

Abstract
Background
Lung function is a long-term predictor of mortality and morbidity.
Objective
We sought to identify single nucleotide polymorphisms (SNPs) associated with lung function.
Methods
We performed a genome-wide association study (GWAS) of forced expiratory volume in 1 second (FEV1), forced vital capacity (FVC), and FEV1/FVC in 1,144 Hutterites aged 6–89 years, who are members of a founder population of European descent. We performed least absolute shrinkage and selection operation (LASSO) regression to select the minimum set of SNPs that best predict FEV1/FVC in the Hutterites and used the GRAIL algorithm to mine the Gene Ontology database for evidence of functional connections between genes near the predictive SNPs.
Results
Our GWAS identified significant associations between FEV1/FVC and SNPs at the THSD4-UACA-TLE3 locus on chromosome 15q23 (P = 5.7x10−8 ~ 3.4x10−9). Nine SNPs at or near four additional loci had P-values < 10−5 with FEV1/FVC. There were only two SNPs with P-values < 10−5 for FEV1 or FVC. We found nominal levels of significance with SNPs at 9 of the 27 previously reported loci associated with lung function measures. Among a predictive set of 80 SNPs, six loci were identified that had a significant degree of functional connectivity (GRAIL P < 0.05), including three clusters of β-defensin genes, two chemokine genes (CCL18 and CXCL12), and TNFRSF13B.
Conclusion
This study identifies genome-wide significant associations and replicates results of previous GWAS. Multimarker modeling implicated for the first time common variation in genes involved in anti-microbial immunity in airway mucosa influences lung function.
Keywords: FEV1/FVC, FEV1, FVC, GWAS, LASSO regression, GRAIL
INTRODUCTION
Chronic lower respiratory diseases are the third leading cause of death In the United States, resulting in 137,082 deaths in 20091. Lung function, as assessed by the spirometric measures of forced expiratory volume in one second (FEV1), forced vital capacity (FVC) and the FEV1 to FVC ratio (FEV1/FVC), is an objective indicator of general respiratory health, as well as an important long-term predictor of morbidity and mortality2–6. Family- and twin-based studies provide consistent evidence of genetic contributions to lung function, with estimates of heritability ranging as high as 85% for FEV1, 91% for FVC, and 45% for the FEV1/FVC ratio7–20.
Recently, genome-wide association studies (GWAS) have begun to shed light on the complex genetic architecture of lung function measures. Two large meta-analyses of lung function GWAS in subjects of European ancestry who participated in the SpiroMeta21 or CHARGE22 consortium reported 11 loci associated with FEV1/FVC or FEV1. A subsequent combined meta-analysis of 48,201 individuals from both consortia reported 16 additional loci influencing lung function23. However, variants at these highly significant loci in the SpiroMeta-CHARGE meta-analysis explained only 3.2% of the variance for FEV1/FVC and 1.5% of the variance for FEV123. Thus, similar to studies of other complex phenotypes, a significant proportion of the heritability remains unexplained by individual variants identified in GWAS24–26.
This missing heritability following GWAS has been attributed to numerous potential causes24–27, many or all of which likely contribute. In particular, the assumptions about the genetic model underlying complex phenotypes that are inherent in standard GWAS approaches may not reflect the true genetic architecture for many phenotypes. GWAS typically assess the effect of each (common) single nucleotide polymorphism (SNP) individually using stringent thresholds of significance. While this strategy has been effective in minimizing false positive associations and capturing the ‘low hanging fruit’, the inability to identify genetic variation that accounts for significant proportions of human phenotypic variation suggests that alternative analytic strategies are required to differentiate the true from false positive associations among the variants with more modest P-values. For example, considering 294,831 SNPs simultaneously in a linear model, Yang et al. found that common SNPs accounted for as much as 45% of the phenotypic variance and 50% of the heritability of height in 3,925 subjects28, compared to only 5% of the variance of height explained by ~50 SNPs that reached genome-wide thresholds of significance in earlier studies29–32.
Here, we conducted a GWAS of lung function phenotypes in members of a founder population, the Hutterites20, 33–34. In addition to loci reported in previous GWAS, multimarker modeling identified a novel set of airway epithelial cell derived host defense genes.
METHODS
The Hutterites
The Hutterites are a young founder population that originated in the South Tyrol in the 16th century and migrated from Europe to the United States in the 1870s35–36. Today more than 40,000 Hutterites live on communal farms (called colonies) in the north central United States and western Canada. We have been conducting genetic studies of complex phenotypes in the Hutterites of South Dakota for over 15 years20, 34, 37–40. Overall, their communal farming lifestyle minimizes environmental heterogeneity. In particular, smoking is prohibited and rare in this population, and air quality is excellent in rural South Dakota (see Table E1 in the Online Repository), eliminating environmental exposures that have profound effects on lung function.
Subjects were recruited for this study if they were (i) at least 6 years of age, (ii) at home on the days of our visit to their colony, and (iii) able to perform spirometry. Participation rates within each colony are typically around 95%, thus minimizing ascertainment biases that could impact our results. The final sample included 1,180 S-leut Hutterites who live on or were visiting one of 10 South Dakota colonies on the days of our visits; 187 individuals (15.8%) were diagnosed with asthma, as previously defined39–40. These subjects are related to each other through multiple lines of descent in a 3,673-person, 13-generation pedigree with 64 founders. Adult participants provided written informed consent for themselves and their children under 18 years old; participants who were under 18 years old provided written assent. These studies were approved by The University of Chicago Institutional Review Board.
Measures of lung function
Spirometry was performed in the Hutterites during two phases of field trips, the first in 1996–1997 and the second in 2006–2009, using identical protocols. Briefly, subjects underwent lung function tests using spirometry in the sitting position while breathing through a mouthpiece and wearing a nose clip in accordance with the American Thoracic Society/European Respiratory Society recommendations73,74. The best FEV1 and FVC were recorded. Of the 1,180 individuals, 335 were studied in phase 1 only, 524 in phase 2 only, and 321 in both phases. For the individuals studied in both phases, we included measurements from the more recent time point only, and excluded 36 individuals (24 used asthma rescue medications before spirometry, 4 had cystic fibrosis, and 8 had poor quality spirometry).
Genotyping and quality control
Hutterite individuals were genotyped with the Affymetrix GeneChip 500k, Genome-Wide SNP 5.0, or Genome-Wide SNP 6.0 arrays (Affymetrix, Santa Clara, CA, USA). An overlapping set of 369,487 autosomal SNPs were present on the 500k, 5.0, and 6.0 arrays; 94,552 of those SNPs were not studied because they were monomorphic (n = 31,246) or had minor allele frequency < 5% (n = 63,306) in the Hutterites. Of the remaining 274,935 SNPs, 28,925 were excluded because they had call rates < 95% (n = 6,456), generated ≥5 Mendelian errors (n = 15,912), or deviated from Hardy-Weinberg expectations at P < 10−3 (correcting for inbreeding and relatedness)41 (n = 6,557), yielding a final set of 246,010 autosomal markers with a median inter-marker spacing of 5.1 kb. The positions of SNPs shown in all figures and tables are based on NCBI release 36 (dbSNP build 129).
Heritability estimates and GWAS in the Hutterites
FEV1, FVC, and FEV1/FVC were transformed to normally distributed z-scores within each phase, and then adjusted for age, sex, age*sex, height, and inbreeding. The residuals of each trait from the two phases were then combined for further analyses. The distributions of these traits by age and sex and the correlations between them are shown in Fig. E1 in the Online Repository. The heritabilities of lung function measures were estimated using variance-component methods, as previously described42. Association testing was performed using a regression-based test for large, complex pedigrees37. Briefly, at each SNP, we used the general two-allele model (GTAM) test of association in the entire pedigree, keeping all inbreeding loops intact; at each SNP we tested an additive model of association. SNP-specific P-values were determined based on Gaussian theory. Genomic inflation was weak or absent (genomic inflation factor λ= 1.10 for FEV1, 1.09 for FVC, and 1.00 for FEV1/FVC). The GWAS P-values for FEV1 and FVC were adjusted using their genomic control43. The Bonferroni corrected genome-wide significance threshold was P < 2.0 x 10−7 (i.e., 0.05/246,010). The proportion of the residual variance explained by each SNP or a group of SNPs was determined by comparing the (RSS) in the regression model to that obtained without a SNP (or a group of SNPs), as implemented in GTAM.
In silico replication
We performed in silico replication of the GWAS results in the Hutterites using available results from the recently published meta-analysis in subjects of European descent in the SpiroMeta (n=20,288)21 and CHARGE (n=20,890)22 consortia. In addition, we investigated in the Hutterites the associations between lung function measures and SNPs at the 27 previously identified lung function associated loci21–23. For the latter, we reported the most significant SNP within 20 kb of the 27 previously reported loci.
Multimarker modeling
To select the minimum set of SNPs that best predict FEV1/FVC in the Hutterites from among SNPs with P < 10−3 in the GWAS, we performed least absolute shrinkage and selection operation (LASSO) regression44–46, as implemented in the R package glmnet47. These studies were conducted in 604 Hutterites without missing genotypes at all 312 SNPs with P < 10−3 (87 SNPs had missing data in at least one individual and were not included in the LASSO regression). Of the 540 subjects that were missing genotypes in these SNPs and not included in the LASSO regression, 261 had no missing genotypes in the 80 SNPs selected by LASSO and were used in subsequent analyses. The minimum set of best predicting SNPs was selected by running a 10-fold cross-validation procedure after choosing the glmnet parameter α= 1.0. The cross-validation procedure selected a LASSO penalty parameter of λ = 3.3 x 10−3 . K-fold cross validation was used to minimize the effects of overfitting the model to our data by randomly dividing the full data set into K-subsamples where K-1 subsamples are used to develop the model and the remaining subsample used for testing the model. LASSO regression uses SNPs as predictors of the phenotype (FEV1/FVC), while minimizing the number of SNPs in the model. Genotypes were coded as 0, 1, 2 doses of the minor allele. Following the 10-fold cross-validation procedure the LASSO regression selected 108 SNPs in the model. However, 28 of these SNPs had negligible effect sizes (absolute value of fixed effect size < .005) and were removed from the model, resulting in a final set of 80 SNPs.
Identifying related sets of genes
To identify related sets of genes and common pathways for genes near the SNPs that best predicted FEV1/FVC, we used the GRAIL algorithm48 to mine the Gene Ontology database. Briefly, GRAIL assesses the degree of relatedness among genes within regions harboring predictive SNPs, selecting the most connected gene that corresponds to one or more SNPs as the likely implicated gene. GRAIL assigns a P-value for each region that reflects the relatedness of the gene(s) in each region to all other regions, correcting for the number of genes in the region.
RESULTS
A total of 1,144 Hutterites (613 females, 53.6%) aged 6–89 years (mean ± SD, 30.6 ± 18.4 years) with both genome-wide genotyping and spirometry phenotypes were included in the GWAS (Table 1). These same data are shown for the non-asthmatic and asthmatic sample subsets in Table E2 in the Online Repository.
Table 1.
Characteristics of the Hutterite sample. Asthma and atopy are defined as described in reference40.
| Males | Females | |||
|---|---|---|---|---|
| 6–17 years | > 17 years | 6–17 years | > 17 years | |
| Sample Size | 180 | 351 | 195 | 418 |
| Number with asthma (%) | 25 (13.9%) | 48 (13.6%) | 38 (19.5%) | 51 (12.2%) |
| Number with atopy (%) | 76 (42.2%) | 191 (54.4%) | 93 (47.7%) | 194 (46.4%) |
| Mean age ± SD (yr) | 11.3 ± 3.2 | 40.1 ± 15.3 | 12.2 ± 2.9 | 39.6 ± 15.7 |
| Mean FEV1 ± SD (L) | 2.60 ± 1.05 | 3.90 ± 0.77 | 2.56 ± 0.78 | 2.94 ± 0.59 |
| Mean FVC ± SD (L) | 3.04 ± 1.29 | 4.91 ± 0.91 | 2.86 ± 0.90 | 3.61 ± 0.70 |
| Mean FEV1/FVC ± SD (%) | 86.8 ± 7.5 | 79.6 ± 7.8 | 90.0 ± 6.5 | 81.5 ± 7.1 |
Heritability of lung function in the Hutterites
The broad (H2) and narrow (h2) heritabilities of lung function measures in the Hutterites were h2 = H2 = 40.2% (SE 5.4%) for FEV1, h2 = 17.8% (SE 3.7%) and H2 = 70.4% (SE 11.2%) for FVC, and h2 = 22.1% (SE 8.0%) and H2 = 91.5% (SE 12.9%) for FEV1/FVC. These estimates indicate that 40.2%, 70.4%, and 91.5% of the phenotypic variances in FEV1, FVC, and the FEV1/FVC ratio, respectively, are attributable to genetic variation in the Hutterites. The heritabilities of FVC and FEV1/FVC included both additive and non-additive (i.e., dominance) genetic variance components, whereas the heritability of FEV1 was comprised entirely of additive genetic variance.
GWAS of lung function traits
We identified genome-wide significant associations between FEV1/FVC and five SNPs at the THSD4-UACA-TLE3 locus on chromosome 15q23 (see Fig. E2a in the Online Repository), replicating results from previous GWAS21, 23. Overall, there were 21 SNPs at this locus with P < 10−5 (see Table E3 in the Online Repository). The most significant SNP at this locus, rs12441227, explained 2.9% of the residual variance in FEV1/FVC in the Hutterites. The evidence for association with SNPs at this locus remained when the individuals with asthma were excluded (Fig. E2d), and when the sample was stratified by age (Table E4).
Nine additional SNPs at four loci had P-values < 10−5 with FEV1/FVC, including SNPs downstream of the C10orf11 gene on chromosome 10q22.3, which was associated with FEV1 in a meta-analysis of lung function GWAS23. When a sub-analysis was performed excluding the Hutterites with asthma, the evidence for association at this locus increased to genome-wide levels of significance (Table E4, Fig. E2f) The evidence for associations with SNPs at three of these loci with P-values < 10−5, CCL23-CCL18 on chromosome 17q12 (Fig. E2b and E2e), PITPNC1 locus on chromosome 17q24.2, and CHAF1B on chromosome 21q22.13, remained in sub-analyses excluding asthmatics. The evidence for association at all locus with P-values < 10−5 was present in subset analyses stratified by age (Table E4). Only two SNPs had P-values < 10−5 in the GWAS for the other two phenotypes: one SNP 7k downstream of the IL37 gene on chromosome 2q13 was associated with FEV1 and one SNP in an intron of ASXL3 on chromosome 18q12.1 was associated with FVC.
The Manhattan and Q-Q plots of P-values for the GWAS of the three phenotypes are shown in Fig. 1; results for all SNPs with P < 10−5 are shown in Table E3 in the Online Repository. The GWAS P-values in the Hutterites for the 27 loci associated with lung function in previous meta-analyses21–23 are shown in Table E5 in the Online Repository. Overall, we found nominal evidence (P < 0.05) of association with at least one of the three phenotypes for 14 SNPs at 9 of the 27 previously reported loci; and five SNPs at an additional five previously reported loci were associated with at least one of the three phenotypes at P < 0.01.
Figure 1.

Manhattan and Q-Q plots of P-values from the GWAS of FEV1/FVC (a, b), FEV1 (c, d), and FVC (e, f). SNPs with P-values < 10−5 are shown in red. The horizontal red line shows the genome-wide significance threshold (P < 2.0 x 10−7).
Multimarker modeling
We assumed that there were additional true associations among the GWAS SNPs that did not reach genome-wide levels of significance because their effects are too small to detect in single SNP analyses, especially in a sample size of ~1,000 subjects. Therefore, to assess a multimarker model of risk including all SNPs with P < 10−3, we performed LASSO regression to identify minimum sets of SNPs that provided the smallest mean square error of FEV1/FVC in the Hutterites. A set of 80 SNPs yielded the best predictive value and were used for further study (see Table E6 in the Online Repository).
First, we assessed the phenotypic effects of these 80 SNPs by binning individuals by the total number of alleles associated with reduced FEV1/FVC that they carried (total possible = 160) and calculated the mean (SE) residual FEV1/FVC for Hutterites in each bin. The mean residual FEV1/FVC decreased with increasing number of ‘low FEV1/FVC alleles’, consistent with an additive genetic architecture (Fig. 2).
Figure 2.
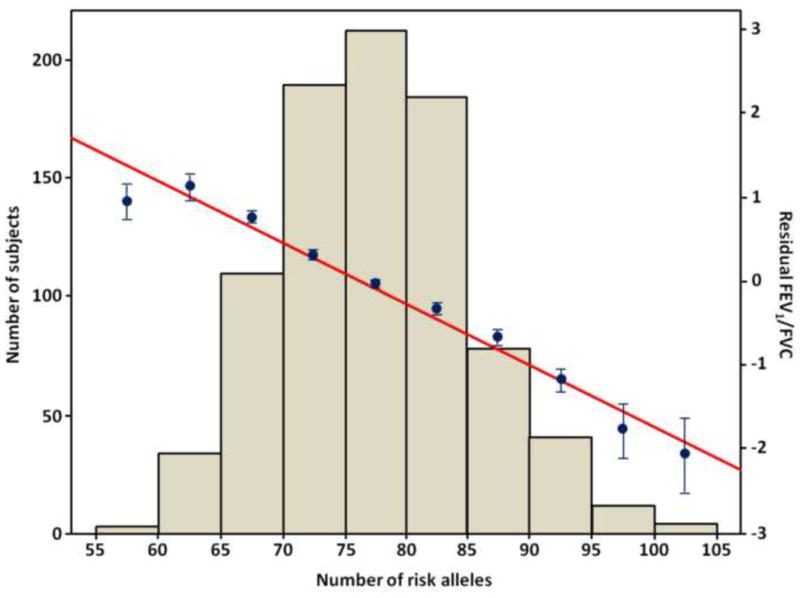
The combined effects of genotypes for 80 SNPs on the residual FEV1/FVC in the Hutterites. Hutterites were binned by their total number of alleles associated with reduced FEV1/FVC (x-axis); the mean (SE) residual FEV1/FVC for each bin is plotted on the right y-axis (blue dots and bars) and the number of subjects in each bin on the left y-axis. The linear regression line through these points is shown in red.
Next, we used the GRAIL algorithm48 to mine the Gene Ontology database for evidence of functional connections between genes near the 80 predictive SNPs. We identified a subset of six SNPs with significantly related genes (GRAIL P < 0.05), including three clusters of β-defensin genes, two chemokine genes (CCL18 and CXCL12), and TNFRSF13B (Table 2 and Fig. 3). Notably, the associated GWAS SNPs at two replicated loci, THSD4-UACA-TLE3 and C10orf11, were not functionally connected to any other genes defined by the 80 SNPs. However, a SNP at the CCL23-CCL18 locus, the second most significant locus in the Hutterite GWAS (see Fig. E3b in the Online Repository), was significantly connected to the β-defensin genes, as well as to CXCL12 and TNFRSF13B in the GRAIL analysis. These six SNPs by themselves explained 5.8% of the residual variance in FEV1/FVC in the Hutterites.
Table 2.
High scoring regions from the GRAIL analysis, sorted by the GRAIL P-value. PGWAS is the P-value from the FEV1/FVC GWAS in the Hutterites; Beta (SE) is that of the predictive SNP in the regression model for the 865 Hutterites; and PGRAIL is the region’s P-value given by GRAIL. The last column shows the candidate gene identified by GRAIL.
| SNP | Chr. | NCBI36 Position | PGWAS | Beta (SE) | PGRAIL | Implicated Gene |
|---|---|---|---|---|---|---|
| rs365548 | 20 | 185618 | 5.61E-04 | 0.003 (0.046) | 6.4E-04 | DEFB129, C20orf96 |
| rs2921026 | 8 | 8384658 | 6.14E-04 | −0.133 (0.046) | 6.6E-04 | DEFB107A |
| rs4815436 | 20 | 25521423 | 9.45E-04 | 0.181 (0.069) | 9.7E-04 | DEFB115, DEFB116, DEFB123, DEFB124 |
| rs854679 | 17 | 31382952 | 3.40E-07 | −0.049 (0.059) | 0.020 | CCL18 |
| rs1570846 | 10 | 43776486 | 7.06E-04 | −0.139 (0.055) | 0.037 | CXCL12 |
| rs7216399 | 17 | 16797303 | 5.96E-04 | −0.065 (0.054) | 0.043 | TNFRSF13B |
Figure 3.
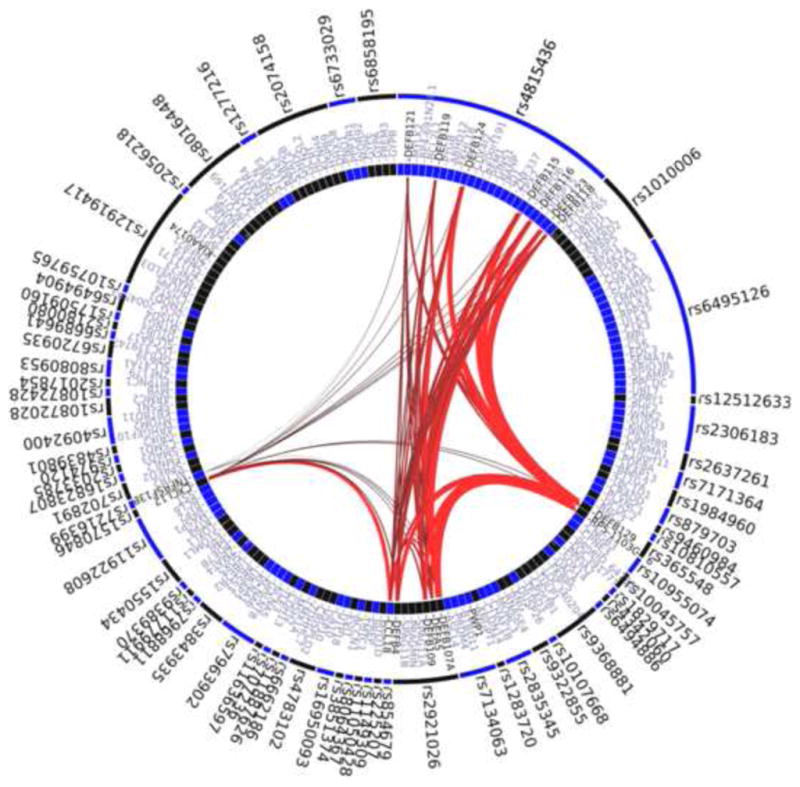
GRAIL functional connections between the 80 predictive SNPs. Six SNPs with no nearby genes defined by GRAIL are not shown. GRAIL identified six pairs of SNPs that implicated the same genes; only one SNP from each these 6 pairs is shown in the figure. The regions (SNPs; outer ring) and genes (inner ring) are optimally ordered to display connections with a minimal number of intersections. Only the genes with PGRAIL-values < 0.05 have connections displayed. The thickness and redness of the connectors reflects the significance of the connections. Three clusters of β-defensin genes are the most connected sets.
DISCUSSION
The success of GWAS for unraveling the genetic architecture of complex phenotypes has been widely debated24–27, 49–51. While there have been many robust associations discovered for a wide spectrum of diseases and phenotypes52, the associated variants typically explain relatively little of the phenotypic variation. Several recent studies have highlighted the importance of approaches that consider multiple variants simultaneously28, 46, 53–56, a more suitable approach if the genetic architecture of common phenotypes is polygenic with many contributing loci with small effects. However, the best way to identify multiple contributing loci is at present unclear.
The GWAS of the FEV1/FVC ratio in the Hutterites revealed two previously reported associations with measures of lung function. Associations with multiple SNPs at the highly replicated locus on 15q2321, 23 reached genome-wide significance in the combined sample, and SNPs at the C10orf11 on chromosome 10q22.323 reached genome-wide significance in the non-asthmatic subset of the Hutterite sample. These results were robust to age, with evidence for association present in both the child and adult subsets of the population. Moreover, we detected nominal levels of significance with SNPs at 9 previously reported loci associated with lung function measures. Together, these results indicate that genes influencing lung function in Europeans and European Americans from the general population also contribute to lung function phenotypes in the Hutterites.
To assess the combined effects of these and other SNPs with less significant evidence of association, we used LASSO regression to select the minimum set of SNPs from among the 312 with P-values < 10−3. The LASSO regression selected 80 independent SNPs as the best predictor of the FEV1/FVC ratio. Consistent with an additive genetic model, the mean phenotypic value decreases with increasing number of “risk” alleles (Fig. 2). Moreover, this approach led to the discovery of additional genes, including three independent clusters of β-defensin genes, two chemokine genes, and a tumor necrosis factor (TNF) family receptor, suggesting an important link between host defense mechanisms and lung function. Defensins are antimicrobial peptides that recruit inflammatory cells and modulate innate and adaptive immune responses, participating in both the promotion and resolution of inflammatory responses57. There are three classes of defensins, but only the β-defensins are specifically expressed in epithelial cells, including those lining the respiratory tract. Genetic studies have implicated the β-defensin genes on chromosome 8p23 in lung function in patients with asthma58, chronic obstructive pulmonary disease (COPD)59 and with cystic fibrosis (CF)60. In particular, DEFB1 mRNA in bronchial epithelial cell biopsies was significantly elevated in COPD patients compared to controls and significantly associated with both reduced FEV1 and the FEV1/FVC ratio in COPD patients and controls59. The results of our studies would further suggest that all three clusters of β-defensin genes on chromosomes 8p23, 20p13, and 20q11 contribute to lung function in healthy, unselected subjects. Chemokines are small proteins that bind to G-protein-coupled receptors and orchestrate the migration of circulating leukocytes to sites of inflammation. CCL18 (also named PARC, pulmonary and activated-regulated cytokine) is constitutively and highly expressed in the human lung61, and can generate regulatory T cells from CD4+CD25− T cells in healthy individuals via direct induction of transforming growth factor β1 (TGF-β1)62. Functional polymorphisms in the promoter of the TGFB1 gene have been associated with airway responsiveness and asthma exacerbations, and haplotypes comprised of polymorphisms and specific coding variants in this gene have been associated with lung function in CF patients63–64, although the exact variants and direction of effect are inconsistent across studies. Moreover, both β-defensin-2 and CCL18 were significantly elevated in peripheral blood from COPD patients compared to smoking and non-smoking controls65, CXCL12 (also name SDF-1, stromal derived growth factor 1) is critical to bone marrow-derived stem cell production, and shows increased expression in bronchial alveolar lavage fluid after bleomycin-induced lung fibrosis in a murine model, and in airway tissues in patients with idiopathic pulmonary fibrosis compared to controls66. The TNFRSF13B gene encodes the transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI), which binds two ligands, BAFF (B-cell activating factor) and APRIL (a proliferating-inducing ligand). It is thought that TACI plays a key role in B cell activation and differentiation into plasma cells. In a recent study, rare mutations in TNFRSF13B were associated with asthma symptoms in Swedish children67. Moreover, BAFF expression in alveolar macrophages was inversely correlated with lung function in COPD patients68. Our study extends the roles of these two chemokines and TNF-family receptor to inter-individual variability in normal lung function.
Despite conducting this study in a relatively small sample (~1,000 Hutterites) and the absence of a major locus influencing variation in lung function compared to other traits (for examples, see references39, 69), we were successful in identifying both genome-wide significant associations with replicated loci on chromosome 15 in the combined sample and on chromosome 10 in the non-asthmatic subset, in addition to a set of novel variants that are highly predictive for lung function in the Hutterites. The power of our study was likely enhanced by the homogeneity of the Hutterite population compared to the larger population samples that have been included in previous studies of lung function21–23. The advantages of this population for genetic studies of complex phenotypes are primarily two-fold. On the one hand, it is possible that there are fewer lung function-associated alleles segregating in the Hutterites due to the population bottleneck that occurred prior to their emigration to the United States35–36. This would result in a simpler genetic architecture due to both overall reduced genetic variation and increased frequencies of some variants with potentially larger phenotypic effects that are rare in other European populations. On the other hand, their communal lifestyle and shared environmental exposures33, which include the absence of exposure to cigarette smoke and air pollution, may have enhanced the effects of genetic variation in general, and on specific pathways in particular, on lung development and subsequent lung function. In this population, exposures are remarkably similar during critical periods of lung development both in utero and in early life. Hutterite women and young children are not directly involved in farming activities, and their homes are generally distant from the agricultural fields and animal barns. Meals are prepared in a communal kitchen, using traditional recipes that are shared among the colonies. There are no pets, televisions, radios, or computers in the homes, and, as a result, Hutterite children spend significant proportions of each day playing outside. Thus, the absence of important environmental exposures that impact lung development and lung function, combined with a shared environment throughout life, not only reduces non-genetic heterogeneity but also allows for the detection of lung function alleles that are not confounded with those related to socioeconomic factors or behavior, such as cigarette smoking, or to ecogenetic pathways that are important in metabolizing inhaled particles. These population characteristics possibly enabled the novel finding in this study of an enrichment of genes involved in anti-microbial immunity in the airways among those associated with lung function.
In summary, this study identifies genome-wide significant associations between lung function and SNPs at the THSD4-UACA-TLE3 locus on chromosome 15q23 and the C10orf11 on chromosome 10, and replicates many other previous GWAS results. Moreover, using LASSO regression, we identified 80 independent SNPs as the best predictor of the FEV1/FVC ratio, with the mean phenotypic value decreases with increasing number of “risk” alleles, consistent with an additive genetic architecture. Of note is that multimarker modeling implicated for the first time common variation in three independent clusters of β-defensin genes, two chemokine genes, and a TNF family receptor that involved in anti-microbial immunity in airway mucosa influences lung function.
Supplementary Material
CLINICAL IMPLICATIONS.
Three independent clusters of β-defensin genes, two chemokine genes (CCL18 and CXCL12), and TNFRSF13B that are involved in anti-microbial immunity in airway mucosa contribute to lung function phenotypes in healthy, unselected subjects.
Acknowledgments
Funding: This work was supported by the National Institutes of Health (grant nos. R01 HL085197 to C.O. and R01 HG002899 to M.A.)
We are grateful to Peter Carbonetto and Xiang Zhou for insightful comments and helpful discussions, Jessica Chong for technical advice, Minsoo Shon for assistance on field trips, and the Hutterites for their continued enthusiasm and participation in our studies.
Abbreviations
- FEV1
forced expiratory volume in one second
- FVC
forced vital capacity
- GWAS
genome-wide association studies
- SNP
single nucleotide polymorphism
- GTAM
general two-allele model
- RSS
residual sum of squares
- LASSO
least absolute shrinkage and selection operation
- TNF
tumor necrosis factor
- COPD
chronic obstructive pulmonary disease
- CF
cystic fibrosis
- PARC
pulmonary and activated-regulated cytokine
- TGF-β1
transforming growth factor β1
- SDF-1
stromal derived growth factor 1
- TACI
transmembrane activator and calcium modulator and cyclophilin ligand interactor
- BAFF
B-cell activating factor
- APRIL
a proliferating-inducing ligand
Footnotes
Conflict of interest: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kochanek KD, Xu J, Murphy SL, Minino AM, Kung HC. Deaths: preliminary data for 2009. Natl Vital Stat Rep. 2011;59:1–51. [PubMed] [Google Scholar]
- 2.Burney PG, Hooper R. Forced vital capacity, airway obstruction and survival in a general population sample from the USA. Thorax. 2011;66:49–54. doi: 10.1136/thx.2010.147041. [DOI] [PubMed] [Google Scholar]
- 3.Weiss ST, Segal MR, Sparrow D, Wager C. Relation of FEV1 and peripheral blood leukocyte count to total mortality. The Normative Aging Study. Am J Epidemiol. 1995;142:493–8. doi: 10.1093/oxfordjournals.aje.a117665. discussion 9–503. [DOI] [PubMed] [Google Scholar]
- 4.Schunemann HJ, Dorn J, Grant BJ, Winkelstein W, Jr, Trevisan M. Pulmonary function is a long-term predictor of mortality in the general population: 29-year follow-up of the Buffalo Health Study. Chest. 2000;118:656–64. doi: 10.1378/chest.118.3.656. [DOI] [PubMed] [Google Scholar]
- 5.Young RP, Hopkins R, Eaton TE. Forced expiratory volume in one second: not just a lung function test but a marker of premature death from all causes. Eur Respir J. 2007;30:616–22. doi: 10.1183/09031936.00021707. [DOI] [PubMed] [Google Scholar]
- 6.Myint PK, Luben RN, Surtees PG, Wainwright NW, Welch AA, Bingham SA, et al. Respiratory function and self-reported functional health: EPIC-Norfolk population study. Eur Respir J. 2005;26:494–502. doi: 10.1183/09031936.05.00023605. [DOI] [PubMed] [Google Scholar]
- 7.Hubert HB, Fabsitz RR, Feinleib M, Gwinn C. Genetic and environmental influences on pulmonary function in adult twins. Am Rev Respir Dis. 1982;125:409–15. doi: 10.1164/arrd.1982.125.4.409. [DOI] [PubMed] [Google Scholar]
- 8.Gibson JB, Martin NG, Oakeshott JG, Rowell DM, Clark P. Lung function in an Australian population: contributions of polygenic factors and the Pi locus to individual differences in lung function in a sample of twins. Ann Hum Biol. 1983;10:547–56. doi: 10.1080/03014468300006771. [DOI] [PubMed] [Google Scholar]
- 9.Lewitter FI, Tager IB, McGue M, Tishler PV, Speizer FE. Genetic and environmental determinants of level of pulmonary function. Am J Epidemiol. 1984;120:518–30. doi: 10.1093/oxfordjournals.aje.a113912. [DOI] [PubMed] [Google Scholar]
- 10.Astemborski JA, Beaty TH, Cohen BH. Variance components analysis of forced expiration in families. Am J Med Genet. 1985;21:741–53. doi: 10.1002/ajmg.1320210417. [DOI] [PubMed] [Google Scholar]
- 11.Beaty TH, Liang KY, Seerey S, Cohen BH. Robust inference for variance components models in families ascertained through probands: II. Analysis of spirometric measures. Genet Epidemiol. 1987;4:211–21. doi: 10.1002/gepi.1370040306. [DOI] [PubMed] [Google Scholar]
- 12.Ghio AJ, Crapo RO, Elliott CG, Adams TD, Hunt SC, Jensen RL, et al. Heritability estimates of pulmonary function. Chest. 1989;96:743–6. doi: 10.1378/chest.96.4.743. [DOI] [PubMed] [Google Scholar]
- 13.Cotch MF, Beaty TH, Cohen BH. Path analysis of familial resemblance of pulmonary function and cigarette smoking. Am Rev Respir Dis. 1990;142:1337–43. doi: 10.1164/ajrccm/142.6_Pt_1.1337. [DOI] [PubMed] [Google Scholar]
- 14.Coultas DB, Hanis CL, Howard CA, Skipper BJ, Samet JM. Heritability of ventilatory function in smoking and nonsmoking New Mexico Hispanics. Am Rev Respir Dis. 1991;144:770–5. doi: 10.1164/ajrccm/144.4.770. [DOI] [PubMed] [Google Scholar]
- 15.McClearn GE, Svartengren M, Pedersen NL, Heller DA, Plomin R. Genetic and environmental influences on pulmonary function in aging Swedish twins. J Gerontol. 1994;49:264–8. doi: 10.1093/geronj/49.6.m264. [DOI] [PubMed] [Google Scholar]
- 16.Chen Y, Horne SL, Rennie DC, Dosman JA. Segregation analysis of two lung function indices in a random sample of young families: the Humboldt Family Study. Genet Epidemiol. 1996;13:35–47. doi: 10.1002/(SICI)1098-2272(1996)13:1<35::AID-GEPI4>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 17.Wilk JB, Djousse L, Arnett DK, Rich SS, Province MA, Hunt SC, et al. Evidence for major genes influencing pulmonary function in the NHLBI family heart study. Genet Epidemiol. 2000;19:81–94. doi: 10.1002/1098-2272(200007)19:1<81::AID-GEPI6>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 18.Palmer LJ, Burton PR, James AL, Musk AW, Cookson WO. Familial aggregation and heritability of asthma-associated quantitative traits in a population-based sample of nuclear families. Eur J Hum Genet. 2000;8:853–60. doi: 10.1038/sj.ejhg.5200551. [DOI] [PubMed] [Google Scholar]
- 19.Palmer LJ, Knuiman MW, Divitini ML, Burton PR, James AL, Bartholomew HC, et al. Familial aggregation and heritability of adult lung function: results from the Busselton Health Study. Eur Respir J. 2001;17:696–702. doi: 10.1183/09031936.01.17406960. [DOI] [PubMed] [Google Scholar]
- 20.Ober C, Abney M, McPeek MS. The genetic dissection of complex traits in a founder population. Am J Hum Genet. 2001;69:1068–79. doi: 10.1086/324025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Repapi E, Sayers I, Wain LV, Burton PR, Johnson T, Obeidat M, et al. Genome-wide association study identifies five loci associated with lung function. Nat Genet. 2010;42:36–44. doi: 10.1038/ng.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hancock DB, Eijgelsheim M, Wilk JB, Gharib SA, Loehr LR, Marciante KD, et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat Genet. 2010;42:45–52. doi: 10.1038/ng.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Artigas MS, Loth DW, Wain LV, Gharib SA, Obeidat M, Tang W, et al. Genome-wide association and large-scale follow up identifies 16 new loci influencing lung function. Nat Genet. 2011 doi: 10.1038/ng.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–53. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maher B. Personal genomes: The case of the missing heritability. Nature. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- 26.McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–69. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 27.Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11:446–50. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–9. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gudbjartsson DF, Walters GB, Thorleifsson G, Stefansson H, Halldorsson BV, Zusmanovich P, et al. Many sequence variants affecting diversity of adult human height. Nat Genet. 2008;40:609–15. doi: 10.1038/ng.122. [DOI] [PubMed] [Google Scholar]
- 30.Lettre G, Jackson AU, Gieger C, Schumacher FR, Berndt SI, Sanna S, et al. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat Genet. 2008;40:584–91. doi: 10.1038/ng.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weedon MN, Lango H, Lindgren CM, Wallace C, Evans DM, Mangino M, et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nat Genet. 2008;40:575–83. doi: 10.1038/ng.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Visscher PM. Sizing up human height variation. Nat Genet. 2008;40:489–90. doi: 10.1038/ng0508-489. [DOI] [PubMed] [Google Scholar]
- 33.Ober C, Cox NJ. The genetics of asthma. Mapping genes for complex traits in founder populations. Clin Exp Allergy. 1998;28(Suppl 1):101–5. doi: 10.1046/j.1365-2222.1998.0280s1101.x. discussion 8–10. [DOI] [PubMed] [Google Scholar]
- 34.Weiss LA, Pan L, Abney M, Ober C. The sex-specific genetic architecture of quantitative traits in humans. Nat Genet. 2006;38:218–22. doi: 10.1038/ng1726. [DOI] [PubMed] [Google Scholar]
- 35.Hostetler JA. Hutterite society. Baltimore: Johns Hopkins University Press; 1974. [Google Scholar]
- 36.Steinberg AG, Bleibtreu HK, Kurczynski TW, Martin AO, EMK . Genetics studies in an inbred human isolate. In: Crow JF, Neel JV, editors. Proceedings of the Third International Congress of Human Genetics. Baltimore: Johns Hopkins University Press; 1967. pp. 267–90. [Google Scholar]
- 37.Abney M, Ober C, McPeek MS. Quantitative trait homozygosity and association mapping and empirical genome-wide significance in large complex pedigrees: Fasting serum insulin level in the Hutterites. Amer J Hum Genet. 2002;70:920–34. doi: 10.1086/339705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cusanovich DA, Billstrand C, Zhou X, Chavarria C, De Leon S, Michelini K, et al. The combination of a genome-wide association study of lymphocyte count and analysis of gene expression data reveals novel asthma candidate genes. Hum Mol Genet. 2012;21:2111–23. doi: 10.1093/hmg/dds021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ober C, Tan Z, Sun Y, Possick JD, Pan L, Nicolae R, et al. Effect of variation in CHI3L1 on serum YKL-40 level, asthma risk, and lung function. NEJM. 2008;358:1682–91. doi: 10.1056/NEJMoa0708801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ober C, Tsalenko A, Parry R, Cox NJ. A second-generation genomewide screen for asthma-susceptibility alleles in a founder population. Am J Hum Genet. 2000;67:1154–62. doi: 10.1016/s0002-9297(07)62946-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bourgain C, Abney M, Schneider D, Ober C, McPeek MS. Testing for hardy-weinberg equilibrium in samples with related individuals. Genetics. 2004;168:2349–61. doi: 10.1534/genetics.104.031617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abney M, McPeek MS, Ober C. Broad and narrow heritabilities of quantitative traits in a founder population. Am J Hum Genet. 2001;68:1302–7. doi: 10.1086/320112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 44.Tibshirani R. Regression shrinkage and selectionv ia the lasso. J R Stat Soc Ser B. 1996;58:267–88. [Google Scholar]
- 45.Guo W, Elston R, Zhu X. Evaluation of a LASSO regression approach on the unrelated samples of Genetic Analysis Workshop 17. BMC Proceedings. 2011;5:S12. doi: 10.1186/1753-6561-5-S9-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu TT, Chen YF, Hastie T, Sobel E, Lange K. Genome-wide association analysis by lasso penalized logistic regression. Bioinformatics. 2009;25:714–21. doi: 10.1093/bioinformatics/btp041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw. 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- 48.Raychaudhuri S, Plenge RM, Rossin EJ, Ng AC, Purcell SM, Sklar P, et al. Identifying relationships among genomic disease regions: predicting genes at pathogenic SNP associations and rare deletions. PLoS Genet. 2009;5:e1000534. doi: 10.1371/journal.pgen.1000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360:1696–8. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 50.Hirschhorn JN. Genomewide association studies--illuminating biologic pathways. N Engl J Med. 2009;360:1699–701. doi: 10.1056/NEJMp0808934. [DOI] [PubMed] [Google Scholar]
- 51.Kraft P, Hunter DJ. Genetic risk prediction--are we there yet? N Engl J Med. 2009;360:1701–3. doi: 10.1056/NEJMp0810107. [DOI] [PubMed] [Google Scholar]
- 52.A catalog of published genome-wide association studies. [Cited 2012 January 11.] Available from http://www.genome.gov/gwastudies/
- 53.Kutalik Z, Whittaker J, Waterworth D, Beckmann JS, Bergmann S. Novel method to estimate the phenotypic variation explained by genome-wide association studies reveals large fraction of the missing heritability. Genet Epidemiol. 2011;35:341–9. doi: 10.1002/gepi.20582. [DOI] [PubMed] [Google Scholar]
- 54.Lee SH, Wray NR, Goddard ME, Visscher PM. Estimating Missing Heritability for Disease from Genome-wide Association Studies. Am J Hum Genet. 2011;88:294–305. doi: 10.1016/j.ajhg.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de los Campos G, Gianola D, Allison DB. Predicting genetic predisposition in humans: the promise of whole-genome markers. Nat Rev Genet. 2010;11:880–6. doi: 10.1038/nrg2898. [DOI] [PubMed] [Google Scholar]
- 56.Makowsky R, Pajewski NM, Klimentidis YC, Vazquez AI, Duarte CW, Allison DB, et al. Beyond missing heritability: prediction of complex traits. PLoS Genet. 2011;7:e1002051. doi: 10.1371/journal.pgen.1002051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tecle T, Tripathi S, Hartshorn KL. Review: Defensins and cathelicidins in lung immunity. Innate Immun. 2010;16:151–9. doi: 10.1177/1753425910365734. [DOI] [PubMed] [Google Scholar]
- 58.Levy H, Raby BA, Lake S, Tantisira KG, Kwiatkowski D, Lazarus R, et al. Association of defensin beta-1 gene polymorphisms with asthma. J Allergy Clin Immunol. 2005;115:252–8. doi: 10.1016/j.jaci.2004.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Andresen E, Gunther G, Bullwinkel J, Lange C, Heine H. Increased expression of beta-defensin 1 (DEFB1) in chronic obstructive pulmonary disease. PLoS One. 2011;6:e21898. doi: 10.1371/journal.pone.0021898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crovella S, Segat L, Amato A, Athanasakis E, Bezzerri V, Braggion C, et al. A polymorphism in the 5′ UTR of the DEFB1 gene is associated with the lung phenotype in F508del homozygous Italian cystic fibrosis patients. Clin Chem Lab Med. 2011;49:49–54. doi: 10.1515/CCLM.2011.023. [DOI] [PubMed] [Google Scholar]
- 61.Hieshima K, Imai T, Baba M, Shoudai K, Ishizuka K, Nakagawa T, et al. A novel human CC chemokine PARC that is most homologous to macrophage-inflammatory protein-1 alpha/LD78 alpha and chemotactic for T lymphocytes, but not for monocytes. J Immunol. 1997;159:1140–9. [PubMed] [Google Scholar]
- 62.Chang Y, de Nadai P, Azzaoui I, Morales O, Delhem N, Vorng H, et al. The chemokine CCL18 generates adaptive regulatory T cells from memory CD4+ T cells of healthy but not allergic subjects. FASEB J. 2010;24:5063–72. doi: 10.1096/fj.10-162560. [DOI] [PubMed] [Google Scholar]
- 63.Bremer LA, Blackman SM, Vanscoy LL, McDougal KE, Bowers A, Naughton KM, et al. Interaction between a novel TGFB1 haplotype and CFTR genotype is associated with improved lung function in cystic fibrosis. Hum Mol Genet. 2008;17:2228–37. doi: 10.1093/hmg/ddn123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Drumm ML, Konstan MW, Schluchter MD, Handler A, Pace R, Zou F, et al. Genetic modifiers of lung disease in cystic fibrosis. N Engl J Med. 2005;353:1443–53. doi: 10.1056/NEJMoa051469. [DOI] [PubMed] [Google Scholar]
- 65.Dickens JA, Miller BE, Edwards LD, Silverman EK, Lomas DA, Tal-Singer R. COPD association and repeatability of blood biomarkers in the ECLIPSE cohort. Respir Res. 2011;12:146. doi: 10.1186/1465-9921-12-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu J, Mora A, Shim H, Stecenko A, Brigham KL, Rojas M. Role of the SDF-1/CXCR4 axis in the pathogenesis of lung injury and fibrosis. Am J Respir Cell Mol Biol. 2007;37:291–9. doi: 10.1165/rcmb.2006-0187OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Janzi M, Melen E, Kull I, Wickman M, Hammarstrom L. Rare mutations in TNFRSF13B increase the risk of asthma symptoms in Swedish children. Genes Immun. 2012;13:59–65. doi: 10.1038/gene.2011.55. [DOI] [PubMed] [Google Scholar]
- 68.Polverino F, Baraldo S, Bazzan E, Agostini S, Turato G, Lunardi F, et al. A novel insight into adaptive immunity in chronic obstructive pulmonary disease: B cell activating factor belonging to the tumor necrosis factor family. Am J Respir Crit Care Med. 2010;182:1011–9. doi: 10.1164/rccm.200911-1700OC. [DOI] [PubMed] [Google Scholar]
- 69.Ober C, Nord AS, Thompson EE, Pan L, Tan Z, Cusanovich D, et al. Genome-wide association study of plasma lipoprotein(a) levels identifies multiple genes on chromosome 6q. J Lipid Res. 2009;50:798–806. doi: 10.1194/jlr.M800515-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.