

Abstract
Despite advances in the treatment of HIV infection, heterosexual transmission of HIV remains high, and vaccines to prevent HIV acquisition have been unfruitful. Vaginal microbicides, on the other hand, have demonstrated considerable potential for HIV prevention, and a variety of compounds have been screened for their activity and safety as anti-HIV microbicides. Among these are the naturally occurring host defense peptides, small peptides from diverse lineages with intrinsic antiviral activity.
Naturally occurring host defense peptides with anti-HIV activity are promising candidates for vaginal microbicide development. Their structural variance and accompanying mechanistic diversity provide a wide range of inhibitors whose antiviral activity can be exerted at nearly every stage of the HIV lifecycle. Additionally, peptide modification has been explored as a method for improving the anti-HIV activity of host defense peptides. Structure- and sequence-based alterations have achieved varying success in improving the potency and specificity of anti-HIV peptides. Overall, peptides have been discovered or engineered to inhibit HIV with therapeutic indices of >1000, encouraging their advancement toward clinical trials.
Here we review the naturally occurring anti-HIV host defense peptides, demonstrating their breadth of mechanistic diversity, and exploring approaches to enhance and optimize their activity in order to expedite their development as safe and effective anti-HIV vaginal microbicides.
Keywords: Defensin, HIV transmission, host defense peptides, microbicide development, modification of peptides
Introduction
The global rate of HIV transmission remains at over two million people annually [1]. Of those infected, a rapidly growing number acquire HIV through male to female intercourse [1]. This is especially a problem in Sub-Saharan Africa and other regions where there is a high incidence of non-consensual intercourse. Here, where females remain at a greater risk, new and affordable methods for preventing vaginal HIV transmission are badly needed. HIV prophylaxis has been attempted by the use of recombinant vaccines and various formulations of microbicides. Despite nearly three decades of such research, no effective vaccine has emerged. For these reasons, improved microbicides are being pursued as a solution that could significantly reduce HIV transmission and ultimately reduce the number of AIDS-associated deaths.
Early attempts at the development of a topical HIV microbicide generally consisted of two major strategies. The first of these approaches was acidic formulations, designed to lower the pH of the vaginal milieu and thereby directly inactivate HIV; the second strategy employed a variety of surfactants, which acted by disrupting the lipid membranes of enveloped viruses such as HIV. While such strategies were shown to be effective in preventing HIV infection in a variety of in vitro studies, these treatments were often neutralized or degraded by the dynamic vaginal environment. In some cases, these treatments proved to be unsafe or even increased the risk of infection due to damage of the vaginal epithelia [2,3].
A safer alternative vaginal microbicide came later in the form of sulfated polyanionic or polysaccharide compounds, which were able to coat mucosal surfaces and protect target cells from infection [4,5]. Two of the more hopeful formulations from this category included Carraguard, a carrageenan based microbicide, and PRO2000, a sulfonated polyanionic compound shown to bind and coat both viral and host proteins. While preclinical data appeared promising, effective in vivo protection could not be achieved in phase III clinical trials [5,6]. With the disappointment of previous attempts at microbicide development, it has become evident that microbicides should work through safe, specific, and potent mechanism-based approaches, rather than the previously attempted non-specific compounds, so as to provide directed protection against viral infection while minimally affecting the contacted tissue.
This directed strategy for microbicide development has begun to yield promising results by implementing microbicide formulations containing the nucleoside analog reverse transcriptase inhibitor, tenofovir [7,8]. The first of these studies was CAPRISA 004, where a tenofovir-containing gel was found to reduce HIV transmission in South African women by as much as 54%, thus demonstrating that a topical microbicide could succeed in significantly preventing HIV infection. Despite these promising results, success is dependent on viral susceptibility to tenofovir and, with the discovery of naturally occurring drug-resistance mutations in chronically infected patients, new microbicides will be required to remain effective against the variety of HIV strains circulating among infected individuals [9].
Though still in early stages of development, naturally occurring anti-HIV peptides are surfacing as potent yet broad-spectrum biomolecules for topical microbicides. These peptides are generally under 50 amino acids, and may inhibit a variety of bacteria, fungi, and viruses. A major advantage of using naturally occurring peptides is the extensive variety of peptides that can be found. Naturally occurring host defense peptides exist across all major lineages and represent one of the most ancient and conserved forms of immunity. Among the hundreds of host defense peptides that have been isolated and characterized, many have demonstrated varying degrees of anti-HIV activity in cell culture and biochemical assays [10]. These peptides vary in size and structure, but can be categorized into three major structural classes; the α-helices, the β-sheets and hairpins, and the closed cyclic peptides. (Table 1)
Table 1. Natural peptides and their analogues inhibit HIV.
| Native Peptide |
Origin or Analogue |
Sequence | IC50 μM |
CC50 μM |
Therapeutic Index |
Mechanism of HIV Inhibition |
Ref. | |
|---|---|---|---|---|---|---|---|---|
| α-helix | cecropin A | Hyalophora cecropia | KWKLFKKIEKVGQNIRDGIIKAGPAVAVVGQATQIAL-NH2 | 2.0-3.0 | 2.0-4.0 | 1.2 | [69] | |
| mellitin | Apis mellifera | GIGAVLKVLTTGLPALISWIKRKRQQ-NH2 | 0.90 | 0.94 | 1.0 | Suppress viral gene expression | [69] | |
| LL-37 | Homo sapiens | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | 20 | >45 | >2.3 | Inhibit Reverse Transcriptase and Protease | [70, 71], A | |
| LL37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | 15 | >130 | >8.3 | B | |||
| LL13-37 | IGKEFKRIVQRIKDFLRNLVPRTES | 7.0 | >130 | >18 | B | |||
| LL17-32 | FKRIVQRIKDFLRNLV | 70 | >130 | >1.8 | B | |||
| LL-37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | 1.6 | 18 | 12 | [72] | |||
| GI-20 | GIKEFKRIVQRIKDFLRNLV | 1.1 | 23 | 21 | [72] | |||
| GI-20Q16 | GIKQFKRIVQRIKDFLRNLV | 0.91 | 14 | 15 | [72] | |||
| dermaseptin S4 | ALWMTLLKKVLKAAAKAALNAVLVGANA | 2.0 | 4.5 | 2.3 | Disrupts Virion Integrity, Attachment, Transcytosis, Dendritic-to-T-Cell transfer | [73], C, D | ||
| dermaseptin S4a | ALWMTLLKKVLKAAAKAALNAVLVGANA-NH2 | 2.0 | 5.6 | 2.8 | C | |||
| dermaseptin K4-S4a | ALWKTLLKKVLKAAAKAALNAVLVGANA-NH2 | 1.4 | 17 | 12 | C | |||
| dermaseptin S4(1-16)a | ALWMTLLKKVLKAAAK-NH2 | >19 | 19 | <1 | C | |||
| dermaseptin K4-S4-(1-16)a | ALWKTLLKKVLKAAAK-NH2 | 28 | >100 | >3.6 | C | |||
| maximin 3 | Bombina maxima | GIGGKILSGLKTALKGAAKELASTYLH | 0.56 | 4.2 | 7.6 | [74] | ||
| lysozyme | Homo sapiens | 148 aa. 14.7kDA | 0.064 | E | ||||
| HL18 | RVVRDPQGIRAWVAWRNR (98-115) | 0.062 | E | |||||
| HL9 | RAWVAWRNR (107-115) | 0.055 | E | |||||
| caerin 1.9 | Litoria chloris | GLFGVLGSIAKHVLPHVVPVIAEKL-NH2 | 1.2 | 20-25 | 19 | Virucidal | F | |
| caerin 1.1 | Litoria caerulea | GLLSVLGSVAKHVLPHVVPVIAEHL-NH2 | 7.8 | >30 | >3.8 | Virucidal | F, G | |
| maculatin 1.1 | Litoria genimaculata | GLFGVLAKVAAHVVPAIAEHF-NH2 | 11 | F, G | ||||
| ponericin L2 | Pachycondyla goeldii | LLKELWTKIKGAGKAVLGKIKGLL-NH2 | 1.4 | 25 | 18 | H, I | ||
| spinigerin | Pseudacanthot ermes spiniger | HVDKKVADKVLLLKQLRIMRLLTRL | 3.1 | >33 | >11 | I, [75] | ||
| β-sheet | gpnp | Cavia porcellus | RRCICTTRTCRFPYRRLGTCIFQNRVYTFCC | 7.9 | >130 | >16 | [76, 77] | |
| rbnp-1 | Oryctolagus cuniculus | VVCACRRALCLPRERRAGFCRIRGRIHPLCCRR | 9.7 | >130 | >13 | [76, 78-80] | ||
| ratnp-4 | Rattus norvegicus | VTCYCRRTRCGFRERLSGACGYRGRIYRLCCR | 7.4 | >130 | >18 | [76, 81] | ||
| hnp1 | Homo sapiens | ACYCRIPACIAGERRYGTCIYQGRLWAFCC | 5-20 | >8.7 | >.70 | Inhibit HIV gene exp., Upregulate CC-Chemokines, Inhibit fusion | [82, 83], J | |
| hnp2 | Homo sapiens | CYCRIPACIAGERRYGTCIYQGRLWAFCC | 5-20 | Upregulate CC-Chemokines, Inhibit Fusion | [83, 84], J | |||
| hnp3 | Homo sapiens | DCYCRIPACIAGERRYGTCIYQGRLWAFCC | 5-20 | J | ||||
| hnp4 | Homo sapiens | VCSCRLVFCRRTELRVGNCLIGGVSFTYCCTR | 2-5 | J | ||||
| hbd2 | Homo sapiens | GIGDPVTCLKSGAICHPVFCPRRYKQIGTCGLPGTKCCKKP | 2.1-9.2 | >9.2 | >1.6 | Inactivate virions, downregulate CXCR4 | K | |
| hbd3 | Homo sapiens | GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKK | 1.7-7.8 | >7.8 | >1.6 | [85], K | ||
| β-turn/hairpin | protegrin 1 | Sus scrofa | RGGRLCYCRRRFCVCVGR-NH2 (6-15, 8-13) | 0.46-10.9 | 4.6-81 | 7.5 | A, L, M, N | |
| PG-1(OH) Type I | RGGRLCYCRRRFCVCVGR-OH (6-8, 13-15) | 1.1 | 7.8 | 7.3 | M | |||
| PG-1(OH) Type II | RGGRLCYCRRRFCVCVGR-OH (6-13, 8-15) | 1.1 | 8.9 | 8.0 | M | |||
| PG-1(OH) Type III | RGGRLCYCRRRFCVCVGR-OH (6-15, 8-13) | 1.4 | 6.8 | 4.7 | M | |||
| 4-Cys(ACM)-PG-I-(OH) | RGGRLCYCRRRFCVCVGR-OH (LINEAR) | 1.6 | 180 | 120 | M | |||
| cPG2 | RGGRLCYCRRRFCVCVGR (CYCLIC) | 12.8 | 140 | 11 | N | |||
| ccPG3 | RGGCLCYCRRRFCVCVCR (CYCLIC, 3 bonds) | 12.2 | 800 | 65 | N | |||
| griffithsin | Griffithsia sp. | 121 aa. 12.7kDa | 3.6E-6-6.3E-6 | >0.78 | >2400 | Binds glycosylated gp120 | O | |
| grifonin-1 | Cha-SC-Chg-R-Chg-RSGSY-Cha-DN-Chg-R-Chg-c-NH2 | 0.19-0.55 | >20 | >54 | P | |||
| polyphemusin II | Limulus polyphemus | RRWCFRVCYKGFCYRKCR-NH2 | 0.42 | 11 | 25 | Binds CXCR4 | Q | |
| T22 | RRWCYRKCYKGYCYRKCR-NH2 | 0.080 | 13 | 170 | Q | |||
| T140 | RR-2Nal-CYRKk-PYR-Cit-CR | 0.0035 | 45 | 13000 | Q | |||
| tachyplesin I | Tachypleus tridentatus | KWCFRVCYRGICYRRCR-NH2 | >20 | 8.4 | <.42 | Binds CXCR4 | Q | |
| VIRIP | Homo sapiens | LEAIPMSIPPEVKFNKPFVF | 15 | >1000 | >68 | Binds Env - gp41 | R | |
| (-L)VIRIP | EAIPMSIPPEVKFNKPFVF | >1000 | >1000 | R | ||||
| VIR-175 | LEAIPMSIPPEFLFGKPFVF | 1.3 | >1000 | >750 | R | |||
| VIR-353 | LEAIPCSIPpCFLFNKPFVF | 0.20 | >1000 | >5000 | R | |||
| alpha-MSH | Homo sapiens | SYSMEHFRWGKPV | 10 | NF-kB Inhibitor | [86, 87], S | |||
| KPV | KPV | 10 | S | |||||
| cyclic | retrocyclin I | RCICGRGICRCICGRGIC | 3.7 | >52 | >14 | Fusion inhibitor -lectin | T, U | |
| RC-101 | RCICGRGICRCICGKGIC | 0.03-4.5 | >260 | >120 | T | |||
| retrocyclin II | RCICGRRICRCICGRGIC | 0.52-1.6 | >15 | >14 | [88] | |||
| rtd1 | Macaca mulatta | GFCRCLCRRGVCRCICTR | 0.45-1.9 | >48 | >41 | [88-91] | ||
| rtd2 | Macaca mulatta | GVCRCLCRRGVCRCICRR | 0.83-3.2 | [88, 90] | ||||
| rtd3 | Macaca mulatta | GFCRCICTRGFCRCICTR | 0.84-2.1 | [88, 90] | ||||
| circulin a | Chassalia parvifolia | NKVCYRNGIPCGESCVWIPCISAALGCSCK | 0.04-0.26 | 0.50 | 3.3 | V, [92] | ||
| circulin b | Chassalia parvifolia | NKVCYRNGVIPCGESCVFIPCISTLLGCSCK | 0.04-0.26 | 0.50 | 3.3 | V, [92] | ||
| kalata b1 | Oldenlandia affinis | WPVCTRNGLPVCGETCVGGTCNTPGCTCS | 0.66 | 12 | 18 | W, [92] | ||
| cycloviolacin Y1 | Viola yedoenis | GGTIFDCGETCFLGTCYTPGCSCGNYGFCYGTN | 1.2 | >40 | 33 | W | ||
| cycloviolacin Y4 | Viola yedoenis | GVPCGESCVFIPCITGVIGCSCSSNVCYLN | 0.12 | 9.3 | 78 | W | ||
| cycloviolacin Y5 | Viola yedoenis | GIPCAESCVWIPCTVTALVGCSCSDKVCYN | 0.04 | 8.7 | 220 | W | ||
| siamycin I | Streptomyces sp. | CLGVGSCNDFAGCGYAIVCFW | 0.05-5.7 | 93-150 | 42.2 | Fusion inhibitor | [93-95] | |
| Other | indolicidin | Bos taurus | LPWKWPWWPWRR-NH2 | 47 | 18.4 | 0.4 | Virucidal, Integrase Inhibition | X |
| gramicidin d | Bacillus brevis | VGAlAvVvWlWlWlW-ETA | 8.2E-6 | 8.2E-3 | 1000 | [96, 97], Y |
A comprehensive compilation of natural peptides that inhibit HIV is shown, along with their structural motif, source, derived analogues, and sequence. For larger proteins whose activity is attributed to a smaller peptide domain, the parent protein and its derivatives are assigned according to the active domain structure. Structural classification is nonexclusive, as some cyclic peptides (e.g. retrocyclins and RTDs) contain β-turn motifs, and β-turns can also be considered β-sheets. Some peptides (e.g. VIRIP) were classified based on structural evidence rather than absolute classification. D-isomer amino acids are shown in lowercase, and non-canonical residues are abbreviated as:
Cha = (L)-Cyclohexylalanine
Chg = (L)-Cyclohexylglycine
Cit = (L)-Citrulline
2Nal = 3-(2-naphthyl)alanine
ETA = ethanolamine
Notable peptide modifications are bolded. For protegrin analogues, bond arrangements are indicated in parentheses. When available, mechanism of inhibition, IC50 and CC50 and therapeutic index are also included. All numbers were rounded to 2 significant figures. For IC50 or CC50 concentrations that were reported as a range, the mid-range value was used for calculating the therapeutic index.
Reference Key: A - Ref [38], B - Ref [37], C - Ref [16], D - Ref [19], E - Ref [18], F - Ref [15], G - Ref 17, H - Ref [14], I - Ref. [40], J - Ref [24], K - Ref. [28], L - Ref. [56], M - Ref. [57], N - Ref. [58], O - Ref. [26], P - Ref. [22], Q - Ref. [45], R - Ref. [30], S - Ref. [43], T - Ref. [29], U - Ref. [23], V - Ref. [13], W - Ref [11], X - Ref. [39], Y - Ref. [12].
Short α-helices make up a large group of peptides, including amphibian dermaseptins, maximins and caerins; mammalian cathelicidins; and insect peptides, such as mellitin. A large family of peptides, collectively known as α-and β-defensins, represents many of the anti-HIV peptides containing β-turns, while the hairpin category is comprised of relatively smaller peptides such as protegrin and polyphemusin. The cyclic structure is less commonly observed and is represented mainly by plant-derived peptides such as the circulins and cycloviolacins, as well as some bacterial peptides such as the antibiotic gramicidin S [11-13]. Another small family of cyclic peptides, the θ-defensins, has been discovered in some non-human primates. However, while characteristics such as charge and structure have been shown to be common across the majority these peptides, structure is not always indicative of how these peptides are able to prevent infections. Anti-HIV activity can be executed by many different specific mechanisms.
The prospect of naturally occurring antiviral peptides as potent anti-HIV compounds has attracted a great deal of research and continues to deliver promising results. Here, we will review the correlation between structure, mechanism, and activity of naturally occurring anti-HIV peptides. Further, we will discuss strategies for the modification of the many characterized anti-HIV peptides and how these approaches enhance their potential for clinical development.
Anti-HIV Mechanisms of Naturally Occurring Peptides
Naturally occurring antiviral peptides have been shown to exert their activity at nearly every stage of the HIV lifecycle. Despite similarities in structure and properties, naturally occurring antiviral peptides have diverse mechanisms targeting specific stages in of HIV infection (Fig. 1). The variety of unique anti-HIV properties displayed by these peptides offers great potential for modification and development into mechanism-based, HIV microbicides.
Fig. (1).
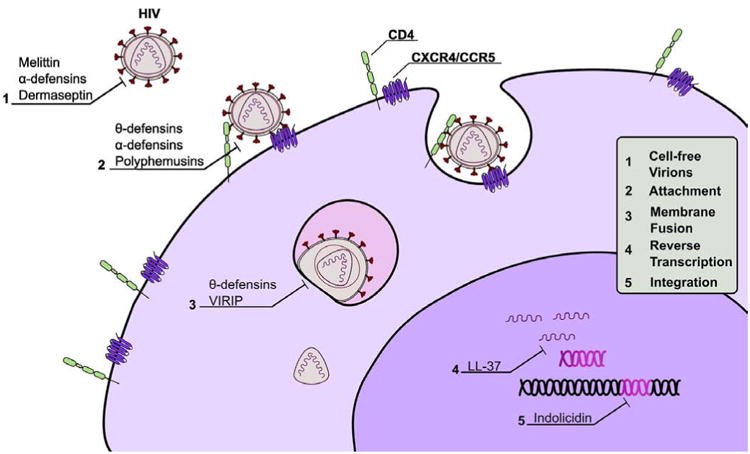
Mechanisms of anti-HIV peptides. Antiviral activity is exerted by representative peptides at the five major stages prior to infection. These stages are: direct inactivation of cell-free virions, attachment of virions to host cells at CD4 and CXCR4/CCR5, fusion of the viral envelope with the host cell membrane (shown here within the endosome), reverse transcription of viral RNA into DNA provirus, and integration of the provirus into the host genome.
A great majority of naturally occurring anti-HIV peptides prevent the initial steps of viral entry into host cells present at the sites of infection. The ability to inhibit HIV at an early stage makes these peptides desirable for development into microbicides. Peptides that inhibit HIV through direct inactivation are often α-helical, and are isolated from arthropod venom or amphibian skin [14-16]. The lytic activity of these peptides against enveloped viruses has been reported as a mechanism for the amphibian-derived caerins and dermaseptins, as well as the active fragment of human lysozyme [17-19]. Here, inhibition of HIV is often attributed to peptide-lipid interactions involving oligomerization of lytic peptides in bilayers and subsequent disruption of membrane integrity [20,21]. Interestingly, peptide-mediated membrane disruption has been shown in biophysical studies to occur through interactions between negatively charged bacterial lipids and cationic host defense peptides [20]. However, when used at concentrations meant to disrupt the host-derived lipids of enveloped viruses, these peptides will disrupt the mammalian cell membranes as well. It is likely for this reason that peptides whose anti-HIV activity remains solely dependent on lipid-peptide interactions, such as honeybee mellitin and various amphibian peptides, possess a low therapeutic index and exhibit a relatively high degree of cytotoxicity (Fig. 2). The observation that many peptides that directly inactivate HIV also maintain a relatively high degree of cytotoxicity suggests that secondary mechanisms, in addition to HIV envelope penetration/disruption, may be necessary for microbicide development. It is therefore understandable that several more HIV-specific mechanisms, in addition to membrane disruption, have also been observed for many anti-HIV peptides, in particular those that directly interfere with the HIV entry process.
Fig. (2).
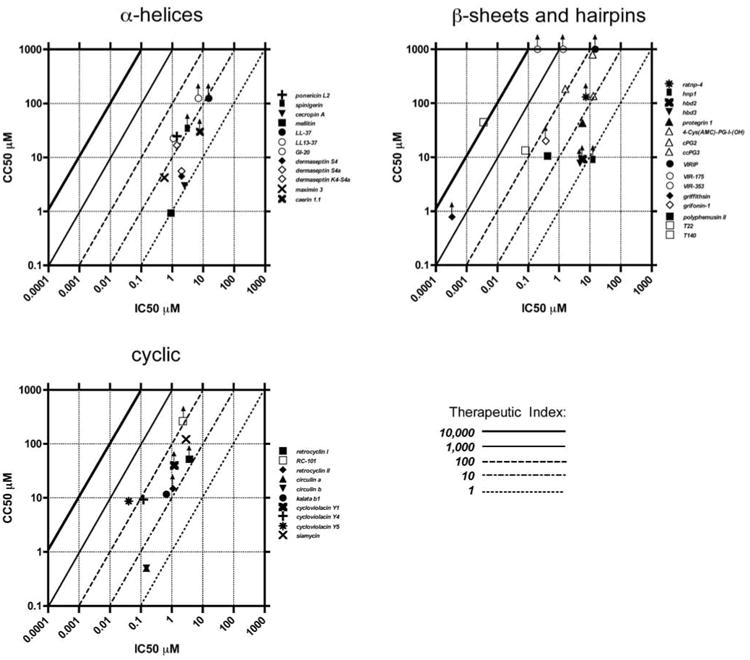
Therapeutic indices of naturally occurring anti-HIV peptides and their analogues. Anti-HIV peptides with available therapeutic indices are plotted as IC50 against CC50. For IC50 or CC50 concentrations that were reported as a range, the mid-range value was plotted. For peptides whose CC50 was not reached experimentally, the highest concentration shown to be nontoxic was used as a CC50 and the plotted symbol is appended with an upward arrow indicating that its CC50 could be higher than the plotted value. Native peptides are closed symbols, and modified analogues are open symbols. Therapeutic index thresholds are indicated as lines at each 10-fold increment.
HIV entry occurs through a well-characterized, yet complex multi-step mechanism. The surface envelope protein, gp120, first engages the host cells by binding to the CD4 receptor present on monocytes, macrophages, dendritic cells, and the CD4+ T-cells. This initial binding brings gp120 into close proximity with one of two major co-receptors recognized by HIV: the chemokine receptors CCR5 and CXCR4. After binding to a co-receptor the viral envelope undergoes a conformational change exposing the fusion protein gp41, which then inserts its N-terminus into the host membrane. At this stage, which has been shown to occur at either the plasma membrane or within endosomes, gp41 mediates the fusion of the host and viral membranes allowing the release of the viral capsid into the host cytosol. This multistep process provides critical points at which infection can be blocked by several anti-HIV peptides.
Peptides shown to bind viral gp120, such as θ-defensins and the Griffithsia-derived grifonin-1, prevent the virus's initial engagement of the host cell [22-24]. This activity may be related to the lectin properties of these molecules combined with the prominent glycosylation of gp120. Lectin activity of naturally occurring antiviral peptides is particularly promising considering the success of several large protein lectins in reducing HIV transmission in organotypic models [25,26]. Another advantage of peptides with lectin activity is that while mutations in envelope proteins may provide resistance to many drugs targeting gp120, molecules with the ability to bind glycosylated proteins should retain their broad-spectrum activity.
HIV co-receptors CXCR4 and CCR5 may also act as targets for antiviral peptides, including horseshoe crab tachyplesins and polyphemusin II and human β-defensins 2 and 3 [27,28]. Polyphemusin II directly binds CXCR4, thus preventing the interaction of HIV gp120 with its host cell target. Beta-defensins 2 and 3 employ a similar mechanism, as they both bind the co-receptor CXCR4, preventing initial HIV engagement of host cells. Additionally, they induce CXCR4 internalization, thus reducing the levels of this co-receptor present on the cell surface.
The final stage of HIV entry is the gp41-mediated fusion of the viral envelope with the host membrane at either the cell surface or within endosomal compartments. An important advantage of a peptide that targets fusion, rather than earlier stages in entry, is that viral tropism and the highly variable gp120 sequence rarely affect anti-HIV activity of such peptides. This is supported by reports that peptides targeting fusion have been shown to be active against a more diverse group of primary HIV isolates of varying subtypes and tropisms [29,30]. HIV fusion is the primary target of the mammalian θ-defensins, in particular the human pseudogene product retrocyclin [31,32]. Theta-defensins likely bind to the C-terminal α-helix of gp41, thereby preventing formation of the 6-helix bundle structure that mediates membrane fusion [32,33]. Research on θ-defensins as fusion inhibitors has focused primarily on retrocyclin and its synthetic analogs. Retrocyclins have been shown to not only be active against a diverse range of subtypes from clinical isolates, but they also have been shown to overcome drug resistance mutations in gp41 through only a two-fold increase in peptide concentration [29,31]. In addition to their anti-HIV activity, retrocyclins are not cytotoxic at relatively high concentrations and do not elicit a host response while remaining active in both organotypic tissue and non-human primate models [34,35]. Another naturally occurring peptide shown to inhibit HIV fusion is a 20-residue cleavage product of human α1-antitrypsin known as VIRus Inhibitory Peptide (VIRIP). This peptide possesses a very specific mechanism whereby it forms a complex with a conserved domain of gp41, thus preventing the initial insertion of gp41 into the host membrane [30]. Where other peptide fusion inhibitors act by binding the N and C terminal heptad repeats of gp41, VIRIP is unique in that it binds to the hydrophobic “fusion peptide” region.
In addition to exhibiting extracellular activity, antiviral mechanisms of some peptides extend to target the early stages of intracellular HIV infection. Such peptides may still serve as effective microbicides if they can prevent successful integration of HIV into the host's genomic DNA. Indolicidin, a peptide of bovine origin, has been shown to prevent infection by specifically inhibiting integration of reverse-transcribed viral DNA into the host genome [36]. Components of the human cathelicidin peptide, LL-37, have also been shown to carry out their activity through inhibition of reverse transcriptase and the viral protease [37]. While mechanisms have been identified for both of these peptides using solely biochemical assays, it is important to note that they also possess anti-HIV activity in infected cell culture experiments [38,39].
While many of these peptides remain in experimental stages, the mechanisms that they employ can all prevent the initial infection that would otherwise lead to acquisition of HIV. Obtaining a mechanistic understanding of these peptides can be useful to develop modifications leading to greater anti-HIV activity, decreased cytotoxicity, and improvement of expression/synthesis. Such modifications will likely be critical for development of naturally occurring peptides as microbicides.
Modification of Naturally Occurring Anti-HIV Peptides
The abundance of host defense peptides with diverse mechanisms of antiviral activity has supported their development as anti-HIV prophylactic microbicides. While such peptides are lauded for their ubiquitous occurrence across diverse lineages and their relatively broad spectrum antimicrobial activity, the challenge for microbicide development is in finding and isolating peptides that demonstrate potent and specific HIV inhibition; these compounds must not demonstrate toxicity to host cells or endogenous flora. The specificity of antiviral compounds is expressed as their therapeutic index, the ratio of their CC50 (concentration at which host cell viability/proliferation is reduced by 50%) to their IC50 (concentration at which viral infection is reduced by 50%) [40]. This is a useful gauge for the clinical potential of anti-HIV peptides. Some antiviral candidates exhibit desirable therapeutic indices as natural isolates (e.g. griffithsin, with an IC50 >1000) [26]. Other compounds may not be as promising in their native form, but they can be drastically improved by engineered modifications to achieve favorable therapeutic indices (e.g. VIRIP, whose therapeutic index was enhanced from >68 to >5000 by sequence optimization) [30].
Various techniques have been explored in an effort to increase the therapeutic index of anti-HIV peptides via sequence-based and structural modifications. Further, several delivery approaches have been employed to improve the application and stability of anti-HIV peptides. In this section we will review the attempts and outcomes of several such modifications in order to evaluate techniques aimed at enhancing the therapeutic index of anti-HIV peptides and their clinical development as topical microbicides.
Many host defense peptides exhibit anti-HIV activity in their native form, whereas other peptides require modifications in order to meet the characteristics of an anti-HIV microbicide candidate. One straightforward technique for antiviral peptide improvement is the identification of active domains. In many cases, the antiviral activity of a large protein is attributed to only a small peptide fragment. Thus, the recombinant expression or synthetic production of the active portion is a practical way to streamline production and delivery of the antiviral peptide. This approach was demonstrated by the identification and isolation of the active portions of human anti-HIV proteins LL-37, lysozyme, and α-melanocyte-stimulating hormone (α-MSH).
The 14.7 kDa host defense protein lysozyme inhibits a wide range of microbes [41]. The protein as a whole exhibits anti-HIV activity at nanomolar concentrations, but this activity can be distilled down to a 9-amino acid peptide sequence, RAWVAWRNR. This peptide adopts an α -helical structure that inhibits HIV infection with approximately the same potency as full-length lysozyme [18]. Similarly, the anti-HIV activity of the human cathelicidin product LL-37 is achieved by a 25-residue region of the C-terminus. This fragment inhibits the activity of HIV reverse transcriptase enzyme with a lower IC50 than the intact LL-37 protein (7μM, down from the original 15 μM) [37]. Thus, isolating the active domain of an antiviral protein can increase its potency.
An extreme instance of active domain isolation is demonstrated by the antiviral peptide α-MSH. Alpha-MSH is a human anti-inflammatory protein expressed by keratinocytes and monocytes, among other cell types. Importantly, microbicides that exhibit anti-inflammatory activity may perform dual functions in preventing HIV transmission. First, they can suppress HIV infection by inhibiting the activation of the proinflammatory transcription complex NF-κB, which otherwise drives replication of HIV by binding the proviral long terminal repeat and promoting transcription of the viral genome [42]. Second, by suppressing inflammation and reducing recruitment of immune cells to mucosal tissues, these peptides minimize the pool of potential CD4+ target cells that could become infected by invading HIV virions. In the case of the antiinflammatory protein α-MSH, antiviral activity can be achieved by only a three amino acid sequence, KPV, which suppresses NF-κB activation and concomitant HIV infection at equimolar concentrations in comparison to the full-length α-MSH protein [43]. These examples illustrate how active domain isolation can simplify antiviral peptide production by focusing expression and isolation to only the relevant domains.
In other cases, antiviral activity is exerted by a more complex structure and cannot be fully recapitulated by a smaller peptide derivative. Such is the case of the small peptide mimetic grifonin-1, which was modeled after the 12.7 kDa antiviral protein griffithsin. Griffithsin is isolated from Griffithsia sp. of red algae, and structural analysis suggests that its antiviral activity is achieved by three glycan-binding motifs that bind the envelope glycoprotein gp120 to inhibit viral attachment to host cells [26,44]. Grifonin-1 is a peptide mimetic of the proteoglycan-binding β-turn domains of griffithsin and was constructed using non-canonical residues to optimize its stability and efficacy. Yet in this instance, the peptide mimetic suffered a 1000-fold decrease in antiviral activity compared to the parent protein when assayed at equimolar concentrations [22]. Still, its therapeutic index remains high, and because of its smaller size, it may prove to possess advantages in delivery over its parent protein griffithsin.
In addition to active domain isolation, intramolecular modifications that alter peptide sequence are often employed in hopes of improving the therapeutic index of anti-HIV peptides. Such modifications have been explored for antiviral peptides isolated from a myriad of sources and exhibiting diverse structures, with varying success. One group of antiviral peptides that has had little success in achieving promising therapeutic indices is comprised of peptides exhibiting direct virucidal activity by membrane lysis. As discussed above, membrane-disrupting anti-HIV peptides have poor therapeutic indices in their native forms. These permeabilizing peptides have been mutated in an effort to increase their therapeutic potential, which has been relatively unsuccessful. This is due to the difficulty of selectively targeting the peptides' lytic activity to viral membranes that are derived from host cell membranes over the host cells themselves. The difficulty of selectively directing antimicrobial peptides to the HIV membrane is evidenced by the peptides dermaseptin S4, the caerins, and indolicidin [15,16,39]. These peptides all execute antiviral activity by disrupting the viral membrane, and all are active in the micromolar range. Yet none of these molecules, nor their derivatives, achieve a therapeutic index of >20, as each induces cytotoxicity near its active concentration. Thus, anti-HIV peptides that function by lysis of viral membranes will likely be limited in therapeutic potential due to their corresponding disruption of host cell membranes.
While lytic peptides are unlikely to achieve specific inhibition of virions over host cells, the majority of anti-HIV peptides exert their inhibitory activity by mechanisms distinct from direct membrane disruption. For example, many antiviral peptides interact with host cell receptors or surface molecules, with viral surface proteins, or they may influence intracellular processes such as receptor trafficking or transcriptional regulation. In the case of such specific interactions, antiviral activity should be individually engineered to enhance peptide affinity for its target molecule. Such an approach was employed for optimization of the horseshoe crab antimicrobial peptide, polyphemusin II.
Polyphemusin II exhibits anti-HIV activity at submicromolar concentrations by binding to the host cell chemokine receptor CXCR4, one of two coreceptors utilized by HIV to enter host cells [27,45]. However the therapeutic index of native polyphemusin II is 25, limiting its potential for development. Yet a peptide with a defined mechanism but undesirable toxicity posed an opportunity for improvement, and through an extensive series of sequence and structural modifications, over 100 analogues of polyphemusin II have been constructed and therapeutic indices of >10,000 have been achieved [45-50]. Notably successful analogues include T22 (therapeutic index = 170) and T140 (therapeutic index = 13,000) [51]. While these modifications have produced potent and specific inhibitors, the prophylactic potential of these peptide analogues is still limited due to their restricted activity against only X4-tropic viruses. This is especially limiting since nearly all HIV founder strains (viral strains responsible for establishing initial infection) utilize the chemokine coreceptor CCR5 rather than CXCR4 to attach and fuse to host cells [52,53].
Yet the survey of sequence substitution and incorporation of non-canonical residues in polyphemusin II analogues paved the way for the optimization of other antiviral peptides. For example, while enantiomeric amino acid residues occur naturally in some antiviral peptides (e.g. gramicidin), they are now also frequently engineered into modified analogues of other antiviral peptides (e.g. analogues grifonin-1, VIR-353 and VIR-449) [22,30,54]. Additionally, synthesized analogues are now often engineered to contain non-canonical residues such as citrulline, naphthylalanine, cyclohexylalanine, and cyclohexylglycine to expand the available repertoire of structural building blocks [22,50]. This has allowed for a broader spectrum of physical characteristics to be incorporated into antiviral peptide analogues, which in turn increases our ability to optimize their anti-HIV activity. In addition to optimizing the therapeutic profile of analogues, incorporation of such non-classical residues may also enhance the stability of microbicides by imparting resistance to stereospecific proteolytic degradation [55].
While sequence substitutions have elucidated some useful trends and approaches, structural modifications aimed at improving antiviral peptides have been more difficult to interpret. As an example, the porcine peptide protegrin has been extensively modified by structural rearrangements that displayed varying success in improving antiviral activity. Protegrin is an 18 amino acid antimicrobial peptide that in its native form contains four cysteines, which are linked by two disulfide bonds to stabilize a β-hairpin structure [56]. Tamamura and colleagues engineered a series of disulfide-linked protegrin variants to construct each possible arrangement of two disulfide linkages between the four cysteines. Surprisingly, these bond rearrangements and accompanying major structural changes did little to alter antiviral activity or cytotoxicity to host cells [57].
On the other hand, Tam and colleagues cyclized protegrin, which slightly enhanced the therapeutic index by subduing cytotoxicity to mammalian cells. However, when a third disulfide bond was incorporated by introducing two additional cysteine residues in the cyclic analogue, the CC50 was improved another 6-fold, resulting in nearly a 9-fold increase in therapeutic index compared to native protegrin [58]. This result suggests that the stabilized hairpin motif might be important for executing anti-HIV activity. Yet, in surprising contrast to the improved therapeutic profile achieved by the stabilized cyclization of protegrin, complete elimination of all disulfide bonds was even more successful in enhancing the therapeutic index of protegrin; a linearized analogue achieved an overall improvement >15-fold compared to native protegrin, arguing that structural rigidity is likely not an important determinant of antiviral activity [57]. Thus, apparently disparate structural alterations achieved similar enhancement of therapeutic potential for the anti-HIV peptide protegrin. The interpretation of these results is confounded by the lack of a recognized antiviral mechanism for protegrin; without understanding the critical molecular interactions that allow protegrin to inhibit HIV, it is increasingly difficult to rationalize and predict the effect of structural rearrangements on the therapeutic profile of antiviral peptides. Protegrin modifications stand in contrast to the engineered analogues of polyphemusin II, which achieved exceptional therapeutic indices on account of a mechanistic understanding of its antiviral activity. These contrasting examples highlight the importance of elucidating the mechanism of viral inhibition in order to successfully design improved anti-HIV peptides.
Through extensive substitutions and rearrangements, a pool of promising antiviral peptides has been assembled, some with therapeutic indices exceeding 1000. Once an antiviral peptide has demonstrated safety and efficacy in vitro, a suitable application system must be formulated for its mucosal delivery to be evaluated in animal and clinical studies. These formulations can be tailored to ensure stability and safety for the intended exposed surfaces (vaginal, penile, rectal), and they have evolved from simple gel and cream suspensions to dissolvable films for mucosal application and vaginal rings for slower release of antimicrobial compounds with fewer reapplications [59]. These formulation options allow microbicides to be designed for immediate application pre-coitus or for continual delivery in the form of long-lasting rings.
Recently, another technique has surfaced in the vaginal microbicide field; the recombinant expression of antimicrobial proteins by transgenic lactobacilli is being explored as a delivery option. Lactobacillus species comprise the majority of the endogenous commensal vaginal microbiota. These probiotic bacteria produce antimicrobial bacteriocins, proteins that have been shown to inhibit urogenital pathogens [60]. Lactobacilli are also known to contribute lactic acid and hydrogen peroxide to the vaginal canal, which can prevent HIV by direct viral inhibition or indirectly by inhibiting pathogenic infections that increase host susceptibility to HIV acquisition [61-63]. In fact, pathogenic conditions in which lactobacilli are depleted from the vaginal lumen are accompanied by increased rates of HIV transmission, such as the microbial shift condition bacterial vaginosis, in which displacement of vaginal lactobacilli is accompanied by a 60% increased rate of HIV infection [64]. As the benefits of these commensal flora have been elucidated, the past decade has seen an increase in studies to supplement the natural antiviral activity of the vaginal microbiome and even to enhance it through recombinant techniques.
To this end, the expression of antiviral peptides by probiotic lactobacilli has been eagerly explored. The transformation of lactobacilli has been improved, and it has been shown that these bacteria can achieve superior folding of recombinant antiviral proteins compared to mammalian expression systems [65]. Furthermore, recombinant fusion inhibitor peptides corresponding to the heptad repeat-2 region of the HIV envelope protein gp41 were expressed by these transgenic bacteria, and they successfully inhibited HIV fusion in vitro [66]. Initial in vivo studies have demonstrated successful colonization of the human vaginal canal when lactobacilli were administered in repeated doses [67]. Most recently, recombinant lactobacilli expressing the antiviral protein cyanovirin-N were administered vaginally to macaques, which successfully prevented vaginal infection by SHIV up to 63% [68]. This rate of inhibition is likely a combined effect of the endogenous protective factors contributed by the lactobacilli in addition to the antiviral protein they were engineered to recombinantly express. Thus, in addition to accomplishing sustained delivery of antiviral compounds, the intravaginal application of transgenic lactobacilli has the dual advantage of also bolstering the endogenous protective barrier of the female reproductive tract. This merits further investigation as a clinical strategy for vaginal microbicide delivery.
Looking ahead, the isolation and optimization of natural peptides with potent and specific anti-HIV activity support their development as prophylactic topical microbicides. Considering the failure of cytotoxic detergent-based microbicides to provide safe and effective protection against HIV, the superior therapeutic indices and diverse mechanisms of action of naturally occurring anti-HIV peptides provide a spectrum of promising antiviral candidates. As experience and understanding of these peptides accumulate, our ability to enhance their activity brings them ever nearer to clinical development as anti-HIV microbicides.
Acknowledgments
The authors would like to thank Michele A. Costello for her helpful revisions of this manuscript.
This work was supported in part by grants AI052017, AI082623, and AI082693 from the National Institutes of Health.
Footnotes
Conflict of Interest: The authors declare no conflicts of interest.
References
- 1.UNAIDS. [cited 2010 Sept 12];Global Report: UNAIDS Report on the Global AIDS Epidemic. [homepage on the Internet]. c2010 [updated 2010. Available from: http://www.unaids.org/globalreport/documents/20101123_GlobalReport_full_en.pdf.
- 2.Mauck C, Ballagh S, Creinin M, et al. Six-day randomized safety trial of intravaginal lime juice. J Acquir Immune Defic Syndr. 2008;49(3):243–50. doi: 10.1097/QAI.0b013e318186eae7. [DOI] [PubMed] [Google Scholar]
- 3.Van Damme L, Ramjee G, Alary M, et al. Effectiveness of COL-1492, a nonoxynol-9 vaginal gel, on HIV-1 transmission in female sex workers: a randomised controlled trial. Lancet. 2002;360(9338):971–7. doi: 10.1016/s0140-6736(02)11079-8. [DOI] [PubMed] [Google Scholar]
- 4.Lederman S, Gulick R, Chess L. Dextran sulfate and heparin interact with CD4 molecules to inhibit the binding of coat protein (gp120) of HIV. J Immunol. 1989;143(4):1149–54. [PubMed] [Google Scholar]
- 5.Pirrone V, Wigdahl B, Krebs F. The rise and fall of polyanionic inhibitors of the human immunodeficiency virus type 1. Antiviral Res. 2011;90(3):168–82. doi: 10.1016/j.antiviral.2011.03.176. [DOI] [PubMed] [Google Scholar]
- 6.McCormack S, Ramjee G, Kamali A, et al. PRO2000 vaginal gel for prevention of HIV-1 infection (Microbicides Development Programme 301): a phase 3, randomised, double-blind, parallel-group trial. Lancet. 2010;376(9749):1329–37. doi: 10.1016/S0140-6736(10)61086-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abdool Karim Q, Abdool Karim S, Frohlich J, et al. Effectiveness and safety of tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science. 2010;329(5996):1168–74. doi: 10.1126/science.1193748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grant R, Lama J, Anderson P, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010;363(27):2587–99. doi: 10.1056/NEJMoa1011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metzner K, Rauch P, Braun P, et al. Prevalence of key resistance mutations K65R, K103N, and M184V as minority HIV-1 variants in chronically HIV-1 infected, treatment-naive patients. J Clin Virol. 2011;50(2):156–61. doi: 10.1016/j.jcv.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 10.Wang G, Li X, Wang Z. APD2: the updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 2009;37:D933–7. doi: 10.1093/nar/gkn823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang C, Colgrave M, Gustafson K, et al. Anti-HIV cyclotides from the Chinese medicinal herb Viola yedoensis. J Nat Prod. 2008;71(1):47–52. doi: 10.1021/np070393g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bourinbaiar A, Krasinski K, Borkowsky W. Anti-HIV effect of gramicidin in vitro: potential for spermicide use. Life Sci. 1994;54(1):PL5–9. doi: 10.1016/0024-3205(94)00579-6. [DOI] [PubMed] [Google Scholar]
- 13.Gustafson K, Sowder R, Henderson L, et al. Circulins A and B: Novel HIV-Inhibitory Macrocyclic Peptides from the Tropical Tree Chassalia parvifolia. J Am Chem Soc. 1994;116:9337–8. [Google Scholar]
- 14.Orivel J, Redeker V, Le Caer J, et al. Ponericins, new antibacterial and insecticidal peptides from the venom of the ant Pachycondyla goeldii. J Biol Chem. 2001;276(21):17823–9. doi: 10.1074/jbc.M100216200. [DOI] [PubMed] [Google Scholar]
- 15.VanCompernolle S, Taylor R, Oswald-Richter K, et al. Antimicrobial peptides from amphibian skin potently inhibit human immunodeficiency virus infection and transfer of virus from dendritic cells to T cells. J Virol. 2005;79(18):11598–606. doi: 10.1128/JVI.79.18.11598-11606.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lorin C, Saidi H, Belaid A, et al. The antimicrobial peptide dermaseptin S4 inhibits HIV-1 infectivity in vitro. Virology. 2005;334(2):264–75. doi: 10.1016/j.virol.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 17.Marcotte I, Wegener K, Lam Y, et al. Interaction of antimicrobial peptides from Australian amphibians with lipid membranes. Chem Phys Lipids. 2003;122(1-2):107–20. doi: 10.1016/s0009-3084(02)00182-2. [DOI] [PubMed] [Google Scholar]
- 18.Lee-Huang S, Maiorov V, Huang P, et al. Structural and functional modeling of human lysozyme reveals a unique nonapeptide, HL9, with anti-HIV activity. Biochemistry. 2005;44(12):4648–55. doi: 10.1021/bi0477081. [DOI] [PubMed] [Google Scholar]
- 19.Zairi A, Tangy F, Bouassida K, Hani K. Dermaseptins and magainins: antimicrobial peptides from frogs' skin-new sources for a promising spermicides microbicides-a mini review. J Biomed Biotechnol. 2009;2009:452567. doi: 10.1155/2009/452567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Epand R, Epand R. Bacterial membrane lipids in the action of antimicrobial agents. J Pept Sci. 2011;17(5):298–305. doi: 10.1002/psc.1319. [DOI] [PubMed] [Google Scholar]
- 21.Lai Y, Gallo R. AMPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 2009;30(3):131–41. doi: 10.1016/j.it.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Micewicz E, Cole A, Jung C, et al. Grifonin-1: A Small HIV-1 Entry Inhibitor Derived from the Algal Lectin, Griffithsin. PLoS ONE. 2010;5(12):e14360. doi: 10.1371/journal.pone.0014360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang W, Cole A, Hong T, Waring A, Lehrer R. Retrocyclin, an antiretroviral theta-defensin, is a lectin. J Immunol. 2003;170(9):4708–16. doi: 10.4049/jimmunol.170.9.4708. [DOI] [PubMed] [Google Scholar]
- 24.Wu Z, Cocchi F, Gentles D, et al. Human neutrophil alpha-defensin 4 inhibits HIV-1 infection in vitro. FEBS Letters. 2005;579(1):162–6. doi: 10.1016/j.febslet.2004.11.062. [DOI] [PubMed] [Google Scholar]
- 25.Balzarini J. Inhibition of HIV entry by carbohydrate-binding proteins. Antiviral Res. 2006;71(2-3):237–47. doi: 10.1016/j.antiviral.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 26.Mori T, O'Keefe B, Sowder R, et al. Isolation and characterization of griffithsin, a novel HIV-inactivating protein, from the red alga Griffithsia sp J Biol Chem. 2005;280(10):9345–53. doi: 10.1074/jbc.M411122200. [DOI] [PubMed] [Google Scholar]
- 27.Murakami T, Zhang T, Koyanagi Y, et al. Inhibitory mechanism of the CXCR4 antagonist T22 against human immunodeficiency virus type 1 infection. J Virol. 1999;73(9):7489–96. doi: 10.1128/jvi.73.9.7489-7496.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quiñones-Mateu M, Lederman M, Feng Z, et al. Human epithelial beta-defensins 2 and 3 inhibit HIV-1 replication. AIDS. 2003;17(16):F39–48. doi: 10.1097/00002030-200311070-00001. [DOI] [PubMed] [Google Scholar]
- 29.Owen S, Rudolph D, Wang W, et al. RC-101, a retrocyclin-1 analogue with enhanced activity against primary HIV type 1 isolates. AIDS Res Hum Retroviruses. 2004;20(11):1157–65. doi: 10.1089/aid.2004.20.1157. [DOI] [PubMed] [Google Scholar]
- 30.Münch J, Ständker L, Adermann K, et al. Discovery and optimization of a natural HIV-1 entry inhibitor targeting the gp41 fusion peptide. Cell. 2007;129(2):263–75. doi: 10.1016/j.cell.2007.02.042. [DOI] [PubMed] [Google Scholar]
- 31.Cole A, Yang O, Warren A, et al. HIV-1 adapts to a retrocyclin with cationic amino acid substitutions that reduce fusion efficiency of gp41. J Immunol. 2006;176(11):6900–5. doi: 10.4049/jimmunol.176.11.6900. [DOI] [PubMed] [Google Scholar]
- 32.Gallo S, Wang W, Rawat S, et al. Theta-defensins prevent HIV-1 Env-mediated fusion by binding gp41 and blocking 6-helix bundle formation. J Biol Chem. 2006;281(27):18787–92. doi: 10.1074/jbc.M602422200. [DOI] [PubMed] [Google Scholar]
- 33.Fuhrman C, Warren A, Waring A, et al. Retrocyclin RC-101 overcomes cationic mutations on the heptad repeat 2 region of HIV-1 gp41. FEBS J. 2007;274(24):6477–87. doi: 10.1111/j.1742-4658.2007.06165.x. [DOI] [PubMed] [Google Scholar]
- 34.Cole A, Patton D, Rohan L, et al. The formulated microbicide RC-101 was safe and antivirally active following intravaginal application in pigtailed macaques. PLoS ONE. 2010;5(11):e15111. doi: 10.1371/journal.pone.0015111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cole A, Herasimtschuk A, Gupta P, et al. The retrocyclin analogue RC-101 prevents human immunodeficiency virus type 1 infection of a model human cervicovaginal tissue construct. Immunology. 2007;121(1):140–5. doi: 10.1111/j.1365-2567.2006.02553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marchand C, Krajewski K, Lee H, et al. Covalent binding of the natural antimicrobial peptide indolicidin to DNA abasic sites. Nucleic Acids Res. 2006;34(18):5157–65. doi: 10.1093/nar/gkl667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wong J, Legowska A, Rolka K, et al. Effects of cathelicidin and its fragments on three key enzymes of HIV-1. Peptides. 2011 doi: 10.1016/j.peptides.2011.04.017. [DOI] [PubMed] [Google Scholar]
- 38.Steinstraesser L, Tippler B, Mertens J, et al. Inhibition of early steps in the lentiviral replication cycle by cathelicidin host defense peptides. Retrovirology. 2005;2:2. doi: 10.1186/1742-4690-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robinson W, McDougall B, Tran D, Selsted M. Anti-HIV-1 activity of indolicidin, an antimicrobial peptide from neutrophils. J Leukoc Biol. 1998;63(1):94–100. doi: 10.1002/jlb.63.1.94. [DOI] [PubMed] [Google Scholar]
- 40.Wang G, Watson K, Peterkofsky A, Buckheit R. Identification of novel human immunodeficiency virus type 1-inhibitory peptides based on the antimicrobial peptide database. Antimicrob Agents Chemother. 2010;54(3):1343–6. doi: 10.1128/AAC.01448-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ganz T. Antimicrobial polypeptides. J Leukoc Biol. 2004;75(1):34–8. doi: 10.1189/jlb.0403150. [DOI] [PubMed] [Google Scholar]
- 42.Nabel G, Baltimore D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature. 1987;326(6114):711–3. doi: 10.1038/326711a0. [DOI] [PubMed] [Google Scholar]
- 43.Barcellini W, Colombo G, La Maestra L, et al. Alpha-melanocyte-stimulating hormone peptides inhibit HIV-1 expression in chronically infected promonocytic U1 cells and in acutely infected monocytes. J Leukoc Biol. 2000;68(5):693–9. [PubMed] [Google Scholar]
- 44.Emau P, Tian B, O'keefe B, et al. Griffithsin, a potent HIV entry inhibitor, is an excellent candidate for anti-HIV microbicide. J Med Primatol. 2007;36(4-5):244–53. doi: 10.1111/j.1600-0684.2007.00242.x. [DOI] [PubMed] [Google Scholar]
- 45.Xu Y, Tamamura H, Arakaki R, et al. Marked increase in anti-HIV activity, as well as inhibitory activity against HIV entry mediated by CXCR4, linked to enhancement of the binding ability of tachyplesin analogs to CXCR4. AIDS Res Hum Retroviruses. 1999;15(5):419–27. doi: 10.1089/088922299311169. [DOI] [PubMed] [Google Scholar]
- 46.DeMarco S, Henze H, Lederer A, et al. Discovery of novel, highly potent and selective beta-hairpin mimetic CXCR4 inhibitors with excellent anti-HIV activity and pharmacokinetic profiles. Bioorg Med Chem. 2006;14(24):8396–404. doi: 10.1016/j.bmc.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 47.Tamamura H, Imai M, Ishihara T, et al. Pharmacophore identification of a chemokine receptor (CXCR4) antagonist, T22 ([Tyr(5,12), Lys7]-polyphemusin II), which specifically blocks T cell-line-tropic HIV-1 infection. Bioorg Med Chem. 1998;6(7):1033–41. doi: 10.1016/s0968-0896(98)00061-3. [DOI] [PubMed] [Google Scholar]
- 48.Tamamura H, Xu Y, Hattori T, et al. A low-molecular-weight inhibitor against the chemokine receptor CXCR4: a strong anti-HIV peptide T140. Bioorg Med Chem. 1998;253(3):877–82. doi: 10.1006/bbrc.1998.9871. [DOI] [PubMed] [Google Scholar]
- 49.Tamamura H, Omagari A, Oishi S, et al. Pharmacophore identification of a specific CXCR4 inhibitor, T140, leads to development of effective anti-HIV agents with very high selectivity indexes. Bioorg Med Chem Lett. 2000;10(23):2633–7. doi: 10.1016/s0960-894x(00)00535-7. [DOI] [PubMed] [Google Scholar]
- 50.Tamamura H, Omagari A, Hiramatsu K, et al. Certification of the critical importance of L-3-(2-naphthyl)alanine at position 3 of a specific CXCR4 inhibitor, T140, leads to an exploratory performance of its downsizing study. Bioorg Med Chem. 2002;10(5):1417–26. doi: 10.1016/s0968-0896(01)00419-9. [DOI] [PubMed] [Google Scholar]
- 51.Tam J, Lu Y. A biomimetic strategy in the synthesis and fragmentation of cyclic protein. Protein Sci. 1998;7(7):1583–92. doi: 10.1002/pro.5560070712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salazar-Gonzalez J, Salazar M, Keele B, et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J Exp Med. 2009;206(6):1273–89. doi: 10.1084/jem.20090378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keele B, Giorgi E, Salazar-Gonzalez J, et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci USA. 2008;105(21):7552–7. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burkhart B, Gassman R, Langs D, et al. Gramicidin D conformation, dynamics and membrane ion transport. Biopolymers. 1999;51(2):129–44. doi: 10.1002/(SICI)1097-0282(1999)51:2<129::AID-BIP3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 55.Owen S, Rudolph D, Wang W, et al. A theta-defensin composed exclusively of D-amino acids is active against HIV-1. J Pept Res. 2004;63(6):469–76. doi: 10.1111/j.1399-3011.2004.00155.x. [DOI] [PubMed] [Google Scholar]
- 56.Kokryakov V, Harwig S, Panyutich E, et al. Protegrins: leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Letters. 1993;327(2):231–6. doi: 10.1016/0014-5793(93)80175-t. [DOI] [PubMed] [Google Scholar]
- 57.Tamamura H, Murakami T, Horiuchi S, et al. Synthesis of protegrin-related peptides and their antibacterial and anti-human immunodeficiency virus activity. Chem Pharm Bull. 1995;43(5):853–8. doi: 10.1248/cpb.43.853. [DOI] [PubMed] [Google Scholar]
- 58.Tam J, Wu C, Yang J. Membranolytic selectivity of cystine-stabilized cyclic protegrins. Eur J Biochem. 2000;267(11):3289–300. doi: 10.1046/j.1432-1327.2000.01359.x. [DOI] [PubMed] [Google Scholar]
- 59.Neff C, Kurisu T, Ndolo T, Fox K, Akkina R. A topical microbicide gel formulation of CCR5 antagonist maraviroc prevents HIV-1 vaginal transmission in humanized RAG-hu mice. PLoS ONE. 2011;6(6):e20209. doi: 10.1371/journal.pone.0020209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Daniele M, Ruiz F, Pascual L, Barberis L. Ureaplasma urealyticum and Mycoplasma hominis Sensitivity to Bacteriocins Produced by Two Lactobacilli Strains. Curr Microbiol. 2011;63(4):360–5. doi: 10.1007/s00284-011-9989-y. [DOI] [PubMed] [Google Scholar]
- 61.Atassi F, Servin A. Individual and co-operative roles of lactic acid and hydrogen peroxide in the killing activity of enteric strain Lactobacillus johnsonii NCC933 and vaginal strain Lactobacillus gasseri KS120.1 against enteric, uropathogenic and vaginosis-associated pathogens. FEMS Microbiol Lett. 2010;304(1):29–38. doi: 10.1111/j.1574-6968.2009.01887.x. [DOI] [PubMed] [Google Scholar]
- 62.Klebanoff S, Coombs R. Viricidal effect of Lactobacillus acidophilus on human immunodeficiency virus type 1: possible role in heterosexual transmission. J Exp Med. 1991;174(1):289–92. doi: 10.1084/jem.174.1.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dimitonova S, Danova S, Serkedjieva J, Bakalov B. Antimicrobial activity and protective properties of vaginal lactobacilli from healthy Bulgarian women. Anaerobe. 2007;13(5-6):178–84. doi: 10.1016/j.anaerobe.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 64.Atashili J, Poole C, Ndumbe P, Adimora A, Smith J. Bacterial vaginosis and HIV acquisition: a meta-analysis of published studies. AIDS. 2008;22(12):1493–501. doi: 10.1097/QAD.0b013e3283021a37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Secchi M, Xu Q, Lusso P, Vangelista L. The superior folding of a RANTES analogue expressed in lactobacilli as compared to mammalian cells reveals a promising system to screen new RANTES mutants. Protein Expr Purif. 2009;68(1):34–41. doi: 10.1016/j.pep.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pusch O, Kalyanaraman R, Tucker L, et al. An anti-HIV microbicide engineered in commensal bacteria: secretion of HIV-1 fusion inhibitors by lactobacilli. AIDS. 2006;20(15):1917–22. doi: 10.1097/01.aids.0000247112.36091.f8. [DOI] [PubMed] [Google Scholar]
- 67.Stapleton A, Au-Yeung M, Hooton T, et al. Randomized, placebo-controlled phase 2 trial of a Lactobacillus crispatus probiotic given intravaginally for prevention of recurrent urinary tract infection. Clin Infect Dis. 2011;52(10):1212–7. doi: 10.1093/cid/cir183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lagenaur L, Sanders-Beer B, Brichacek B, et al. Prevention of vaginal SHIV transmission in macaques by a live recombinant Lactobacillus. Mucosal Immunol. 2011 doi: 10.1038/mi.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wachinger M, Kleinschmidt A, Winder D, et al. Antimicrobial peptides melittin and cecropin inhibit replication of human immunodeficiency virus 1 by suppressing viral gene expression. J Gen Virol. 1998;79(Pt 4):731–40. doi: 10.1099/0022-1317-79-4-731. [DOI] [PubMed] [Google Scholar]
- 70.Dürr U, Sudheendra U, Ramamoorthy A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim Biophys Acta - Biomembranes. 2006;1758(9):1408–25. doi: 10.1016/j.bbamem.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 71.Johansson J, Gudmundsson G, Rottenberg M, Berndt K, Agerberth B. Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J Biol Chem. 1998;273(6):3718–24. doi: 10.1074/jbc.273.6.3718. [DOI] [PubMed] [Google Scholar]
- 72.Wang G, Watson KM, Buckheit RW., Jr Anti-human immunodeficiency virus type 1 activities of antimicrobial peptides derived from human and bovine cathelicidins. Antimicrob Agents Chemother. 2008;52(9):3438–40. doi: 10.1128/AAC.00452-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mor A, Nicolas P. Isolation and structure of novel defensive peptides from frog skin. Eur J Biochem. 1994;219(1-2):145–54. doi: 10.1111/j.1432-1033.1994.tb19924.x. [DOI] [PubMed] [Google Scholar]
- 74.Lai R, Zheng Y, Shen J, et al. Antimicrobial peptides from skin secretions of Chinese red belly toad Bombina maxima. Peptides. 2002;23(3):427–35. doi: 10.1016/s0196-9781(01)00641-6. [DOI] [PubMed] [Google Scholar]
- 75.Landon C, Meudal H, Boulanger N, Bulet P, Vovelle F. Solution structures of stomoxyn and spinigerin, two insect antimicrobial peptides with an alpha-helical conformation. Biopolymers. 2006;81(2):92–103. doi: 10.1002/bip.20370. [DOI] [PubMed] [Google Scholar]
- 76.Nakashima H, Yamamoto N, Masuda M, Fujii N. Defensins inhibit HIV replication in vitro. AIDS. 1993;7(8):1129. doi: 10.1097/00002030-199308000-00019. [DOI] [PubMed] [Google Scholar]
- 77.Selsted M, Harwig S. Purification, primary structure, and antimicrobial activities of a guinea pig neutrophil defensin. Infect Immun. 1987;55(9):2281–6. doi: 10.1128/iai.55.9.2281-2286.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ganz T, Rayner J, Valore E, et al. The structure of the rabbit macrophage defensin genes and their organ-specific expression. J Immunol. 1989;143(4):1358–65. [PubMed] [Google Scholar]
- 79.Selsted M, Brown D, DeLange R, Lehrer R. Primary structures of MCP-1 and MCP-2, natural peptide antibiotics of rabbit lung macrophages. J Biol Chem. 1983;258(23):14485–9. [PubMed] [Google Scholar]
- 80.Selsted M, Brown D, DeLange R, Harwig S, Lehrer R. Primary structures of six antimicrobial peptides of rabbit peritoneal neutrophils. J Biol Chem. 1985;260(8):4579–84. [PubMed] [Google Scholar]
- 81.Eisenhauer P, Harwig S, Szklarek D, et al. Purification and antimicrobial properties of three defensins from rat neutrophils. Infect Immun. 1989;57(7):2021–7. doi: 10.1128/iai.57.7.2021-2027.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chang T, Francois F, Mosoian A, Klotman M. CAF-mediated human immunodeficiency virus (HIV) type 1 transcriptional inhibition is distinct from alpha-defensin-1 HIV inhibition. J Virol. 2003;77(12):6777–84. doi: 10.1128/JVI.77.12.6777-6784.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Furci L, Sironi F, Tolazzi M, Vassena L, Lusso P. Alpha-defensins block the early steps of HIV-1 infection: interference with the binding of gp120 to CD4. Blood. 2007;109(7):2928–35. doi: 10.1182/blood-2006-05-024489. [DOI] [PubMed] [Google Scholar]
- 84.Guo C, Tan N, Song L, Douglas S, Ho W. Alpha-defensins inhibit HIV infection of macrophages through upregulation of CC-chemokines. AIDS. 2004;18(8):1217–8. doi: 10.1097/00002030-200405210-00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Klüver E, Schulz-Maronde S, Scheid S, et al. Structure-activity relation of human beta-defensin 3: influence of disulfide bonds and cysteine substitution on antimicrobial activity and cytotoxicity. Biochemistry. 2005;44(28):9804–16. doi: 10.1021/bi050272k. [DOI] [PubMed] [Google Scholar]
- 86.Hruby V, Sharma S, Toth K, et al. Design, synthesis, and conformation of superpotent and prolonged acting melanotropins. Ann N Y Acad Sci. 1993;680:51–63. doi: 10.1111/j.1749-6632.1993.tb19674.x. [DOI] [PubMed] [Google Scholar]
- 87.Catania A, Cutuli M, Garofalo L, et al. The neuropeptide alpha-MSH in host defense. Ann N Y Acad Sci. 2000;917:227–31. doi: 10.1111/j.1749-6632.2000.tb05387.x. [DOI] [PubMed] [Google Scholar]
- 88.Wang W, Owen S, Rudolph D, et al. Activity of alpha- and theta-defensins against primary isolates of HIV-1. J Immunol. 2004;173(1):515–20. doi: 10.4049/jimmunol.173.1.515. [DOI] [PubMed] [Google Scholar]
- 89.Tang Y, Yuan J, Osapay G, et al. A cyclic antimicrobial peptide produced in primate leukocytes by the ligation of two truncated alpha-defensins. Science. 1999;286(5439):498–502. doi: 10.1126/science.286.5439.498. [DOI] [PubMed] [Google Scholar]
- 90.Tran D, Tran P, Tang Y, et al. Homodimeric theta-defensins from rhesus macaque leukocytes: isolation, synthesis, antimicrobial activities, and bacterial binding properties of the cyclic peptides. J Biol Chem. 2002;277(5):3079–84. doi: 10.1074/jbc.M109117200. [DOI] [PubMed] [Google Scholar]
- 91.Tran D, Tran P, Roberts K, et al. Microbicidal properties and cytocidal selectivity of rhesus macaque theta defensins. Antimicrob Agents Chemother. 2008;52(3):944–53. doi: 10.1128/AAC.01090-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Derua R, Gustafson K, Pannell L. Analysis of the disulfide linkage pattern in circulin A and B, HIV-inhibitory macrocyclic peptides. Biochem Biophys Res Commun. 1996;228(2):632–8. doi: 10.1006/bbrc.1996.1708. [DOI] [PubMed] [Google Scholar]
- 93.Lin P, Samanta H, Bechtold C, et al. Characterization of siamycin I, a human immunodeficiency virus fusion inhibitor. Antimicrob Agents Chemother. 1996;40(1):133–8. doi: 10.1128/aac.40.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tsunakawa M, Hu S, Hoshino Y, et al. Siamycins I and II, new anti-HIV peptides: I. Fermentation, isolation, biological activity and initial characterization. J Antibiot. 1995;48(5):433–4. doi: 10.7164/antibiotics.48.433. [DOI] [PubMed] [Google Scholar]
- 95.Constantine K, Friedrichs M, Detlefsen D, et al. High-resolution solution structure of siamycin II: novel amphipathic character of a 21-residue peptide that inhibits HIV fusion. J Biomol NMR. 1995;5(3):271–86. doi: 10.1007/BF00211754. [DOI] [PubMed] [Google Scholar]
- 96.Dubos R. Studies on a Bactericidal Agent Extracted from a Soil Bacillus: I. Preparation of the Agent. its Activity in vitro J Exp Med. 1939;70(1):1–10. doi: 10.1084/jem.70.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Koeppe R, Providence L, Greathouse D, et al. On the helix sense of gramicidin A single channels. Proteins. 1992;12(1):49–62. doi: 10.1002/prot.340120107. [DOI] [PubMed] [Google Scholar]