

Abstract
Eukaryotic cells are composed of an intricate system of internal membranes that are organized into different compartments—including the endoplasmic reticulum (ER), the nuclear envelope, the Golgi complex (GC), lysosomes, endosomes, caveolae, mitochondria, and peroxisomes—that perform specialized tasks within the cell. The localization and dynamics of intracellular compartments are now being studied in living cells because of the availability of green fluorescent protein (GFP)-fusion proteins and recent advances in fluorescent microscope imaging systems. Results using these techniques are revealing how intracellular compartments maintain their steady-state organization and distributions, how they undergo growth and division, and how they transfer protein and lipid components between themselves through the formation and trafficking of membrane transport intermediates. This article describes methods using GFP-fusion proteins to visualize the behavior of organelles and to track membrane-bound transport intermediates moving between them. Practical issues related to the construction and expression of GFP-fusion proteins are discussed first. These are essential for optimizing the brightness and expression levels of GFP-fusion proteins so that intracellular membrane-bound structures containing these fusion proteins can be readily visualized. Next, techniques for performing time-lapse imaging using a confocal laser-scanning microscope (CLSM) are detailed, including the use of photobleaching to highlight organelles and transport intermediates. Methods for the acquisition and analysis of data are then discussed. Finally, commonly used and exciting new approaches for perturbing membrane traffic are outlined.
INSTRUMENTATION
Eukaryotic cells have internal membranes that are organized into different compartments and that perform specialized tasks within the cell (see Figs. 1 and 2). To image organelle dynamics and membrane-trafficking processes, it is useful to have a microscope system that permits the use of multiple fluorescent markers and high-resolution images. Confocal microscopy meets these requirements. In addition, most newer CLSMs permit selective photobleaching, which can be extremely useful for imaging trafficking processes. To image adherent monolayers of mammalian cells, a Plan-Neo ×40 numerical aperture (NA) 1.3 oil and a Plan-Apo ×63 NA 1.4 oil objective provide excellent resolution and efficient light collection from a thin confocal slice of a sample. For imaging organelles in thicker samples, such as Drosophila embryos, a ×63 NA 1.2 water objective can increase the available working distance.
FIGURE 1.
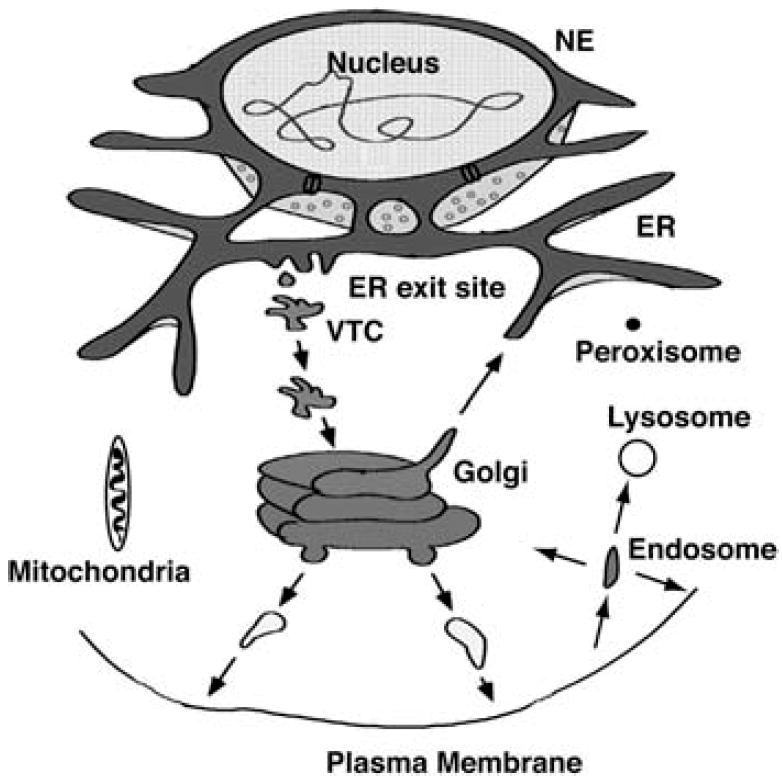
Illustration of the secretory pathway and associated organelles. Most membrane and secretory proteins are cotranslationally translocated into the endoplasmic reticulum (ER). In addition to protein processing and secretion, the ER forms the nuclear envelope (NE). Newly synthesized proteins are sorted at ER exit sites and enter vesicles to traffic to intermediate compartments (VTCs), in which proteins are sorted for movement forward to the GC or retrieval to the ER. Within the cisternal stacks of the GC, proteins are continually and selectively sorted toward the trans-Golgi network (TGN) or sent back to earlier compartments. At the TGN, proteins are sorted for trafficking to the plasma membrane, endosomes, or lysosomes. From the plasma membrane, proteins can be recycled through endosomes back to the plasma membrane or to the TGN or sorted to late endosomes and potentially to lysosomes for protein degradation. Protein trafficking to mitochondria and peroxisomes appears to be mediated posttranslationally in the absence of vesicular traffic. Mitochondria are important for ATP production and lipid synthesis. Peroxisomes play a role in lipid metabolism and help reduce oxidative radicals.
FIGURE 2.
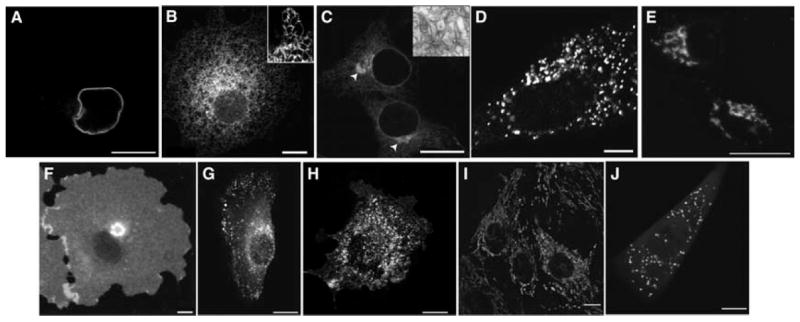
Fluorescence distributions of organelle markers in mammalian cells. (A) Nuclear envelope highlighted in COS-7 cells expressing Lamin B receptor GFP. (B) COS-7 cell expressing the ER marker GFP-cytochrome b(5), which labels a mesh-like network (inset, detail of a typical ER network). (C) COS-7 cell overexpressing cytochrome p450-mGFP, which induces proliferation of anastomosing smooth ER (white arrows and transmission EM [inset], courtesy of Maura Francolini). (D) Normal rat kidney (NRK) cell expressing the ER exit site marker Sec13-YFP. (E) MDCK (Madin–Darby canine kidney) cells expressing the GC marker GalT-GFP. (F) MDCK cell expressing the plasma membrane marker GPI–YFP. (G) NRK cell expressing the endosome marker CFP-Rab5. (H) Mouse mammary PyMT tumor cell expressing the caveolae marker Cav1-mRFP. (I) MDCK cells stained with the mitochondrial dye MitoTracker. (J) HeLa cell expressing the peroxisome marker DsRed-SKL. Scale bars, 10 μm.
Trafficking events within cells often require very fast image acquisition. The newer ultrafast CLSMs such as the Zeiss LSM5 LIVE, the PerkinElmer spinning disk, and the Nikon LiveScan SFC now permit confocal image-acquisition rates up to 180 frames per second (fps) per 512 × 512 field. Whereas vesicular trafficking studies frequently use particle-tracking software to follow individual vesicles, it is now possible to image fast enough that little ambiguity exists between the identities of vesicles between frames (Fig. 3). A protocol is available for Time-Lapse Imaging of Membrane Traffic in Living Cells (Snapp and Lajoie 2011a).
FIGURE 3.

High-speed imaging of vesicular traffic. A COS-7 cell expressing VSV-G-mCherry imaged with a Zeiss DuoScan at a single Airy unit resolution at 10 fps. Only the GC and individual trafficking vesicles are visible. Over a period of 20 sec, the pattern of vesicles changes. At slow imaging speeds, it is not easy to keep track of individual vesicles, and particle-tracking algorithms are frequently necessary to determine the path of each vesicle. In contrast, with high-speed imaging, all of the images can be combined into a maximum intensity projection, and all of the vesicle positions are superimposed to create clear paths for each vesicle. The length of each path can be measured and compared relative to time to determine the velocity of movement for each vesicle. Scale bar, 15 μm.
Temperature control is essential for maintaining cellular processes and to stabilize microscope focus. In addition, some experiments require temperature blocks or the maintenance of nonpermissive temperatures. Environmental chambers or less expensive options, such as the Model ASI 400 Air Stream Stage Incubator (Nevtek, Burnsville, Virginia), provide sufficient environmental homeostasis to permit cells to pass through mitosis. A temperature probe, such as a Thermolyne Pyrometer (Cole- Parmer, Vernon Hills, Illinois), should be used to confirm that the proper temperature is maintained at the coverslip or chamber. For tighter temperature control, our laboratory uses thermal collars (available from Bioptechs, Inc.) around the objectives.
SETTING UP THE IMAGING CHAMBER
Cells expressing GFP-fusion proteins must be grown on or attached to a glass coverslip before imaging. It is important to use the correct glass coverslip thickness for imaging cells (i.e., no. 1.5 glass coverslips for ×63 and ×100 objectives) to avoid significant spherical aberration of the image. After growing the cells on a coverslip, the coverslip must be mounted into a chamber that contains buffered imaging medium. A simple inexpensive chamber can be built by punching a hole through a silicon rubber sheet (as described in Fig. 4).
FIGURE 4.
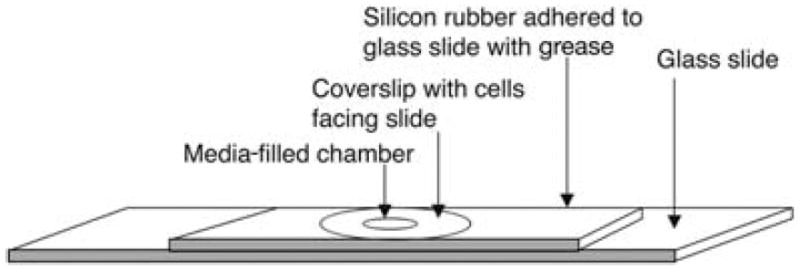
Rubber gasket imaging chamber construction. A rubber sheet with a hole is sealed to a glass slide with grease, and the hole is filled with imaging medium. The coverslip is then inverted (with attached cells placed cell side down) and pressed onto the hole, allowing the cells to face the medium. The coverslip adheres to the gasket by capillary action. Excess liquid from the top of the coverslip is wicked away with absorbant tissue.
Commercially available chambers (Lab-Tek chambers, Nunc, Rochester, New York, or MatTek dishes, MatTek Corporation, Ashland, Massachusetts) can also be used. These have a glass coverslip for a base with individual plastic chambers adhered to the coverslip. Alternatively, a microslide-containing chamber with a plastic bottom of high optical quality can be used for high-resolution microscopy (Integrated BioDiagnostics [ibidi], Munich, Germany). These chambers permit drugs or other compounds to be added during the experiments. These chambers, however, are moderately expensive and can only be used on inverted microscopes. To prevent evaporation and alkalinization of the media, the top cover of the chamber can be sealed onto the chamber using petroleum jelly or silicon grease. Alternatively, the media-filled well can be overlaid with mineral oil.
Cells grown in suspension will adhere to coverslips or Lab-Tek chambers that have been precoated for 15 min with concentrated poly-d-lysine (5–10 mg/mL in phosphate-buffered saline [PBS]). After coating, the coverslip or chamber is washed twice in PBS. The coating lasts for up to 1 wk. Suspension cells must be washed three times in PBS before allowing them to adhere to the poly-d-lysine-coated surface. Incubate cells on the surface for 2–5 min, and wash away nonadherent cells with PBS (wash twice without incubation). Finally, immerse cells in imaging buffer.
CONSTRUCTING AND EXPRESSING ORGANELLE-TARGETED GFP-FUSION PROTEINS
In live cell studies (i.e., during time-lapse imaging or serial sectioning to reconstruct three-dimensional images) of membrane-bound organelles and their trafficking pathways, multiple images of the same cell will be collected. Therefore, the brightest, most stable GFP variants available should be used in constructing a fusion protein. The enhanced green fluorescent protein (EGFP) variants Venus, Cerulean, and mCherry fit this criterion (Shaner et al. 2007).
Because GFP variants can dimerize at sufficiently high concentrations (Zacharias et al. 2002), which can perturb membrane structure (Snapp et al. 2003) or lead to false-positive interactions in cyan fluorescent protein–yellow fluorescent protein (CFP–YFP) fluorescence resonance energy transfer (FRET) experiments (Zacharias et al. 2002), the use of monomeric GFP variants is recommended (Snapp et al. 2003). Similarly, monomerized red fluorescent proteins (RFPs) are recommended (Shaner et al. 2007).
In addition to the intrinsic brightness of the GFP molecule, the GFP-fusion protein expression level should be optimized to produce a bright intracellular signal. The use of vectors with strong promoters to enhance transcription levels and the use of proper codons to optimize translation is essential for increasing expression levels of the fusion protein and thereby enhancing its overall brightness in the cell. This is particularly important when cellular autofluorescence makes it difficult to distinguish GFP-fusion protein-derived fluorescence from background.
A variety of procedures can be used to enhance the expression level and brightness of the fusion protein. However, the investigator should be cautious of overexpression artifacts such as protein aggregation or saturation of protein-targeting machinery, which lead to inappropriate localization. Another method for increasing fusion protein brightness is to construct it with double the GFP molecules in tandem (Zaal et al. 1999).
Once a GFP-fusion protein is optimally expressed within cells, the next goal is to determine whether it has targeted correctly. The simplest way to accomplish this is to compare its distribution to the parent molecule detected with fluorescent-labeled antibodies in nonexpressing cells that have been fixed and permeabilized. If the overall pattern of the two fluorescent signals is similar, then addition of the GFP tag to the parent protein has not interfered with its targeting. In the event that the two patterns are dissimilar, then it is possible that addition of the tag mistargets the protein or that the expression is so high that the machinery for targeting the fusion protein has been saturated. In the latter case, lowering the expression level of the fusion protein by decreasing the amount of DNA used during transfection, or selecting stable cell lines or less active promoters should help. It is also possible that the antibody staining does not properly reflect the parent protein distribution because the epitope recognized by the antibody is masked in some way during fixation.
The preservation of parental function by the GFP-fusion protein may be showed by showing that the fusion protein can rescue a phenotype in cells with mutations or deletions of the parent molecule or incorporation of the fusion protein into a functional macromolecular structure. When a GFP-fusion protein fails to either target or function in a similar way as the parent molecule, one solution is to change the position in which the GFP is attached to the parent molecule. For example, if the GFP is initially placed at the amino terminus, try it at the carboxyl terminus, and vice versa.
USE OF PHOTOBLEACHING TO HIGHLIGHT TRANSPORT INTERMEDIATES
When expressing GFP-fusion proteins in cells, specific organelles or structures may appear substantially brighter than other labeled structures, or two fluorescently labeled organelles in close proximity may not be easily resolved. To visualize dimmer or closely associated structures, use either inverse fluorescence recovery after photobleaching (IFRAP) or selective photobleaching. A CLSM capable of photobleaching discrete regions of interest (ROIs) is required for both of these methods. In IFRAP, the area surrounding a specific organelle is photobleached to visualize trafficking of molecules out of this structure. An example is shown in Fig. 5A, in which IFRAP revealed that glycosyl phosphatidylinositol–green fluorescent protein (GPI–GFP) molecules localized in the GC underwent rapid export and transport to the plasma membrane. In IFRAP, the protected organelle fluorescence intensity should not be saturating.
FIGURE 5.
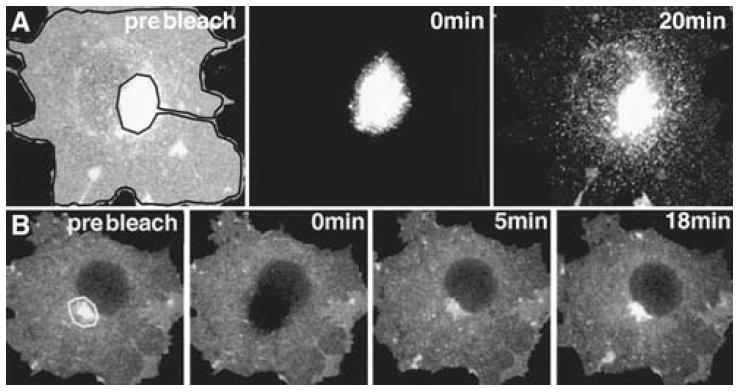
IFRAP and selective photobleaching. (A) A cell expressing a protein (GPI–GFP) that localizes and traffics between the GC and the plasma membrane is shown in the prebleach image. The fluorescence of the entire cell within the black outline is then photobleached (IFRAP), and trafficking of carriers out of the GC to the plasma membrane can be visualized. (B) Movement of fluorescent protein into the GC can be visualized when the GC fluorescence within the white outline is photobleached and the whole cell is monitored over time.
Selective photobleaching can reveal dim structures masked by bright organelles and can be used to visualize trafficking or flux through an organelle. For example, the GC (Nichols et al. 2001) expressing a GFP-fusion protein can be photobleached, and then recovery of fluorescence into the photobleached organelle can be imaged for both qualitative and quantitative analyses (see Fig. 5B). For selective photobleaching, an ROI appropriate to the organelle or structure of interest must be defined. Imaging conditions must be determined that enable photobleaching of either the structure of interest or around the structure of interest to occur while bleaching of the overall cell is avoided. When bleaching a bright structure to visualize dimmer structures, the conditions need to be set such that the dim structure(s) is sufficiently bright but not saturating. The bright structure can be saturated, as it will be photobleached. Photobleaching Regions of Living Cells to Monitor Membrane Traffic (Snapp and Lajoie 2011b) describes how to photobleach selected cellular regions using a CLSM.
USE OF PHOTOACTIVATABLE GFP
To follow a population of proteins over time, biochemists typically perform pulse-labeling experiments. Similar reagents are now available for live cell imaging. Photoactivatable and photoswitchable proteins such as PA-GFP, PS-CFP2, and mEos are fluorescent protein variants that can be optically marked to highlight proteins, organelles, or cells (Shaner et al. 2007). Each protein has a different activation protocol. Activating Photoactivatable Proteins with Laser Light to Visualize Membrane Systems and Membrane Traffic in Living Cells (Snapp and Lajoie 2011c) describes the steps for using one of the first photoactivatable proteins, PA-GFP (Patterson and Lippincott-Schwartz 2002).
ANALYSIS OF DATA
Membrane Movement
Time-lapse imaging data allow the movement of fluorescent objects to be studied. To analyze the path and velocity of an object, one must first convert the distance between two image locations in pixels to actual distance. The calibration can be performed by imaging a calibration grid using bright-field illumination and then imaging with the same objective lens and camera system used for fluorescence imaging. The distance traversed by an object of interest between two successive frames can then be calculated. Dividing this distance by the elapsed time between the frames gives the velocity. The path followed by the object can be determined by plotting the x and y coordinates at each time point.
This procedure can be simplified by writing a simple macro for ImageJ (http://rsbweb.nih.gov/ij/). The aim of the macro is to allow one to use a computer mouse to click on the object of interest in the first frame in a stack and then again in subsequent frames. As output, it should produce a text file with complete distance and velocity information in a form that is readable by a graphing program. It also should be able to produce a graph that describes the path followed by the object of interest.
Calculation of Changes in Protein Concentration
It is often useful to determine whether the number of GFP-fusion proteins that are associated with an intracellular structure changes over time because of transport or sorting processes. To accomplish this, start by identifying the object of interest and a nearby region that gives an appropriate estimate of the background contribution of fluorescence intensity of the object. Measure the average total intensity of the two regions in each frame of the time-lapse sequence. Make sure that the object of interest does not move out of the plane of focus during the experiment and that the background does not change in unexpected ways. Next, subtract background intensity from the intensity associated with the object of interest. If the total intensities are measured, the two regions should contain identical numbers of pixels or the background intensity should be appropriately normalized to account for the difference in the areas. Plot the raw values, the background values, and the background-corrected values as a function of time on a graph.
It is also useful to plot total fluorescence associated with the images as a function of time on a log-linear plot. This will reveal the extent of photobleaching, which should be a simple exponential decay. Any problems owing to focal drift, which will tend to show up as an abrupt or oscillating change in total fluorescence, can easily be determined.
MEMBRANE TRAFFICKING AND ORGANELLE REAGENTS
Drugs
A number of pharmacological reagents are available for studying organelle distribution and trafficking. In this section, several common drugs and their targets are described. The drugs are readily available from general suppliers including Sigma-Aldrich, Roche, and Calbiochem. All drugs should be aliquoted and stored for single use or made freshly, as noted.
Note that several of the listed drugs are toxic. Be sure to read the material safety data sheet, and follow safe handling precautions.
Brefeldin A
Brefeldin A (BFA) is a fungal metabolite that disrupts Arf1 function, causing COPI to be released from the GC, followed by a rapid redistribution of the GC into the ER (Sciaky et al. 1997). To disrupt the GC, cells are treated with 5 μg/mL of BFA (prepared as a 5 mg/mL stock solution in ethanol and stored at −20°C) in imaging buffer for 15–30 min at 37°C. BFA can be washed out by changing the imaging medium three times in rapid succession. The GC reforms over the course of 1 h (Lippincott-Schwartz et al. 1989).
Microtubule Disruptors: Nocodazole and Colchicine
Many of the secretory and endocytic organelles (including the ER, GC, endosomes, and lysosomes) associate with the microtubule cytoskeleton. Thus, disrupting microtubules can profoundly affect the distribution of these organelles. After microtubule disruption, the GC, for example, changes its distribution from a juxtanuclear area to hundreds of small elements localized adjacent to ER exit sites (Fig. 6) (Cole et al. 1996). To acutely disrupt microtubules, cells are incubated on ice for 10 min in the imaging chamber immersed in imaging medium with the disrupting drug. The drugs prevent microtubules from repolymerizing. Nocodazole (prepared as a 5 mg/mL stock solution in dimethylsulfoxide [DMSO] and stored at −20°C) is used at 5 μg/mL. Colchicine (prepared as a 50 mm stock solution in distilled H2O and stored at −20°C) is used at 1 mm.
FIGURE 6.

The distribution of the GC in formaldehyde-fixed HeLa cells stained with an antimannosidase II antibody and a fluorescent secondary antibody. In untreated cells, the GC is compact and restricted to a perinuclear localization. After 1 h of microtubule disruption with nocodazole treatment, the GC becomes redistributed throughout the cell at ER exit sites.
Actin-Depolymerizing Drugs
The movement of some organelles, such as melanosomes (Wu et al. 1997), and the formation of phagosomes are actin-dependent processes. To depolymerize actin, use cytochalasin B (prepared as a 10 mm stock solution in DMSO and stored at −20°C) at 1–20 μm for 15–60 min in imaging media at 37°C or latrunculin A (prepared as a 10 mg/mL stock solution in DMSO or ethanol and stored at −20°C) at 0.2–10 μg/mL in imaging medium for 1–12 h at 37°C.
Aluminum Fluoride
Aluminum fluoride (AlF) treatment causes persistent activation of heterotrimeric G proteins (Gilman 1987) and induces binding of peripheral coat proteins to GC membranes. The latter effect prevents trafficking of proteins through the secretory pathway. When applied directly to cells for 10 min, AlF will block ER to GC transport and GC to plasma membrane trafficking (Hirschberg et al. 1998). AlF is prepared fresh as a mixture of 60 μm AlCl3 and 20 mm NaF in imaging medium. Cells are incubated with the drug for 30 min to 3 h.
ATP Depletion
Many organelle functions depend on adenosine-5′-triphosphate (ATP). The one most relevant to membrane trafficking is that ATP depletion inhibits cytoskeletal motor proteins and vesicular transport. To minimize effects owing to cytotoxicity, cells should not be depleted of ATP for longer than 45 min. To deplete ATP, cells are incubated in growth medium lacking glucose (i.e., depletion medium). The medium should contain 10% dialyzed serum, 2 mm glutamine, 50 mm 2-deoxyglucose (prepared as a 1 m stock in distilled H2O and stored at 4°C), and 0.02% sodium azide (NaN3, prepared as a 1 m solution in distilled H2O and stored at room temperature). Depletion medium should be prepared fresh. Most cellular ATP is depleted within 15 min.
Protein Synthesis Inhibitors
To block new synthesis of all proteins in eukaryotes, cycloheximide (prepared as a 10 mg/mL solution in distilled H2O and stored at −20°C) can be used at 10–150 μg/mL in imaging buffer at 37°C depending on the cell type. Alternatively, puromycin (prepared as a 100 mm solution in distilled H2O and stored at −20°C), which is a structural analog of aminoacyl-tRNA, can be used at 200–1000 μm and incubated in imaging buffer for 10 min at 37°C.
Small-Molecule Compounds
Insights into membrane-trafficking events benefit from new tools to dissect molecular mechanisms of the various pathways. Small molecules (carbon-based compounds with molecular mass ≤500) have the ability to bind proteins and acutely inhibit their normal functions (Kawasumi and Ngheim 2007). The use of small molecules can provide greater flexibility than genetic methods and avoid the complications of cell adaptation to chronic loss of a protein function. High-throughput screening of libraries of chemical compounds led to the discovery of several small molecules that target, for example, microtubules (Gerdes and Katsanis 2005) or endocytic transport (Saenz et al. 2007). The variety of small molecules available to the scientific community is constantly increasing, and investigators are encouraged to scan the literature regularly for new compounds that might be useful for their research programs.
Temperature Blocks
Within the secretory pathway, it has been shown that distinct steps are differentially sensitive to temperature reduction. Thus, incubation at 20°C leads to accumulation of secretory proteins in the TGN, whereas incubation at 15°C leads to accumulation of these proteins in vesicular transport carriers (VTCs) (Griffiths et al. 1989).
Cells are incubated in imaging medium in an incubator with the appropriate level of CO2 (i.e., 5%) for 1–3 h at 15°C to block protein traffic at ER exit sites. When cells are removed from the incubator, the block can be released, and trafficking can be monitored. Cells can be imaged and manipulated at 15°C (with an objective heater or by combining a blower apparatus with a container of dry ice and blowing low-temperature air at the imaging chamber) or fixed for immunofluorescence or other imaging studies. Similarly, incubating cells at 20°C will block protein trafficking in the cis and medial GC.
Small Interfering RNA
Use of small interfering RNA (siRNA) molecules is commonly used to silence the expression of specific proteins. This method enables the inhibition of biological processes. SiRNA-mediated knockdown of various proteins involved in the secretory or endocytic pathways is a valuable method for understanding membrane trafficking within the cell. It is important to remember that protein turnover is highly variable and that the incubation period following transfection with siRNA required for significant knockdown will depend on the protein of interest. In addition, response to protein knockdownmay differ between cell types based on the capacity of the cell to adapt to the reduced expression of the targeted protein. An optimal strategy is to use both pharmacologic treatments and siRNA to determine both acute and adaptive cellular responses to the loss of activity of a trafficking effector protein.
Dyes
A wide variety of organelle-specific fluorescent dyes is available for live cell imaging. For example, dyes are available for mitochondria (MitoTracker) (Fig. 2I), lysosomes (LysoTracker), the GC (Bodipy ceramide), the ER (ER tracker), plasma membrane (FM 1-43 FX), and endosomes (rhodaminetransferrin). For a more extensive list, see the Molecular Probes/Invitrogen catalog or website (http://www.probes.com). These dyes often can be imaged in conjunction with a GFP-fusion protein. Unlike GFP, these dyes should not be photobleached (i.e., a fluorescence recovery after photobleaching [FRAP] experiment) because photobleaching of the dyes can release cytotoxic free radicals. An additional caveat is that some dyes label multiple organelles. For example, DiOC6(3) preferentially labels mitochondria at low concentrations, but at higher concentrations, it labels the ER as well. Follow the protocol supplied by the manufacturer when using one of these dyes. Changing the buffers or the dye concentration may result in nonspecific labeling.
ts045 Vesicular Stomatitis Virus Glycoprotein–GFP
One of the most popular proteins for studying membrane trafficking is the ts045 temperaturesensitive vesicular stomatitis virus glycoprotein (VSVG) (Presley et al. 1997). The protein is a glycosylated single-transmembrane-spanning protein that, on proper folding and trimerization, traffics to the plasmamembrane (Bergmann 1989). At the nonpermissive temperature of 40°C, the protein misfolds, associates with chaperones, and is retained in the ER. It is possible to accumulate the protein to high levels within the ER by incubating at 40°C. Then, on reducing the temperature to 32°C, the movement of the protein out of the ER and through successive secretory compartments can be imaged (Fig. 7) (Hirschberg et al. 1998).
FIGURE 7.
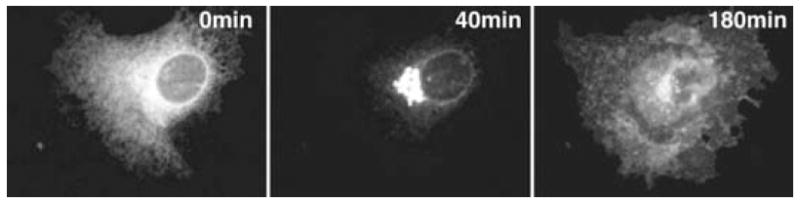
The movement of ts045 VSVG–GFP through the secretory pathway of a COS-7 cell can be visualized over time. VSVG–GFP accumulates in the ER at the nonpermissive temperature of 40°C (0 min). After shifting to the permissive temperature of 32°C, the majority of the protein has trafficked to the GC after 40 min. By 180 min, most of the protein is on the plasma membrane and endosomes.
Fluorescent Protein Alternatives
Fluorescent protein fusions are powerful tools for imaging proteins in live cells. However, they have some limitations. The relatively large size of fluorescent proteins may disrupt the localization or the functions of a protein of interest (Andresen et al. 2004). An alternative to fluorescent proteins is the use of fluorescent dyes that bind short peptides on recombinant proteins. For example, the FlAsH-EDT2 and ReAsH-EDT2 dyes (Invitrogen) become fluorescent when they bind proteins containing the tetracysteine motif Cys-Cys-Pro-Gly-Cys-Cys. These small peptide tags enable protein detection in live samples and are suitable for applications such as monitoring protein turnover and trafficking in live cells. However, the cysteines in the peptide make this labeling method inappropriate for proteins in the oxidizing environment of the lumina of secretory organelles.
References
- Andresen M, Schmitz-Salue R, Jakobs S. Short tetracysteine tags to β-tubulin demonstrate the significance of small labels for live cell imaging. Mol Biol Cell. 2004;15:5616–5622. doi: 10.1091/mbc.E04-06-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann JE. Using temperature-sensitive mutants of VSV to study membrane protein biogenesis. Methods Cell Biol. 1989;32:85–110. doi: 10.1016/s0091-679x(08)61168-1. [DOI] [PubMed] [Google Scholar]
- Cole NB, Sciaky N, Marotta A, Song J, Lippincott-Schwartz J. Golgi dispersal during microtubule disruption: Regeneration of Golgi stacks at peripheral endoplasmic reticulum exit sites. Mol Biol Cell. 1996;7:631–650. doi: 10.1091/mbc.7.4.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes JM, Katsanis N. Small molecule intervention in microtubule-associated human disease. Hum Mol Genet. 2005;14:R291–R300. doi: 10.1093/hmg/ddi269. [DOI] [PubMed] [Google Scholar]
- Gilman AG. G proteins: Transducers of receptor-generated signals. Annu Rev Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- Griffiths G, Fuller SD, Hollinshead M, Pfeiffer S, Simons K. The dynamic nature of the Golgi complex. J Cell Biol. 1989;108:277–297. doi: 10.1083/jcb.108.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschberg K, Miller CM, Ellenberg J, Presley JF, Siggia ED, Phair RD, Lippincott-Schwartz J. Kinetic analysis of secretory protein traffic and characterization of Golgi to plasma membrane transport intermediates in living cells. J Cell Biol. 1998;143:1485–1503. doi: 10.1083/jcb.143.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasumi M, Nghiem P. Chemical genetics: Elucidating biological systems with small-molecule compounds. J Invest Dermatol. 2007;127:1577–1584. doi: 10.1038/sj.jid.5700853. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Yuan LC, Bonifacino JS, Klausner RD. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: Evidence for membrane cycling from Golgi to ER. Cell. 1989;56:801–813. doi: 10.1016/0092-8674(89)90685-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki A, Sawano A, Kogure T. Lighting up cells: Labelling proteins with fluorophores. Nat Cell Biol Sep. 2003;(Suppl):S1–S7. [PubMed] [Google Scholar]
- Nichols BJ, Kenworthy A, Polishchuk RS, Lodge R, Roberts TH, Hirschberg K, Phair RD, Lippincott-Schwartz J. Rapid cycling of lipid raft markers between the cell surface and Golgi complex. J Cell Biol. 2001;153:529–542. doi: 10.1083/jcb.153.3.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson GH, Lippincott-Schwartz J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJM, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- Saenz JB, Doggett TA, Haslam DB. Identification and characterization of small molecules that inhibit intracellular toxin transport. Infect Immun. 2007;75:4552–4561. doi: 10.1128/IAI.00442-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciaky N, Presley J, Smith C, Zaal KJM, Cole N, Moreira JE, Terasaki M, Siggia E, Lippincott-Schwartz J. Golgi tubule traffic and the effects of brefeldin A visualized in living cells. J Cell Biol. 1997;139:1137–1155. doi: 10.1083/jcb.139.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Patterson GH, Davidson MW. Advances in fluorescent protein technology. J Cell Sci. 2007;120:4247–4260. doi: 10.1242/jcs.005801. [DOI] [PubMed] [Google Scholar]
- Snapp EL, Lajoie P. Time-lapse imaging of membrane traffic in living cells. Cold Spring Harb Protoc. 2011a doi: 10.1101/pdb.prot066555. [DOI] [PubMed] [Google Scholar]
- Snapp EL, Lajoie P. Photobleaching regions of living cells to monitor membrane traffic. Cold Spring Harb Protoc. 2011b doi: 10.1101/pdbprot066563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapp EL, Lajoie P. Activating photoactivatable proteins with laser light to visualize membrane systems and membrane traffic in living cells. Cold Spring Harb Protoc. 2011c doi: 10.1101/pdb.prot066571. [DOI] [PubMed] [Google Scholar]
- Snapp E, Hegde R, Colombo S, Borgese N, Francolini M, Lippincott-Schwartz J. Formation of stacked cisternae by low affinity protein interactions. J Cell Biol. 2003;163:257–269. doi: 10.1083/jcb.200306020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Bowers B, Wei Q, Kocher B, Hammer JA., 3rd Myosin V associates with melanosomes in mouse melanocytes: Evidence that myosin V is an organelle motor. J Cell Sci. 1997;110:847–859. doi: 10.1242/jcs.110.7.847. [DOI] [PubMed] [Google Scholar]
- Zaal KJM, Smith CL, Polishchuk RS, Altan N, Cole NB, Ellenberg J, Hirschberg K, Presley JF, Roberts TH, Siggia E, et al. Golgi membranes are absorbed into and reemerge from the ER during mitosis. Cell. 1999;99:589–601. doi: 10.1016/s0092-8674(00)81548-2. [DOI] [PubMed] [Google Scholar]
- Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipidmodified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]