

Abstract
Aims: Connective tissue growth factor (CTGF/CCN2) is a developmental gene upregulated in pathological conditions, including cardiovascular diseases, whose product is a matricellular protein that can be degraded to biologically active fragments. Among them, the C-terminal module IV [CCN2(IV)] regulates many cellular functions, but there are no data about redox process. Therefore, we investigated whether CCN2(IV) through redox signaling regulates vascular responses. Results: CCN2(IV) increased superoxide anion (O2•−) production in murine aorta (ex vivo and in vivo) and in cultured vascular smooth muscle cells (VSMCs). In isolated murine aorta, CCN2(IV), via O2•−, increased phenylephrine-induced vascular contraction. CCN2(IV) in vivo regulated several redox-related processes in mice aorta, including increased nonphagocytic NAD(P)H oxidases (Nox)1 activity, protein nitrosylation, endothelial dysfunction, and activation of the nuclear factor-κB (NF-κB) pathway and its related proinflammatory factors. The role of Nox1 in CCN2(IV)-mediated vascular responses in vivo was investigated by gene silencing. The administration of a Nox1 morpholino diminished aortic O2•− production, endothelial dysfunction, NF-κB activation, and overexpression of proinflammatory genes in CCN2(IV)-injected mice. The link CCN2(IV)/Nox1/NF-κB/inflammation was confirmed in cultured VSMCs. Epidermal growth factor receptor (EGFR) is a known CCN2 receptor. In VSMCs, CCN2(IV) activates EGFR signaling. Moreover, EGFR kinase inhibition blocked vascular responses in CCN2(IV)-injected mice. Innovation and Conclusion: CCN2(IV) is a novel prooxidant factor that in VSMCs induces O2•− production via EGFR/Nox1 activation. Our in vivo data demonstrate that CCN2(IV) through EGFR/Nox1 signaling pathway induces endothelial dysfunction and activation of the NF-κB inflammatory pathway. Therefore, CCN2(IV) could be considered a potential therapeutic target for redox-related cardiovascular diseases. Antioxid. Redox Signal. 22, 29–47.
Introduction
Reactive oxygen species (ROS) are a class of molecules derived from the metabolism of oxygen and include free radical and nonradical species that are generally capable of oxidizing molecular targets. Physiological levels of ROS are cellular modulators of signaling pathways, regulating many cellular processes. However, increased ROS production (oxidative stress), exceeding the basal antioxidant defenses, promotes cellular damage through the oxidation of cellular membranes, DNA, or proteins (43, 64). At vascular level, redox signaling plays a physiological role in the regulation of cell growth, proliferation, apoptosis, migration, matrix deposition, modulation of endothelial function, and vascular tone (66, 69). Increased ROS production contributes to the development of cardiovascular diseases, including hypertension, atherosclerosis, diabetes, cardiac hypertrophy, heart failure, ischemia-reperfusion injury, and stroke (29, 54, 65, 66). The family of nonphagocytic NAD(P)H oxidases (Nox) proteins, which consists of seven members (Nox1–5 and dual oxidases 1 and 2), is the most prominent and well-studied source of signaling ROS (29, 66). Increased Nox expression and activity has been described in cardiovascular disease. Thus, ROS derived from vascular Nox mediates vascular damage caused by angiotensin II (AngII) and transforming growth factor-β (TGF-β) (5, 6).
Innovation.
Our findings expand the known profibrogenic CCN2 actions and identify CCN2 as a true cytokine with an active role in the activation of the inflammatory nuclear factor-κB (NF-κB) pathway, mediated through EGFR/Nox1 activation and superoxide anion (O2•−) production in vascular cells (Fig. 10). This new role supports the idea that modulation of the oxidative stress–inflammatory signaling elicited by CCN2 could be an additional therapeutic option for inflammatory cardiovascular diseases.
Connective tissue growth factor (CTGF/CCN2), a member of the CCN protein family, is a matricellular protein implicated in several biological processes, such as cell proliferation, survival, angiogenesis, and migration (11, 47, 50). CCN2 is a developmental gene re-expressed in several pathological conditions, including cardiovascular diseases like heart failure, pulmonary hypertension, vascular remodeling, and atherosclerosis (8, 28, 44, 50). In vascular cells, CCN2 is a downstream mediator of AngII and TGF-β profibrotic actions (56, 57). In patients with myocardial fibrosis and chronic heart failure, circulating CCN2 has been suggested as a risk biomarker for cardiac dysfunction (26). Therapeutic approaches that selectively block CCN2 activity were beneficial in experimental models of fibrosis, including the liver, lung, and kidney (18, 48, 49), pulmonary vascular remodeling, and aortic restenosis (28, 71). In contrast, cardiac CCN2 overexpression conferred cardioprotection in AngII-infused mice, ischemia reperfusion injury, and after myocardial infarction (1, 17, 39, 45), showing that CCN2 exerts protective effects in some pathological settings. However, there are no studies evaluating the direct effect of CCN2 on the vascular wall.
Although many studies have shown that CCN2 is a redox-regulated gene (33, 46, 57), there are no data about the direct effect of CCN2 on the regulation of redox processes. CCN2 contains four functional modules that can be cleaved by proteases, and each one elicits different functional cellular responses (11). These degradation fragments can be detected in biological fluids. Among them, the C-terminal module IV [CCN2 (IV)] shares many responses with full-length CCN2, including regulation of fibrotic-related events, angiogenesis, cell migration, inflammatory responses, and proliferation, whereas the N-terminal domain exerted none of these effects (32, 35, 51, 52, 58, 59, 70). For these reasons, our aim was to evaluate whether CCN2(IV), could regulate Nox1 activity and redox signaling in vascular smooth muscle cells (VSMCs) in vitro and in vivo and its relation to functional responses.
Results
CCN2 overexpression is linked to ROS production in experimental vascular damage
In several cardiovascular pathologies, induction of CCN2 expression has been described (12, 50); however, its direct relation to oxidative stress has not been investigated. Therefore, we first evaluated whether CCN2 induction and elevated ROS production were presented in aortic VSMCs in a vascular disease model. The systemic infusion of AngII in rodents is a model of vascular damage characterized by inflammation and fibrosis (4, 15, 55, 57, 61, 69). Different groups have shown the involvement of redox process in AngII-induced vascular damage (5, 36, 69). In AngII-infused mice, increased aortic CCN2 gene and protein expression was observed after 7 days (Fig. 1). CCN2 production was mainly located in VSMCs, as observed by confocal microscopy (Fig. 1A). ROS production was evaluated in serial sections by dihydroethidium (DHE) staining. Colocalization of CCN2 induction with elevated ROS production was detected (Fig. 1A), supporting the hypothesis that CCN2 could be linked to ROS production in vascular damage.
FIG. 1.
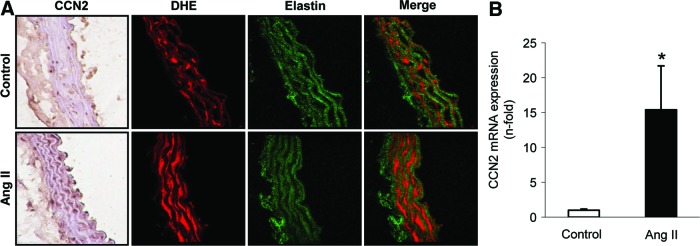
Angiotensin II infusion increased CCN2 expression and O2•− production in the vascular wall. (A) Representative CCN2 inmunostaining and fluorescent confocal photomicrographs of O2•−, measured as DHE staining in aortic serial sections from saline and AngII-infused mice after 7 days. (B) Gene expression of CCN2 was evaluated by real-time PCR. Data are expressed as mean±SEM of fold increase saline-infused control of five to six animals per group. *p<0.05 versus saline-infused mice. AngII, angiotensin II; DHE, dihydroethidium; O2•−, superoxide anion; PCR, polymerase chain reaction. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
CCN2(IV) increases O2•− production in isolated mice aorta
To demonstrate whether CCN2(IV) could directly regulated redox processes, we first evaluated the effect of CCN2(IV) in isolated aorta. CCN2(IV) binding to aorta ex vivo was visualized by confocal microscopy. After adding labeled CCN2(IV)-Cy5 to mouse aortic segments, the red immunofluorescent signal was found in VSMCs at 5 and 10 min, indicating that CCN2(IV) can go through the endothelium and binds to VSMCs (Fig. 2A).
FIG. 2.

The C-terminal module IV [CCN2(IV)] increases Nox1 activity and O2•− production in isolated mice aorta. (A) Representative confocal photomicrographs of CCN2(IV)-Cy5 binding to VSMCs in the aortic wall (as red points in the merge). (B–D) Aortic segments were incubated 60 min with 50 ng/ml CCN2(IV), and in some points, samples were also preincubated for 60 min with 1 μM ML-171. (B) Confocal photomicrographs of O2•−, measured as DHE staining. Elastin autofluorescence shows the structure of the artery, clearly indicating that localization of O2•− production was mainly found in the VSMCs (media layer). The arrows show O2•− production in endothelial cells (upper panel). Tiron was used as a control. Quantification of O2•− production in the media layer is shown in the lower panel. (C) Chromatogram of O2•− production by HPLC analysis (upper panel). O2•− production was evaluated by an increase in 2-OH-E+ generation by HPLC analysis of DHE fluorescence in aortic samples (lower panel). (D) NAD(P)H oxidase activity was measured in total cellular extracts by an enzymatic assay. Data expressed as mean±SEM of fold-change over control of at least three independent experiments. *p<0.05 versus control. 2-OH-E+, 2-hydroxyethidium; HPLC, high-performance liquid chromatography; Nox, nonphagocytic NAD(P)H oxidases; VSMCs, vascular smooth muscle cells. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Next, we determined whether CCN2(IV) could induce superoxide anion (O2•−) production. For these experiments, mouse aorta was isolated and incubated with CCN2(IV). DHE-derived red fluorescence was increased in CCN2(IV)-treated aorta compared with untreated ones, mainly located in VSMCs (Fig. 2B), but DHE staining is also present in endothelial cells (ECs). The O2•− scavenger Tiron reduced CCN2(IV)-induced DHE staining, therefore suggesting local O2•− production (Fig. 2B). To demonstrate more specifically the O2•− production, high-performance liquid chromatography (HPLC) measurements were performed since this technique detects 2-hydroxyethidium (2-OH-E+), a specific product of superoxide oxidation of DHE (12). The HPLC chromatogram of acetonitrile-extracted mice aortas showed both the 2-OH-E+ and the ethidium peaks. In CCN2(IV)-treated samples, a significant increase was observed in the 2-OH-E+ peak compared with controls (Fig. 2C). Treatment with the selective Nox1 pharmacological inhibitor ML-171 (14) decreased CCN2(IV)-induced O2•− production (Fig. 2C). Finally, Nox enzymatic activity was determined by the lucigenin method, showing a marked increase in Nox activity in total extracts of CCN2(IV)-treated aortas, which was diminished by ML-171 pretreatment to values similar to control-untreated aortas (Fig. 2D). Our data clearly show that CCN2(IV) increased O2•− production in mice aorta and suggest that Nox1 is involved in this process.
CCN2(IV) increases ROS production in cultured murine VSMCs
Stimulation of cultured murine VSMCs with CCN2(IV) increased O2•− production in a dose- and time-dependent manner (Fig. 3A, B). CCN2(IV) increased O2•− production after 60 min, with a maximal response at 50 ng/ml, therefore that time point and concentration were used for the following experiments. By L-012 chemiluminescence, increased ROS production by CCN2(IV) stimulation was confirmed (Fig. 3C). In addition, in total cellular extracts of CCN2(IV)-treated cells, Nox enzymatic activity, determined by lucigenin-enhanced chemiluminescence, was also increased compared with untreated cells (Fig. 3D). These data clearly show that CCN2(IV) increased O2•− production in cultured VSMCs.
FIG. 3.

CCN2(IV) increases Nox1 activity and O2•− production in cultured murine VSMCs. (A) Concentration–response curve and (B) time course of O2•− production by CCN2(IV) in murine VSMCs. A representative fluorescent confocal photomicrographs of O2•− measured as DHE staining production is shown in upper panel and quantification in lower panel. (C) O2•− production in mice VSMC measured as luminescence of L-012. (D) NAD(P)H oxidase activity was measured in total cellular extracts by an enzymatic assay. Data expressed as mean±SEM of fold-change over control of at least three to four independent experiments. *p<0.05 versus control. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
CCN2(IV) via redox process increases phenylephrine-induced vascular contractile responses
ROS like O2•− modulates the vascular tone (38). Therefore, the potential effect of CCN2(IV) in vascular function and the role of O2•− in this effect were evaluated in isolated mouse aortic segments. CCN2(IV) increased contractile responses to phenylephrine (Phe) that were significantly inhibited by preincubation with the Nox1 inhibitor ML-171, whereas no effect was found in control arteries (Fig. 4A). Similar results were observed with the antioxidant apocynin (Fig. 4A). Our findings suggest that CCN2(IV) via redox-mediated processes regulates vascular contraction.
FIG. 4.
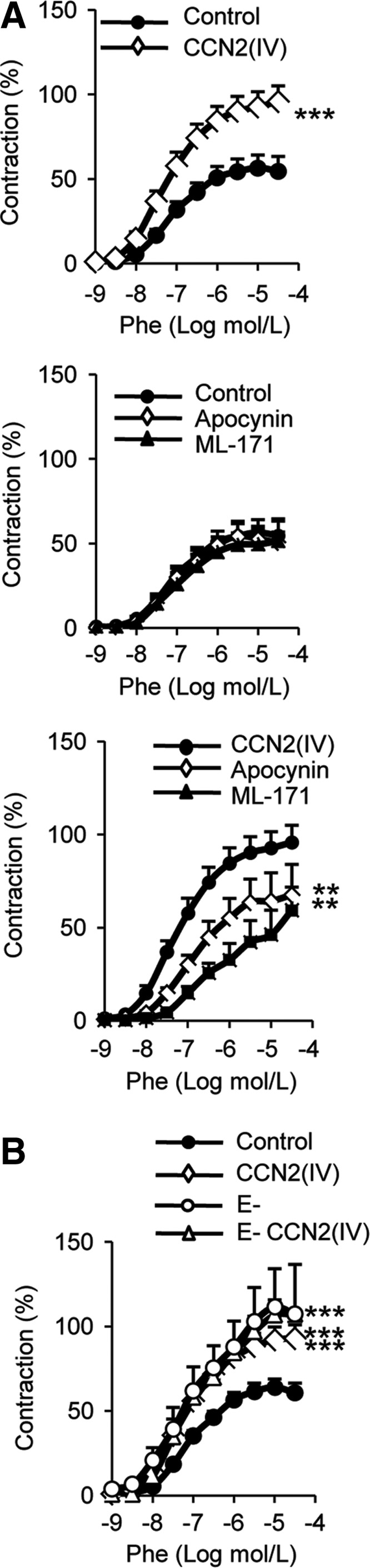
CCN2(IV) increases phenylephrine vascular contractile response. (A) Concentration–response curve to Phe (1 nM–30 μM) in aortic segments treated 1 h with 50 ng/ml CCN2(IV), in the absence or in presence of apocynin (0.3 mM) or ML-171 (1 μM). (B) Concentration–response curve to Phe (1 nM–30 μM) in aortic segments treated 1 h with 50 ng/ml CCN2(IV), in the presence or absence (E−) of endothelium. Data expressed as mean±SEM of four to eight experiments. **p<0.01, ***p<0.001 versus each control situation. Phe, phenylephrine.
To evaluate the contribution of endothelium in CCN2(IV) responses, vascular reactivity was determined in endothelium-denuded aortic rings. Endothelial removal increased the contractile response to phenylephrine in mice aorta and abolished the effect of CCN2(IV) on contraction (Fig. 4B), suggesting that a potential endothelium-derived mediator, probably O2•−, is involved in CCN2(IV) effects on vascular function. In agreement, in cultured ECs, CCN2(IV) increased O2•− production (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/ars).
CCN2(IV) administration in vivo regulates redox processes and induces a proinflammatory response in murine aorta
To investigate the in vivo effect of CCN2(IV) in redox processes in the aorta, an experimental model of intraperitoneal administration of CCN2(IV) in C57BL/6 mice was performed. Superoxide production was higher in the aorta of CCN2(IV)-injected mice than in saline-injected ones, as determined by HPLC analysis (Fig. 5A). Aortic Nox activity was elevated in CCN2(IV)-injected aorta samples (Fig. 5B), but no changes were found in Nox1 gene or protein levels between the groups (Fig. 5C, D).
FIG. 5.

CCN2(IV) administration into normal mice increases oxidative stress. C57BL/6 mice received a single i.p. injection of recombinant CCN2(IV) (2.5 ng/g body weight) or saline and were sacrificed after 24 h. Measurements were performed in aortic samples from saline and CCN2(IV)-injected mice. (A) O2•− production was evaluated by an increase in 2-OH-E+ generation by HPLC analysis of DHE fluorescence. A representative HPLC chromatogram (left) and the quantification of the data (right). The arrows show 2-OH-E+ peak. (B) NAD(P)H oxidase activity was evaluated by the lucigenin method. (C, D) Nox1 gene and protein expression was determined by real-time PCR and Western blot, respectively. (E) NO production was analyzed by diaminofluorescein fluorescence in aortic samples. (F) Protein nitrosylation levels were evaluated in paraffin-embedded aortic sections. (G) eNOS protein expression was analyzed by Western blot. (H) iNOS gene expression was performed by real-time PCR. (I) Concentration–response curve to ACh (1 nM–10 μM) in aortic segments. Data are expressed as mean±SEM of fold increase over saline of 8–10 animals per group. *p<0.05 versus saline. **p<0.01 versus control. ACh, acetylcholine; eNOS, endothelial NO synthase; iNOS, inducible NO synthase; NO, nitric oxide. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
The vasodilator nitric oxide (NO), which is formed by the endothelial, inducible, and neuronal NO synthases (eNOS, iNOS, and nNOS), is an important modulator of vascular tone (40). In aortas from CCN2(IV)-injected mice, acetylcholine (ACh)-induced NO production was not modified (Fig. 5E). In addition, in CCN2(IV)-injected mice, aortic eNOS was not changed (Fig. 5F), whereas iNOS was upregulated compared with saline-injected mice (Fig. 5G). O2•− can react with NO, diminishing NO bioavailability and increasing peroxynitrite levels, a potent oxidant that leads to the nitration of tyrosine resides in tissue proteins. Accordingly, in CCN2(IV)-injected mice increased aortic nitrosylated protein levels were found (Fig. 5H), suggesting increased peroxynitrite formation. All together, these data show that CCN2(IV) acts as a novel prooxidant factor.
Next, we further evaluated the main features of our acute model of CCN2(IV)-induced vascular damage. One of the earliest responses to vascular injury is endothelial dysfunction (61). In the aorta of CCN2(IV)-injected mice, a decrease in the endothelium-dependent relaxation to ACh was found compared to control (Fig. 5I), suggesting that CCN2(IV) caused an impaired vasomotor response. Although the pathophysiology of cardiovascular disease is multifactorial, many evidences indicate the importance of the inflammatory process (61). Next, we investigate whether CCN2(IV) could contribute to vascular inflammation by the early upregulation of key proinflammatory factors involved in cardiovascular diseases (61). CCN2(IV) treatment increased the aortic gene expression of several proinflammatory cytokines, including interleukin-6 (IL-6) and tumor necrosis factor α (TNFα), the adhesion molecule intercellular adhesion molecule 1 (ICAM-1), and the chemokines monocyte chemotactic protein-1 (MCP-1) and regulated on activation normal T cell expressed and secreted (RANTES) (Supplementary Fig. S2A). IL-6 was also increased at the protein level, as observed by enzyme-linked immunosorbent assay (ELISA) (Supplementary Fig. S2B). In chronic vascular inflammatory disorders, such as atherosclerosis, infiltration of immune cells in the vascular wall has been described (61). However, no infiltrating inflammatory cells were found in mice aorta of CCN2(IV) at 24 h (data not shown), may be due to the early time point of this study. In several vascular diseases, such as hypertension, the accumulation of extracellular matrix proteins in the vascular wall has been described (56, 61). After 24 h of CCN2(IV) injection, there were no changes in the expression of profibrotic-related genes, including TGF-β and plasminogen activator inhibitor-1 (PAI-1) (Supplementary Fig. S2A), or in aortic collagen content (data not shown) compared with saline-injected mice.
Activation of nuclear factor-κB (NF-κB) is an important molecular mechanism involved in the regulation of the inflammatory response in cardiovascular diseases (55, 68). An initial step on NF-κB activation is the phosphorylation of the p65 NF-κB subunit on Ser536 that has been involved in the NF-κB transcriptional control of proinflammatory genes (10). Aortic Western blot showed increased phosphorylated-p65 NF-κB subunit levels (p-p65) compared with saline-injected mice (Supplementary Fig. S2C). Thus, immunohistochemistry of aortic sections from CCN2(IV)-injected mice showed elevated p-p65 staining mainly located in VSMCs (Supplementary Fig. S2D), showing that CCN2(IV) in vivo activates the NF-κB pathway.
These data suggest that CCN2(IV) in vivo induced a vascular proinflammatory response characterized by local activation of the NF-κB pathway, overexpression of proinflammatory and redox factors, and endothelial dysfunction.
CCN2(IV) by a redox-mediated process regulates the NF-κB pathway and downstream proinflammatory genes in murine aorta
To evaluate the in vivo contribution of NF-κB activation to CCN2(IV)-induced proinflammatory factors, mice were pretreated with the NF-κB inhibitor parthenolide 24 h before CCN2(IV) administration. Parthenolide significantly diminished CCN2(IV)-induced aortic NF-κB activation, as assessed by p65 NF-κB phosphorylation on Ser536, and upregulation of proinflammatory factors (Fig. 6). As a control, some animals were injected with 0.05% dimethyl sulfoxide (DMSO) (parthenolide vehicle), showing no difference with saline-injected mice (data not shown). NF-κB is a redox-regulated transcription factor (41, 60). In mice, treatment with the antioxidant apocynin inhibited CCN2(IV)-induced aortic NF-κB activation to values similar to control mice and significantly downregulated proinflammatory genes (Fig. 6). These data suggest that NF-κB is an important intracellular signaling pathway involved in CCN2(IV)-induced vascular proinflammatory responses, regulated by redox mechanisms.
FIG. 6.

CCN2(IV) activates NF-κB via redox process in murine aorta. C57BL/6 mice received a single i.p. injection of 2.5 ng/g body weight recombinant CCN2(IV) or saline and were sacrificed after 24 h (n=10 mice per group). Some mice were pretreated with parthenolide (3.5 mg/kg of body weight; n=9) or apocynin (50 g/kg of body weight; n=8) 24 h before CCN2(IV) administration. NF-κB activation was determined by evaluation of phosphorylated p65 NF-κB immunostaining in paraffin-embedded aortic sections. (A) A representative immunostained section (left) and the quantification (right) of the immunostaining. (B) Gene expression of the proinflammatory factors (IL-6, MCP-1, and RANTES) was evaluated by real-time PCR in aorta from saline and CCN2(IV)-injected mice. Data are expressed as mean±SEM of fold increase over saline of 8–10 animals per group. *p<0.05 versus saline. †p<0.05 versus CCN2(IV). IL-6, interleukin-6; MCP-1, monocyte chemotactic protein-1; NF-κB, nuclear factor-κB; RANTES, regulated on activation normal T cell expressed and secreted. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
CCN2(IV) activates the NF-κB pathway and regulates proinflammatory genes in cultured murine VSMCs
To demonstrate further, the direct effect of CCN2(IV) on vascular inflammation and activation of NF-κB pathway in vitro studies were performed. In murine VSMCs, CCN2(IV) induced a rapid activation of NF-κB, observed by increased phosphorylation of p65 NF-κB subunit, maximal at 50 ng/ml after 60 min (Fig. 7A, B). To evaluate the potential role of Nox1 in NF-κB activation, we tested whether knockdown of the membrane-associated p22phox subunit (29, 66), or Nox1, could block CCN2(IV) responses. Transfection with a specific small interfering RNA molecule (siRNA) targeting p22phox or Nox1, but not with a nonspecific scramble siRNA, abolished the CCN2(IV)-mediated p65 phosphorylation (Fig. 7C, D). CCN2(IV)-induced p65 phosphorylation was also diminished by ML-171 and the antioxidant apocynin (Supplementary Fig. S3). Activation of NF-κB consists of translocation of the p65/p50 active NF-κB complex into the nucleus where it binds to specific DNA sequences to regulate gene transcription (60). In VSMCs, CCN2(IV) induced p65 nuclear translocation, which was markedly diminished by ML-171 and apocynin and by p22phox gene silencing (Fig. 7E, F). Preincubation of VSMCs with the NF-κB pharmacological inhibitors, parthenolide and BAY 11-7082, or the antioxidant apocynin diminished CCN2(IV)-induced upregulation of several proinflammatory genes, including ICAM-1, IL-6, and RANTES (Supplementary Fig. S3). These data suggest that CCN2(IV) may regulate the NF-κB pathway and related proinflammatory genes through Nox1 activation.
FIG. 7.

CCN2(IV) activates NF-κB via Nox1 in VSMCs. (A) Time course of phosphorylation of p65 NF-κB subunit (p-p65) in mice VSMCs treated with 50 ng/ml CCN2(IV). (B) Concentration–response curve [CCN2(IV) 1–100 ng/ml; 60 min]. (C, D) VSMCs were transfected with p22phox, Nox1 siRNA, or scramble siRNA before CCN2(IV) stimulation for 60 min. Figure shows a representative Western blot, and the quantification of p-p65 protein levels expressed as mean±SEM of fold-change over control of eight (A), four (B), five (C), and three (D) independent experiments. *p<0.05 versus control. †p<0.05 versus CCN2(IV). (E) Cells were preincubated 60 min with apocynin (0.3 mM) or ML-171 (1 μM) or (F) transfected with p22phox before CCN2(IV) stimulation for 60 min. Localization of p65 NF-κB subunit was evaluated by confocal microscopy. (E, F) A representative image of three independent experiments. siRNA, small interfering RNA molecule. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
In vivo Nox1 gene silencing blocked CCN2(IV)-induced vascular responses
To clearly demonstrate whether Nox1 was involved in CCN2(IV)-observed vascular responses, Nox1 gene silencing in vivo was performed. For these experiments, mice were pretreated with a Nox1 morpholino before CCN2(IV) administration. Gene silencing of Nox1 by morpholino significantly diminished Nox1 levels in mice aorta (Fig. 8A) and other tissues (data not shown) and blocked aortic O2•− production (Fig. 8B), clearly showing that CCN2(IV) induces O2•− production via Nox1.
FIG. 8.

In vivo Nox1 gene silencing blocked CCN2(IV)-induced vascular responses. C57BL/6 mice received a single i.p. injection of recombinant CCN2(IV) (2.5 ng/g body weight) or vehicle and were sacrificed after 24 h. Some mice were pretreated with a Nox1 morpholino (10 mg/kg, retroorbital injection) 24 h before CCN2(IV) administration. (A) Nox1 protein expression was evaluated by Western blot. (B) O2•− production was determined by an increase in 2-OH-E+ generation by HPLC analysis of DHE fluorescence. (C) Protein nitrosylation and phosphorylated p65 NF-κB subunit (p-p65) levels were evaluated by immunostaining in paraffin-embedded aortic sections. (D) Levels of phosphorylated IκBα protein were evaluated in aortic protein extracts by Western blot. (E) Gene expression was evaluated by real-time PCR in aortic samples. Data are expressed as mean±SEM of fold increase over saline of 8–10 animals per group. *p<0.05 versus saline. †p<0.05 versus CCN2(IV). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
In the aorta of Nox1 morpholino-treated CCN2(IV)-injected mice, phosphorylated p65 NF-κB subunit levels were diminished to control values (Fig. 8C). Activation of the canonical NF-κB pathway involves the phosphorylation of the inhibitory IκBα subunit, dissociation from NF-κB complex, and subsequent proteasomal-mediated degradation in the cytosol (60). In the aorta of CCN2(IV)-injected mice, increased cytosolic levels of IκBα phosphorylated at Ser32 were observed, which was diminished to control levels in response to morpholino treatment (Fig. 8D). Moreover, in morpholino-treated CCN2(IV)-injected mice, proinflammatory genes were downregulated to values similar to vehicle-treated mice (Fig. 8E). These data clearly demonstrate that Nox1 is involved in the activation of NF-κB and downstream proinflammatory factors caused by CCN2(IV) in aorta.
In addition, Nox1 gene silencing diminished other CCN2(IV)-induced vascular responses, including protein nitrosylation (Fig. 8C) and endothelial dysfunction (Table 1).
Table 1.
Effect of Treatments on Acetylcholine Vasorelaxation in Mice Aorta In Vivo
| ACh relaxation (%) | |
|---|---|
| Saline-injected mice | 118.8±7.3 |
| CCN2(IV)-injected mice | 78.5±8.1a |
| CCN2(IV)-injected mice treated with Nox1 morpholino | 131.7±11.6b |
| CCN2(IV)-injected mice treated with erlotinib | 127.7±19.6b |
p<0.05 saline versus CCN2(IV).
p<0.05 versus CCN2(IV); n=6 per group.
ACh, acetylcholine; Nox, nonphagocytic NAD(P)H oxidases.
CCN2(IV) via EGFR pathway activation regulates vascular responses
We have recently described that CCN2 is a novel ligand of the epidermal growth factor receptor (EGFR) (51). CCN2 binds to EGFR via the C-terminal module, as shown by the Biacore studies. Moreover, in cell-based assays and in in vivo studies, we have demonstrated that EGFR is a functional receptor of CCN2(IV) in the kidney (51). Therefore, we investigated whether EGFR is also involved in CCN2(IV) responses in the vasculature.
In cultured VSMCs, CCN2(IV) activates EGFR pathway, as shown by increased EGFR phosphorylation on tyrosine 1068 (Fig. 9A). Moreover, immunohistochemistry studies showed an increase in phosphorylated-EGFR staining in aorta of CCN2(IV)-injected mice compared with controls (Fig. 9B). To evaluate the role of EGFR pathway activation in CCN2(IV)-induced vascular responses, mice were pretreated with erlotinib, a specific EGFR kinase inhibitor. Erlotinib blocked the EGFR pathway activation (Fig. 9B) and significantly diminished the CCN2(IV)-induced O2•− production (Fig. 9C), NF-κB activation (Fig. 9B), and upregulation of proinflammatory genes (Fig. 9D). In addition, endothelial dysfunction caused by CCN2(IV) was also significantly decreased by EGFR kinase inhibition (Table 1).
FIG. 9.

CCN2(IV) via EGFR pathway activation regulates vascular responses. (A) CCN2(IV) increased EGFR phosphorylation in VSMCs. Time course of EGFR activation in VSMC were treated with CCN2(IV) 50 ng/ml. Figure shows a representative Western blot, and the data are expressed as mean±SEM of fold increase over control of five independent experiments. *p<0.05 versus control. (B–D) C57BL/6 mice received a single i.p. injection of recombinant CCN2(IV) (2.5 ng/g body weight) or saline and were sacrificed after 24 h. Some mice were pretreated with erlotinib (40 mg/kg/i.p) 24 h before CCN2(IV) administration (B). O2•− production was determined by an increase in 2-OH-E+ generation by HPLC analysis of DHE fluorescence (C). Phosphorylated levels of EGFR or p65 NF-κB subunit (p-p65) were evaluated by immunostaining in paraffin-embedded aortic sections. (D) Gene expression was evaluated by real-time PCR in aortic samples. Data are expressed as mean±SEM of fold increase over saline of 8–10 animals per group. *p<0.05 versus saline. †p<0.05 versus CCN2(IV). EGFR, epidermal growth factor receptor. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Discussion
Our data show, for the first time to our knowledge, that CCN2(IV) regulates redox signaling in the vasculature and activates the NF-κB pathway and downstream proinflammatory genes. Cardiovascular diseases are characterized by increased oxidative stress and inflammation (2, 5, 15, 61). Thus, our data suggest that CCN2(IV) could contribute to vascular inflammatory damage through the regulation of redox signaling (Fig. 10) and could be a good therapeutic target for inflammatory cardiovascular diseases.
FIG. 10.

Mechanisms involved on CCN2(IV) responses in VSMCs. CCN2(IV) binds to EGFR and activates Nox1 leading to O2•− production. CCN2(IV) through this signaling mechanism increases protein nitrosylation and endothelial dysfunction. In addition, O2•−, acting as a second messenger, activates the NF-κB pathway and induces its related proinflammatory genes.
CCN2 overexpression has been found in many redox-sensitive diseases, including human atherosclerosis, diabetes, and cardiac hypertrophy, as well as in experimental models of redox-related vascular damage (11, 44, 50, 57). Thus, in patients with thoracic aortic aneurysm, ROS accumulation correlates with media layer degeneration and increased CCN2 (3). Now, we demonstrate that CCN2(IV) induces O2•− production in VSMCs and ECs both in vivo and ex vivo. Interestingly, in the experimental model of AngII infusion, we have found colocalization of CCN2 expression and ROS production, mainly in VSMCs, suggesting that there might be a relationship between CCN2, redox processes, and AngII-induced vascular damage.
There are few data about the relationship between CCN proteins and oxidative stress. CCN2 is a redox-regulated gene (33, 46, 57). In VSMCs, antioxidants, including Nox inhibitors and O2•− scavengers, inhibited CCN2 overexpression induced by factors involved in vascular damage, including AngII and endothelin-1 (56). Moreover, hydrogen peroxide can directly increase CCN2 expression (46). In human skin fibroblasts, CCN1 induced ROS generation by a process regulated by a Rac1/5-lipoxygenase/mitochondria-dependent mechanism, whereas Nox1 is dispensable for CCN1-induced ROS accumulation (7). In hepatic stellate cells, CCN1 also induced ROS via Rac/Nox1 (25). In skin fibroblasts cells, CCN2 increased the ROS production linked to Fas-mediated apoptosis; however, the mechanism involved was not evaluated (22). Now, we have found that CCN2(IV) induced O2•− production via Nox1 in mice aorta and in VSMCs. Interestingly, in cultured ECs, CCN2(IV) also induced O2•− production. There are a number of potential candidates responsible for redox signaling in these cells, including Nox1, Nox2, Nox4, uncoupled eNOS, and xanthine oxidase (43, 67), which remain to be determined.
ROS overproduction participates in the alterations of vascular tone associated with vascular diseases, such as hypertension, diabetes, and atherosclerosis (5, 61, 65). Interestingly, we describe here that the activation of redox signaling by CCN2(IV) was linked to direct effects on the vascular wall. CCN2(IV) increased phenylephrine contraction and induced endothelial dysfunction via Nox1 activation as demonstrated by pharmacological and genetic Nox1 deletion, thus demonstrating the contribution of O2•− to the vascular dysfunction induced by CCN2(IV). The exact mechanisms responsible of this endothelial dysfunction require further investigation. However, a plausible explanation relies on the reaction between iNOS-derived NO due to increased iNOS expression, and the increased O2•− formation leading to the generation of the highly prooxidant peroxynitrite. Peroxynitrite can induce endothelial dysfunction in a number of pathologies, including hypertension, diabetes, septic shock, or atherosclerosis by mechanisms dependent on its cytotoxic effects, which may cause oxidative damage to proteins, lipids, and DNA, or even by affecting the production of protective endothelial mediators, such as prostacyclin (40, 43). In support of this possibility, in CCN2(IV)-treated mice aorta, increased nitrotyrosine staining was found. Our data indicated that CCN2(IV) could regulate the arterial tone via redox mechanisms and therefore contribute to vascular disease.
Traditionally, CCN2 was considered mainly a profibrotic mediator. However, recent studies support the novel concept of CCN2 as a proinflammatory cytokine (27). Thus, CCN2 acts as a chemotactic factor for mononuclear cells (8) and regulates adhesion and migration (27). CCN2(IV) induces the differentiation of human CD4 T cells to Th17 lymphocytes (52). In cultured cells, both CCN2 and CCN2(IV) upregulate proinflammatory genes in cardiomyocytes (70), pancreatic stellate cells (24), and renal cells (59, 72). In the kidney, CCN2(IV) contributes to renal inflammation (52, 59). We have found that CCN2(IV) administration in mice induced a local proinflammatory response in the aorta characterized by the overexpression of the cytokines IL-6 and TNFα, the adhesion molecule ICAM-1, and the chemokines MCP-1 and RANTES. Moreover, the stimulation of cultured VSMCs with CCN2(IV) also upregulated these proinflammatory mediators. These data suggest that CCN2(IV) activates early events involved in the vascular inflammatory response, including endothelial dysfunction, activation of adhesion molecules, and production of proinflammatory cytokines. Our data extend the concept of CCN2 as an immune modulator to the vascular tissue.
Among the intracellular signaling mechanisms involved in the regulation of the immune and inflammatory responses, NF-κB has a special interest (55, 60, 68). Our studies clearly demonstrate that CCN2(IV) activates NF-κB in VSMCs in vivo and in vitro. NF-κB is a transcription factor that regulates the expression of key inflammatory mediators (60). A correlation between NF-κB and upregulation of chemokines has been described in human biopsies of patients with diverse cardiovascular diseases and in experimental models of vascular damage (34, 37, 55, 61, 68), similar to our current observation of CCN2(IV)-induced vascular damage in vivo. Moreover, pharmacological NF-κB inhibition by parthenolide markedly diminished CCN2(IV)-induced proinflammatory responses in vivo in the aorta and in cultured VSMCs. The beneficial effects of this NF-κB inhibitor were previously shown in experimental models of renal damage and atherosclerosis (16, 34). Our data demonstrate a novel mechanism, the activation of NF-κB pathway, involved in CCN2(IV) induction of vascular proinflammatory factors. Interestingly, CCN1 had no effect on p65 phosphorylation or NF-κB-dependent transcription (7), showing that both CCN proteins regulate redox processes, but the downstream mechanisms differ.
ROS can interact with NF-κB signaling pathways in many ways. A variety of enzymes involved in ROS metabolism are regulated by NF-κB. Thus, the enzymes involved in the generation of ROS, such as Nox2, xanthine oxidase, or cyclooxygenase, or in the degradation of ROS, such as catalase, superoxide dismutase (SOD), or heme oxygenase-1, are regulated by NF-κB. On the other hand, ROS can activate the NF-κB pathway (41). Here, we described that both in vivo and in vitro pharmacological blockade or p22phox/Nox1 gene silencing blocked CCN2(IV)-induced NF-κB activation and related proinflammatory gene upregulation in mice aorta and/or in VSMCs. These results show that in the vasculature, CCN2(IV) activates Nox1 and O2•− production, leading to NF-κB activation and upregulation of downstream NF-κB-controlled proinflammatory factors.
EGFR signaling blockade has emerged as a novel therapeutic option in cancer. EGFR regulates cell growth, angiogenesis, and migration (62) and has been involved in the pathogenesis of atherosclerosis, hypertension, and type 2 diabetes (13, 23, 38). EGFR can be activated by direct binding of specific ligands or it can be transactivated by extracellular stimuli, such as agonists of G protein-coupled receptors (including AngII), cytokines, integrins, and other physical stimuli (62). EGFR transactivation by AngII leads to oxidative stress and a proinflammatory phenotype (63). We have recently described that CCN2 is a novel EGFR ligand and through this signaling pathway contributes to renal damage (51). At vascular level, EGFR is expressed in VSMCs and ECs and these cells can produce EGFR ligands, including CCN2. Our data clearly show that direct activation of EGFR by CCN2(IV) increased O2•− formation and activated the NF-κB pathway linked to proinflammatory genes induction.
There are few studies evaluating the in vivo contribution of CCN2 to vascular damage. CCN2 gene expression in VSMCs and ECs is strongly upregulated by various inflammatory cytokines, including AngII or endothelin-1 (53, 57). In a rat carotid angioplasty model, CCN2, via Smad3, regulated adaptive remodeling and neointimal hyperplasia by inducing VSMCs proliferation (28). CCN2, via ERK activation, stimulated VSMCs transdifferentiation into osteoblasts (21). Several in vivo studies have demonstrated that CCN2 alone is not sufficient to cause ongoing fibrotic changes (42), as we have observed in response to CCN2(IV) administration in mice aorta. In normal human blood vessels, CCN2 mRNA was not detected. By contrast, CCN2 overexpression has been described in human atherosclerotic lesions, mainly in the plaque shoulder and colocalized with inflammatory infiltration (8, 44). However, there are no studies about CCN2 blockade on experimental atherosclerosis. Our data suggest that CCN2 via redox process and upregulation of proinflammatory factors could participate in vascular damage.
Previous reports present some contradictory data between in vitro and in vivo studies in the cardiovascular system. In rat neonatal cardiomyocytes, both full-length CCN2 and CCN2(IV) increased cell surface area by an Akt-dependent process (19). However, in transgenic mice with cardiac-restricted CCN2 overexpression, ischemia-reperfusion-induced infarct size was markedly diminished, suggesting that CCN2 confers cardioprotection by inhibition of GSK-3β activities, activation of phospho-Smad2, and reprogramming gene expression (1). In this transgenic mice, preserved cardiac function under AngII-induced pressure overload conditions were also observed (45). Additionally, several studies in models of experimental fibrosis have found beneficial effects using blockers of CCN2, by antisense oligonucleotides or gene silencing (18, 31, 71). However, there are no studies addressing the role of CCN2 in inflammatory cardiovascular disease. Our in vivo model shows that CCN2(IV) activates early signals involved in vascular inflammation, including endothelial dysfunction and NF-κB-related proinflammatory factors; however, its role in sustained vascular inflammation has not been demonstrated, and therefore, future research is warranted to address this issue.
Materials and Methods
Cell culture
VSMCs were isolated from male mice aortas (3- to 4-month-old C57BL/6 mice) treated with 2 mg/ml collagenase II (Sigma Chemical Co, St Louis, MO) for 20 min and adventitia was carefully removed. Then, VSMCs were obtained by the explant method (53). Cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 20% fetal bovine serum (FBS), 1% l-glutamine 200 mM, 100 U/ml penicillin, and 100 U/ml streptomycin (all reagents obtained from Lonza, Verviers, Belgium). VSMCs from passages 2 to 7 were used showing 99% positive immunostaining for smooth muscle α-actin (data not shown). ECs used were the pancreatic cell line Mile Sven 1, obtained from American Type Culture Collection (ATCC CRL-2279; Barcelona, Spain). ECs were grown in the DMEM medium supplemented with 5% FBS, 2% l-glutamine 200 mM, 100 U/ml penicillin, and 100 U/ml streptomycin. For experiments, cells at 80% confluence were growth-arrested by serum starvation for 24 h.
Reagents
Recombinant CCN2(IV) was from PeproTech EC Ltd. (London, United Kingdom), and angiotensin II was from Tocris (Ellisville, MO). The pharmacological inhibitors were from Sigma Chemical Co: the NF-κB inhibitors, parthenolide and BAY 11-7082, the antioxidant apocynin (4′-hydroxy-3′methoxyacetophenone), or ML-171, a specific Nox1 inhibitor. In parallel experiments, no cell toxicity was found at the doses of CCN2(IV) used (data not shown, evaluated by MTT assays in VSMCs). None of the inhibitors were toxic at the dose used (data not shown).
Design of the experimental models
Studies were performed in adult male C57BL/6 mice (9–12 weeks old, 20 g; Harlan InterfaunaIbérica, S.A., Barcelona, Spain) and maintained at the local animal facilities, with free access to food and water, normal light–dark cycles and under special pathogen-free conditions. All the procedures on animals were performed according to the European Community and Instituto de Investigación Sanitaria Fundación Jiménez Díaz Animal Research Ethical Committee guidelines.
Experimental model of AngII-induced vascular damage
Systemic infusion of AngII (1000 ng/kg/min, subcutaneously, ALZET micro-osmotic pumps 2001; Alza Corp., Palo Alto, CA) was carried out into C57BL/6 mice for 14 days (15). As control, saline infusion was carried out.
Experimental model of CCN2(IV)-induced vascular damage
Mice received a single intraperitoneal injection of CCN2(IV) (2.5 ng/g of body weight, dissolved in saline; see Supplementary Materials and Methods section for further information) and were studied after 24 h. A control saline-injected group was also studied (n=10 mice per group). The purity of CCN2(IV) (endotoxin levels <0.01) was also evaluated by Malditoff (data not shown).
The following treatments were performed, starting 24 h before CCN2(IV) administration. To study the NF-(B pathway, parthenolide, a sesquiterpene lactone that inhibits NF-(B activation by preventing I(Bα degradation (20), was used. Mice were treated with parthenolide (3.5 mg/kg of body weight; i.p. daily) or its vehicle (0.05% DMSO; n=9 mice per group), based on our previous experience (16, 34). To evaluate the role of redox processes, mice were treated with the antioxidant apocynin (50 mg/g of body weight, i.p. daily dissolved in saline; n=8 mice) (36). The EGFR pathway was studied using the EGFR kinase inhibitor erlotinib (40 mg/kg/day, i.p. n=9 mice per group, Vichem) (51). Nox1 was blocked by gene silencing, using a Nox1 targeting vivo-morpholino oligonucleotide (5′-ACCAGCCAGTTTCCCATTGTCAAAT-3′) 10 mg/kg, retro-orbital injection, according to the manufacturer (GeneTools LLC, Philomath, OR) (9).
Tissue preparation
Mice were sacrificed under anesthesia (Isofluorane; Abbott Laboratories, Madrid, Spain) and aortas were dissected free of fat and connective tissue. Aortic segments were processed in different conditions depending on the particular experiment. For confocal microscopy evaluation of O2•− production, segments were placed in the cold Krebs–Henseleit solution (KHS in mM: 115 NaCl, 25 NaHCO3, 4.7 KCl, 1.2 MgSO4.7H2O, 2.5 CaCl2, 1.2 KH2PO4, 11.1 glucose, and 0.01 Na2EDTA) containing 30% sucrose for 20 min, transferred to a cryomold containing a Tissue Tek OCT-embedding medium (Sakura Finetek Europe BV, Zoeterwoude, The Netherlands) and then immediately frozen in liquid nitrogen for storage at −80°C. For HPLC measurement of O2•− production, segments were equilibrated in KHS, incubated with DHE (5 μM, 30 min, 37°C), and kept at −80°C. For vascular reactivity, NO production or NAD(P)H oxidase activity, aortic segments were preincubated for 60 min at 37°C in KHS-HEPES (in mM: NaCl 115; HEPES 20; CaCl2 1.2; KCl 4.6; MgSO4 1; KH2PO4 0.4; NaHCO3 5; glucose 5.5; Na2HPO4 0.15; pH 7.4). For immunohistochemistry, arterial segments were buffered in formalin and paraffin-embedded. For protein or gene expression studies, segments were immediately frozen in liquid nitrogen and kept at −80°C. All the evaluations were carried out in aortic segments of at least six mice per group.
Confocal microscopy analysis of CCN2(IV) binding
Isolated aortic rings were incubated with Cy5-labbeled CCN2(IV) for different times. Then, the rings were incubated in KHS-HEPES buffer containing 30% sucrose for 20 min before OCT-embedded. Images of aortic sections were captured using a Leica TCS SP5 confocal microscope, equipped with a×63 oil immersion objective. Fluorophore Cy-5-emitted fluorescence was monitored with a 550±20 nm band-pass filter or with a 670 nm long-pass filter, and 4′,6-diamidino-2-phenylindole (DAPI) was excited using a DIODE laser (51).
Vascular reactivity
Vascular reactivity was studied in a wire myograph by isometric tension recording (36). Ex vivo experiments were performed in intact or in endothelium-denuded aorta (endothelium was removed by mechanical scrape) from control mice. Contractility of segments was tested by an initial exposure to a high-K+ solution (K+-KHS, 120 mM). The presence of endothelium was determined by the ability of 10 μM ACh to relax arteries precontracted with Phe at ∼50% K+-KHS contraction. Afterward, concentration–response curves to ACh and Phe were performed in each segment. Vasoconstrictor responses were expressed as a percentage of the tone generated by K+-KHS.
Measurement of O2•− production by DHE immunostaining
The oxidative fluorescent dye DHE (Invitrogen, Groningen, The Netherlands) was used to evaluate the production of O2•− in situ by confocal microscopy (36). Depending on the tissue, different preparation was carried out. For ex vivo experiments, mouse aortic segments were equilibrated in KHS-HEPES, pretreated or not with ML-171 (1 μM, 1 h), before incubation with CCN2(IV) (50 ng/ml, 60 min, 37°C in a cell chamber), and the OCT-embedded. Afterward, the aortic section was equilibrated in KHS-HEPES (30 min, 37°C), incubated in fresh buffer containing DHE (5 μM, 30 min, 37°C), and visualized. For in vitro studies, VSMCs and ECs (seeded in 24-well multidish over glass coverslips) were used. After stimulation, cells were equilibrated in KHS-HEPES, incubated in DHE/fresh buffer, fixed in 4% paraformaldehyde (PFA) during 20 min, washed with PBS, and mounted in ProLong® Gold Antifade Reagent (Invitrogen). In all studies, fluorescence was detected with a fluorescent laser scanning confocal microscope (excitation at 540 nm and emission 610 nm; Leica Microsystems, Wetzlar, Germany). Elastin layer was captured by autofluorescence (excitation at 488 nm). As a negative control of the staining, sample were preincubated with the O2•− scavenger Tiron (100 μM). Control and stimulated cells were imaged in parallel keeping the same imaging settings of the microscope. The quantitative analysis of O2•− production was performed with MetaMorph Image Analysis Software (Molecular Devices Corporation, Downingtown, PA). The integrated optical density was calculated in four areas per samples for each experimental condition.
Measurement of O2•− production by high-performance liquid chromatography
Aortic samples were homogenized in acetonitrile (300 μl), sonicated, centrifuged (12,000 rpm, 15 min at 4°C), and the supernatant was collected and dried. Pellet was resuspended in Krebs-HEPES-DPTA 25 μM, and 5 μl was used for protein determination. Samples (4 μg) were filtered (0.22 μm) and analyzed by HPLC (Agilent Technologies 1200 series, Santa Clara, CA) using a 5 μm C-18 reverse-phase column (Kinetex 150×4.6 mm; Phenomenex, Torrance, CA) and a gradient of solutions A (pure acetonitrile) and B (water/10% acetonitrile/0.1% trifluoroacetic acid, v/v/v) at a flow rate of 0.4 ml/min and run (30). Ethidium and 2-OH-E+ were monitored by fluorescence detection with excitation at 480 nm and emission at 580 nm. The 2-OH-E+ peak reflects the amount of O2•− formed in the tissue during the incubation per microgram of protein. The increase of 2-OH-E+ peak was represented by an increase (n-fold) versus control. To optimize the HPLC analysis, 5 μM DHE was incubated with xanthine/xanthine oxidase (0–50 μM/0.1 U/ml) in KHS-HEPES containing 100 μM diethylenetriamine pentaacetic acid (KHS-HEPES/DTPA) at 37°C for 30 min. Chromatograms are shown in Supplementary Figure S4.
NAD(P)H oxidase activity
The lucigenin-enhanced chemiluminescence assay was used to determine the Nox activity. For ex vivo experiments, mouse aortic segments were incubated with CCN2(IV) (50 ng/ml, 60 min, 37°C) alone or preincubated with ML-171. Cell growth-arrested VSMCs, growing in a 60-mm tissue culture dish, were incubated with 50 ng/ml CCN2(IV). Mouse aortic segments from CCN2(IV)-treated and control mice were incubated in KHS (37°C). Tissues and cells were homogenized and lysed in buffer (in mM: KH2PO4 50, ethyleneglycoltetraacetic acid 1, sucrose 150, pH 7.4). The reaction was started by adding 0.1 mM NAD(P)H, 5 μM lucigenin, and assay phosphate buffer. The luminescence was measured in a plate luminometer (AutoLumat LB 953, Berthold, Germany). Buffer blank was subtracted from each reading. As loading control, protein concentration was determined by the BCA method (Pierce Thermo Scientific, Rockford, IL), and luminescence was normalized by protein concentration.
L-012 chemiluminescence
Serum-starved VSMCs and/or ECs were harvested and resuspended in OPTI-MEM® I (Gibco, Life Technologies, Paisley, United Kingdom) into six-well plates. After 1 h of incubation with 50 ng/ml CCN2(IV), cells were scrapped and superoxide was detected using L-012 (400 μM). Luminescence was measured using a Microplate Reader. The specificity of L-012 for O2•− was confirmed by the addition of SOD (150 U/ml), data not shown (9). Protein concentration was determined by the BCA method, and luminescence was normalized by protein concentration.
Gene expression studies
Total mRNA was obtained by the TRIzol method (Invitrogen, Life Technologies, Philadelphia, PA). Two micrograms of total RNA was reverse-transcribed into cDNA using TaqMan Reverse Transcription Reagents kit (Applied Biosystems, Life Technologies, Paisley, United Kingdom). Multiplex RT-PCR was performed using fluorogenic (FAM or VIC) primers designed by the Assay-on-Demand™ mouse gene expression products (Applied Biosystems): RANTES Mm01302428_m1, IL-6 Mm00446190_m1, ICAM-1 Mm00516023_m1, MCP-1 Mm00441242_m1, TNFα Mm0043258_m1, TGF-β Mm001178819_m1, PAI-1 Mm00435860_m1, p22phox Mm00514478_m1, Nox1 Mm00549170_m1, and iNOs Mm01309901_m1. Data were normalized with 18s eukaryotic ribosomal RNA expression (4310893E). Results were expressed in copy numbers, calculated relative to unstimulated cells or control mice, after normalization against 18s using the ABIPrism 7500 Fast sequence detection PCR system software (Applied Biosystems) (16).
In vitro siRNA approach
In VSMCs, gene silencing was performed using either a predesigned siRNA corresponding to p22phox (CYBA silencer 5 ng/ml) or Nox1 (Nox1 silencer 5 ng/ml) (Ambion, Life Technologies, Paisley, United Kingdom) or its corresponding scramble siRNA. Subconfluent cells were transfected in the Opti-MEM reduced serum medium (Gibco) for 24 h with 5 ng/ml siRNA using 50 nM Lipofectamine RNAiMAX (Invitrogen). Then, cells were incubated in the medium with 20% FBS for 24 h, following by 24 incubation in the serum-free medium. The efficacy of p22phox/Nox1 silencing was confirmed by gene expression. In all experiments, controls were untransfected cells (treated only with lipofectamine vehicle), cells transfected with scramble siRNA and with target siRNA, both treated or not with CCN2(IV).
Protein studies
Protein extracts from cultured cells and murine aorta were obtained by homogenization and centrifugation (53). Antibodies used were (dilution): phosphorylated-NF-κB p65 (1/2000; Cell signaling, Danvers, MA), eNOS (1/1000; Transduction Laboratories, BD Biosciences, Madrid, Spain), phosphorylated-EGFR (p-EGFR on Tyr 1068; 1/250; Calbiochem, Millipore, Billerica, MA), phosphorylated-IκBα (1/1000, sc-371; Santa Cruz Biotechnology, Inc., Heidelberg, Germany). To evaluate equal loading, membranes were stained with an anti-total NF-κB p65 antibody (1/2500, sc-372; Santa Cruz Biotechnology), GAPDH (1/10,000; Millipore), or β-actin (1/5000, sc-47778; Santa Cruz Biotechnology). Autoradiographs were scanned using Gel DOC™ EZ Imager (Bio-Rad, Hercules, CA) and quantified with Image Lab 3.0 software (Bio-Rad).
IL-6 levels were analyzed in aortic murine tissue by an ELISA kit (BD Biosciences). Cytokine level was quantified by comparison with a standard curve, and the data were expressed as fold-change over the mean value of control mice levels.
Immunofluorescence staining of NF-κB subunits
VSMCs were seeded in 24-well Multidish over glass coverslips. After experiments, cells were fixed in 4% PFA, treated with 0.1% Triton-X100, blocked with 4% BSA in TBS, incubated overnight with NF-κB p65 antibody (1/200; Santa Cruz Biotechnology), followed by AlexaFluor®488 conjugated antibody (1/300; Invitrogen). Nuclei were contrasted with DAPI (Sigma Chemical Co). The absence of primary antibody was the negative control (data not shown). Samples were mounted in Mowiol 40–88 (Sigma Chemical Co) and examined by a laser scanning confocal microscope (oil immersion objective 40×, zoom 2) (Leica Microsystems). Experiments were repeated with three different cell culture preparations.
Immunohistochemistry
Paraffin-embedded sections were stained using standard histology procedures. Immunostaining was carried out in 3-μm-thick tissue sections that were deparaffinized and antigen retrieved using the PT Link system (Dako Diagnósticos S.A., Barcelona, Spain) with sodium citrate buffer (10 mM) adjusted to pH 6. Endogenous peroxidase was blocked and aorta sections were incubated with the primary antibody overnight at 4°C. After washing, slides were treated with the corresponding anti-IgG biotinylated-conjugated secondary antibody (Amersham Bioscience, Amersham, United Kingdom) followed by the avidin–biotin–peroxidase complex, and 3,3′-diaminobenzidine as chromogen (Dako Diagnósticos S.A.). Sections were counterstained with Carazzi's hematoxylin and mounted with DPX. Primary antibodies used were NF-κB p-p65 subunit (1/50; Cell Signaling), p-EGFR (1/200; Cell Signaling), 3-Nitrotyrosin (1/1000; Abcam, Cambridge, United Kingdom), and CCN2 (1/500; Sigma Chemical Co). The specificity was checked by omission of primary antibodies and use of nonimmune sera (data not shown). Images were obtained with the Nikon Eclipse E400 microscope and analyzed by Image Pro-plus (Media Cybernetics, Inc., Rockville, MD). All samples were evaluated in a blinded manner. For each mouse, the mean score value was obtained by evaluating four different high-power fields (40×) per section.
NO release
Aortic segments were incubated in KHS-HEPES (60 min, 37°C), followed by incubation with 4,5-diaminofluorescein (DAF-2; 2 μM, 45 min). Then, the medium was collected to measure basal NO release. Afterward, these segments were incubated with phenylephrine (1 μM, 1 min) and ACh (10 μM, 1 min) and the medium was collected to measure agonist-induced NO release. The fluorescence was measured using a spectrofluorometer (excitation at 492 nm and emission at 515 nm; FLUOstar OPTIMA; BMG Labtech, Ortenberg, Germany). NO-induced release was calculated by subtracting basal NO release from that evoked by ACh. Blank samples with the segment-free medium were used to correct background emission. The amount of NO released was expressed as arbitrary units/mg tissue.
Statistical analysis
Results are expressed as mean±SEM. Differences between agonist-treated groups and controls were assessed by the Mann–Whitney test. p<0.05 was considered significant. Vascular reactivity was analyzed by two-way ANOVA followed by the Bonferroni's post hoc test. Statistical analysis was conducted using the SPSS statistical software (version 11.0, Chicago, IL).
Supplementary Material
Abbreviations Used
- 2-OH-E+
2-hydroxyethidium
- ACh
acetylcholine
- AngII
angiotensin II
- BCA
bicinchoninic acid
- BSA
bovine serum albumin
- CCN1
cysteine-rich protein 61
- CCN2(IV)
C-terminal module IV of CCN2
- CD4
cluster of differentiation 4
- CTGF/CCN2
connective tissue growth factor
- DAF-2
4,5-diaminofluorescein
- DAPI
4′,6-diamidino-2-phenylindole
- DHE
dihydroethidium
- DMEM
Dulbecco's modified Eagle's medium
- DMSO
dimethyl sulfoxide
- DPX
Distrene, Plasticiser, Xylene
- DTPA
diethylenetriamine pentaacetic acid
- EC
endothelial cells
- EGFR
epidermal growth factor receptor
- ELISA
enzyme-linked immunosorbent assay
- eNOS
endothelial NO synthase
- ERK
extracellular signal-regulated kinases
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GSK-3β
glycogen synthase kinase-3β
- HPLC
high-performance liquid chromatography
- ICAM-1
intercellular adhesion molecule 1
- IL-6
interleukin-6
- IκBα
nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha
- iNOS
inducible NO synthase
- KHS
Krebs–Henseleit solution
- MCP-1
monocyte chemotactic protein-1
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NF-κB
nuclear factor-κB
- nNOS
neuronal NO synthase
- NO
nitric oxide
- Nox
nonphagocytic NAD(P)H oxidases
- O2•−
superoxide anion
- PAI-1
plasminogen activator inhibitor-1
- PBS
phosphate buffer solution
- PCR
polymerase chain reaction
- PFA
paraformaldehyde
- Phe
phenylephrine
- RANTES
regulated on activation normal T cell expressed and secreted
- ROS
reactive oxygen species
- RT-PCR
reverse transcription–polymerase chain reaction
- siRNA
small interfering RNA molecule
- SOD
superoxide dismutase
- TBS
Tris-buffered saline
- TGF-β
transforming growth factor-β
- Th17
T helper cells producing interleukin 17
- TNFα
tumor necrosis factor α
- VSMCs
vascular smooth muscle cells
Acknowledgments
We thank Susana Carrasco and Ma Mar Gonzalez Garcia-Parreño at the IIS-Fundación Jiménez Díaz for their help in immunohistochemical and immunofluorescence procedures, respectively. This work was supported by grants from the Instituto de Salud Carlos III (ISCIII-RETIC REDINREN RD06/0016, RD012/0021, RECAVA RD12/0042/0024, PI11/01854, PI10/00072, PI13/01488), Comunidad de Madrid (Fibroteam: S2010/BMD-2321), the Hungarian Research and Technological Innovation Fund (KMR_12-1-2012-0074). Kidney Connect FP7-HEALTH-2013-INNOVATION-1 (HEALTH-F2-2013-602422), ePredice FP7-HEALTH-2011(279074), and DiabetesCancerConnect (PIE13/00051). Programa Intensificación Actividad Investigadora (ISCIII/Agencia Laín-Entralgo/CM) to A.O. Ministerio de Economia y Competitividad (SAF 2009-07201, 2012-36400). A.M.B. was supported by the Ramon y Cajal Program (RYC-2010-06473). A.B.G.R. was supported by the Sara Borrell Program (CD11/00165).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Ahmed MS, Gravning J, Martinov VN, von Lueder TG, Edvardsen T, Czibik G, Moe IT, Vinge LE, Øie E, Valen G, and Attramadal H. Mechanisms of novel cardioprotective functions of CCN2/CTGF in myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 300: H1291–H1302, 2011 [DOI] [PubMed] [Google Scholar]
- 2.Al Ghouleh I, Khoo NK, Knaus UG, Griendling KK, Touyz RM, Thannickal VJ, Barchowsky A, Nauseef WM, Kelley EE, Bauer PM, Darley-Usmar V, Shiva S, Cifuentes-Pagano E, Freeman BA, Gladwin MT, and Pagano PJ. Oxidases and peroxidases in cardiovascular and lung disease: new concepts in reactive oxygen species signaling. Free Radic Biol Med 51: 1271–1288, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Branchetti E, Poggio P, Sainger R, Shang E, Grau JB, Jackson BM, Lai EK, Parmacek MS, Gorman RC, Gorman JH, Bavaria JE, and Ferrari G. Oxidative stress modulates vascular smooth muscle cell phenotype via CTGF in thoracic aortic aneurysm. Cardiovasc Res 100: 316–324, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Briones AM, Rodríguez-Criado N, Hernanz R, García-Redondo AB, Rodrigues-Díez RR, Alonso MJ, Egido J, Ruiz-Ortega M, and Salaices M. Atorvastatin prevents angiotensin II-induced vascular remodeling and oxidative stress. Hypertension 54: 142–149, 2009 [DOI] [PubMed] [Google Scholar]
- 5.Briones AM. and Touyz RM. Oxidative stress and hypertension: current concepts. Curr Hypertens Rep 12: 135–142, 2010 [DOI] [PubMed] [Google Scholar]
- 6.Chan EC, Peshavariya HM, Liu GS, Jiang F, Lim SY, and Dusting GJ. Nox4 modulates collagen production stimulated by transforming growth factor β1 in vivo and in vitro. Biochem Biophys Res Commun 430: 918–925, 2013 [DOI] [PubMed] [Google Scholar]
- 7.Chen CC, Young JL, Monzon RI, Chen N, Todorović V, and Lau LF. Cytotoxicity of TNFalpha is regulated by integrin-mediated matrix signaling. EMBO J 26: 1257–1267, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cicha I, Yilmaz A, Klein M, Raithel D, Brigstock DR, Daniel WG, Goppelt-Struebe M, and Garlichs CD. Connective tissue growth factor is overexpressed in complicated atherosclerotic plaques and induces mononuclear cell chemotaxis in vitro. Arterioscler Thromb Vasc Biol 25: 1008–1013, 2005 [DOI] [PubMed] [Google Scholar]
- 9.Csányi G1, Yao M, Rodríguez AI, Al Ghouleh I, Sharifi-Sanjani M, Frazziano G, Huang X, Kelley EE, Isenberg JS, and Pagano PJ. Thrombospondin-1 regulates blood flow via CD47 receptor-mediated activation of NADPH oxidase 1. Arterioscler Thromb Vasc Biol 32: 2966–2973, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cui R, Tieu B, Recinos A, Tilton RG, and Brasier AR. RhoA mediates angiotensin II-induced phospho-Ser536 nuclear factor kappaB/RelA subunit exchange on the interleukin-6 promoter in VSMCs. Circ Res 99: 723–730, 2006 [DOI] [PubMed] [Google Scholar]
- 11.De Winter P, Leoni P, and Abraham D. Connective tissue growth factor: structure-function relationships of a mosaic, multifunctional protein. Growth Factors 26: 80–91, 2008 [DOI] [PubMed] [Google Scholar]
- 12.Dikalov S, Griendling KK, and Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension 49: 717–727, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dreux AC, Lamb DJ, Modjtahedi H, and Ferns GA. The epidermal growth factor receptors and their family of ligands: their putative role in atherogenesis. Atherosclerosis 186: 38–53, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Drummond GR, Selemidis S, Griendling KK, and Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov 10: 453–471, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esteban V, Ruperez M, Rodriguez-Vita J, Sanchez-López E, Mezzano S, Plaza JJ, Egido J, and Ruiz-Ortega M. Effect of simultaneous blockade of AT1 and AT2 receptors on the NF-κB pathway and renal inflammatory response. Kidney Int 64: S33–S38, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Esteban V, Lorenzo O, Rupérez M, Suzuki Y, and Ruiz-Ortega M. Angiotensin II, via AT1 and AT2 receptors and NF-kappaB pathway, regulates the inflammatory response in unilateral ureteral obstruction. J Am Soc Nephrol 15: 1514–1529, 2004 [DOI] [PubMed] [Google Scholar]
- 17.Gravning J, Ørn S, Kaasbøll OJ, Martinov VN, Manhenke C, Dickstein K, Edvardsen T, Attramadal H, and Ahmed MS. Myocardial connective tissue growth factor (CCN2/CTGF) attenuates left ventricular remodeling after myocardial infarction. PLoS One 7: e52120, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hao C, Xie Y, Peng M, Ma L, Zhou Y, Zhang Y, Kang W, Wang J, Bai X, Wang P, and Jia Z. Inhibition of connective tissue growth factor suppresses hepatic stellate cell activation in vitro and prevents liver fibrosis in vivo. Clin Exp Med 14: 141–150, 2014 [DOI] [PubMed] [Google Scholar]
- 19.Hayata N, Fujio Y, Yamamoto Y, Iwakura T, Obana M, Takai M, Mohri T, Nonen S, Maeda M, and Azuma J. Connective tissue growth factor induces cardiac hypertrophy through Akt signaling. Biochem Biophys Res Commun 37: 274–278, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Hehner SP, Hofmann TG, Droge W, and Schmitz ML. The antiinflammatorysesquiterpene lactone Parthenolide inhibits NF-kappa B by targeting the I kappa B kinase complex. J Immunol 163: 5617–5623, 1999 [PubMed] [Google Scholar]
- 21.Huang J, Huang H, Wu M, Li J, Xie H, Zhou H, Liao E, and Peng Y. Connective tissue growth factor induces osteogenic differentiation of vascular smooth muscle cells through ERK signaling. Int J Mol Med 32: 423–429, 2013 [DOI] [PubMed] [Google Scholar]
- 22.Juric V, Chen CC, and Lau LF. Fas-mediated apoptosis is regulated by the extracellular matrix protein CCN1 (CYR61) in vitro and in vivo. Mol Cell Biol 29: 3266–3279, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kagiyama S, Eguchi S, Frank GD, Inagami T, Zhang YC, and Phillips MI. Angiotensin II-induced cardiac hypertrophy and hypertension are attenuated by epidermal growth factor receptor antisense. Circulation 106: 909–912, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Karger A, Fitzner B, Brock p, Sparmann G, Emmrich J, Liebe S, and Jaster R. Molecular insights into connective tissue growth factor action in rat pancreatic stellate cells. Cell Signal 20: 1865–1872, 2008 [DOI] [PubMed] [Google Scholar]
- 25.Kim KH, Chen CC, Monzon RI, and Lau LF. Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol Cell Biol 33: 2078–2090, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koitabashi N, Arai M, Niwano K, Watanabe A, Endoh M, Suguta M, Yokoyama T, Tada H, Toyama T, Adachi H, Naito S, Oshima S, Nishida T, Kubota S, Takigawa M, and Kurabayashi M. Plasma connective tissue growth factor is a novel potential biomarker of cardiac dysfunction in patients with chronic heart failure. Eur J Heart Fail 10: 373–379, 2008 [DOI] [PubMed] [Google Scholar]
- 27.Kular L, Pakradouni J, Kitabgi P, Laurent M, and Martinerie C. The CCN family: a new class of inflammation modulators? Biochimie 93: 377–388, 2011 [DOI] [PubMed] [Google Scholar]
- 28.Kundi R, Hollenbeck ST, Yamanouchi D, Herman BC, Edlin R, Ryer EJ, Wang C, Tsai S, Liu B, and Kent KC. Arterial gene transfer of the TGF-beta signalling protein Smad3 induces adaptive remodelling following angioplasty: a role for CTGF. Cardiovasc Res 84: 326–335, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lassègue B, San Martín A, and Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res 110: 1364–1390, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laurindo FR, Fernandes DC, and Santos CX. Assessment of superoxide production and NADPH oxidase activity by HPLC analysis of dihydroethidium oxidation products. Methods Enzymol 441: 237–260, 2008 [DOI] [PubMed] [Google Scholar]
- 31.Leask A. CCN2/decorin interactions: a novel approach to combating fibrosis? J Cell Commun Signal 5: 249–250, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu BC, Zhang JD, Zhang XL, Wu GQ, and Li MX. Role of connective tissue growth factor (CTGF) module 4 in regulating epithelial mesenchymal transition in HK-2 cells. Clin Chim Acta 373: 144–150, 2006 [DOI] [PubMed] [Google Scholar]
- 33.Liu X, Gai Y, Liu F, Gao W, Zhang Y, Xu M, and Li Z. Trimetazidine inhibits pressure overload-induced cardiac fibrosis through NADPH oxidase-ROS-CTGF pathway. Cardiovasc Res 88: 150–158, 2010 [DOI] [PubMed] [Google Scholar]
- 34.López-Franco O, Hernández-Vargas P, Ortiz-Muñoz G, Sanjuán G, Suzuki Y, Ortega L, Blanco J, Egido J, and Gómez-Guerrero C. Parthenolide modulates the NF-kappaB-mediated inflammatory responses in experimental atherosclerosis. Arterioscler Thromb Vasc Bio 26: 1864–1870, 2006 [DOI] [PubMed] [Google Scholar]
- 35.Markiewicz M, Nakerakanti SS, Kapanadze B, Ghatnekar A, and Trojanowska M. Connective tissue growth factor (CTGF/CCN2) mediates angiogenic effect of S1P in human dermal microvascular endothelial cells. Microcirculation 18: 1–11, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martínez-Revelles S, Avendaño MS, García-Redondo AB, Alvarez Y, Aguado A, Pérez-Girón JV, García-Redondo L, Esteban V, Redondo JM, Alonso MJ, Briones AM, and Salaices M. Reciprocal relationship between reactive oxygen species and cyclooxygenase-2 and vascular dysfunction in hypertension. Antioxid Redox Signal 18: 51–65, 2013 [DOI] [PubMed] [Google Scholar]
- 37.Martín-Ventura JL, Blanco-Colio LM, Gómez-Hernández A, Muñoz-García B, Vega M, Serrano J, Ortega L, Hernández G, Tuñón J, and Egido J. Intensive treatment with atorvastatin reduces inflammation in mononuclear cells and human atherosclerotic lesions in one month. Stroke 36: 1796–1800, 2005 [DOI] [PubMed] [Google Scholar]
- 38.Matrougui K. Diabetes and microvascular pathophysiology: role of epidermal growth factor receptor tyrosine kinase. Diabetes Metab Res Rev 26: 13–16, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moe IT, Pham TA, Hagelin EM, Ahmed MS, and Attramadal H. CCN2 exerts direct cytoprotective actions in adult cardiac myocytes by activation of the PI3-kinase/Akt/GSK-3β signaling pathway. J Cell Commun Signal 7: 31–47, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moncada S. and Higgs EA. Nitric oxide and the vascular endothelium. Handb Exp Pharmacol (176 Pt 1): 213–254, 2006 [DOI] [PubMed] [Google Scholar]
- 41.Morgan MJ. and Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res 21: 103–115, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mori T, Kawara S, Shinozaki M, Hayashi N, Kakinuma T, Igarashi A, Takigawa M, Nakanishi T, and Takehara K. Role and interaction of connective tissue growth factor with transforming growth factor-beta in persistent fibrosis: a mouse fibrosis model. J Cell Physiol 181: 153–159, 1999 [DOI] [PubMed] [Google Scholar]
- 43.Münzel T, Gori T, Bruno RM, and Taddei S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur Heart J 31: 2741–2748, 2010 [DOI] [PubMed] [Google Scholar]
- 44.Oemar BS, Werner A, Garnier JM, Do DD, Godoy N, Nauck M, März W, Rupp J, Pech M, and Lüscher TF. Human connective tissue growth factor is expressed in advanced atherosclerotic lesions. Circulation 9: 831–839, 1997 [DOI] [PubMed] [Google Scholar]
- 45.Panek AN, Posch MG, Alenina N, Ghadge SK, Erdmann B, Popova E, Perrot A, Geier C, Dietz R, Morano I, Bader M, and Ozcelik C. Connective tissue growth factor overexpression in cardiomyocytes promotes cardiac hypertrophy and protection against pressure overload. PLoS One 4: e6743, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park SK, Kim J, Seomun Y, Choi J, Kim DH, Han IO, Lee EH, Chung SK, and Joo CK. Hydrogen peroxide is a novel inducer of connective tissue growth factor. Biochem Biophys Res Commun 284: 966–971, 2001 [DOI] [PubMed] [Google Scholar]
- 47.Perbal B. CCN proteins: multifunctional signaling regulators. Lancet 363: 62–64, 2004 [DOI] [PubMed] [Google Scholar]
- 48.Phanish MK, Winn SK, and Dockrell ME. Connective tissue growth factor-(CTGF, CCN2)-a marker mediator and therapeutic target for renal fibrosis. Nephron Exp Nephrol 114: e83–e92, 2010 [DOI] [PubMed] [Google Scholar]
- 49.Ponticos M, Holmes AM, Shi-wen X, Leoni P, Khan K, Rajkumar VS, Hoyles RK, Bou-Gharios G, Black CM, Denton CP, Abraham DJ, Leask A, and Lindahl GE. Pivotal role of connective tissue growth factor in lung fibrosis: MAPK-dependent transcriptional activation of type I collagen. Arthritis Rheum 60: 2142–2155, 2009 [DOI] [PubMed] [Google Scholar]
- 50.Ponticos M. Connective tissue growth factor (CCN2) in blood vessels. Vascul Pharmacol 58: 189–193, 2013 [DOI] [PubMed] [Google Scholar]
- 51.Rayego-Mateos S, Rodrigues-Díez R, Morgado-Pascual JL, Rodrigues Díez RR, Mas S, Lavoz C, Alique M, Pato J, Keri G, Ortiz A, Egido J, and Ruiz-Ortega M. Connective tissue growth factor is a new ligand of epidermal growth factor receptor. J Mol Cell Biol 5: 323–335, 2013 [DOI] [PubMed] [Google Scholar]
- 52.Rodrigues-Díez R, Rodrigues-Díez RR, Rayego-Mateos S, Suarez-Alvarez B, Lavoz C, Stark Aroeira L, Sánchez-López E, Orejudo M, Alique M, Lopez-Larrea C, Ortiz A, Egido J, and Ruiz-Ortega M. The C-terminal module IV of connective tissue growth factor is a novel immune modulator of the Th17 response. Lab Invest 93: 812–824, 2013 [DOI] [PubMed] [Google Scholar]
- 53.Rodriguez-Vita J, Ruiz-Ortega M, Rupérez M, Esteban V, Sanchez-López E, Plaza JJ, and Egido J. Endothelin-1, via ETA receptor and independently of transforming growth factor-beta, increases the connective tissue growth factor in vascular smooth muscle cells. Circ Res 97: 125–134, 2005 [DOI] [PubMed] [Google Scholar]
- 54.Ruiz-Ortega M. and Ortiz A. Angiotensin II and reactive oxygens pecies. Antioxid Redox Signal 7: 1258–1260, 2005 [DOI] [PubMed] [Google Scholar]
- 55.Ruiz-Ortega M, Esteban V, Rupérez M, Sánchez-López E, Rodríguez-Vita J, Carvajal G, and Egido J. Renal and vascular hypertension-induced inflammation: role of angiotensin II. Curr Opin Nephrol Hypertens 15: 159–166, 2006 [DOI] [PubMed] [Google Scholar]
- 56.Ruiz-Ortega M, Rodríguez-Vita J, Sanchez-Lopez E, Carvajal G, and Egido J. TGF-beta signaling in vascular fibrosis. Cardiovasc Res 74: 196–206, 2007 [DOI] [PubMed] [Google Scholar]
- 57.Rupérez M, Lorenzo O, Blanco-Colio LM, Esteban V, Egido J, and Ruiz-Ortega M. Connective tissue growth factor is a mediator of angiotensin II-induced fibrosis. Circulation 108: 1499–1505, 2003 [DOI] [PubMed] [Google Scholar]
- 58.Sánchez-López E, Rodriguez-Vita J, Cartier C, Rupérez M, Esteban V, Carvajal G, Rodrígues-Díez R, Plaza JJ, Egido J, and Ruiz-Ortega M. Inhibitory effect of interleukin-1 on angiotensin II-induced connective tissue growth factor and type IV collagen production in cultured mesangial cells. Am J Physiol Renal Physiol 294: F149–F160, 2008 [DOI] [PubMed] [Google Scholar]
- 59.Sánchez-López E, Rayego S, Rodrigues-Díez R, Rodriguez JS, Rodrigues-Díez R, Rodríguez-Vita J, Carvajal G, Aroeira LS, Selgas R, Mezzano SA, Ortiz A, Egido J, and Ruiz-Ortega M. CTGF promotes inflammatory cell infiltration of the renal interstitium by activating NF-kappaB. J Am Soc Nephrol 20: 1513–1526, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sanz AB, Sanchez-Niño MD, Ramos AM, Moreno JA, Santamaria B, Ruiz-Ortega M, Egido J, and Ortiz A. NF-kappaB in renal inflammation. J Am Soc Nephrol 21: 1254–1262, 2010 [DOI] [PubMed] [Google Scholar]
- 61.Savoia C. and Schiffrin EL. Vascular inflammation in hypertension and diabetes: molecular mechanisms and therapeutic interventions. Clin Sci (Lond) 112: 375–384, 2007 [DOI] [PubMed] [Google Scholar]
- 62.Schreier B, Gekle M, and Grossmann C. Role of epidermal growth factor receptor in vascular structure and function. Curr Opin Nephrol Hypertens 23: 113–121, 2014 [DOI] [PubMed] [Google Scholar]
- 63.Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, and Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res 91: 406–413, 2002 [DOI] [PubMed] [Google Scholar]
- 64.Sirker A, Zhang M, and Shah AM. NADPH oxidases in cardiovascular disease: insights from in vivo models and clinical studies. Basic Res Cardiol 106: 735–747, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tousoulis D, Kampoli AM, and Stefanadis C. Diabetes mellitus and vascular endothelial dysfunction: current perspectives. Curr Vasc Pharmacol 10: 19–32, 2012 [DOI] [PubMed] [Google Scholar]
- 66.Touyz RM, Briones AM, Sedeek M, Burger D, and Montezano AC. NOX1 isoforms and reactive oxygen species in vascular health. Mol Interv 11: 27–35, 2011 [DOI] [PubMed] [Google Scholar]
- 67.Van Buul JD, Fernandez-Borja M, Anthony EC, and Hordijk PL. Expression and localization of NOX2 and NOX4 in primary human endothelial cells. Antioxid Redox Signal 7: 308–317, 2005 [DOI] [PubMed] [Google Scholar]
- 68.Van der Heiden K, Cuhlmann S, Luong le A, Zakkar M, and Evans PC. Role of nuclear factor kappaB in cardiovascular health and disease. Clin Sci (Lond) 118: 593–605, 2010 [DOI] [PubMed] [Google Scholar]
- 69.Virdis A, Duranti E, and Taddei S. Oxidative stress and vascular damage in hypertension: role of angiotensin II. Int J Hypertens 916310, 2011. DOI: 10.4061/2011/916310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang X, McLennan SV, Allen TJ, and Twigg SM. Regulation of pro-inflammatory and pro-fibrotic factors by CCN2/CTGF in H9c2 cardiomyocytes. J Cell Commun Signal 4: 15–23, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang R, Xu YJ, Liu XS, Zeng DX, and Xiang M. Knockdown of connective tissue growth factor by plasmid-based short hairpin RNA prevented pulmonary vascular remodeling in cigarette smoke-exposed rats. Arch Biochem Biophys 508: 93–100, 2011 [DOI] [PubMed] [Google Scholar]
- 72.Wu S-H, Lu C, Dong L, and Chen Z-Q. Signal transduction involved in CTGF-induced production of chemokines in mesangial cells. Growth Factors 26: 192–200, 2008 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.