed] [Google Scholar]
- 104.Nakamura K, Yamagishi SI, Matsui T, Adachi H, Takeuchi M, Imaizumi T. Serum levels of soluble form of receptor for advanced glycation end products (sRAGE) are correlated with AGEs in both diabetic and non-diabetic subjects. Clin Exp Med. 2007;7:188–190. doi: 10.1007/s10238-007-0146-7. [DOI] [PubMed] [Google Scholar]
- 105.Yamagishi S, Adachi H, Nakamura K, Matsui T, Jinnouchi Y, Takenaka K, Takeuchi M, Enomoto M, Furuki K, Hino A, et al. Positive association between serum levels of advanced glycation end products and the soluble form of receptor for advanced glycation end products in nondiabetic subjects. Metabolism. 2006;55:1227–1231. doi: 10.1016/j.metabol.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 106.Nakamura K, Yamagishi S, Adachi H, Kurita-Nakamura Y, Matsui T, Yoshida T, Imaizumi T. Serum levels of sRAGE, the soluble form of receptor for advanced glycation end products, are associated with inflammatory markers in patients with type 2 diabetes. Mol Med. 2007;13:185–189. doi: 10.2119/2006-00090.Nakamura. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nakamura K, Yamagishi S, Adachi H, Matsui T, Kurita-Nakamura Y, Takeuchi M, Inoue H, Imaizumi T. Circulating advanced glycation end products (AGEs) and soluble form of receptor for AGEs (sRAGE) are independent determinants of serum monocyte chemoattractant protein-1 (MCP-1) levels in patients with type 2 diabetes. Diabetes Metab Res Rev. 2008;24:109–114. doi: 10.1002/dmrr.766. [DOI] [PubMed] [Google Scholar]
- 108.Yamagishi S, Imaizumi T. Serum levels of soluble form of receptor for advanced glycation end products (sRAGE) may reflect tissue RAGE expression in diabetes. Arterioscler Thromb Vasc Biol. 2007;27:e32; author reply e33–e34. doi: 10.1161/ATVBAHA.107.139923. [DOI] [PubMed] [Google Scholar]
- 109.Yamagishi S, Matsui T, Nakamura K. Kinetics, role and therapeutic implications of endogenous soluble form of receptor for advanced glycation end products (sRAGE) in diabetes. Curr Drug Targets. 2007;8:1138–1143. doi: 10.2174/138945007782151298. [DOI] [PubMed] [Google Scholar]
- 110.Yoshida T, Yamagishi S, Matsui T, Nakamura K, Ueno T, Takeuchi M, Sata M. Telmisartan, an angiotensin II type 1 receptor blocker, inhibits advanced glycation end-product (AGE)-elicited hepatic insulin resistance via peroxisome proliferator-activated receptor-gamma activation. J Int Med Res. 2008;36:237–243. doi: 10.1177/147323000803600204. [DOI] [PubMed] [Google Scholar]
- 111.Yamagishi S, Nakamura K, Matsui T. Potential utility of telmisartan, an angiotensin II type 1 receptor blocker with peroxisome proliferator-activated receptor-gamma (PPAR-gamma)-modulating activity for the treatment of cardiometabolic disorders. Curr Mol Med. 2007;7:463–469. doi: 10.2174/156652407781387073. [DOI] [PubMed] [Google Scholar]
- 112.Marx N, Walcher D, Ivanova N, Rautzenberg K, Jung A, Friedl R, Hombach V, de Caterina R, Basta G, Wautier MP, et al. Thiazolidinediones reduce endothelial expression of receptors for advanced glycation end products. Diabetes. 2004;53:2662–2668. doi: 10.2337/diabetes.53.10.2662. [DOI] [PubMed] [Google Scholar]
- 113.Yoshida T, Yamagishi S, Nakamura K, Matsui T, Imaizumi T, Takeuchi M, Makino T, Shimizu T, Sata M. Atorvastatin inhibits advanced glycation end products (AGE)-induced C-reactive expression in hepatoma cells by suppressing reactive oxygen species generation. Vascular Dis Prevent. 2007;4:213–216. [Google Scholar]
- 114.Kimura Y, Hyogo H, Yamagishi S, Takeuchi M, Ishitobi T, Nabeshima Y, Arihiro K, Chayama K. Atorvastatin decreases serum levels of advanced glycation endproducts (AGEs) in nonalcoholic steatohepatitis (NASH) patients with dyslipidemia: clinical usefulness of AGEs as a biomarker for the attenuation of NASH. J Gastroenterol. 2010;45:750–757. doi: 10.1007/s00535-010-0203-y. [DOI] [PubMed] [Google Scholar]
- 115.Vilà L, Rebollo A, Ađalsteisson GS, Alegret M, Merlos M, Roglans N, Laguna JC. Reduction of liver fructokinase expression and improved hepatic inflammation and metabolism in liquid fructose-fed rats after atorvastatin treatment. Toxicol Appl Pharmacol. 2011;251:32–40. doi: 10.1016/j.taap.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 116.Patel R, Baker SS, Liu W, Desai S, Alkhouri R, Kozielski R, Mastrandrea L, Sarfraz A, Cai W, Vlassara H, et al. Effect of dietary advanced glycation end products on mouse liver. PLoS One. 2012;7:e35143. doi: 10.1371/journal.pone.0035143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Leung C, Herath CB, Jia Z, Goodwin M, Mak KY, Watt MJ, Forbes JM, Angus PW. Dietary glycotoxins exacerbate progression of experimental fatty liver disease. J Hepatol. 2014;60:832–838. doi: 10.1016/j.jhep.2013.11.033. [DOI] [PubMed] [Google Scholar]
- 118.Sato T, Wu X, Shimogaito N, Takino J, Yamagishi S, Takeuchi M. Effects of high-AGE beverage on RAGE and VEGF expressions in the liver and kidneys. Eur J Nutr. 2009;48:6–11. doi: 10.1007/s00394-008-0753-4. [DOI] [PubMed] [Google Scholar]
- 119.Niwa T, Nomura T, Sugiyama S, Miyazaki T, Tsukushi S, Tsutsui S. The protein metabolite hypothesis, a model for the progression of renal failure: an oral adsorbent lowers indoxyl sulfate levels in undialyzed uremic patients. Kidney Int Suppl. 1997;62:S23–S28. [PubMed] [Google Scholar]
- 120.Ueda S, Yamagishi S, Takeuchi M, Kohno K, Shibata R, Matsumoto Y, Kaneyuki U, Fujimura T, Hayashida A, Okuda S. Oral adsorbent AST-120 decreases serum levels of AGEs in patients with chronic renal failure. Mol Med. 2006;12:180–184. doi: 10.2119/2005-00034.Ueda. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Choei H, Sasaki N, Takeuchi M, Yoshida T, Ukai W, Yamagishi S, Kikuchi S, Saito T. Glyceraldehyde-derived advanced glycation end products in Alzheimer’s disease. Acta Neuropathol. 2004;108:189–193. doi: 10.1007/s00401-004-0871-x. [DOI] [PubMed] [Google Scholar]
- 122.Sato T, Shimogaito N, Wu X, Kikuchi S, Yamagishi S, Takeuchi M. Toxic advanced glycation end products (TAGE) theory in Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2006;21:197–208. doi: 10.1177/1533317506289277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Takeuchi M, Yamagishi S. Possible involvement of advanced glycation end-products (AGEs) in the pathogenesis of Alzheimer’s disease. Curr Pharm Des. 2008;14:973–978. doi: 10.2174/138161208784139693. [DOI] [PubMed] [Google Scholar]
- 124.Abe R, Yamagishi S. AGE-RAGE system and carcinogenesis. Curr Pharm Des. 2008;14:940–945. doi: 10.2174/138161208784139765. [DOI] [PubMed] [Google Scholar]
- 125.Sakuraoka Y, Sawada T, Okada T, Shiraki T, Miura Y, Hiraishi K, Ohsawa T, Adachi M, Takino J, Takeuchi M, et al. MK615 decreases RAGE expression and inhibits TAGE-induced proliferation in hepatocellular carcinoma cells. World J Gastroenterol. 2010;16:5334–5341. doi: 10.3748/wjg.v16.i42.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Takino J, Yamagishi S, Takeuchi M. Glycer-AGEs-RAGE signaling enhances the angiogenic potential of hepatocellular carcinoma by upregulating VEGF expression. World J Gastroenterol. 2012;18:1781–1788. doi: 10.3748/wjg.v18.i15.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kan H, Yamagishi SI, Ojima A, Fukami K, Ueda S, Takeuchi M, Hyogo H, Aikata H, Chayama K. Elevation of Serum Levels of Advanced Glycation End Products in Patients With Non-B or Non-C Hepatocellular Carcinoma. J Clin Lab Anal. 2014:Epub ahead of print. doi: 10.1002/jcla.21797. [DOI] [PMC free article] [PubMed] [Google Scholar]