

Abstract
Recent evidence indicates that metastatic capacity is an inherent feature of breast tumours and not a rare, late acquired event. This has led to new models of metastasis. The interpretation of expression-profiling data in the context of these new models has identified the cofilin pathway as a major determinant of metastasis. Recent studies indicate that the overall activity of the cofilin pathway, and not that of any single gene within the pathway, determines the invasive and metastatic phenotype of tumour cells. These results predict that inhibitors directed at the output of the cofilin pathway will have therapeutic benefit in combating metastasis.
Tumour cell motility is the hallmark of invasion and an essential step in metastasis1,2. The identification of molecular pathways that contribute to tumour cell motility and invasion is essential for understanding how motility is initiated in tumour cells and how the tumour microenvironment contributes to cell migration. The identification of the molecular pathways of tumour cell invasion will provide new diagnostic approaches and targets for the treatment of metastatic cancer. Recent studies using new technologies, including high-density microarray-based expression profiling, intravital imaging and the collection of invasive tumour cells from live tumours, have started to extend the traditional model of metastasis and supply new diagnostic and therapeutic markers of metastatic disease.
According to the traditional model of metastasis, metastases result from a process similar to Darwinian evolution whereby natural selection works on individual tumour cells to select for stable genetic changes. The cells so selected are very rare, and the metastatic cells that arise from this progressive selection of stable genetic mutations within the primary tumour cause metastasis late in tumour progression3. However, studies of mammary tumours in mice4–7, expression profiling of both whole human breast tumours8,9 and the invasive sub-population of tumour cells isolated from rat and mouse mammary tumours10–12 suggest that the metastatic ability of breast tumours is encoded throughout the bulk of the primary tumour, involves transient changes in gene expression and is acquired at much earlier stages of tumour progression than postulated by the traditional model. These results suggest that a Darwinian-like evolution is accompanied by, and may contribute to, microenvironment-induced transient changes in gene expression that support the invasive and metastatic phenotype. These results have led to new models where the microenvironment initiates the expression of genes that induce cell motility, invasion and metastasis11,13. In the ‘tumour microenvironment invasion model’ it is proposed that oncogenic mutations in tumour cells lead to microenvironments that are potentially encoded throughout the bulk of the tumour. Examples of microenvironments that are correlated with increased invasion and metastasis are increased microvascular density14 and macrophage infiltration15. These microenvironments might be induced by the expression of hypoxia-induced growth factors like vascular endothelial growth factor (VEGF) by macrophages16, and the expression of colony stimulating factor 1 (CSF1) by tumour cells15. These microenvironments are speculated to elicit transient and epigenetic changes in gene expression in tumour and stromal cells. These changes in gene expression would lead to changes in cell–cell interactions and the migration of a subpopulation of tumour cells within the primary tumour that are migration competent. In the ‘integrative model of breast cancer metastasis’, oncogenic mutations occur in breast stem cells to generate a tumour associated with a poor prognosis. Under the influence of stromal cells the population of breast cancer stem cells acquires the ability to migrate and metastasize13. In both models migration competence requires the activation of the motility cycle, the first step of which is actin polymerization, which drives the formation of cell protrusions that are used to adhere to the extracellular matrix, define the direction of migration and initiate cell crawling. Cofilin and its regulatory proteins (the cofilin pathway, recently comprehensively reviewed in REFS 17,18) are involved in the initiation of the early steps in the motility cycle, and evidence has emerged that the expression of certain genes of the cofilin pathway are altered in invasive tumour cells.
A pattern of changes in gene expression that is consistently observed in mammary tumour cell lines, the invasive population of mammary tumour cells isolated from primary tumours, and in whole mammary tumours, is clustered in the cofilin pathway11 (TABLE 1). These genes are coordinately regulated in invasive tumour cells during cell migration in vivo, suggesting that the cofilin pathway has a direct role in determining the invasive and metastatic phenotype10,12,17,18.
Table 1.
Range of expression in tumour cells and their correlated phenotypes
| Changes in expression | Cancer cell lines or tissue | Prognostic and/or histological information | Refs |
|---|---|---|---|
| Total cofilin | |||
| Highly expressed* | C6 rat glioblastoma cell line | Highly invasive | 72 |
| Highly expressed‡ | A549 human lung cancer cells, adenocarcinoma | Undergoing epithelial–mesenchymal transition | 73 |
| Highly expressed‡ | Human pancreatic cancer cell lines: EPP85-181RDB and EPP85-181RNOV, derived from the adenocarcinoma of the pancreas (EPP85-181P) | Multidrug-resistant subline | 74 |
| Highly expressed§ | MTLn3 rat mammary carcinoma | Invasive subpopulation | 10 |
| Highly expressed‡ | Human breast cancer MDA-MB-435S | Superinvasive population | 104 |
| Highly expressed‡ | Oral squamous cellular carcinoma (OSCC) | Tumour species from patients with | 76 |
| Highly expressed‡ | Renal tumour tissue | Conventional renal cell carcinomas (RCCs) | 77 |
| Highly expressed§ | Ovarian cancer | Carcinoma, obtained from postmenopausal women | 78 |
| Downregulated‡ | MHCC97-H hepatocellular carcinoma (HCC) cells | High metastatic potential | 79 |
| Downregulated‡ | Ovarian surface epithelium (OSE) | Derived from women with a family history of ovarian and/or breast cancer and mutations in the BRCA1 tumour- suppressor gene | 80 |
| Phosphorylated cofilin | |||
| Decreased|| | T-cell lymphoma cells (Jurkat), cells from carcinomas of the cervix (HeLa), colon (KM12), liver (HepG2) and kidney (COS1) | Tumorigenic | 75 |
| LIM kinase 1 | |||
| Moderate|| | Human breast cancer MCF-7 cells and prostate cancer LNCaP cells | Low invasive | 84 |
| Highly expressed|| | Prostate cancer PC-3 cells, breast cancer MDA-MB-231 cells | Highly invasive | 84 |
| Highly expressed | Melanoma, ovarian carcinoma, lung, breast and prostate | Highly invasive human tumour | 84 |
| Highly expressed|| | LNCaP and M21 prostate cancer cell lines | Tumorigenic | 83 |
| Highly expressed|| | PC3, DU145 and M12 prostate cancer cell lines | Metastatic | 83 |
| Highly expressed¶ | Cancerous glands in prostatic epithelium | Cancerous | 83 |
| Upregulated§ | MTLn3 rat mammary carcinoma | Invasive subpopulation | 10 |
| Upregulated§ | PyMT mouse mammary tumour | Invasive subpopulation | 12 |
| Slingshot 1 (SSH) | |||
| Upregulated§ | PyMT mouse mammary tumour | Invasive subpopulation | 12 |
| Coordinated overexpression of cofilin and LIMK | |||
| Both upregulated | Invasive tumour cells isolated from rat mammary tumours | Highly invasive | 10 |
| Coordinated overexpression of SSH and LIMK | |||
| Both upregulated | Invasive tumour cells isolated from mouse PyMT mammary tumours | Highly invasive | 12 |
Changes in gene expression were identified using the following technologies:
SAGE (serial analysis of gene expression);
2-DE (two-dimensional gel electrophoresis) and/or 2DLC (two-dimensional liquid chromatography) and mass spectrometry;
cDNA microarrays;
western blotting;
immunohistochemistry.
The cofilin pathway is composed of a group of kinases and phosphatases that regulate cofilin and coordinately initiate actin polymerization and cell motility in response to stimuli in the microenvironment of mammary tumours (FIG. 1). Such stimuli include epidermal growth factor (EGF), transforming growth factor-α (TGFα), stromal cell-derived factor 1 (SDF1) and heregulin, which have been implicated in the stimulation of cell migration and correlated with progression in various tumours. Cofilin is then regulated by four independent processes. First, the phosphorylation of cofilin on serine 3 by LIM kinase 1 (LIMK1) and its related kinases (LIMK2, the skeletal muscle-specific kinases Nik related kinase (NRK, also known as NESK) and testicular protein kinase 1 (TESK1) and TESK2) regulates cofilin by inhibiting its actin-binding activity19–22; second, the dephosphorylation of serine 3 of cofilin by phosphatase types 1, 2A and 2B, slingshot (SSH) and chronophin phosphatases results in the activation of actin binding by cofilin23–26. Third, actin binding by cofilin is inhibited by binding to phosphatidylinositol-4,5-bisphosphate (PIP2)27,28, and cofilin activity is dependent on phospho lipase Cγ (PLCγ)-mediated hydrolysis of PIP2 (REFS 29,30); and fourth, changes in pH over the physiological range (6.8–7.4) as mediated by the Na-H exchanger protein can activate the severing activity of cofilin when it is in the dephosphorylated state31–34. Furthermore, in an additional level of regulation, LIMKs are activated by phosphorylation by p21-activated kinase 1 (PAK1), PAK4 and Rho-dependent protein kinase (ROCK1)31,105, and inhibited by dephosphorylation by SSH1 (REF. 106), thus potentiating the dephosphorylation of cofilin (FIG. 1).
Figure 1. The EGF-regulated cofilin pathway.

The cofilin pathway is activated in tumour cells by stimuli in the microenvironment, such as epidermal growht factor (EGF), detected by the EGF receptor (EGFR) and an unidentified subunit of the ErbB family, which could be either ERBB2 or ERBB3. In animal models ERBB2 has been shown to have a large potentiating effect on EGF-stimulated protrusion and cell migration in mammary tumours100. EGFR heterodimers, with either ERBB2 or ERBB3, can activate phosphatidylinositol 3-kinase (PI3K) and then a group of small G-proteins (Rho, CDC42 (cell division cycle 42) and Rac) and their cofilin-regulating kinases (ROCK1 and Pak). These kinases stimulate LIM kinase (LIMK) to phosphorylate cofilin (P-cofilin), thereby inactivating it. And phosphatases such as slingshot (SSH), chronophin, type 1, 2A and 2B phosphatases, can dephosphorylate cofilin upon activation making it potentially active (if not bound to phosphatidylinositol-4,5-bisphosphate (PIP2) and if the pH is above 7.0). Although SSH can be activated by F-actin binding and phosphorylation by PAK4 in vitro106, the mechanism for chronophin activation, is currently unknown. In addition, EGFR–ERBB2 can activate phospholipase Cγ (PLCγ)101 which is proposed to activate cofilin by the hydrolysis of PIP2 (REF. 29), thereby releasing cofilin from the cell membrane. Cofilin activated by either path severs mother filaments (older pre-existing filaments) to produce free barbed ends leading to the elongation of newly polymerized actin filaments that are preferred for dendritic nucleation by the ARP2/3 complex (BOX 1) and G-actin resulting from the depolymerization of pointed ends produced by the same severing reaction. In invasive tumour cells, subunits of the ARP2/3 complex are coordinately overexpressed along with genes of the cofilin pathway10,12, thereby potentially increasing the synergistic interaction between the cofilin and ARP2/3 complex to cause dendritic nucleation, which pushes on the cell membrane to cause cell protrusion45,47. The ARP2/3 complex is activated by members of the WASP (Wiskott-Aldrich syndrome protein) family, including WAVE2 and NWASP. NWASP is known to be activated in invadopodia38,102 under the regulation of CDC42 and Rho and/or Src-mediated phosphorylation. The cofilin pathway is coordinately regulated in invasive tumour cells during cell migration in vivo (genes that are upregulated in rat mammary tumours are highlighted in bold), suggesting that the cofilin pathway (which is shown in red boxes and circles) has a direct role in determining the invasive and metastatic phenotype.
Actin polymerization can generate forces that drive the formation of membrane protrusions. These forces underlie alterations in cellular morphology, protrusion, migration and chemotaxis that occur during morphogenesis35. The force of actin polymerization that drives cell protrusion depends mainly on the formation of free barbed actin filament ends that can elongate by actin monomer (actin monomers are known as G-actin) addition to form growing actin filaments (actin filaments are termed F-actin) that push against cell membranes (BOX 1). This causes organelle trafficking inside the cell and protrusion of the cell membrane36. The appearance of free barbed filament ends coupled with further remodelling of the actin filament-containing cytoskeleton can generate protrusive structures such as lamellipodia, invadopodia and filopodia that initiate cell movements and determine cell polarity37. These types of cell protrusions are essential path-finding structures in chemotaxis, cell migration and invasion38. The cofilin pathway has emerged recently as a central player in the generation of free barbed ends39 and actin filament-turnover40 in various motile cells, including mammary carcinoma cells38,39, fibroblasts40, Dictyostelium discoideum41 and glioblastoma cells42 during the formation of these path-finding structures.
Box 1. Actin filament dynamics.
Actin filaments hydrolyse ATP bound to the actin subunits resulting in filaments that are composed of ADP-containing (green) and ATP-containing (red) subunits. The ATP subunits are biased to the preferred growing end (barbed end) of the filament, as indicated in the initial mother filament shown in the figure90. Cofilin severing increases both the number of barbed and pointed ends, as it does not remain bound to the barbed ends of filament fragments following the severing of the initial mother filament. This results in a large increase in net actin polymerization if actin subunits are available to polymerize onto the barbed and pointed ends of the filament fragments (green regions of filaments indicated by a star). The barbed ends have a higher affinity for G-actin than the pointed ends and tend to elongate rapidly, whereas pointed ends depolymerize slowly under physiological conditions. In the absence of actin subunits the fragments will depolymerize from both pointed and barbed ends (not shown). In the presence of the G-actin-binding protein profilin, actin subunits are speculated to only add to barbed ends, resulting in net polymerization from barbed ends and depolymerization from pointed ends (not shown)17. The ARP2/3 complex is a seven subunit complex of proteins that, when activated by WASP (Wiskott-Aldrich syndrome protein) family proteins, will bind to the side of an actin filament to nucleate new daughter filaments as branches from the side of the mother filaments91. This is called dendritic nucleation. Activated ARP2/3 complex preferentially binds to the side of mother filaments that have recently polymerized from ATP-containing G-actin (red regions of filaments), and cofilin supplies these new filaments by severing to form new barbed ends that support their polymerization45.
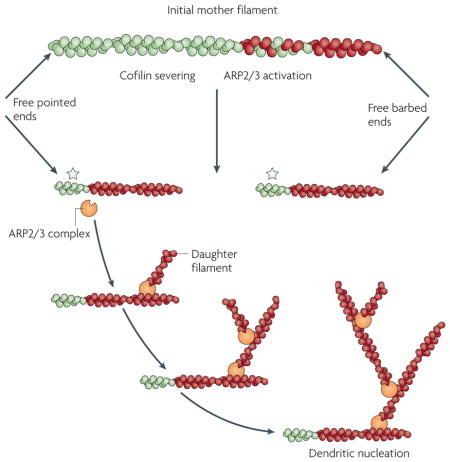
Biochemistry of the cofilin pathway
Cofilin belongs to a family of related proteins with similar biochemical activities called the actin depolymerizing factor (ADF)/cofilin family. Unicellular organisms such as yeasts usually have only one ADF/cofilin-type protein, whereas multicellular organisms typically have several isoforms. In some cultured mammalian cell lines40 and invasive mammary tumour cells10,43, cofilin 1 is the most abundant isoform, whereas ADF is expressed at much lower levels (5%). Therefore, for this Review ‘cofilin’ will refer to cofilin 1, the most abundant isoform found in invasive mammary tumour cells10,12.
Cofilin is a small ubiquitous protein (~19 kDa) that is able to bind both G-actin (monomeric) and F-actin (filamentous actin). The use of light microscopy to directly observe the interaction between cofilin and actin filaments that are immobilized by cross linking to a substratum has clarified how cofilin can both increase the number of free barbed ends for polymerization and increase the rate of actin depolymerization (hence replenishing G-actin in the cell)40,44–46. These studies have shown that nM concentrations of cofilin can sever actin filaments efficiently, thereby generating free actin filament barbed and pointed ends that are available for polymerization or depolymerization depending on the free G-actin concentration and the availability of barbed-end-capping proteins44–46 (BOX 1). As the concentration of cofilin required for severing as measured in these studies is 100 × lower than previously estimated from experiments in bulk solution, cofilin is a much more powerful severing protein than was previously suspected45,46. These results have allowed a direct analysis of the effect of cofilin on the off rate constant for actin depolymerization. Previously it was proposed from experiments in bulk solution that cofilin increases the off rate of actin monomer depolymerization from the pointed end of actin filaments. However, when the number of new barbed and pointed ends generated by cofilin severing were accounted for in the depolymerization rate of actin filaments it was found that there is no significant increase in off rate per filament end and no bias for depolymerization to the pointed end44. The direct observation of the rate of filament depolymerization at pointed ends by total internal reflection microscopy supports this conclusion and shows that cofilin does not elevate the off rate above that of ADP–actin monomers usually seen in the absence of cofilin46. This greatly simplifies our understanding of the mechanism of how cofilin affects actin filament dynamics. That is, both the polymerization and depolymerization of actin filaments observed in the presence of cofilin can be explained by the severing of actin filaments by cofilin without the need to invoke either an increase in off rate or bias in depolymerization at the pointed filament end.
More remarkable still are the findings that cofilin can both nucleate the assembly of actin filaments directly when at μM concentrations46 and greatly amplify the dendritic nucleation activity of the actin-related protein 2 and 3 (ARP2/3) complex at nM concentrations45 (BOX 1). The latter occurs because the severing of mother filaments by cofilin produces newly polymerized actin filaments that are preferred for dendritic nucleation by the ARP2/3 complex45,47. Both results are supported by kinetic simulations indicating that cofilin will cause large increases in actin polymerization when the capping of barbed ends is regulated46,48 (FIG. 2). These results indicate that cofilin is a much more powerful nucleation factor than previously estimated from experiments in bulk solution.
Figure 2. The spatial and temporal localization of cofilin activity in response to EGF stimulation.

a | In vivo studies in tumour cells indicate that EGF-stimulated phospholipase Cγ (PLCγ) hydrolyses phosphatidylinositol-4,5-bisphosphate (PIP2), causing the release of active cofilin locally from its complex with PIP2 in the plasma membrane. This activates cofilin asymmetrically inside the tumour cell to generate free barbed ends adjacent to the cell membrane facing the source of epidermal growth factor (EGF). Transient cofilin activity occurs as the result of the near simultaneous activation of the molecules within the cofilin pathway that both activate cofilin (PLCγ, slingshot (SSH) and chronophin) and inhibit cofilin (LIMK) by the EGF receptor. As a result, cofilin severs filaments locally to start polymerization and cell protrusion, and global LIMK activity inactivates cofilin that diffuses from the initial site of activation, thereby spatially sharpening cofilin activity. The signalling pathways from the receptor through G-proteins and LIMK are the same as in FIG. 1 but redrawn here to show how LIMK captures cofilin that diffuses from the site of activation. This results in the sharpened localization of cofilin-dependent free barbed end production and the initiation of directional cell motility and chemotaxis. b | The cofilin activity cycle fits a local excitation global inhibition (LEGI) model of chemotaxis. Cofilin is activated asymmetrically inside the cell in response to the gradient in EGF detected by cell surface receptors from the front to the back of the cell. The asymmetry of cofilin activation inside the cell may follow the slope of the gradient of EGF. However, this shallow gradient in cofilin activation is locally sharpened by the global stimulation of LIMK activity, which is postulated to inactivate cofilin throughout the cell, resulting in a remnant of cofilin activity only on the side of the cell facing the EGF source (front). This is shown by the two lines indicating the cofilin activation gradient (sloping line) and global LIMK activity (horizontal line) resulting from the same stimulation with a shallow EGF gradient, thereby compressing the effective cofilin activity between them to sharpen cofilin activity to the side of the cell facing the EGF source.
Cofilin is regulated by the four independent processes described above. However, the local activation of a photosensitive caged cofilin shows that the local activation of cofilin in mammary tumour cells generates free actin filament barbed ends, initiates actin polymerization, induces localized protrusion of the cell membrane and determines the direction of cell migration39. Cofilin activation is essential for the formation of stable invadopods, which are used during the migration of invasive tumour cells, linking cofilin to tumour cell invasion49. Therefore, the local activation of cofilin by any of the mechanisms described above — dephosphorylation, PIP2 hydrolysis and increasing pH — could in principle initiate directional tumour cell motility and invasion.
However, the initiation of cofilin activation in response to EGF in invasive mammary tumour cells is not coupled to cofilin dephosphorylation50, so models in which this coupling is assumed51 may not apply to invasion in mammary tumours. A word of caution is appropriate here. It is sometimes assumed that the amount of dephosphorylated cofilin in a cell is a direct measure of cofilin activity. However, given that there are four regulatory mechanisms for cofilin activity that appear to be uncoupled, the activity status of cofilin in a cell cannot be assessed by measuring the ratio of dephosphorylated cofilin to the total cofilin present. Studies in mammary tumour cells suggest that the EGF-stimulated and PLCγ-mediated hydrolysis of PIP2 can activate cofilin in mammary tumour cells29, and this is uncoupled from dephosphorylation50. In fact, during stimulation with EGF the levels of phosphorylated cofilin in mammary tumour cells increase29,50. In this case, the release of cofilin locally from its complex with PIP2 results in its activation, at the same time as the phosphorylation of cofilin globally throughout the cell by LIMK1 leads to a net increase in phosphorylated cofilin, sharpening the cofilin activity asymmetrically inside the tumour cell (FIG. 2). This is proposed to result in the initiation of directional cell motility and chemotaxis30,39. This balance between phosphorylation-independent cofilin activation and phosphorylation-dependent inhibition, which is essential for chemotaxis, could be incorrectly interpreted as a requirement for the inhibition of cofilin for chemotaxis if only the phosphorylation status of cofilin is measured at the time of stimulation.
Cell biology of the cofilin pathway
There is general agreement that cofilin activity is required for cell motility in vitro. However, how cofilin functions in cell motility and which parts of the motility cycle are affected by cofilin activity is complex and requires a careful cell-type-specific analysis. In mammary tumour cells, cofilin activity is observed to occur as a transient activity 60 seconds after stimulation of mammary tumour cells with EGF, and is responsible for the early transient of actin filament barbed end formation29,50 (FIG. 3). This early transient of barbed ends is the output of the cofilin pathway29 and this output is positively correlated with the invasive and metastatic activity of mammary tumour cells in vivo12,43.
Figure 3. Cofilin activity is required for the early barbed end transient responsible for the initiation of chemotaxis.

a | Stimulation of mammary tumour cells with epidermal growth factor (EGF) results in two transients of barbed end formation (b), which are localized at the cell membrane103. The first transient is required for initiating actin polymerization and protrusion towards the source of EGF, resulting in chemotaxis, and the second transient is required for sustaining the cell protrusions required for cell migration29,30. Two transients of barbed end formation have been observed in chemotactic cells (Dictyostelium, mammalian fibroblasts and macrophages) in response to various chemoattractants, indicating that this sequence of events is conserved in crawling chemotactic cells. c | In mammary tumour cells the first, but not the second, transient requires both phospholipase Cγ (PLCγ) and cofilin activity, implicating the cofilin pathway in chemotaxis (the red line shows the effect of inhibiting either PLCγ or cofilin activity)29,30. d | Cofilin-dependent free barbed ends are sharply localized in vivo in response to a gradient of EGF. The first transient of free barbed ends (stained red) 60 seconds after the introduction of a micropipette source of EGF are found predominantly on the side of the cell facing the source of EGF (right hand image; *marks the position of EGF-filled micropipette) compared with the more uniform distribution of free barbed ends in unstimulated cells (0 seconds). Part a of the figure is taken from REF. 103 and reproduced with permission of the Company of Biologists; parts b and c of the figure are reproduced from REF. 29 © (2004) The Rockefeller University Press; part d is taken from REF 30 © (2006) Elsevier Science. au, arbitrary units.
The transient cofilin activity occurs as a result of the near simultaneous activation, by the EGF receptor (EGFR), of the molecules within the cofilin pathway that both activate cofilin (PLCγ, SSH and chronophin) and inhibit cofilin (LIMK) (FIGS 1,2). In mammary tumour cells the activation and inhibition of cofilin must be balanced for transient cofilin activity to occur, be spatially localized30 and for protrusion to occur optimally29,30. Too much or too little cofilin activity inhibits protrusion, chemotaxis and motility. In addition, the spatial localization of this transient activity is required for directional cell migration and chemotaxis by tumour cells in response to EGF30 (FIGS 2,3). The spatial localization of the transient cofilin activity is postulated to result from a local excitation global inhibition (LEGI)-type model involving the local excitation (activation) of cofilin and its global inhibition by LIMK1 (REF. 30) (FIG. 2). LEGI models have been used successfully to fit the chemotactic behaviour of other cell types such as Dictyostelium, and explain how a chemotactic signal can be locally amplified52. In mammary tumour cells part of the amplifier is the balance between the PLCγ-mediated activation of cofilin and LIMK1-mediated inhibition of cofilin, and this balance is disrupted by either the microinjection of constitutively active cofilin mutant S3A, which cannot be phosphorylated by LIMK1, the overexpression of constitutively active LIMK1, which phosphorylates all of the cofilin in the cell, or the suppression of expression of LIMK1, in which case cofilin phosphorylation does not occur. All of these manipulations disrupt the balance between cofilin and LIMK1, thereby disrupting the early transient generation of barbed ends thereby inhibiting protrusion29 and chemotaxis to EGF30.
The results with mammary tumour cells are consistent with the findings that the unbalanced activation or inhibition of cofilin in various cell types is found to alter protrusion and motility. However, the literature is sometimes confusing. One source of confusion is that the phenotype observed in various cell types upon manipulating the expression of cofilin depends on the effect of the manipulation on the output of the cofilin pathway, i.e. the early barbed end transient-initiating movement, but the barbed end transients are usually not measured, so it is difficult to know if the manipulation is increasing or decreasing cofilin pathway output. For example, the moderate (twofold–fourfold) overexpression of cofilin at the protein level increases the velocity of cell migration in Dictyostelium41 and human glioblastoma cells42, but higher levels of expression are reported to inhibit cell motility53. Second, the cell behaviour chosen for analysis is often different, resulting in apparently conflicting conclusions. For example, the inhibition of cofilin activity in mammalian cell lines with either small interfering RNA (siRNA)40 or the expression of constitutively active LIMK domain54 inhibits cell motility, but is reported to increase lamellipod size in Drosophila melanogaster S2 cells55. Expression of the wild-type or the non-phosphorylatable cofilin mutant S3A increases melanoma cell migration on vitronectin56, but S3A is reported to inhibit chemotaxis in mammary tumour cells30. Because of the complex and transient regulation of the cofilin pathway, the careful analysis of the output of the pathway in each cell type for each manipulation, together with comparisons of the same compartments and cell behaviours between cell types, is required to define conclusions that can be generalized for all cells.
The cofilin pathway in morphogenesis
The proper functioning of the cofilin pathway is required for normal cell migration and polarity during morphogenesis. In lower eukaryotes such as Caenorhabditis elegans, different isoforms of cofilin are required for distinct steps in morphogenesis, including blastocyst positioning and body wall formation57. In Drosophila embryos, cofilin is required for cell migration during ovary development and oogenesis58, as well as in maintaining planar cell polarity in the wing, eye, and other epithelia59. Furthermore, flies that harbour mutations in genes that encode proteins involved in regulating cofilin phosphorylation provide further evidence for the role of the cofilin pathway in the regulation of Drosophila morphogenesis (BOX 2).
Box 2. Linking cofilin to Drosophila melanogaster morphogenesis.
During Drosophila morphogenesis, the disruption of cofilin alleles by transposon mutagenesis caused an abnormal actin cytoskeleton characterized by large F-actin aggregates in primary spermatocytes and at the contractile ring during cytokinesis92, as well as the follicular epithelium during ovarian development58. Loss-of-function mutations in genes that regulate cofilin phosphorylation, such as the cofilin phosphatase slingshot and the cofilin kinase LIM kinase (LIMK), further support a role for cofilin in regulating actin dynamics during Drosophila morphogenesis. Wing hair and bristle morphogenesis depend on the polymerization and bundling of actin filaments. Not surprisingly, transposon mutagenesis of the slingshot allele reduced the pool of activated cofilin, resulting in the increased accumulation of F-actin and the disruption of proper wing hair and bristle development23. In the nervous system, deletion of LIMK by transposon mutagenesis leads to aberrant development at the neuromuscular junction. LIMK mutants had enlarged terminals and increased bouton (axon synaptic terminals) numbers, whereas the overexpression of active LIMK caused stunted terminals and reduced boutons. This overgrowth of the neuromuscular junction suggests that the normal function of LIMK is to suppress synaptic sprouting93. LIMK deletion mutants also abolish the ability of p21-activated kinase (Pak) to alter glomerular morphology and the development of antennal lobe synapses. Overexpression of LIMK causes ectopic glomeruli that can be suppressed by the co-expression of active cofilin93, supporting the idea that LIMK negatively regulates cofilin function by serine 3 phosphorylation. During photoreceptor cell differentiation in the developing eye, cofilin is a downstream target of Mbt (a Pak homologue) signalling. Constitutive activation of Mbt alters the actin cytoskeleton as well as adherens junction (cell–cell junctions in epithelial tissue) organization94. Gain- and loss-of-function mutations in Cdi (serine/ threonine kinase centre divider), a testicular protein kinase 1 (TESK1) homologue, also caused alterations in actin organization and adherens junctions95. Furthermore, slingshot deletion mutants that are predicted to antagonize Cdi or LIMK also suppress proper photoreceptor development95,96. Therefore, the regulation of the phosphorylation state of cofilin has a crucial role during various cellular and morphogenetic events in Drosophila.
There is accumulating evidence that the cofilin pathway also has an essential role during mammalian morphogenesis. Although mutant mice that lack cofilin 1 (the non-muscle isoform) are indistinguishable in their gross morphology from control mice embryos at E9.5, no embryos were found at term, suggesting that cofilin 1 mutants are embryonic lethal60. Before stage E9.5, cofilin might not be essential for the extensive morphogenetic movements during gastrulation, raising the possibility that additional cofilin isoforms such as ADF might compensate in this type of cell migration, as evidenced by a threefold–fourfold overexpression of the cofilin isoform ADF compared with that in wild-type control embryos. However, following gastrulation ADF overexpression is not sufficient to compensate for the loss of non-muscle cofilin. Defects in neural crest polarization and migration were observed in non-muscle cofilin mutant embryos that lead to a failure in closure of the neural tube. Differences in the regulation of the various isoforms of the cofilin family by cofilin-directed kinases, phosphatases, pH and PIP2 binding have not been routinely detected. However, ADF is more sensitive to changes in pH than cofilin61. In addition, in the mouse and C. elegans, the non-muscle isoforms cofilin 1 and ADF are more efficient at depolymerizing F-actin in vitro than the muscle isoform of cofilin62,63. These findings suggest that specific cofilin isoforms may be crucial to the changes in cell shape and migration that occur during different stages of embryonic development and morphogenesis.
In mammals, two different LIMK isoforms, LIMK1 and 2, have been described that can phosphorylate cofilin downstream of Rho-family GTPases20,64,65. LIMKs can be activated upstream by Pak66 as well as ROCK65,67 and MRCK (myotonic dystrophy kinase-related CDC42-binding kinase)68. Normal central nervous system development may depend in part on LIMK1, as its deletion has been associated with the human genetic disease Williams syndrome, which shows similarities to the neurodevelopmental abnormalities seen in Limk1 knockout mice69. Williams syndrome is a developmental disorder characterized by several behavioural and neurological abnormalities, including deficits in visuospatial cognition. Patients with Williams syndrome possess a heterozygous deletion in a region on chromosome 7q11.23 that contains approximately 20 genes, including LIMK1. However, owing to the number of genes involved in this deletion, it is unclear if any of these developmental defects can be assigned to LIMK1 deletion alone. In addition, there are no conclusive reports linking Williams syndrome with increased tumorigenesis and progression. Limk1 knockout mice show significant abnormalities in synaptic function, specifically in the morphogenesis of the dendritic spine, a structure highly enriched in F-actin. Limk1 knockout mice, which contain decreased levels of phosphorylated cofilin, also show deficits in spatial learning and increased hippocampal long-term potentiation69, as well as impaired excitatory synaptic function of the hippocampus69. Spatial learning deficits in these mice suggest that LIMK1 has a crucial role in dendritic spine morphogenesis and brain function, which might lead to impaired visuospatial cognition as well as other behavioural and neurological symptoms observed in patients with Williams syndrome. Although Limk2 knockout mice showed little change in the level of phosphorylated cofilin and minimal abnormalities in synaptic function in the hippocampus70 or significant alterations in postnatal growth or development71, defects in testicular germ cell spermatogenesis potential, viability and reduced testes size were observed71. The fact that cofilin deficiency is embryonic lethal whereas Limk1 or Limk2 knockouts are viable indicates that phosphorylation-independent mechanisms of cofilin regulation such as pH changes or PIP2 binding may be important during morphogenesis and development. In addition, other kinases usually restricted to the testes (TESK1 and 2) and skeletal muscle (NRK) might compensate under these extreme conditions.
The cofilin pathway in tumour invasion
As expected from studies that showed the cofilin pathway is essential for cell motility and morphogenesis in vitro and in vivo, the cofilin pathway has also been implicated in tumour cell invasion and metastasis (TABLE 1). Cofilin is overexpressed in the highly invasive C6 rat glioblastoma cell line72, A549 human lung cancer cells73 and human pancreatic cancer cells74. The amount of phosphorylated cofilin is decreased in cell lines derived from T-cell lymphoma (Jurkat) and carcinomas from the cervix (HeLa), colon (KM12), liver (HepG2) and kidney (COS1)75. The spontaneous overexpression of cofilin has been detected in the invasive subpopulation of tumour cells in mammary tumours of rats10. Furthermore, increased levels of cofilin expression is detected in clinical tumour samples of oral squamous-cell carcinoma76, renal cell carcinoma77 and ovarian cancer78 using proteomic and cDNA microarray approaches. Expression of the wild-type or the non-phosphorylatable cofilin mutant S3A increases melanoma cell invasion through a reconstituted basement membrane56. However, several studies have found that cofilin is downregulated in cancer and that overexpression is antagonistic to invasion. The downregulation of cofilin is observed in MHCC97-H hepatocellular carcinoma (HCC) cells with high meta-static potential79, and in ovarian surface epithelium (OSE) cells derived from women with a family history of ovarian and/or breast cancer and mutations in the BRCA1 tumour-suppressor gene80. The regulated overexpression of cofilin inhibits the invasiveness of human lung cancer H1299 cells by disrupting the actin cytoskeleton at the leading edge of the cell53. Clearly these studies do not agree as to the affect of cofilin expression level on tumour invasion. Therefore, it is unlikely that the expression status of cofilin alone is sufficient to determine the motility and invasion status of carcinoma cells. As noted above, the balanced contribution of cofilin and other molecules in the cofilin pathway, including LIMK1, is required for chemotaxis and motility in tumour cells. Without information about the activity status of the cofilin pathway in each of these cases it is difficult to predict or explain the outcome of changing the activity of a single component of the pathway.
LIMK1 has been shown to affect cell motility and invasion, but initial reports were inconsistent about whether the overexpression of LIMK1 increases or decreases invasion. The expression of a constitutively active LIMK1 that increases the amount of phospho-cofilin in vivo inhibits actin polymerization and motility in response to EGF in mammary carcinoma cells54. In addition, the overexpression of LIMK1 inhibits cell motility in neuroblastoma cell lines81 and Ras-transformed fibroblasts82, whereas dominant-negative LIMK1 increases cell motility in neuroblastoma cell lines81. However, the inhibition of LIMK1 expression by siRNA inhibits the motility of Jurkat T cells51, and the overexpression of LIMK1 in prostate epithelial cells increases their invasiveness in vitro83 and the formation of osteolytic lesions in nude mice84. Furthermore, a reduction in LIMK1 expression in meta-static prostate cell lines decreases their ability to invade matrigel in vitro83. Clearly, altering the expression and activity of LIMK1 has major effects on tumour cell motility and invasion. However, the studies on LIMK1, like those on cofilin, do not agree as to the affects of increased LIMK1 expression on tumour invasion, again supporting the need to investigate the output of the cofilin pathway as an essential part of studies in which the activity of either cofilin, LIMK1 or other proteins within the pathway are manipulated.
Insights into the source of the inconsistencies in the literature about how cofilin and LIMK1 affect tumour cell invasion have emerged from the microarray-based expression profiling of invasive tumour cells collected from mammary tumours of rats and mice10,12. In these experiments the expression of genes in the invasive subpopulation of tumour cells was compared to that in the average tumour cell isolated from the primary tumour. It was found that in rat mammary tumours derived from the orthotopic injection of mammary tumour cell lines, invasive tumour cells have increased transient expression of both cofilin and LIMK1 (REFS 10,43). Considering either cofilin or LIMK1 in isolation would have led to the paradoxical conclusion that LIMK1 overexpression, which leads to the inhibition of cofilin activity, is correlated with increased invasion at the same time as cofilin overexpression, which leads to an increase in cofilin activity, is also correlated with increased invasion. However, when the activity of the cofilin pathway was measured in these invasive tumour cells as the EGF-stimulated early transient of free barbed ends (FIG. 3), it was found to be significantly increased compared with non-invasive tumour cells from the same tumour with normal levels of LIMK1 and cofilin expression43. Furthermore, increasing the expression of LIMK1 alone in these mammary tumour cells inhibited the cofilin pathway and inhibited invasion and metastasis43. In mice with polyoma middle T oncogene (PyMT)-derived mammary tumours, invasive cells have increased expression of both LIMK1 and SSH, but not cofilin. Again, measurement of the output of the cofilin pathway in these PyMT-tumour derived invasive tumour cells showed that it is increased compared with non-invasive tumour cells from the same tumour12. Therefore, consideration of the cofilin pathway as a whole must be done to predict the effect of manipulating any single component of the pathway and requires that the inhibitory (LIMK1) and stimulatory (cofilin and SSH) branches of the pathway be viewed as a balance. If both branches are increased together, the paired overexpression of inhibitory and stimulatory components might increase the activity of the cofilin pathway overall to increase cell migration and chemotaxis (see the ‘cell biology of the cofilin pathway’ section). Another important implication of these results in rats and mice is that looking at the expression status of a single gene can be misleading when interpreting phenotype, as it is the collective activity of multiple genes of the pathway that determines the integrated output of the pathway and therefore phenotype.
The cofilin pathway tumour metastasis
In mammary tumours in particular, the microenvironment for metastasis depends heavily on macrophages, which provide signals for angiogenesis, tumour growth, tumour cell chemotaxis and invasion15. The communication between macrophages and tumour cells in rats and mice is paracrine, involving the production of EGF by macrophages and CSF1 by tumour cells to stimulate tumour cell and macrophage chemotaxis and invasion, respectively7,85. Therefore, in mammary tumours, tumour cell invasion and metastasis are under the control of macrophage-supplied EGF7,85. As discussed above, in mammary tumour cells chemotaxis to EGF is regulated by the cofilin pathway and involves the balance between cofilin and LIMK activities to produce a transient and spatially localized amplification of the EGF gradient in the form of free actin filament barbed ends (FIG. 3, BOX 3). Therefore, metastatic potential in mammary tumours is predicted to depend in part on the output of the cofilin pathway. This hypothesis was tested by preparing rat mammary tumour cells that otherwise had normal levels of cofilin expression but that either overexpressed LIMK1 (which suppressed the output of the cofilin pathway in the presence of normal cofilin levels) or that expressed dominant-negative LIMK domain (which increased the output of the cofilin pathway)43. When these cells were injected into mammary glands to form tumours, the metastatic potential of the mammary tumours was directly related to the output of the cofilin pathway. Animals with tumours containing cells in which cofilin pathway activity was suppressed (LIMK1 overexpression) showed decreased invasion and metastasis and were associated with increased survival; whereas tumours derived from cells with increased cofilin pathway activity (LIMK dominant negative) showed increased invasion and metastasis and were associated with decreased survival. Of particular interest was the finding that tumours derived from tumour cells in which cofilin was overexpressed along with LIMK1 had significantly increased invasion in vivo compared with the level of invasion in tumours with LIMK1 overexpression but normal levels of cofilin expression43. These results show that the effect of LIMK1 expression on invasion, intravasation and metastasis of cancer cells can be simply explained by considering the regulation and activity status of the cofilin pathway as a whole. More generally these results emphasize that it is the pathway of which the altered gene product is a part that determines the invasive and metastatic phenotype of a tumour, not the individual gene. This is consistent with our knowledge of how signalling pathways, but not necessarily single genes within, affect tumorigenesis, progression86 and migration87.
Box 3. The ADF and cofilin family of proteins.
The actin depolymerizing factor (ADF)/cofilin family of proteins are expressed in all eukaryotes examined to date. Unicellular eukaryotes such as Saccharomyces cerevisiae, Acanthamoeba castellanii and Dictyostelium discoideum possess a single ADF or cofilin gene. Multicellular organisms typically have several ADF/ cofilin isoforms. Caenorhabditis elegans expresses two ADF/cofilins, unc-60A and unc-60B, whereas Drosophila melanogaster expresses a single copy, twinstar (for a complete phylogenetic review of ADF/ cofilins see REF. 18). In general, mammals have many isoforms classified as ADFs and cofilins based on sequence homology and biological activity. All mammals are predicted to have a single ADF isoform expressed most strongly in epithelia and neurons, and two cofilin isoforms, a non-muscle type cofilin 1 expressed in most embryonic and adult cells, and a muscle-type cofilin 2 expressed almost exclusively in skeletal muscle62. Based on studies in various mouse cells, cofilin 1 is the predominant isoform in non-muscle cells, expressed at 6–11 times greater molar amounts than ADF40. Wang et al.10 has shown that the cofilin 1 isoform is selectively upregulated in invasive mammary tumour cells in rats.
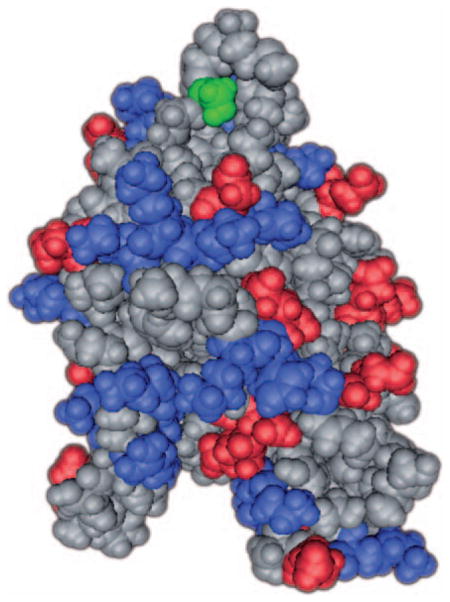
All of the homologues of ADF and cofilin are believed to share a common structure, as shown in this this space-filling model. This model has been based on the findings of three papers97–99. The serine 3 phosphorylation site is highlighted in green, G-actin binding in red and F-actin and PIP2 binding, which overlap, in blue.
Future directions
The concept that metastasis is encoded throughout the bulk of the primary tumour has focused attention on the contribution of the microenvironment to metastasis8,9. In many tumours, inflammation contributes to the progression to malignancy88. In mammary tumours in particular, macrophages provide signals to tumour cells for chemotaxis and cell motility that lead to invasion and metastasis. In this context macrophages help provide the microenvironment for metastasis in a similar way to the function of macrophages during normal mammary morphogenesis89. This suggests that metastasis may be an unregulated repetition of morphogenesis15. Therefore, many of the genes involved in epithelial cell migration during mammary morphogenesis are likely to be hard at work during mammary tumour metastasis. Current results indicate that many of the genes whose expression status is upregulated during morphogenesis and metastasis are clustered in the cofilin pathway and its downstream effectors10–12. This makes the cofilin pathway a potential target for anti-metastatic therapy. Although methodical studies of the effect of cofilin activity on metastasis have only been done on mammary tumours, it is likely that the cofilin pathway also has a major role in the invasion and metastasis of other types of cancer given the fact that cofilin is essential for cell motility and cell viability in many tissue types, and the correlation of expression of cofilin pathway genes with metastatic phenotype in various cancers (TABLE 1). In rat and mouse mammary tumour models nine genes of the cofilin pathway are overexpressed, making many combinations of ways to increase the output of the cofilin pathway. Only one pair of genes (LIMK1 and cofilin) have been tested for their effects on the activity of the cofilin pathway, and the results show that cofilin pathway output is predictive for metastatic potential43. Therefore, inhibitors directed at the output of the cofilin pathway and not just the activities of single genes are more likely to have an inhibitory affect on metastasis. In addition, it is not possible to inhibit cofilin alone, as this is lethal and will also kill normal cells. In mammary tumour cells, inhibitors of the stimulatory branch of the cofilin pathway directed at PLCγ1, SSH and chronophin, which could be used to turn down the activity of the pathway enough to inhibit chemotaxis, might have therapeutic benefit without killing normal cells. There are no such inhibitors available at this time. Another approach would be to inhibit signals from stromal cells that activate the cofilin pathway in tumour cells. This would have the advantage of being more specific for tumour cells in the metastatic microenvironment. To design inhibitors of stromal cell contributions to the activation of the cofilin pathway in human breast tumours, and any other type of tumour, it will be necessary to describe the microenvironment of metastasis that activates the cofilin pathway, including the stromal cells involved. In mammary tumours of rats and mice the stromal cell essential for this activation is the macrophage. However, this analysis has not been done using animal models of other types of tumours, so the stromal cells that stimulate tumour cell migration in other types of tumours are not precisely known.
Altering the expression of genes in experimental animal tumours, as has been done for cofilin and LIMK1 in mammary tumours43, will be necessary to determine which combinations of genes must be altered in expression or activity status in order to suppress the output of the cofilin pathway and its initiation of tumour cell migration. Based on these results the rational design of inhibitors of the cofilin pathway for human breast tumours and other tumour types can then proceed. Measuring the output of the cofilin pathway directly in living invasive tumour cells isolated from tumours will be necessary to assess the efficacy of the inhibitors. New technologies for intravital imaging, invasive tumour cell collection and expression profiling, and for measuring cofilin pathway activity, make this analysis, as well as the testing of inhibitors of the cofilin pathway, possible.
At a glance.
A pattern of changes in gene expression clustered in the cofilin pathway is consistently observed in mammary tumours and cells derived from them.
The cofilin pathway has emerged as having a central role in the generation of free actin filament ends resulting in actin filament remodelling by polymerization and depolymerization. Filament remodelling is essential during the formation and retraction of path-finding structures used in the chemotaxis, cell migration and invasion of tumour cells.
The spatial localization of cofilin activity is required for chemotaxis by tumour cells in response to epidermal growth factor, and fits a local excitation global inhibition (LEGI)-type model of chemotaxis.
A balance of the stimulatory and inhibitory branches of the cofilin pathway must be achieved for protrusion, cell migration and chemotaxis to occur optimally. Too much or too little activity will inhibit all of these essential steps in motility and invasion.
As there are four regulatory mechanisms for cofilin activity which seem to be uncoupled, the activity status of cofilin in a cell cannot be assessed by measuring the ratio of dephosphorylated cofilin to the total cofilin present.
An important implication of recent studies of the cofilin pathway is that looking at the expression status of a single gene can be misleading when interpreting phenotype, as it is the collective activity of multiple genes of the pathway that determines the integrated output of the pathway and therefore phenotype.
The rational design of inhibitors of the cofilin pathway is possible. Measuring the output of the cofilin pathway directly in living cells isolated from invasive tumours will be necessary to assess the efficacy of the inhibitors. New technologies for intravital imaging, invasive tumour cell collection and expression profiling, and for measuring cofilin pathway activity, make inhibitor design and testing possible.
Acknowledgments
This work was supported by grants CA100324 and GM38511.
Glossary
- Motility cycle
The motility cycle consists of a minimum of four steps starting with protrusion, which is essential for determining subsequent cell direction. Protrusion is followed by the adhesion of the new protrusion, contraction and tail retraction. See reference 35 for more details
- Lamellipodium
A 1–5 μm wide cytoplasmic projection at the leading edge of the cell that contains a dendritic network of actin filaments (see box 1). The force of actin polymerization extends the lamellapodium forward and advances the cell front, setting the direction of cell migration
- Invadopodium
A cytoplasmic projection from tumour cells into the extracellular matrix, which contain a core of actin filaments. Invadopodia can secrete proteases that degrade the extracellular matrix and whose formation is associated with increased tumour cell invasiveness
- Filopodium
A finger-shaped cytoplasmic projection that can extend from the leading edge of migrating cells. Filopodia contain actin filaments that are crosslinked into bundles by actin-binding proteins such as fimbrin
- Off rate constant
Also known as the dissociation constant (Kd), for actin filaments it measures the rate of dissociation of actin monomers from free filament ends
- Caged
A protein or compound conjugated with a chromophore that allows for the controlled photorelease of a biologically active protein or compound with high temporal and spatial precision
- Vitronectin
An abundant adhesive glycoprotein found in blood plasma and the extracellular matrix. Vitronectin contains an RGD sequence that is a binding site for membrane-bound integrins, which serve to anchor cells to the extracellular matrix
- Neural crest
A component of the ectoderm that is found between the neural tube and the epidermis of an embryo. Shortly after neural tube formation, neural crest cells migrate and give rise to neurons and glia of the peripheral nervous system, skeletal and smooth muscle, and other specialized cells
- Neural tube
A developmental precursor of the central nervous system that will form the mature brain and spinal cord
- Visuospatial cognition
The ability to distinguish the orientation of objects in space
- for example
depth perception
- Dendritic spine
A small (<1 μm) membranous extension that protrudes from a dendrite and forms one half of a synapse. Changes in dendritic spine density underlie many brain functions, including long-term memory and learning
- Osteolytic
Having the property of osteolysis, which is defined as the active resorption or dissolution of bone tissue as part of normal bone remodelling and some disease processes
- Paracrine
Signalling between two different types of cells through secreted molecules
Footnotes
Competing interests statement
The authors declare no competing financial interests.
DATABASES
The following terms in this article are linked online to:
Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene
ARP2 | ARP3 | BRCA1 | cofilin 1 | CSF1 | EGF | EGFR | LIMK1 | LIMK2 | MRCK | NRK | PAK1 | PAK4 | PLCγ | ROCK1 | SDF1 | TESK1 | TESK2 | TGFα | VEGF
OMIM: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM
Williams syndrome: http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=194050
Access to this links box is available online.
References
- 1.Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411:375–379. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- 2.Condeelis J, Segall JE. Intravital imaging of cell movement in tumours. Nature Rev Cancer. 2003;3:921–930. doi: 10.1038/nrc1231. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 4.Mantovani A, Giavazzi R, Alessandri G, Spreafico F, Gerattini S. Characterization of tumor lines derived from spontaneous metastases of transplanted murine sarcoma. Eur J Cancer. 1981;17:71–76. doi: 10.1016/0014-2964(81)90213-9. [DOI] [PubMed] [Google Scholar]
- 5.Giavazzi R, Alessandri G, Spreafico F, Garattini S, Mantovani A. Metastasizing capacity of tumour cells from spontaneous metastases of transplanted murine tumours. Br J Cancer. 1980;42:462–472. doi: 10.1038/bjc.1980.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Milas L, Peters LJ, Ito H. Spontaneus metastasis: random or selective? Clin Exp. 1983;1:309–315. doi: 10.1007/BF00121193. [DOI] [PubMed] [Google Scholar]
- 7.Wyckoff J, et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004;64:7022–7029. doi: 10.1158/0008-5472.CAN-04-1449. First description of the paracrine loop between tumour cells and macrophages and its behavioural consequences in vivo. [DOI] [PubMed] [Google Scholar]
- 8.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nature Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 9.van ‘t Veer LJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. Expression profiling study that identified metastatic potential as an early property encoded throughout the tumour as inconsistent with a simple Darwinian evolution model. [DOI] [PubMed] [Google Scholar]
- 10.Wang W, et al. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res. 2004;64:8585–8594. doi: 10.1158/0008-5472.CAN-04-1136. First description of an invasion signature including the cofilin pathway. [DOI] [PubMed] [Google Scholar]
- 11.Wang W, et al. Tumor cells caught in the act of invading: their strategy for enhanced cell motility. Trends Cell Biol. 2005;15:138–145. doi: 10.1016/j.tcb.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 12.Wang W, et al. Coordinated regulation of pathways for enhanced cell motility and chemotaxis is conserved in rat and mouse mammary tumors. Cancer Res. 2007;64:3505–3511. doi: 10.1158/0008-5472.CAN-06-3714. [DOI] [PubMed] [Google Scholar]
- 13.Weigelt B, Peterse J, van’t Veer L. Breast cancer metastasis: markers and models. Nature Rev Cancer. 2005;5:591–602. doi: 10.1038/nrc1670. First description of the integrated model of breast cancer metastasis. [DOI] [PubMed] [Google Scholar]
- 14.Leek RD, Harris AL. Tumor-associated macrophages in breast cancer. J Mammary Gland Biol Neoplasia. 2002;7:177–189. doi: 10.1023/a:1020304003704. [DOI] [PubMed] [Google Scholar]
- 15.Condeelis J, Pollard J. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 16.Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J Clin Invest. 2004;114:623–633. doi: 10.1172/JCI22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ono S. Mechanism of depolymerization and severing of actin filaments and its significance in cytoskeletal dynamics. Int Rev Cytol. 2007;258:1–82. doi: 10.1016/S0074-7696(07)58001-0. A comprehensive description of the cofilin family and the activities of its members. [DOI] [PubMed] [Google Scholar]
- 18.Maciver SK, Hussey P. The ADF/cofilin family: actin remodeling proteins. Genome Biology. 2002;3 doi: 10.1186/gb-2002-3-5-reviews3007. reviews 3007. A description of the cofilin family and the evolutionary relatedness of its members. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okano I, et al. Identification and characterization of a novel family of serine/threonine kinases containing two N-terminal LIM motifs. J Biol Chem. 1995;270:31321–31330. doi: 10.1074/jbc.270.52.31321. [DOI] [PubMed] [Google Scholar]
- 20.Yang N, et al. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 1998;393:809–812. doi: 10.1038/31735. Initial description of the connection between cofilin and LIM kinase. [DOI] [PubMed] [Google Scholar]
- 21.Toshima J, et al. Cofilin phosphorylation by protein kinase testicular protein kinase 1 and its role in integrin-mediated actin reorganization and focal adhesion formation. Mol Biol Cell. 2001;12:1131–1145. doi: 10.1091/mbc.12.4.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakano K, et al. Cofilin phosphorylation and actin polymerization by NRK/NESK, a member of the germinal center kinase family. Exp Cell Res. 2003;287:219–227. doi: 10.1016/s0014-4827(03)00136-8. [DOI] [PubMed] [Google Scholar]
- 23.Niwa R, Nagata-Ohashi K, Takeichi M, Mizuno K, Uemura T. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell. 2002;108:233–246. doi: 10.1016/s0092-8674(01)00638-9. Identification of slingshot and its action on cofilin. [DOI] [PubMed] [Google Scholar]
- 24.Meberg PJ, et al. Actin depolymerizing factor and cofilin phosphorylation dynamics: response to signals that regulate neurite extension. Cell Motil Cytoskeleton. 1998;39:172–190. doi: 10.1002/(SICI)1097-0169(1998)39:2<172::AID-CM8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 25.Ambach A, et al. The serine phosphatases PP1 and PP2A associate with and activate the actin-binding protein cofilin in human T lymphocytes. Eur J Immunol. 2000;30:3422–3431. doi: 10.1002/1521-4141(2000012)30:12<3422::AID-IMMU3422>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 26.Gohla A, Birkenfeld J, Bokoch GM. Chronophin, a novel HAD-type serine protein phosphatase, regulates cofilin-dependent actin dynamics. Nature Cell Biol. 2005;7:21–29. doi: 10.1038/ncb1201. [DOI] [PubMed] [Google Scholar]
- 27.Yonezawa N, Homma Y, Yahara I, Sakai H, Nishida E. A short sequence responsible for both phosphoinositide binding and actin binding activities of cofilin. J Biol Chem. 1991;266:17218–17221. [PubMed] [Google Scholar]
- 28.Yonezawa N, Nishida E, Iida K, Yahara I, Sakai H. Inhibition of the interactions of cofilin, destrin, and deoxyribonuclease I with actin by phosphoinositides. J Biol Chem. 1990;265:8382–8386. [PubMed] [Google Scholar]
- 29.Mouneimne G, et al. Phospholipase C and cofilin are required for carcinoma cell directionality in response to EGF stimulation. J Cell Biol. 2004;166:697–708. doi: 10.1083/jcb.200405156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mouneimne D, et al. Spatial and temporal control of cofilin activity is required for directional sensing during chemotaxis. Curr Biol. 2006;16:2193–2205. doi: 10.1016/j.cub.2006.09.016. Showed that cofilin is required for directional sensing during the chemotaxis of breast tumour cells. [DOI] [PubMed] [Google Scholar]
- 31.Bamburg JA, Wiggan O. ADF/cofilin and actin dynamics in disease. Trends Cell Biol. 2002;12:598–605. doi: 10.1016/s0962-8924(02)02404-2. [DOI] [PubMed] [Google Scholar]
- 32.Patel H, Barber D. A developmentally regulated Na-H exchanger in Dyctyostelium discoideum is necessary for cell polarity during chemotaxis. J Cell Biol. 2005;169:321–329. doi: 10.1083/jcb.200412145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernstein BW, et al. Intracellular pH modulation of ADF/cofilin proteins. Cell Motil Cytoskeleton. 2000;47:319–336. doi: 10.1002/1097-0169(200012)47:4<319::AID-CM6>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 34.Srivastava J, Barber D, Jacobson M. Intracellular pH sensors: design principles and functional significance. Physiology. 2006;22:30–39. doi: 10.1152/physiol.00035.2006. [DOI] [PubMed] [Google Scholar]
- 35.Mitchison TJ, Cramer LP. Actin-based cell motility and cell locomotion. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- 36.Mogilner A, Oster G. Polymer motors: pushing out the front and pulling up the back. Curr Biol. 2003;13:R721–R733. doi: 10.1016/j.cub.2003.08.050. [DOI] [PubMed] [Google Scholar]
- 37.dos Remedios C, et al. Actin binding proteins: regulation of cytoskeletal microfilaments. Physiol Rev. 2003;83:433–473. doi: 10.1152/physrev.00026.2002. [DOI] [PubMed] [Google Scholar]
- 38.Yamaguchi H, et al. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell Biol. 2005;168:441–452. doi: 10.1083/jcb.200407076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghosh M, et al. Cofilin promotes actin polymerization and defines the direction of cell motility. Science. 2004;304:743–746. doi: 10.1126/science.1094561. Showed the consequences of cofilin activation in vivo. [DOI] [PubMed] [Google Scholar]
- 40.Hotulainen P, Paunola E, Vartiainen MK, Lappalainen P. Actin-depolymerizing factor and cofilin-1 play overlapping roles in promoting rapid F-actin depolymerization in mammalian nonmuscle cells. Mol Biol Cell. 2005;16:649–664. doi: 10.1091/mbc.E04-07-0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aizawa H, Sutoh K, Yahara I. Overexpression of cofilin stimulates bundling of actin filaments, membrane ruffling, and cell movement in Dictyostelium. J Cell Biol. 1996;132:335–344. doi: 10.1083/jcb.132.3.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yap CT, Simpson TI, Pratt T, Price DJ, Maciver SK. The motility of glioblastoma tumour cells is modulated by intracellular cofilin expression in a concentration-dependent manner. Cell Motil Cytoskeleton. 2005;60:153–165. doi: 10.1002/cm.20053. [DOI] [PubMed] [Google Scholar]
- 43.Wang W, et al. The activity status of cofilin is directly related to invasion, intravasation, and metastasis of mammary tumors. J Cell Biol. 2006;173:395–404. doi: 10.1083/jcb.200510115. Analysis of the consequences of cofilin pathway activity status on metastasis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ichetovkin I, Han J, Pang KM, Knecht DA, Condeelis JS. Actin filaments are severed by both native and recombinant dictyostelium cofilin but to different extents. Cell Motil Cytoskeleton. 2000;45:293–306. doi: 10.1002/(SICI)1097-0169(200004)45:4<293::AID-CM5>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 45.Ichetovkin I, Grant W, Condeelis J. Cofilin produces newly polymerized actin filaments that are preferred for dendritic nucleation by the Arp2/3 complex. Curr Biol. 2002;12:79–84. doi: 10.1016/s0960-9822(01)00629-7. [DOI] [PubMed] [Google Scholar]
- 46.Adrianantoandro E, Pollard T. Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/Cofilin. Mol Cell. 2006;24:13–23. doi: 10.1016/j.molcel.2006.08.006. Direct analysis of the rate constants for actin polymerization and depolymerization, showing that cofilin does not alter off rate. [DOI] [PubMed] [Google Scholar]
- 47.Des Marais V, Macaluso F, Condeelis J, Bailly M. Synergistic interaction between the Arp2/3 complex and cofilin drives stimulated lamellipod extension. J Cell Sci. 2004;117:3499–3510. doi: 10.1242/jcs.01211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carlsson A. Stimulation of actin polymerization by filament severing. Biophys J. 2006;90:413–422. doi: 10.1529/biophysj.105.069765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamaguchi H, Wyckoff J, Condeelis J. Cell migration in tumors. Curr Opin Cell Biol. 2005;17:559–564. doi: 10.1016/j.ceb.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 50.Song X, et al. Initiation of cofilin activity in response to EGF is uncoupled from cofilin phosphorylation and dephosphorylation in carcinoma cells. J Cell Sci. 2006;119:2871–2881. doi: 10.1242/jcs.03017. [DOI] [PubMed] [Google Scholar]
- 51.Nishita M, et al. Spatial and temporal regulation of cofilin activity by LIM kinase and Slingshot is critical for directional cell migration. J Cell Biol. 2005;171:349–359. doi: 10.1083/jcb.200504029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Devreotes P, Janetopoulos C. Eukaryotic chemotaxis: distinctions between directional sensing and polarization. J Biol Chem. 2003;278:20445–20448. doi: 10.1074/jbc.R300010200. [DOI] [PubMed] [Google Scholar]
- 53.Lee YJ, Mazzatti DJ, Yun Z, Keng PC. Inhibition of invasiveness of human lung cancer cell line H1299 by over-expression of cofilin. Cell Biol Int. 2005;29:877–883. doi: 10.1016/j.cellbi.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 54.Zebda N, et al. Phosphorylation of ADF/cofilin abolishes EGF-induced actin nucleation at the leading edge and subsequent lamellipod extension. J Cell Biol. 2000;151:1119–1128. doi: 10.1083/jcb.151.5.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iwasa J, Mullins RD. Spatial and temporal relationships between actin-filament nucleation, capping, and disassembly. Curr Biol. 2007;17:395–406. doi: 10.1016/j.cub.2007.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dang D, Bamburg JR, Ramos DM. αvβ3 integrin and cofilin modulate K1735 melanoma cell invasion. Exp Cell Res. 2006;312:468–477. doi: 10.1016/j.yexcr.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 57.Ono K, Parast M, Alberico C, Benian GM, Ono S. Specific requirement for two ADF/cofilin isoforms in distinct actin-dependent processes in Caenorhabditis elegans. J Cell Sci. 2003;116:2073–2085. doi: 10.1242/jcs.00421. [DOI] [PubMed] [Google Scholar]
- 58.Chen J, et al. Cofilin/ADF is required for cell motility during Drosophila ovary development and oogensis. Nature Cell Biol. 2001;3:204–209. doi: 10.1038/35055120. [DOI] [PubMed] [Google Scholar]
- 59.Blair A, et al. Twinstar, the Drosophila homolog of cofilin/ADF, is required for planar cell polarity patterning. Development. 2006;133:1789–1797. doi: 10.1242/dev.02320. [DOI] [PubMed] [Google Scholar]
- 60.Gurniak CB, Perlas E, Witke W. The actin depolymerizing factor n-cofilin is essential for neural tube morphogenesis and neural crest cell migration. Dev Biol. 2005;278:231–241. doi: 10.1016/j.ydbio.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 61.Hayden SM, Miller PS, Brauweiler A, Bamburg JR. Analysis of the interactions of actin depolymerizing factor with G- and F-actin. Biochemistry. 1993;32:9994–10004. doi: 10.1021/bi00089a015. [DOI] [PubMed] [Google Scholar]
- 62.Vartiainen MK, et al. The three mouse ADF/cofilins evolved to fulfill cell type specific requirements for actin dynamics. Mol Biol Cell. 2002;13:183–194. doi: 10.1091/mbc.01-07-0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ono S, Benian G. Two C. elegans ADF/cofilin proteins, encoded by the unc-60 gene, differentially regulate actin filament dynamics. J Biol Chem. 1998;273:3778–3783. doi: 10.1074/jbc.273.6.3778. [DOI] [PubMed] [Google Scholar]
- 64.Arber S, et al. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–809. doi: 10.1038/31729. Initial description of the connection between cofilin and LIM kinase. [DOI] [PubMed] [Google Scholar]
- 65.Sumi T, Matsumoto K, Nakamura T. Specific activation of LIM Kinase 2 via phosphorylation of threonine 505 by ROCK, a Rho-dependent protein kinase. J Biol Chem. 2001;276:670–676. doi: 10.1074/jbc.M007074200. [DOI] [PubMed] [Google Scholar]
- 66.Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nature Cell Biol. 1999;1:253–259. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- 67.Ohashi K, et al. Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J Biol Chem. 2000;275:3577–3582. doi: 10.1074/jbc.275.5.3577. [DOI] [PubMed] [Google Scholar]
- 68.Sumi T, Matsumoto K, Shibuya A, Nakamura T. Activation of LIM kinases by myotonic dystrophy kinase-related Cdc42-binding kinase α. J Biol Chem. 2001;276:23092–23096. doi: 10.1074/jbc.C100196200. [DOI] [PubMed] [Google Scholar]
- 69.Meng Y, et al. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron. 2002;35:121–133. doi: 10.1016/s0896-6273(02)00758-4. [DOI] [PubMed] [Google Scholar]
- 70.Meng Y, et al. Regulation of ADF/cofilin phosphorylation and synaptic function by LIM-kinase. Neuropharmacology. 2004;47:746–754. doi: 10.1016/j.neuropharm.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 71.Takahashi H, Koshimizu U, Miyazaki J, Nakamura T. Impaired spermatogenic ability of testicular germ cells in mice deficient in the LIM-kinase 2 gene. Dev Biol. 2002;241:259–272. doi: 10.1006/dbio.2001.0512. [DOI] [PubMed] [Google Scholar]
- 72.Gunnersen JM, Spirkoska V, Smith PE, Danks RA, Tan SS. Growth and migration markers of rat C6 glioma cells identified by serial analysis of gene expression. Glia. 2000;32:146–154. [PubMed] [Google Scholar]
- 73.Keshamouni VG, et al. Differential protein expression profiling by iTRAQ-2DLC-MS/MS of lung cancer cells undergoing epithelial-mesenchymal transition reveals a migratory/invasive phenotype. J Proteome Res. 2006;5:1143–1154. doi: 10.1021/pr050455t. [DOI] [PubMed] [Google Scholar]
- 74.Sinha P, et al. Increased expression of epidermal fatty acid binding protein, cofilin, and 14–3-3-σ (stratifin) detected by two-dimensional gel electrophoresis, mass spectrometry and microsequencing of drug-resistant human adenocarcinoma of the pancreas. Electrophoresis. 1999;20:2952–2960. doi: 10.1002/(SICI)1522-2683(19991001)20:14<2952::AID-ELPS2952>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 75.Nebl G, Meuer SC, Samstag Y. Dephosphorylation of serine 3 regulates nuclear translocation of cofilin. J Biol Chem. 1996;271:26276–26280. doi: 10.1074/jbc.271.42.26276. [DOI] [PubMed] [Google Scholar]
- 76.Turhani D, Krapfenbauer K, Thurnher D, Langen H, Fountoulakis M. Identification of differentially expressed, tumor-associated proteins in oral squamous cell carcinoma by proteomic analysis. Electrophoresis. 2006;27:1417–1423. doi: 10.1002/elps.200500510. [DOI] [PubMed] [Google Scholar]
- 77.Unwin RD, et al. Proteomic changes in renal cancer and co-ordinate demonstration of both the glycolytic and mitochondrial aspects of the Warburg effect. Proteomics. 2003;3:1620–1632. doi: 10.1002/pmic.200300464. [DOI] [PubMed] [Google Scholar]
- 78.Martoglio AM, et al. Changes in tumorigenesis- and angiogenesis-related gene transcript abundance profiles in ovarian cancer detected by tailored high density cDNA arrays. Mol Med. 2000;6:750–765. [PMC free article] [PubMed] [Google Scholar]
- 79.Ding SJ, et al. Proteome analysis of hepatocellular carcinoma cell strains, MHCC97-H and MHCC97-L, with different metastasis potentials. Proteomics. 2004;4:982–994. doi: 10.1002/pmic.200300653. [DOI] [PubMed] [Google Scholar]
- 80.Smith-Beckerman DM, et al. Proteome changes in ovarian epithelial cells derived from women with BRCA1 mutations and family histories of cancer. Mol Cell Proteomics. 2005;4:156–168. doi: 10.1074/mcp.M400157-MCP200. [DOI] [PubMed] [Google Scholar]
- 81.Meyer G, Kim B, van Golen C, Feldman EL. Cofilin activity during insulin-like growth factor I-stimulated neuroblastoma cell motility. Cell Mol Life Sci. 2005;62:461–470. doi: 10.1007/s00018-004-4456-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sahai E, Olson MF, Marshall CJ. Cross-talk between Ras and Rho signalling pathways in transformation favours proliferation and increased motility. EMBO J. 2001;20:755–766. doi: 10.1093/emboj/20.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Davila M, Frost AR, Grizzle WE, Chakrabarti R. LIM kinase 1 is essential for the invasive growth of prostate epithelial cells: implications in prostate cancer. J Biol Chem. 2003;278:36868–36875. doi: 10.1074/jbc.M306196200. [DOI] [PubMed] [Google Scholar]
- 84.Yoshioka K, Foletta V, Bernard O, Itoh K. A role for LIM kinase in cancer invasion. Proc Natl Acad Sci USA. 2003;100:7247–7252. doi: 10.1073/pnas.1232344100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Goswami S, et al. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 2005;65:5278–5283. doi: 10.1158/0008-5472.CAN-04-1853. [DOI] [PubMed] [Google Scholar]
- 86.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nature Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 87.Kumar N, Wolf-Yadlin A, White F, Lauffenburger D. Modeling HER2 effects on cell behavior from mass spectrometry phosphotyrosine data. PLoS Compt Biol. 2007;3:e4. doi: 10.1371/journal.pcbi.0030004. Systems analysis showing that pathways but not single genes determine the migratory phenotype. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ingman WV, Wyckoff J, Gouon-Evans V, Condeelis J, Pollard JW. Macrophages promote collagen fibrillogenesis around terminal end buds of developing mammary gland. Dev Dyn. 2006;235:3222–3229. doi: 10.1002/dvdy.20972. [DOI] [PubMed] [Google Scholar]
- 90.Pollard TD, Blanchoin L, Mullins RD. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu Rev Biophys Biomol Struct. 2000;29:545–576. doi: 10.1146/annurev.biophys.29.1.545. [DOI] [PubMed] [Google Scholar]
- 91.Pollard TD, Beltzner CC. Structure and function of the Arp2/3 complex. Curr Opin Struct Biol. 2002;12:768–774. doi: 10.1016/s0959-440x(02)00396-2. [DOI] [PubMed] [Google Scholar]
- 92.Gunsalus KC, et al. Mutations in twinstar, a Drosophila gene encoding a cofilin/ADF homologue, result in defects in centrosome migration and cytokinesis. J Cell Biol. 1995;131:1243–1259. doi: 10.1083/jcb.131.5.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ang LH, et al. Lim kinase regulates the development of olfactory and neuromuscular synapses. Dev Biol. 2006;293:178–190. doi: 10.1016/j.ydbio.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 94.Menzel N, Schneeberger D, Raabe T. The Drosophila p21 activated kinase Mbt regulates the actin cytoskeleton and adherens junctions to control photoreceptor cell morphogenesis. Mech Dev. 2007;124:78–90. doi: 10.1016/j.mod.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 95.Sese MC, et al. The Cdi/TESK1 kinase is required for Sevenless signaling and epithelial organization in the Drosophila eye. J Cell Sci. 2006;119:5047–5056. doi: 10.1242/jcs.03294. [DOI] [PubMed] [Google Scholar]
- 96.Rogers EM, Hsiung F, Rodrigues AB, Moses K. Slingshot cofilin phosphatase localization is regulated by receptor tyrosine kinases and regulates cytoskeletal structure in the developing Drosophila eye. Mech Dev. 2005;122:1194–1205. doi: 10.1016/j.mod.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 97.Lappalainen P, Kessels M, Cope M, Drubin DG. The ADF homology (ADF-H) domain: a highly exploited actin-binding module. Mol Biol Cell. 1998;9:1951–1959. doi: 10.1091/mbc.9.8.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ojala PJ, Paavilainen V, Lappalainen P. Identification of yeast cofilin residues specific for actin monomer and PIP2 binding. Biochemistry. 2001;40:15562–15569. doi: 10.1021/bi0117697. [DOI] [PubMed] [Google Scholar]
- 99.Pope BJ, Zierler-Gould K, Kuhne R, Weeds A, Ball L. Solution structure of human cofilin: actin binding, ph sensitivity, and relationship to actin-depolymerizing factor. J Biol Chem. 2004;279:4840–4848. doi: 10.1074/jbc.M310148200. [DOI] [PubMed] [Google Scholar]
- 100.Xue W, et al. Epidermal growth factor receptor overexpression results in increased tumor cell motility in vivo coordinately with enhanced intravasation and metastasis. Cancer Res. 2005;66:192–197. doi: 10.1158/0008-5472.CAN-05-1242. [DOI] [PubMed] [Google Scholar]
- 101.Marone R, et al. Memo mediates ErbB2-driven cell motility. Nature Cell Biol. 2004;23:515–522. doi: 10.1038/ncb1134. [DOI] [PubMed] [Google Scholar]
- 102.Lorenz M, Yamaguchi H, Wang Y, Singer RH, Condeelis J. Imaging sites of N-wasp activity in lamellipodia and invadopodia of carcinoma cells. Curr Biol. 2004;14:697–703. doi: 10.1016/j.cub.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 103.Chan AY, et al. EGF stimulates an increase in actin nucleation and filament number at the leading edge of the lamellipod in mammary adenocarcinoma cells. J Cell Sci. 1998;111:199–211. doi: 10.1242/jcs.111.2.199. [DOI] [PubMed] [Google Scholar]
- 104.Dowling P, et al. Proteomic analysis of isolated membrane fractions from superinvasive cancer cells. Biochim Biophys Acta. 2007;1774:93–101. doi: 10.1016/j.bbapap.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 105.Dan C, Kelly A, Bernard O, Minden A. Cytoskeletal changes regulated by the PAK4 serine/ threonine kinase are mediated by LIM kinase 1 and cofilin. J Biol Chem. 2001;276:32115–32121. doi: 10.1074/jbc.M100871200. [DOI] [PubMed] [Google Scholar]
- 106.Sousairajah J, et al. Interplay between components of a novel LIM kinase–slingshot phosphatase complex regulates cofilin. EMBO J. 2005;24:473–486. doi: 10.1038/sj.emboj.7600543. [DOI] [PMC free article] [PubMed] [Google Scholar]