

Abstract
Shiga toxin (Stx) is the primary cause of severe host responses including renal and central nervous system (CNS) disease in Shiga toxin-producing E. coli (STEC) infections. The interaction of Stx with different eukaryotic cell types is described. Host responses to Stx and bacterial lipopolysaccharide (LPS) are compared as related to the features of the STEC-associated Hemolytic Uremic Syndrome (HUS). Data derived from animal models of HUS and CNS disease, in vivo, and eukaryotic cells, in vitro, are evaluated in relation to HUS disease of humans.
I. Activities of Stx and LPS in renal disease
1. Shiga toxin actions
It is generally accepted that all actions of Shiga toxin (Stx) depend on its interaction with the receptor, globotriaosylceramide (Gb3) on eukaryotic cells. While alternative receptors for Stx have been postulated, no definitive data have been forthcoming in support. Stx holotoxin is internalized by receptor-mediated endocytosis, retrograde transported via the Golgi apparatus and processed through in the endoplasmic reticulum, and released into the cytoplasm where it enzymatically inactivates ribosomes and inhibits protein synthesis (Fig. 1). However, it is important to note that in addition to Stx holotoxin, the B-subunit alone can interact with Gb3 in a physiologically meaningful manner where it activates signal transduction pathways in target cells (Fig. 1)[1]. An additional, but unexplained anomaly is the interaction of Stx with eukaryotic cells in a Gb3-independent manner that leads to induction of cytokines by these cells [2]. As shown in Figure 1, intracellular responses to Stx are diverse, including inhibition of protein synthesis, activation of cellular stress responses, and induction of cytokines and chemokines. It is likely that these different schemes take place in cell-specific activities during Shiga toxin E. coli (STEC) infections in humans culminating in typical hemolytic uremic syndrome (HUS) disease. As depicted, it is clear that in some cases Stx can result in activation of p38 MAP kinase as well as apoptotic and necrotic cell death (Fig. 1). The topic of HUS renal disease has been reviewed recently [3–5].
Figure 1.

Schema: Shiga toxin interaction with eukaryotic cells.
2. Cell types responsive to Stx
The high number of Stx-sensitive cell types makes more difficult identification of more important events responsible for HUS. Renal microvascular endothelial cells are generally accepted to be the primary target of Stxs in HUS. Data in support of this concept comes from many sources, most notably autopsy kidney pathology samples showing swollen and detached endothelial cells accompanied by thrombi [6]. Such human renal microvascular endothelial cells were also shown to be very sensitive to Stxs, in vitro [7]. However, other cells which comprise the human renal glomerulus are also sensitive to Stx including podocytes and mesangial cells [8, 9]. In addition, extraglomerular epithelial cell types of the human kidney have been postulated to be targets of Stx, including proximal tubule and collecting duct cells [8, 10, 11]. Cell types in the blood circulation which may be key to development of HUS and which are sensitive to Stx include platelets, neutrophils, and monocytes [12–16].
In summary, most, if not all, of the cell types mentioned may well have a role in STEC related kidney disease and typical HUS. The relative importance and role of these cell types in STEC HUS remains to be determined. For example, it is not clear which of the renal cell types are actually responsible for renal failure in STEC HUS, although apoptosis of tubules appears to be a common feature [8, 17]. The relative contributions in HUS disease of renal microvascular coagulation and thrombosis (i.e. endothelial cells), imbalance of fluid and electrolytes (i.e. nephron tubules), and altered filtration barrier function (i.e. endothelial and podocyte cells) has yet to be elucidated for typical HUS. If in vitro cell culture studies are pertinent to HUS in patients, the sensitivity (LD50) of human renal cells to Stx2 (endothelial, 0.1 pM > podocyte, 0.5 pM ≫ proximal tubule, 10 pM) suggests the renal filtration barrier is at considerable risk [8].
3. Inflammatory cells, chemokines, and renal thrombosis
A primary feature in the renal pathology of STEC HUS is microvascular coagulation and thrombosis. In humans and in a murine model of HUS, the Interaction of Stx and LPS with circulating cells and resident renal cells appears to have a causal role in microvascular thrombosis [18, 19]. In a series of studies in the Stx/LPS murine model of HUS, a pathway leading to fibrin deposition was revealed (Fig. 2). LPS-activation of cells such as endothelial and renal tubule cells elicited chemokines (MCP-1, MIP-1alpha, RANTES) known as chemoattractants for monocyte/macrophage cells and co-activators of platelets. In this response, Stx enhances the effects of, but does not replace LPS. The response was associated with renal fibrin deposition [12, 20]. In the murine model, simultaneous neutralization of these three chemokines inhibited LPS/Stx-induced monocyte accumulation and fibrin deposition in the kidneys [20]. Further, administration of adenosine A2a receptor (A2aR) agonists to Stx/LPS mice also reduced monocyte and fibrin accumulation in the kidneys. As shown (Fig. 3), A2aR agonists act as anti-inflammatory agents in monocytes, platelets, and endothelial cells [21]. Taken together these studies indicate that both LPS and Stx are required for maximal renal fibrin deposition and that platelets may be required. Because mice deficient in MCP-1 have sharply reduced platelet deposition after exposure to Stx/LPS, we have suggested that this chemokine serves as a co-activator of platelets in typical HUS (Keepers, unpublished data). The primary activators of platelet activation are thrombin or adenosine diphosphate (ADP). Our renal gene array analysis of the LPS response in mice indicated that LPS strongly elicited fibrinogen mRNA, the precursor of fibrin (Obrig, unpublished data). In addition, it is noteworthy that selective elimination of monocytes from mice prior to the above studies had no effect on the ability of Stx/LPS to elicit renal fibrin deposition suggesting the chemokines are being generated from other cell types such as renal tubules [20]. Important conclusions from the murine HUS model are that LPS, not Stx, is the initial primary elicitor of renal coagulation and thrombosis, but Stx, not LPS, is the lethal agent of STEC.
Figure 2. Proposed pathways of Stx and LPS actions in mice.

Data derived from a Stx/LPS murine model of HUS indicate that LPS is the primary elicitor of fibrin deposition in kidneys. This pathway requires chemokines and platelets, but is not responsible for renal failure. Stx is responsible for renal failure in this murine model in a process which involves non-endothelial renal cell types.
Figure 3. Anti-inflammatory actions of adenosine in HUS.

Data derived from a Stx/LPS murine model of HUS suggest adenosine A2a receptor agonist, i.e. adenosine, effectively blocks the actions of LPS (enhanced by Stx2) at the level of different renal cell types to prevent platelet activation and coagulation.
In the murine Stx/LPS model of HUS, monocyte migration into the kidneys was restricted to the extra-glomerular space in contrast to polymorphonuclear leukocytes (PMN) which in addition migrated into the glomeruli. The latter may be important in humans because neutrophilia has been implicated as a primary risk factor for HUS disease and increased neutrophil migration into the kidneys was a key observation in HUS renal biopsies [22, 23]. In the murine model of HUS, the neutrophil chemotactic factors CXCL1 (KC) and CXCL2 (MIP-2) were induced in the kidneys by LPS [15]. The induction was at the transcriptional level and was enhanced by Stx2. Administration of neutralizing antibodies for these neutrophil chemotactic factors prevented the movement of neutrophils into the kidneys. It was also demonstrated that VCAM-1 was induced in the kidneys simultaneously with CXCL-1 and CXCL-2 in response to Stx2/LPS in mice (Fig. 5). VCAM-1 is known to assist movement of neutrophils across the endothelium and appeared to exhibit this function for neutrophils in the Stx2/LPS murine model of HUS. However, the relative importance of renal neutrophils in Stx-induced renal failure has yet to be determined in mice and humans.
Figure 5. Neutrophil-endothelial cell interactions in HUS.

In the Stx2/LPS murine model of HUS, analysis of renal gene activation and neutrophil infiltration into kidneys demonstrates a concomitant increase in PMNs and VCAM-1 expression, suggesting a mechanism of PMN-endothelial association.
4. Renal gene array analysis of murine responses to Stx2 and LPS
Much information is now available regarding the biological effects of Stx2 and LPS on kidneys in the murine HUS model. The following is a synopsis of the more pertinent gene microarray data obtained from temporal studies of the murine renal responses to Stx2, LPS, or Stx2/LPS [19]. Based on the total of both up- and down-regulated genes, five-times more renal genes responded to LPS than to Stx2 over the 72h time course. Response to LPS was mostly early, while Stx2 responses occurred later in the 72h time course. These results are more meaningful when viewed in the larger picture of HUS disease where renal failure occurs later in the time course in both mice and humans. It should be emphasized that Stx2, rather than LPS, is the lethal factor in the murine HUS model. The gene array data revealed different roles for LPS and Stx2 in the renal physiological responses. LPS responses were mostly inflammatory, stress related, or cell defensive in nature. In contrast, Stx2 responses were related to cell repair and involved cell proliferation and differentiation or cell cycle control genes. An interesting finding was that renal genes down-regulated by Stx2 included membrane transporters which appeared to signal a protective survival mode and slowing of cell metabolism.
The renal genes most up-regulated by Stx2 or LPS are depicted in Fig. 4. As expected from the inflammatory responses described above, LPS induced a number of chemokine genes which code for chemotactic factors for monocytes and neutrophils. These tend to be ‘immediate’ response genes which attract monocytes and neutrophils into the kidneys and set the stage for a broad inflammatory response in the kidneys. Such LPS ‘immediate’ response genes are mentioned in the literature in descriptions of typical HUS, i.e. MCP-1, MIP-2alpha, and the murine IL-8 mimic, KC. It was also observed that IP-10 (CXCL10) was induced by LPS as well as by Stx2, albeit in early and late parts of the HUS disease time course, respectively. Related to renal coagulation and thrombosis in HUS, LPS induced a set of fibrinogen genes ‘late’ in the time course of the murine model of HUS concomitant with the appearance of fibrin deposition and coagulation in the renal microvasculature of HUS (Fig. 4). These data agree with our observation that LPS is responsible, in part, for fibrin deposition in the Stx2/LPS murine model of HUS [19]. Amyloid protein which has been reported to be a Stx-sensitizing factor in HUS is induced at the mRNA level by LPS in mice as shown in Fig. 4 as a renal ‘late’ gene product [24]. More recently, complement has been identified as a factor that may contributes to renal failure in atypical HUS.
Figure 4. Renal gene activation in the Stx/LPS murine model.

Shown are the 10 most up-regulated genes in the temporal response of mice to either LPS or Stx2. Gene microarrays were employed to analyze kidney gene activation over a 72h response of C57BL/6 mice to 300 ug/kg LPS or 100 ng/kg Stx2.
Products of some of the genes shown in Fig. 4 have been examined by investigators as potential biomarkers for diagnostic purposes. For example, IP-10 has been identified as a urine biomarker for other kidney diseases such as lupus nephritis [25, 26]. Lipocalin 2 (NGAL), an LPS-induced ‘early’ gene (Fig. 4) is a common urine biomarker for numerous renal diseases including STEC-HUS [27].
5. How valid is the murine model of HUS for translation to the human disease?
A large volume of data exists for mouse models of Stx-HUS [28]. The two common experimental approaches for these murine models are either oral infection with STEC or injection with purified Stx plus or minus LPS [17, 19, 29, 30]. In virtually all cases these are lethality models within 4 to 12 days after exposure to the agents and are accompanied by renal damage. Where examined these murine models usually exhibit the three hallmarks of HUS; thrombocytopenia, hemolytic anemia, and renal failure. However, every animal model has its limitations, and for the murine models of HUS, the renal microvascular endothelial cells do not express Gb3 and are resistant to Stx action. This is important if one believes that the primary target of Stx is the renal microvascular endothelium. Indeed, human renal endothelial cells, in vitro, are very sensitive to Stx, and the pathology of human kidneys in HUS describes swollen and detached glomerular endothelial cells. But, it is surprising why such human glomerular endothelium is not killed by Stx in HUS kidneys. This suggests either a more indirect action of Stx in human HUS or dominant survival activities are activated within the endothelium after exposure to Stx. An alternative explanation is that the primary target of Stx in human kidneys is not the endothelium, but rather glomerular podocytes and extra-glomerular tubules along the nephron. Support for this exists for HUS in mice and humans where urine specific gravity changes, chemokines are increased in the urine, and biomarkers of damaged podocytes and tubule cells are detected.
Mouse models have been helpful in separating the actions of Stx and LPS in HUS. In general, and as described above, LPS is the primary inducer of cytokines and chemokines where Stx enhances the activity of LPS. The complexity of inflammation in HUS is critical, but really has yet to be fully delineated in murine models and in human HUS. The murine model mirrors typical HUS of humans as resting platelets are resistant to Stx and require pre-activation with LPS [19]. However, it is most important to reiterate that Stx, not LPS is responsible for the renal failure in typical HUS. In conclusion, the murine responses to Stx and LPS include most of the features of STEC-HUS in humans.
II. Activities of Stx in CNS disease
1. CNS symptoms of animal models
In either an oral inoculation of Shiga toxin-producing E. coli (STEC) model or purified Shiga toxin (Stx) injection animal model, the most common and most frequently reported central nervous system (CNS) impairment is paralysis of extremities. Most frequently, the hind legs are affected first followed by the fore-legs. Other symptoms include anorexia, lethargy, ataxic gait, recumbency (the affected animals lose strength required to hold their body in an upright position), convulsions, seizure, coma and death.
STEC oral administration animal models are summarized in Table 1. The oral inoculation models of STEC that describes CNS symptoms are limited to pig and mouse. Pigs develop “edema disease” with Stx2e-producing E. coli and present CNS symptoms (Table 3). Experimentally, edema disease-like state is reproducible with Stx2-producing E. coli that has been isolated from human patients. CNS symptoms are only seen in Stx2-(both Stx2 and Stx2e) producers, but not in non-Stx2 producers. This indicates a strong association of Stx2 to CNS impairment.
Table 1.
STEC oral administration model with CNS descriptions
| Ref. | animal | E. coli strain | CNS (a) | Histopathology (b) (c) (d) | IHC(e)/TUNEL (f) (b)(c) | Model notes | Other assays |
|---|---|---|---|---|---|---|---|
| [31] | pig | RCH/86 (Stx2+) | yes | HE: CL cap, small inf, small hrrg, fib in sub and cap | n.d. | gnotobiotic (caesarian section derived) | n.a. |
| [32] | pig | 86-24 (Stx2+) | yes | Gross: MO and CL hrrg and nec PAS: MO, CL, and Sc, cap swl nec, peri deposits |
n.d. | Suckling (colostrum provided) | n.a. |
| [32] | pig | 87-23 Stx (−) | no | No lesion | n.d. | Suckling (colostrum provided) | n.a. |
| [33] | pig | S1191 (Stx2e+) M112 (Stx2e+) |
yes | EM: myo and cap nec not apop, mono apop | TUNEL + myo in MO (5/11 pigs) | 3-w-o | n.a. |
| [33] | pig | Strain 123 (non-pathogenic E. coli) | no | No lesion | N.d. | 3-w-o | n.a. |
| [34] | pig | sakai | yes | LFB: mye deg, hrrg, pyk and prolif cap, peri ede | n.d. | neonatal | n.a. |
| [35] | ICR mouse | E32511/HSC (Stx2c+) (i) | n.d. | EM: cap ede in CR ctx, mye deg HE: hrrg and ede in CR ctx only in CNS symptom (+) mice |
Immuno EM-DAB (g): Stx2+ in CR ctx pyr and deg mye | Sm, MMC (h) | Tracer (i.v.) detected in cap and deg mye |
| [29] | C3H/HeN mouse | 86-24, 86BL or 134 (Stx2+) | yes | n.d. | n.d. | fasted | n.a. |
| [29] | C3H/HeN mouse | 87-23, 87BL (Stx2−) | yes | n.d. | n.d. | fasted | n.a. |
| [29] | C3H/HeJ mouse | 86-24, 86BL or 134 (Stx2+) | yes (biph asic) | n.d. | n.d. | fasted | n.a. |
| [29] | C3H/HeJ mouse | 87-23, 87BL (Stx2−) | no | n.d. | n.d. | fasted | n.a. |
| [36] | C57BL/6 mouse | N-9 (Stx1+/Stx2+) | n.d. | HE: infilt, hrrg, cap with fib in CR ctx LFB: No deg mye in hippo |
Anit-Stx + hippo | PCM (j) | n.a. |
| [37] | IQI mouse | EDL931 (Stx1+/Stx2+) | yes | HE: ede, fib in cap, neu deg, cap prolif | n.d. | gnotobiotic | Brain TNFa increased |
| [55] | C57BL/6 mouse | Smr N-9 (Stx1+/Stx2+) | n.d. | n.d. | TUNEL + hippo neu during CNS symptom (+) | PCM | Serum Stx ↑, TNFa ↑, IL10 ↑ TLC-anti-PkMab (k) brain + |
| [38] | IQI mouse | O157:H7 strain 6 (Stx1+/Stx2+) | yes | HE: CR ctx and CL neu nec and slight loss of Purkinje | n.d. | gnotobiotic | n.d. |
| [56] | ICR mouse | E32511/HSC (Stx2c+) | yes | n.d. | GFAP (m) ↑, AQP4↓, casp3↑(n) neu cer Sc ventral and MO dorsal | Sm, MMC | ISH (l) Gb3 synthase |
Detailed CNS symptoms are summarized in Table 3.
Histopathology analysis keys are Gross (gross observation in non-stained tissue), HE (hematoxylin-esosin stain that stains cytoplasm in pink and nucleus blue, light microscopic findings (LM)), PAS (Periodic acid-Schiff stain that detects polysaccharides, glycoproteins and glycolipid, LM), LFB (Luxol fast blue stain that stains myelin in blue, LM), EM (electron microscopic findings)
CNS regions and cell type abbreviations are CR (cerebrum), ctx (cortex), hippo (hippocampus), str (striatum), CL (cerebellum), MO (medulla oblongata), Sc (spinal cord), cer (cervical), tho (thoracic), lum (lumbaris), sub (subarachinoid space), BS (brain stem is used where midbrain, pons or medulla oblongata are not specified), Histopathologic feature abbreviations are cap (endothelial cells or capillaries), inf (infarction), hrrg (hemorrhage), fib (fibrin deposition), nec (necrosis), swl (swelling), peri (perivascular), myo (myocytes), apop (apoptotic), mono (monocytes), mye (myelin), deg (degeneration), pyk (pyknotic nuclei), prolif (proliferation/hyperplasia), ede (edema), pyr (pyramidal neuron), inflt (infiltration of blood cells to parenchyma), neu (neuron), Purkinje (Purkinje cells are large neurons in CL)
Histopathologic feature abbreviations are cap (endothelial cells or capillaries), inf (infarction), hrrg (hemorrhage), fib (fibrin deposition), nec (necrosis), swl (swelling), peri (perivascular), myo (myocytes), apop (apoptotic), mono (monocytes), mye (myelin), deg (degeneration), pyk (pyknotic nuclei), prolif (proliferation/hyperplasia), ede (edema), pyr (pyramidal neuron), inflt (infiltration of blood cells to parenchyma), neu (neuron), Purkinje (Purkinje cells are large neurons in CL)
IHC = immunohistochemistry, immunodetection of the target in the tissue sections
TUNEL = terminal deoxynucleotidyl transferase dUTP nick end labeling detects DNA fragmentation that is a hallmark of apoptosis.
Immuno-EM-DAB: immunodetection of the target with 3,3′-diaminobenzidine (DAB) deposition by EM
Sm =streptomycin, MMC = mitomycin C
Smr, MMCr
PCM = protein calorie malnutrition
TLC-anti-PkMab (thin layer chromatography with anti-Pk monoclonal antibody detectin)
ISH = in situ hybridization
GFAP = glial fibrillary acidic protein, an astrocyte marker, an increase of GFAP suggests astrogliosis.
IHC for activated (cleaved) caspase-3
Table 3.
Observed CNS symptoms in animal models (a)
| ref. | animal | model | ANOX | LTHG | HL para | FL para | ATX | RCM (b) | CV/TR | SZR | coma | death | other |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [46] | Baboon | Stx1 | n.d. (b) | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | yes | |
| [67] | Baboon | Stx1 | yes | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 3/6 (50%) | yes | yes | |
| [31] | Pig | STEC | yes | yes | yes | n.d. | yes | yes | yes | yes | yes | yes | |
| [71] | Pig | STEC | n.d. | n.d. | n.d. | n.d. | yes | yes | n.d. | n.d. | n.d. | yes | Diarrhea then CNS+ |
| [32] | Pig | STEC | n.d. | n.d. | yes | yes | n.d. | yes | yes | n.d. | n.d. | yes | paddling |
| [39] | Pig | Stx2e iv | yes | n.d. | n.d. | n.d. | yes | n.d. | yes | n.d. | yes | yes | Paddling, extensor rigidity, dyspnea |
| [40] | pig | Sup Stx2e iv | n.d. | n.d. | n.d. | n.d. | yes | yes | yes | n.d. | yes | yes | Paddling, extensor rigidity |
| [33] | Pig | STEC | n.d. | n.d. | n.d. | n.d. | Yes (1/11) | Yes (1/11) | n.d. | n.d. | n.d. | n.d. | |
| [41] | Rabbit | Stx1 | yes | yes | yes | n.d. | yes | n.d. | n.d. | n.d. | n.d. | yes | |
| [42] | Rabbit | Stx1 iv | yes | yes | yes | yes | n.d. | yes | No | n.d. | n.d. | Yes | Ruffle fur, rapid respiration |
| [52] | Rabbit | Stx2 iv | n.d. | n.d. | Yes (50%) | Yes (50%) | Yes (33%) | n.d. | Yes (50%) | n.d. | n.d. | Yes (50%) | Opisthotonic posture |
| [47] | Rabbit | Stx2 iv and it | yes | yes | yes | yes | n.d. | yes | n.d. | n.d. | n.d. | yes | |
| [68] | Rabbit | Stx2 iv | yes | n.d. | yes | yes | n.d. | n.d. | n.d. | n.d. | n.d. | yes | |
| [54] | Rabbit | Stx2 iv | yes | n.d. | yes | yes | n.d. | n.d. | n.d. | n.d. | n.d. | Yes | dyspnea |
| [43] | Rabbit | Stx2 iv | yes | n.d. | Yes (83.3%) | n.d. | Yes (83.3%) | n.d. | n.d. | n.d. | n.d. | yes | |
| [48] | Rabbit | Stx2 iv | n.d. | n.d. | Yes (25%) | n.d. | n.d. | n.d. | Yes (25%) | n.d. | n.d. | n.d. | |
| [57] | Rat | Stx2 i.c.v. | n.d. | Yes | yes | n.d. | n.d. | n.d. | n.d. | yes | n.d. | yes | crawling |
| [35] | Mouse | STEC | n.d. | yes | yes | yes | n.d. | n.d. | n.d. | n.d. | n.d. | death | Deformity of backbone, Loss of pain |
| [29] | Mouse | STEC | n.d. | n.d. | yes | yes | yes | n.d. | yes | n.d. | yes | yes | Jerky rhythmic motion |
| [36] | Mouse | STEC | yes | yes | yes | n.d. | n.d. | n.d. | (yes) (c) | n.d. | n.d. | yes | Ruffled fur, jerky rhythmic motion |
| [37] | Mouse | STEC | yes | yes | yes | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | Yes | |
| [44] | Mouse | Stx2 iv | n.d. | n.d. | yes | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | yes | |
| [44] | Mouse | Stx2+LPS iv | n.d | n.d | n.d | n.d | n.d | n.d | yes | Yes | n.d | yes | |
| [38] | Mouse | STEC | yes | yes | yes | n.d. | n.d. | n.d. | yes | n.d. | n.d. | yes | Ruffled fur |
| [53] | Mouse | Stx2 ip | n.d. | yes | yes | n.d. | yes | n.d. | yes | Yes | n.d. | yes | Retain sense (pain) |
| [56] | Mouse | STEC | n.d. | n.d. | yes | n.d. | n.d. | n.d. | yes | n.d. | n.d. | yes | Spinal deformity |
Abbreviations for CNS symptoms are ANOX (anorexia), LTHG (lethargy), HL para (hind-leg paralysis), FL para (fore-leg paralysis), ATX (ataxic gait), RCM (recumbency, difficulty holding body upright by itself), CV/TR (convulsions/tremors), SZR (seizure).
Lateral, sternal or dorsal recumbency; the animal is lying down with leaning on its side, abdomen or back, having a difficulty holding its body upright.
N.d. = not described
Shivering
Lipopolysaccharide (LPS) is an outer membrane component of Gram negative bacteria and a strong inflammation inducer. The involvement of LPS in STEC-associated CNS symptoms was tested by using LPS non-responder mouse C3H/HeJ [29]. C3H/HeJ did present CNS symptoms when given Stx2-producer E. coli, but did not when Stx-non-producer was inoculated. This again suggests a strong involvement of Stx in CNS symptoms. The difference between LPS-responder mouse (C3H/HeN) and C3H/HeJ in CNS symptoms was that C3H/HeN showed a progressive time course of CNS symptoms whereas C3H/HeJ showed ‘biphasic’ response in that they developed milder CNS symptoms and recovered once, but then progressed to a severe form of CNS impairment. This suggests that even though Stx2 may be the central cause of CNS symptoms, addition of LPS response may contribute to the progress of the disease.
To further study the action of Stx2 in CNS disease, different animals were tested with purified Stx2. Stx2 injection animal models with CNS complications are summarized in Table 2. Also, LPS involvement or contribution to Stx2-associated CNS disease was tested in some reports. The reproducible results of hind leg paralysis and high frequency of convulsions and seizures with purified Stx confirms the central role of the toxin in STEC-associated CNS disease. Human STEC patients present various CNS symptoms that range from eye involvement (diplopia, hallucinations and cortical blindness), behavioral changes (hyperactivity, distractibility, irritability and altered sensorium), posturing/coordination difficulties (poor fine-motor coordination, hemiplegia, ataxia and clumsiness) and severe symptoms as seizures, dysregulation of breathing, alteration in consciousness such as coma. Within these varieties of symptoms, ataxia or hemiparesis resembles Stx-associated animal CNS symptoms. Also, it is notable in human patients, seizures are a frequent observation. This resemblance between patients and animal models of STEC/Stx suggest to us there is a great possibility that analyzing these animal models may give us some clues to define the mechanisms of CNS impairment in Stx-associated disease.
Table 2.
Shiga toxin and/or LPS administration model with CNS descriptions
| Ref. | Animal (a) | toxin | CNS (b) | Gb3/Stx binding | Histopathology (c) (d) (e) | imaging | IHC (f) | Injection route (g) | Other assays |
|---|---|---|---|---|---|---|---|---|---|
| [66] | baboon (h) | Stx1 | n.d. | n.d. | EM: mye deg, peri ede, large neu and glia deg, cap normal | n.d. | n.d. | i.v. | |
| [67] | baboon | Stx1 | yes | n.d. | n.d. | n.d. | n.d. | i.v. | |
| [39] | weaned YL pigs | Stx2e | yes | n.d. | Gross: Sc ede, CL ede and hrrg HE: CL hrrg but not ede or cap nec, no lesions in CR, BS, thalamus, |
n.d. | n.d. | i.v. | |
| [40] | weaned YL pigs | Sup (i) Stx2e | yes | n.d. | HE: CL, sub ede, hrrg, cap nec HE: MB, fib nec, peri eos |
n.d. | n.d. | i.v. | |
| [41] | NZW rabbit | Stx1 | yes | TLC-Stx1 over lay of CL, BS and Sc (+) at LacCer (j) position | HE: BS, Sc, CL cap narrowed, peri ede, cap damage and fib, Purkinje decreased | n.d. | n.d. | i.v. | |
| [42] | NZW rabbit | Stx1 | yes | 125I-Stx1 tissue distribution high in cecum, brain, small intestine, colon, Sc | HE: cerv Sc hrrg, inf, ede, fib in cap, lum Sc fib cap, pyk cap | n.d. | Anti-Stx (+)in Sc cap | i.v. | |
| [52] | JW rabbit | Stx2 | yes | n.d. | EM: mye deg, axo normal | MRI: V3 (24 h), later BS, cc, lateral amy, CL vermis | Immuno EM-DAB (k): anti-Stx2 (+) at luminal side of cap, deg mye | i.v. | Tracer |
| [47] | JW rabbit | Stx2 | Yes | n.d. | CR ctx, CL ctx, Sc, pyk neu, str neu not affected, inf in MB and CL | n.d. | Anti-Stx2 IHC (+)cap and sub | i.v. and i.t. | Stx2 ↑ in CSF |
| [65] | JW rabbit | Stx2 | n.d. | n.d. | n.d. | MRI: CL at 82 h | n.d. | i.v. | Stx2 ↑ in CSF |
| [65] | JW rabbit | Stx2 | n.d. | n.d. | n.d. | MRI: the rear of CL (contact w CSF) at 48 h | n.d. | i.t. | n.d. |
| [68] | JW rabbit | Stx2 | yes | n.d. | n.d. | MRI: BS and cerv Sc dorsal close to death | n.d. | i.v. | Barorefl ex function |
| [69] | JW rabbit | Stx1 | n.d. | n.d. | n.d. | MRI: MB, BS and Sc ede, cerv Sc dorsal hematoma | n.d. | i.v. | Stx1 ↑ slightly in CSF |
| [54] | JW rabbit young | Stx2 | yes | n.d. | HE: inf CL, myo thickening, fib, pyk and fragmented, pons myo nec | n.d. | Anti-Stx2 IHC(+) in pons cap and myo, Anti-ssDNA IHC small number cap (+),IHC caspase-3 &-9 small number (+) | i.v. | |
| [43] | JW rabbit | Stx2 | yes | Anti-Gb3 (+) in cap lum Sc | HE: inf lum Sc with hrrg and fib cap, str neu pyk, hippos pyr neu apop | n.d. | TUNEL + in hippo (DG, CA1, CR ctx neu, pons glia, cap IHC: Ib4↑ (microglia activated) in lum Sc and thalamus | i.v. | qRT-PCR: TNFa↑ and IFNb ↑ |
| [70] | JW rabbit | Stx2 | n.d. | n.d. | n.d. | MRI: Gd leak (↑permeability) | n.d. | i.v. | |
| [48] | DB rabbit | Stx2 | yes | n.d. | HE: neu deg in the BS | n.d. | n.d. | i.v. | |
| [49] | SD rat | Stx2 | n.d. | n.d. | n.d. | n.d. | Anti-Stx IHC (+)peri | i.p. | |
| [49] | SD rat | Stx2 | n.d. | n.d. | EM: irregular shape neu, mye deg, hypertrophic axo, astro phago mye, neu apop, deg, vacuol, peri astro ede, non-peri astro gliosis, oligo pathologic | n.d. | Anti-Stx2 (+) in neu, immunoEM-DAB: anti-Stx2 (+) in neu fibers and astro nucleus | i.c.v. | |
| [58] | SD rat | Stx2 | n.d. | n.d. | n.d. | n.d. | Stx2 (+) in anterior hippo astro, ips hippo Stx2(+)astro and neu, cont hippo Stx2(+)neuropils | i.c.v. | Str and CR ctx neu↓, cap↓ NADPH-d/NOS activity |
| [57] | SD rat | Stx2 | yes | Anti-Gb3↑ CA1, str neu | n.d. | n.d. | Anti-Stx2↑ MAP2 ↑ CA1 neu. Anti-bax ↑neu (inner ctx, CA1, subV dorsal str, hypothala mic peri | i.c.v. | |
| [50] | SD rat | Sup Stx2 | n.d. | n.d. | Nissle: neu pyk hypertrohy axo, ede ctx, subV CL. | n.d. | Anti-AQP4↓ cp | i.p. | |
| [35] | ICR mouse | Stx2 | yes | n.d. | n.d. | n.d. | Immune-EM-DAB: anti-Stx2 (+) mye deg, lyso pyr ctx | i.p. | Tracer |
| [44] | C57BL/6 mouse | Stx2 | yes | n.d. | HE: pyk oligo astro nuclei, hrrg sub | n.d. | n.d. | i.v. | |
| [44] | C57BL/6 mouse | Stx2 + LPS | yes | n.d. | HE: hrrg BS sub severe than Stx2 alone | n.d. | n.d. | i.v. | |
| [45] | ICR mouse | Stx2 | n.d. | n.d. | HE: congestion CL and hippo, hrrg CL, neu and glia normal | n.d. | anti-Stx2 IHC (+) in RBC and cap CL, MB and thalamus, anti-Stx2 IHC (−) in neu, glia | i.v. | |
| [63] | C57BL/6 a4galt−/− | Stx1 and Stx2 | Not suscep tible to Stx1 | Anti-Gb3 became negative in cap | n.d. | n.d. | n.d. | Not specified | |
| [53] | C57BL/6 mouse | Stx2 | yes | Anti-Gb3 (+) in neu mouse and human Sc, human cap | EM: glia lamellipodia-like foot process interrupts synapse at motor neu of lum Sc | Immuno-gold EM (l): anti-Gb3 and anti-Stx2 double positive in motor neu of lum Sc | i.p. | ||
| [51] | NIH mouse | Stx2 | n.d. | n.d. | EM: neu, astro and peri ede, synaptic disruption, oligo defect | i.v. | Behavioral motor test + | ||
| [61] | Human | n.a. | n.a. | DRG (m), Stx1 binding (+) neu and cap | n.a. | n.a. | n.a. | n.a. | |
| [61] | Rabbit | n.a. | n.a. | DRG, Stx1 binding (+) neu and cap | n.a. | n.a. | n.a. | n.a. | |
| [61] | rat | n.a. | n.a. | DRG, Stx1 binding (+) neu | n.a. | n.a. | n.a. | n.a. | |
| [62] | Human | n.a. | n.a. | DRG, anti-Gb3 and Stx1 binding (+) neu and cap | n.a. | n.a. | n.a. | n.a. | |
| [62] | rabbit | n.a. | n.a. | DRG, anti-Gb3 and Stx1 binding (+) neu and cap | n.a. | n.a. | n.a. | n.a. | |
| [62] | Rat | n.a. | n.a. | DRG, anti-Gb3 and Stx1 binding (+) neu | n.a. | n.a. | n.a. | n.a. | |
| [62] | mouse | n.a. | n.a. | DRG, anti-Gb3 and Stx1 binding (+) neu | n.a. | n.a. | n.a. | n.a. | |
| [60] | C57BL/6 mouse | n.a. | n.a. | Anti-Gb3 (+) neu at olf, CR ctx, str, hippo, hypothalamus, CVOs, CL, MO, Sc V3 ependyma | n.a. | n.a. | n.a. | n.a. |
Animal keys: YL (Yorkshire-Landrace), NZW (New Zealand White), JW (Japanese White), DB (Dutch Belted), SD (Spraque-Dawley)
Detailed CNS symptoms are summarized in Table 3.
Histopathology analysis keys are Gross (gross observation in non-stained tissue), HE (hematoxylin-esosin stain that stains cytoplasm in pink and nucleus blue, light microscopic findings (LM)), PAS (Periodic acid-Schiff stain that detects polysaccharides, glycoproteins and glycolipid, LM), LFB (Luxol fast blue stain that stains myelin in blue, LM), EM (electron microscopic findings)
CNS regions and cell type abbreviations are CR (cerebrum), ctx (cortex), hippo (hippocampus), DG (dentate gyrus), str (striatum and other basal ganglia), CL (cerebellum), MO (medulla oblongata), MB (midbrain), BS (brain stem is used where midbrain, pons or medulla oblongata are not specified), Sc (spinal cord), cerv (cervical), tho (thoracic), lum (lumbaris), sub (subarachinoid space),, Histopathologic feature abbreviations are cap (endothelial cells or capillaries), inf (infarction), hrrg (hemorrhage), fib (fibrin deposition), nec (necrosis), swl (swelling), peri (perivascular), myo (myocytes), apop (apoptotic), mono (monocytes), mye (myelin), deg (degeneration), pyk (pyknotic nuclei), prolif (proliferation/hyperplasia), ede (edema), pyr (pyramidal neuron), inflt (infiltration of blood cells to parenchyma), neu (neuron), Purkinje (Purkinje cells are large neurons in CL), V3 (third ventricle), cc (corpus callosum), amy (amygdala), ips (ipsilateral, injection side of brain), cont (contlateral, opposite of injection side), subV (subventricular region), cp (choroid plexus), CVO (circumventricular organs)
Histopathologic feature abbreviations are cap (endothelial cells or capillaries), inf (infarction), hrrg (hemorrhage), fib (fibrin deposition), nec (necrosis), swl (swelling), peri (perivascular), myo (myocytes), apop (apoptotic), mono (monocytes), mye (myelin), deg (degeneration), pyk (pyknotic nuclei), prolif (proliferation/hyperplasia), ede (edema), pyr (pyramidal neuron), inflt (infiltration of blood cells to parenchyma), neu (neuron), Purkinje (Purkinje cells are large neurons in CL), glia (glial cells such as astrocytes, microglia and oligodendrocytes), eos (eosinophilic globules, deposits), axo (axon, axoplasm), astro (astrocyte), oligo (oligodendrocyte), phago (phagocytosis), lyso (lysosome), RBC (red blood cells)
IHC = immunohistochemistry, immunodetection of the target in the tissue sections
Injection route abbreviations: i.v. (intravenous), i.t. (intrathecal, injection from cysterna magna that makes it possible to inject into cerebrospinal fluid (CSF), i.p. (intraperitoneal), i.c.v. (intracerebroventricular injection that inject solution directly into CNS parenchyma of cerebral cortex/ventricle).
Baboon in this chart is Papio c. cynocephalus, or Papio c. Anubis
Sup = E. coli culture supernatant
LacCer = lactsylceramide, adding galastose to LacCer completes Gb3.
Immuno-EM-DAB: immunodetection of the target with 3,3′-diaminobenzidine (DAB) deposition by EM
Immunogold EM: immunodetection of the target with 5–10 nm gold particle allows precise localization as well as double labeling.
DRG = dorsal root ganglion, a peripheral nervous system structure consists of sensory neurons and other cell types.
2. CNS histopathology of animal models
In animal models with STEC oral inoculation which describe CNS symptoms, most exhibit defective capillaries (pig: [31–34], mouse: [35, 36]). Those capillary lesions are mostly related to endothelial cell weakening that appears as hemorrhage, with leaked red blood cells in parenchyma. Non-capillary components in the parenchyma such as neurons and myelin defects were seen in some mouse STEC models [35, 37, 38], but not others [36]. In purified Stx2 injection models, similar lesions involving capillary/endothelial cells were found in pig [39, 40], rabbit [41–43] and mouse [44, 45]. In contrast, other models did not have these lesions, but rather lesions related to neuronal degeneration (baboon: [46], rabbit: [43, 47, 48], rat: [49, 50], mouse: [51]) or myelin degeneration (baboon: [46], rabbit: [52], rat: [49]). Also, some reports showed normal appearance of neurons (rabbit: [47]striatal neurons, mouse: [53] lumbar spinal cord neurons). As all models exhibit similar CNS symptoms such as hind leg paralysis, the difference in histopathological lesions may be due to involvement of different parts of CNS, different time points in the disease, or species specific sensitivities. The mechanism of inducing CNS symptoms may be weakening of endothelial cells/capillary composition caused neurotoxicity, or direct effect of Stx in neuronal toxicity. The observation of lamellipodia-like processes of glial origin interrupting synaptic connections at the lumbar spinal cord interneuron to motor neuron may explain the resulting hind leg paralysis (mouse: [53]). A similar observation is reported in a rat model of striatum neurons [51].
3. CNS molecular physiology of animal models
Molecular marker analysis in STEC or Stx animal models suggests possible mechanisms for Stx-associated CNS impairment.
The apoptotic nature of Stx-associated lesions has been described. TUNEL stain detects fragmented DNA and therefore is often used as an apoptotic assay. Capillaries (pig:[33], rabbit:[54] [43]), neurons (mouse:[55], rabbit: [43]), and glial cells (rabbit:[43]) have been detected as TUNEL positive. Activated caspase-3 targeted IHC has been used for another marker of apoptotic cells. Neurons (mouse: [56]) and capillaries (rabbit: [54]) have been detected positive. Another pro-apoptotic marker, bax, was found increased in rat neurons [57]. Along with EM observation (rat: [49]), some neurons and capillary cells (endothelial cells and/or pericytes) undergo apoptosis, but some appear as necrotic (rabbit: [33]). Careful and detailed information of which area of CNS and what types of cells in that area present apoptotic features may help elucidate these conflicting results.
AQP4 is mostly expressed in astrocyte foot processes that have a direct contact to capillaries in the CNS. The reduction of AQP4 suggests that there is alteration in astrocytic foot process, which is important to strengthen the BBB. AQP4 expression decreased in Stx2 injected rat [50] and STEC infected mouse [56], while astrocytic activation marker glial fibrillary acidic protein (GFAP) increased. This suggests Stx-associated astrocyte activation that may participate in weakening the BBB.
An increase in TNFα in STEC inoculated mouse [37] and Stx2 injected rabbit [43] brain along with serum TNFα increase in STEC inoculated rabbit [55], suggests Stx-associated inflammation in the CNS.
Ca2+ imaging and electrophysiological study are useful tool to assess direct physiological action of Stx in fresh brain slices. Our group showed Stx2-associated neuronal glutamate release in mouse brain slice (cerebral cortex) indirectly by recording intracellular Ca2+ in astrocyte [53]. Recently, it is shown that Stx2 induces depolarization of neurons in the thalamic area of female rat [58].
4. Receptor Gb3 expression in animal central and peripheral nervous systems (CNS, PNS)
Shiga toxin receptor localization in the animal nervous system has been described for different species. There are three ways to localize Shiga toxin receptor. Firstly, is to perform anti-Stx immunodetection in tissues of STEC infected or Stx injected animals (rabbit: [42, 47, 52], rat: [49, 59], mouse: [35, 36]). Secondly, is to incubate a naïve tissue section with Stx followed by anit-Stx immunodetection (pig [60]). Thirdly, is to recognize globotriaosylceramide (Gb3) as a Stx receptor with anti-Gb3 immunodetection in tissues. Detecting anti-Gb3 immunoreaction in the naïve tissue gives us basal expression level and cell types that would be influenced by Stx initially in the course of disease. These include neurons in the mouse spinal cord [53] and other regions of CNS [61]. In the Stx-administered tissue, it may or may not indicate the spontaneous Stx receptor expression but certainly indicate cell types responsive to Stx. The cell types that are positive in either of the analyses above often include small vessel endothelial cells (rabbit: [42, 43, 47, 52, 62, 63], mouse: [45, 64]), neurons (rat: [49, 57, 59], mouse: [35, 45, 53, 61]) and glial cells (rat:[49, 57, 59], mouse: [45, 61]). Miyatake and colleagues compared the peripheral nervous system (dorsal root ganglion) of different species with the same method and found that human and rabbit expressed Stx receptor in endothelial cells and neurons, whereas rat and mouse expression was restricted to neurons [62, 63]. Our group reported that throughout the mouse CNS, the only non-neuronal cell type to exhibit anti-Gb3 immunoreactivity was the third ventricle ependymal cell [61]. Studies have suggested, in the naïve state, human and rabbit express Stx receptor in their vessels as well as neurons and rodents appear to express Gb3 mainly in neurons. However, it was shown that Stx receptors in the rat CNS are induced by Stx administration [57]. Among different species, the receptor expression patterns in different regions of CNS, the cell types and the amount expressed may be different, however, all models present with common CNS impairment such as hind leg paralysis. This may be interpreted as expression of Stx receptor in endothelial cells is not necessary for toxin to be able to internalize into the CNS parenchyma to have an effect.
In 2006, Okuda et al reported [64] a4galt knockout mouse that lacks Gb3 synthase (alpha 1,4-galactosyltransferase) and therefore produces no Gb3. In this mouse, originally Gb3 positive vessels lost their anti-Gb3 immunoreactivity, and became Stx resistant. Gb3 synthase probe has been applied for an in situ hybridization in the mouse [56] and rat [58] CNS. While metabolic pathway enzymes such as Gb3 synthase, a glycosyltransferase, adds the terminal galactose to complete Gb3, other glycosyltransferases in the pathway are unique in each step of glycolipid synthesis, and there are catabolic pathway enzymes as well (see Fig. 6). All these enzymes participate in determining the amount of Gb3 in the cell. Measuring these Gb3-associated enzymes may give us more insight into Shiga toxin receptor regulation.
Figure 6. Metabolic and catabolic pathway enzymes for Gb3 synthesis.
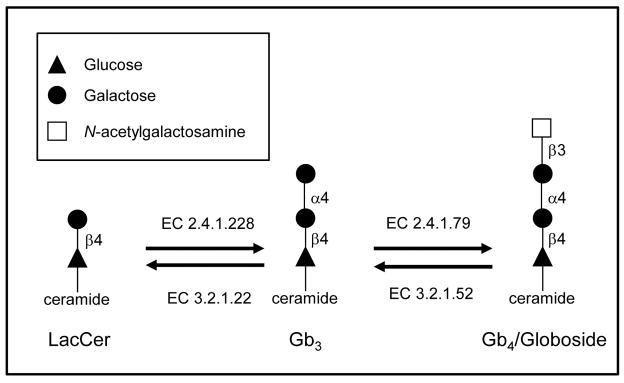
A part of Gb3 synthesis pathway is shown. From lactosylceramide (LacCer) to Gb3, alpha 1, 4-galactosyltransferase (EC 2.4.1.228) adds a galactose to LacCer to produce Gb3. Likewise, UDP-GalNAc: beta 1,3-galactosaminyltransferase (EC 2.4.1.79) works on Gb3 to make Gb4. In the catabolic pathway, beta-hexosaminidase (EC 3.2.1.52) degrades Gb4 to Gb3, and alpha-galactosidase (EC 3.2.1.22) makes LacCer from Gb3.
5. Discussion about how Shiga toxin enters CNS of animals
Purified Stx peripheral injection (intraperitoneal/i.p. or i.v.) is able to induce CNS impairment similar to STEC oral infection suggesting that there is a direct effect of Stx on CNS parenchymal cells. The rat model of intraventricular purified Stx2 injection in which purified Stx2 is inoculated directly into CNS parenchyma also induces similar CNS symptoms such as lethargy, hind leg weakness or paralysis [57]. These results suggest that Stx released from STEC would internalize into the blood and then transfer to CNS parenchyma and assert its toxicity.
The route and CNS region of Stx permeabilization is of great interest in order to explain which part of the CNS is most likely influenced by Stx. Stx injected via i.v. has been detected in cerebrospinal fluid (CSF) (rabbit: [47, 65]). This suggests there is translocation of Stx from blood to CSF. A reduction of AQP1 in choroid plexus in rat with Stx (i.p.) suggests that there is weakening of the blood-CSF barrier in this location that may allow Stx to enter CSF from the blood. The ependymal cells lining at the third ventricle are a border between CSF and CNS parenchyma. Our group showed in mouse CNS that ependymal cells at the third ventricle are expressing Gb3 in a naïve state [61]. The tracer horse radish peroxidase (HRP) that is injected intrathecally (i.t.) into CSF crossed and entered ependymal cells and parenchyma (rabbit: [52]), and also magnetic resonance imaging showed the third ventricle area with a bright signal that is an indication of leakiness into the fluid in this area. Taken together, it is reasonable to think that Stx utilizes blood-CSF barrier penetration as one of the routes into CNS parenchyma. On the other hand, Stx injected via i.p. was detected in the perivascular area in rat [49], and blood-brain barrier (BBB) weakening was suggested by the reduction of AQP4 (rat: [50], mouse: [56]), and also by tracer HRP (i.v.) detection in parenchyma (mouse: [35]). These results suggest that Stx can also use the BBB crossing route to enter the CNS. An important fact to note is that purified Stx by itself, without any other bacterial component, can enter CNS and assert its toxicity regardless of differences in receptor expressing cell types among different species.
References
- 1.Ohmura M, Yamamoto M, Tomiyama-Miyaji C, Yuki Y, Takeda Y, Kiyono H. Nontoxic Shiga toxin derivatives from Escherichia coli possess adjuvant activity for the augmentation of antigen-specific immune responses via dendritic cell activation. Infect Immun. 2005;73(7):4088–97. doi: 10.1128/IAI.73.7.4088-4097.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tesh VL, Ramegowda B, Samuel JE. Purified Shiga-like toxins induce expression of proinflammatory cytokines from murine peritoneal macrophages. Infect Immun. 1994;62(11):5085–94. doi: 10.1128/iai.62.11.5085-5094.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Obrig TG. Escherichia coli Shiga Toxin Mechanisms of Action in Renal Disease. Toxins (Basel) 2010;2(12):2769–2794. doi: 10.3390/toxins2122769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Obrig TG, Karpman D. Shiga toxin pathogenesis: kidney complications and renal failure. Curr Top Microbiol Immunol. 2012;357:105–36. doi: 10.1007/82_2011_172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karpman D, Sartz L, Johnson S. Pathophysiology of typical hemolytic uremic syndrome. Semin Thromb Hemost. 2010;36(6):575–85. doi: 10.1055/s-0030-1262879. [DOI] [PubMed] [Google Scholar]
- 6.Habib R, Mathieu H, Royer P. Hemolytic-uremic syndrome of infancy: 27 clinical and anatomic observations. Nephron. 1967;4(3):139–72. doi: 10.1159/000179580. [DOI] [PubMed] [Google Scholar]
- 7.Obrig TG, Louise CB, Lingwood CA, Boyd B, Barley-Maloney L, Daniel TO. Endothelial heterogeneity in Shiga toxin receptors and responses. J Biol Chem. 1993;268(21):15484–8. [PubMed] [Google Scholar]
- 8.Psotka MA, Obata F, Kolling GL, Gross LK, Saleem MA, Satchell SC, Mathieson PW, Obrig TG. Shiga toxin 2 targets the murine renal collecting duct epithelium. Infect Immun. 2009;77(3):959–69. doi: 10.1128/IAI.00679-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simon M, Cleary TG, Hernandez JD, Abboud HE. Shiga toxin 1 elicits diverse biologic responses in mesangial cells. Kidney Int. 1998;54(4):1117–27. doi: 10.1046/j.1523-1755.1998.00085.x. [DOI] [PubMed] [Google Scholar]
- 10.Shibolet O, Shina A, Rosen S, Cleary TG, Brezis M, Ashkenazi S. Shiga toxin induces medullary tubular injury in isolated perfused rat kidneys. FEMS Immunol Med Microbiol. 1997;18(1):55–60. doi: 10.1111/j.1574-695X.1997.tb01027.x. [DOI] [PubMed] [Google Scholar]
- 11.Hughes AK, Stricklett PK, Kohan DE. Cytotoxic effect of Shiga toxin-1 on human proximal tubule cells. Kidney Int. 1998;54(2):426–37. doi: 10.1046/j.1523-1755.1998.00015.x. [DOI] [PubMed] [Google Scholar]
- 12.Ghosh SA, Polanowska-Grabowska RK, Fujii J, Obrig T, Gear AR. Shiga toxin binds to activated platelets. J Thromb Haemost. 2004;2(3):499–506. doi: 10.1111/j.1538-7933.2004.00638.x. [DOI] [PubMed] [Google Scholar]
- 13.Karpman D, Manea M, Vaziri-Sani F, Stahl AL, Kristoffersson AC. Platelet activation in hemolytic uremic syndrome. Semin Thromb Hemost. 2006;32 (2):128–45. doi: 10.1055/s-2006-939769. [DOI] [PubMed] [Google Scholar]
- 14.Fernandez GC, Rubel C, Dran G, Gomez S, Isturiz MA, Palermo MS. Shiga toxin-2 induces neutrophilia and neutrophil activation in a murine model of hemolytic uremic syndrome. Clin Immunol. 2000;95(3):227–34. doi: 10.1006/clim.2000.4862. [DOI] [PubMed] [Google Scholar]
- 15.Roche JK, Keepers TR, Gross LK, Seaner RM, Obrig TG. CXCL1/KC and CXCL2/MIP-2 are critical effectors and potential targets for therapy of Escherichia coli O157:H7-associated renal inflammation. Am J Pathol. 2007;170(2):526–37. doi: 10.2353/ajpath.2007.060366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foster GH, Armstrong CS, Sakiri R, Tesh VL. Shiga toxin-induced tumor necrosis factor alpha expression: requirement for toxin enzymatic activity and monocyte protein kinase C and protein tyrosine kinases. Infect Immun. 2000;68(9):5183–9. doi: 10.1128/iai.68.9.5183-5189.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eaton KA, Friedman DI, Francis GJ, Tyler JS, Young VB, Haeger J, Abu-Ali G, Whittam TS. Pathogenesis of renal disease due to enterohemorrhagic Escherichia coli in germ-free mice. Infect Immun. 2008;76(7):3054–63. doi: 10.1128/IAI.01626-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stahl AL, Sartz L, Nelsson A, Bekassy ZD, Karpman D. Shiga toxin and lipopolysaccharide induce platelet-leukocyte aggregates and tissue factor release, a thrombotic mechanism in hemolytic uremic syndrome. PLoS One. 2009;4(9):e6990. doi: 10.1371/journal.pone.0006990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keepers TR, Psotka MA, Gross LK, Obrig TG. A murine model of HUS: Shiga toxin with lipopolysaccharide mimics the renal damage and physiologic response of human disease. J Am Soc Nephrol. 2006;17(12):3404–14. doi: 10.1681/ASN.2006050419. [DOI] [PubMed] [Google Scholar]
- 20.Keepers TR, Gross LK, Obrig TG. Monocyte chemoattractant protein 1, macrophage inflammatory protein 1 alpha, and RANTES recruit macrophages to the kidney in a mouse model of hemolytic-uremic syndrome. Infect Immun. 2007;75(3):1229–36. doi: 10.1128/IAI.01663-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7(9):759–70. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walters MD, Matthei IU, Kay R, Dillon MJ, Barratt TM. The polymorphonuclear leucocyte count in childhood haemolytic uraemic syndrome. Pediatr Nephrol. 1989;3(2):130–4. doi: 10.1007/BF00852893. [DOI] [PubMed] [Google Scholar]
- 23.Inward CD, Howie AJ, Fitzpatrick MM, Rafaat F, Milford DV, Taylor CM. Renal histopathology in fatal cases of diarrhoea-associated haemolytic uraemic syndrome. British Association for Paediatric Nephrology. Pediatr Nephrol. 1997;11(5):556–9. doi: 10.1007/s004670050337. [DOI] [PubMed] [Google Scholar]
- 24.Griener TP, Strecker JG, Humphries RM, Mulvey GL, Fuentealba C, Hancock RE, Armstrong GD. Lipopolysaccharide renders transgenic mice expressing human serum amyloid P component sensitive to Shiga toxin 2. PLoS One. 2011;6(6):e21457. doi: 10.1371/journal.pone.0021457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kawachi H, Han GD, Miyauchi N, Hashimoto T, Suzuki K, Shimizu F. Therapeutic targets in the podocyte: findings in anti-slit diaphragm antibody-induced nephropathy. J Nephrol. 2009;22(4):450–6. [PubMed] [Google Scholar]
- 26.Das L, Brunner HI. Biomarkers for renal disease in childhood. Curr Rheumatol Rep. 2009;11(3):218–25. doi: 10.1007/s11926-009-0030-4. [DOI] [PubMed] [Google Scholar]
- 27.Trachtman H, Christen E, Cnaan A, Patrick J, Mai V, Mishra J, Jain A, Bullington N, Devarajan P. Urinary neutrophil gelatinase-associated lipocalcin in D+HUS: a novel marker of renal injury. Pediatr Nephrol. 2006;21(7):989–94. doi: 10.1007/s00467-006-0146-y. [DOI] [PubMed] [Google Scholar]
- 28.Mohawk KL, O’Brien AD. Mouse models of Escherichia coli O157:H7 infection and shiga toxin injection. J Biomed Biotechnol. 2011;2011:258185. doi: 10.1155/2011/258185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karpman D, Connell H, Svensson M, Scheutz F, Alm P, Svanborg C. The role of lipopolysaccharide and Shiga-like toxin in a mouse model of Escherichia coli O157:H7 infection. J Infect Dis. 1997;175(3):611–20. doi: 10.1093/infdis/175.3.611. [DOI] [PubMed] [Google Scholar]
- 30.Wadolkowski EA, Sung LM, Burris JA, Samuel JE, O’Brien AD. Acute renal tubular necrosis and death of mice orally infected with Escherichia coli strains that produce Shiga-like toxin type II. Infect Immun. 1990;58 (12):3959–65. doi: 10.1128/iai.58.12.3959-3965.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tzipori S, Chow CW, Powell HR. Cerebral infection with Escherichia coli O157:H7 in humans and gnotobiotic piglets. J Clin Pathol. 1988;41 (10):1099–103. doi: 10.1136/jcp.41.10.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dean-Nystrom EA, Pohlenz JF, Moon HW, O’Brien AD. Escherichia coli O157:H7 causes more-severe systemic disease in suckling piglets than in colostrum-deprived neonatal piglets. Infection & Immunity. 2000;68(4):2356–8. doi: 10.1128/iai.68.4.2356-2358.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matise I, Sirinarumitr T, Bosworth BT, Moon HW. Vascular ultrastructure and DNA fragmentation in swine infected with Shiga toxin-producing Escherichia coli. Vet Pathol. 2000;37(4):318–27. doi: 10.1354/vp.37-4-318. [DOI] [PubMed] [Google Scholar]
- 34.Shringi S, Garcia A, Lahmers KK, Potter KA, Muthupalani S, Swennes AG, Hovde CJ, Call DR, Fox JG, Besser TE. Differential virulence of clinical and bovine-biased enterohemorrhagic Escherichia coli O157:H7 genotypes in piglet and Dutch belted rabbit models. Infect Immun. 2012;80 (1):369–80. doi: 10.1128/IAI.05470-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fujii J, Kita T, Yoshida S, Takeda T, Kobayashi H, Tanaka N, Ohsato K, Mizuguchi Y. Direct evidence of neuron impairment by oral infection with verotoxin-producing Escherichia coli O157:H- in mitomycin-treated mice. Infect Immun. 1994;62(8):3447–53. doi: 10.1128/iai.62.8.3447-3453.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kurioka T, Yunou Y, Kita E. Enhancement of Susceptibility to Shiga Toxin-Producing Escherichia coli O157:H7 by Protein Calorie Malnutrition in Mice. Infect Immun. 1998;66(4):1726–1734. doi: 10.1128/iai.66.4.1726-1734.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Isogai E, Isogai H, Kimura K, Hayashi S, Kubota T, Fujii N, Takeshi K. Role of tumor necrosis factor alpha in gnotobiotic mice infected with an Escherichia coli O157:H7 strain. Infect Immun. 1998;66(1):197–202. doi: 10.1128/iai.66.1.197-202.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taguchi H, Takahashi M, Yamaguchi H, Osaki T, Komatsu A, Fujioka Y, Kamiya S. Experimental infection of germ-free mice with hyper-toxigenic enterohaemorrhagic Escherichia coli O157:H7, strain 6. J Med Microbiol. 2002;51(4):336–43. doi: 10.1099/0022-1317-51-4-336. [DOI] [PubMed] [Google Scholar]
- 39.MacLeod DL, Gyles CL, Wilcock BP. Reproduction of edema disease of swine with purified Shiga-like toxin-II variant. Vet Pathol. 1991;28(1):66–73. doi: 10.1177/030098589102800109. [DOI] [PubMed] [Google Scholar]
- 40.Gannon VP, Gyles CL, Wilcock BP. Effects of Escherichia coli Shiga-like toxins (verotoxins) in pigs. Can J Vet Res. 1989;53(3):306–12. [PMC free article] [PubMed] [Google Scholar]
- 41.Zoja C, Corna D, Farina C, Sacchi G, Lingwood C, Doyle MP, Padhye VV, Abbate M, Remuzzi G. Verotoxin glycolipid receptors determine the localization of microangiopathic process in rabbits given verotoxin-1. J Lab Clin Med. 1992;120(2):229–38. [PubMed] [Google Scholar]
- 42.Richardson SE, Rotman TA, Jay V, Smith CR, Becker LE, Petric M, Olivieri NF, Karmali MA. Experimental verocytotoxemia in rabbits. Infect Immun. 1992;60(10):4154–67. doi: 10.1128/iai.60.10.4154-4167.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takahashi K, Funata N, Ikuta F, Sato S. Neuronal apoptosis and inflammatory responses in the central nervous system of a rabbit treated with Shiga toxin-2. J Neuroinflammation. 2008;5:11. doi: 10.1186/1742-2094-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sugatani J, Igarashi T, Munakata M, Komiyama Y, Takahashi H, Komiyama N, Maeda T, Takeda T, Miwa M. Activation of coagulation in C57BL/6 mice given verotoxin 2 (VT2) and the effect of co-administration of LPS with VT2. Thromb Res. 2000;100(1):61–72. doi: 10.1016/s0049-3848(00)00305-4. [DOI] [PubMed] [Google Scholar]
- 45.Nishikawa K, Matsuoka K, Kita E, Okabe N, Mizuguchi M, Hino K, Miyazawa S, Yamasaki C, Aoki J, Takashima S, Yamakawa Y, Nishijima M, Terunuma D, Kuzuhara H, Natori Y. A therapeutic agent with oriented carbohydrates for treatment of infections by Shiga toxin-producing Escherichia coli O157:H7. Proc Natl Acad Sci U S A. 2002;99(11):7669–74. doi: 10.1073/pnas.112058999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taylor C, Williams J, Lote C, Howie A, et al. A laboratory model of toxin-induced hemolytic uremic syndrome. Kid Intern. 1999;55:1367–1374. doi: 10.1046/j.1523-1755.1999.00387.x. [DOI] [PubMed] [Google Scholar]
- 47.Mizuguchi M, Tanaka S, Fujii I, Tanizawa H, Suzuki Y, Igarashi T, Yamanaka T, Takeda T, Miwa M. Neuronal and vascular pathology produced by verocytotoxin 2 in the rabbit central nervous system. Acta Neuropathol (Berl) 1996;91(3):254–62. doi: 10.1007/s004010050423. [DOI] [PubMed] [Google Scholar]
- 48.Garcia A, Marini RP, Catalfamo JL, Knox KA, Schauer DB, Rogers AB, Fox JG. Intravenous Shiga toxin 2 promotes enteritis and renal injury characterized by polymorphonuclear leukocyte infiltration and thrombosis in Dutch Belted rabbits. Microbes Infect. 2008;10(6):650–6. doi: 10.1016/j.micinf.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 49.Goldstein J, Loidl CF, Creydt VP, Boccoli J, Ibarra C. Intracerebroventricular administration of Shiga toxin type 2 induces striatal neuronal death and glial alterations: an ultrastructural study. Brain Res. 2007;1161:106–15. doi: 10.1016/j.brainres.2007.05.067. [DOI] [PubMed] [Google Scholar]
- 50.Lucero MS, Mirarchi F, Goldstein J, Silberstein C. Intraperitoneal administration of Shiga toxin 2 induced neuronal alterations and reduced the expression levels of aquaporin 1 and aquaporin 4 in rat brain. Microb Pathog. 2012;53(2):87–94. doi: 10.1016/j.micpath.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 51.Tironi-Farinati C, Geoghegan PA, Cangelosi A, Pinto A, Loidl CF, Goldstein J. A translational murine model of sub-lethal intoxication with Shiga toxin 2 reveals novel ultrastructural findings in the brain striatum. PLoS One. 2013;8(1):e55812. doi: 10.1371/journal.pone.0055812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fujii J, Kinoshita Y, Kita T, Higure A, Takeda T, Tanaka N, Yoshida S. Magnetic resonance imaging and histopathological study of brain lesions in rabbits given intravenous verotoxin 2. Infect Immun. 1996;64(12):5053–60. doi: 10.1128/iai.64.12.5053-5060.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Obata F, Tohyama K, Bonev AD, Kolling GL, Keepers TR, Gross LK, Nelson MT, Sato S, Obrig TG. Shiga toxin 2 affects the central nervous system through receptor globotriaosylceramide localized to neurons. J Infect Dis. 2008;198(9):1398–406. doi: 10.1086/591911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mizuguchi M, Sugatani J, Maeda T, Momoi T, Arima K, Takashima S, Takeda T, Miwa M. Cerebrovascular damage in young rabbits after intravenous administration of Shiga toxin 2. Acta Neuropathol (Berl) 2001;102(4):306–12. doi: 10.1007/s004010100384. [DOI] [PubMed] [Google Scholar]
- 55.Kita E, Yunou Y, Kurioka T, Harada H, Yoshikawa S, Mikasa K, Higashi N. Pathogenic mechanism of mouse brain damage caused by oral infection with Shiga toxin-producing Escherichia coli O157:H7. Infect Immun. 2000;68 (3):1207–14. doi: 10.1128/iai.68.3.1207-1214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Amran MY, Fujii J, Suzuki SO, Kolling GL, Villanueva SY, Kainuma M, Kobayashi H, Kameyama H, Yoshida S. Investigation of Encephalopathy Caused by Shiga Toxin 2c-Producing Escherichia coli Infection in Mice. PLoS One. 2013;8(3):e58959. doi: 10.1371/journal.pone.0058959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tironi-Farinati CLC, Boccoli J, Parma Y, Fernandez-Miyakawa ME, Goldstein J. Intracerebroventricular Shiga toxin 2 increases the expression of its receptor globotriaosylceramide and causes dendritic abnormalities. Journal of Neuroimmunology. 2010;222(1–2):48–61. doi: 10.1016/j.jneuroim.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 58.Meuth SG, Gobel K, Kanyshkova T, Ehling P, Ritter MA, Schwindt W, Bielaszewska M, Lebiedz P, Coulon P, Herrmann AM, Storck W, Kohmann D, Muthing J, Pavenstadt H, Kuhlmann T, Karch H, Peters G, Budde T, Wiendl H, Pape HC. Thalamic involvement in patients with neurologic impairment due to Shiga toxin 2. Ann Neurol. 2013;73(3):419–29. doi: 10.1002/ana.23814. [DOI] [PubMed] [Google Scholar]
- 59.Boccoli J, Loidl CF, Lopez-Costa JJ, Creydt VP, Ibarra C, Goldstein J. Intracerebroventricular administration of Shiga toxin type 2 altered the expression levels of neuronal nitric oxide synthase and glial fibrillary acidic protein in rat brains. Brain Res. 2008;1230:320–33. doi: 10.1016/j.brainres.2008.07.052. [DOI] [PubMed] [Google Scholar]
- 60.Winter KR, Stoffregen WC, Dean-Nystrom EA. Shiga toxin binding to isolated porcine tissues and peripheral blood leukocytes. Infect Immun. 2004;72(11):6680–4. doi: 10.1128/IAI.72.11.6680-6684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Obata F, Obrig T. Distribution of Gb(3) Immunoreactivity in the Mouse Central Nervous System. Toxins (Basel) 2010;2(8):1997–2006. doi: 10.3390/toxins2081997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ren J, Utsunomiya I, Taguchi K, Ariga T, Tai T, Ihara Y, Miyatake T. Localization of verotoxin receptors in nervous system. Brain Res. 1999;825(1–2):183–8. doi: 10.1016/s0006-8993(99)01196-8. [DOI] [PubMed] [Google Scholar]
- 63.Utsunomiya I, Ren J, Taguchi K, Ariga T, Tai T, Ihara Y, Miyatake T. Immunohistochemical detection of verotoxin receptors in nervous system. Brain Res Brain Res Protoc. 2001;8(2):99–103. doi: 10.1016/s1385-299x(01)00091-5. [DOI] [PubMed] [Google Scholar]
- 64.Okuda T, Tokuda N, Numata S, Ito M, Ohta M, Kawamura K, Wiels J, Urano T, Tajima O, Furukawa K. Targeted disruption of Gb3/CD77 synthase gene resulted in the complete deletion of globo-series glycosphingolipids and loss of sensitivity to verotoxins. J Biol Chem. 2006;281(15):10230–5. doi: 10.1074/jbc.M600057200. [DOI] [PubMed] [Google Scholar]
- 65.Fujii J, Kinoshita Y, Yamada Y, Yutsudo T, Kita T, Takeda T, Yoshida S. Neurotoxicity of intrathecal Shiga toxin 2 and protection by intrathecal injection of anti-Shiga toxin 2 antiserum in rabbits. Microb Pathog. 1998;25(3):139–46. doi: 10.1006/mpat.1998.0220. [DOI] [PubMed] [Google Scholar]
- 66.Taylor CM, Williams JM, Lote CJ, Howie AJ, Thewles A, Wood JA, Milford DV, Raafat F, Chant I, Rose PE. A laboratory model of toxin-induced hemolytic uremic syndrome. Kidney Int. 1999;55(4):1367–74. doi: 10.1046/j.1523-1755.1999.00387.x. [DOI] [PubMed] [Google Scholar]
- 67.Siegler RL, Pysher TJ, Lou R, Tesh VL, Taylor FB., Jr Response to Shiga toxin-1, with and without lipopolysaccharide, in a primate model of hemolytic uremic syndrome. Am J Nephrol. 2001;21(5):420–5. doi: 10.1159/000046288. [DOI] [PubMed] [Google Scholar]
- 68.Yamada Y, Fujii J, Murasato Y, Nakamura T, Hayashida Y, Kinoshita Y, Yutsudo T, Matsumoto T, Yoshida S. Brainstem mechanisms of autonomic dysfunction in encephalopathy-associated Shiga toxin 2 intoxication. Ann Neurol. 1999;45(6):716–23. [PubMed] [Google Scholar]
- 69.Fujii J, Kinoshita Y, Yutsudo T, Taniguchi H, Obrig T, Yoshida SI. Toxicity of Shiga toxin 1 in the central nervous system of rabbits. Infect Immun. 2001;69(10):6545–8. doi: 10.1128/IAI.69.10.6545-6548.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fujii J, Kinoshita Y, Matsukawa A, Villanueva SY, Yutsudo T, Yoshida S. Successful steroid pulse therapy for brain lesion caused by Shiga toxin 2 in rabbits. Microb Pathog. 2009;46(4):179–84. doi: 10.1016/j.micpath.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 71.Tzipori S, Gunzer F, Donnenberg MS, de Montigny L, Kaper JB, Donohue-Rolfe A. The role of the eaeA gene in diarrhea and neurological complications in a gnotobiotic piglet model of enterohemorrhagic Escherichia coli infection. Infect Immun. 1995;63(9):3621–7. doi: 10.1128/iai.63.9.3621-3627.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]