

Abstract
Different toxic agents have a varying potential to induce the production of the proinflammatory chemokine, CXCL8 (interleukin [IL]-8), in lung cells. A critical question is which mechanisms determine the magnitude and persistence of the CXCL8 responses to different stimuli. To approach this, we compared the potential of the phorbol ester, 12-O-tetradecanoylphorbol-13-acetate (TPA), and sodium fluoride (NaF) to induce CXCL8 responses in A549 cells, with emphasis on the importance of nuclear factor kappa B (NF-κB)- and mitogen-activated protein kinase (MAPK) signaling. Notably, TPA induced a greater release of CXCL8 than did NaF. Furthermore, TPA induced a strong, rapid, but transient upregulation of CXCL8 messenger (m)RNA, whereas NaF induced a weaker, more delayed, but persistent upregulation. With respect to signaling, TPA led to an early, strong, and relatively transient extracellular signal-regulated kinase (ERK)1/2 phosphorylation, and a less marked and even more transient phosphorylation of c-jun-N-terminal kinases (JNK1/2) and p38. In contrast, NaF elicited a lower, but relatively sustained increase in phosphorylation of ERK1/2, and a marked phosphorylation of p38 and JNK1/2, with the JNK1/2 response as most transient. Only ERK1/2 inhibition affected the TPA response, whereas inhibition of all the three MAPK cascades reduced NaF-induced CXCL8 release. TPA also induced an early, marked phosphorylation/translocation of p65 (NF-κB), whereas NaF induced slower, less pronounced effects on p65. The CXCL8 responses by TPA and NaF were reduced by p65-siRNA. In conclusion, all MAPK cascades were involved in NaF-induced CXCL8 release, whereas only ERK1/2 activation was involved in response to TPA. Furthermore, NF-κB activation appeared to be indispensable for CXCL8 induction. The early response, magnitude, and persistency of MAPK and NF-κB signaling seemed to be critical determinants for the potential to induce CXCL8. These findings underscore that a strong, rapid, and relatively transient activation of ERK1/2 in combination with NF-kB may be sufficient for a strong induction of CXCL8, which may exceed the effects of a more moderate ERK1/2 activation in combination with activation of p38, JNK1/2, and NF-κB.
Keywords: TPA, sodium fluoride, CXCL8, MAPK, NF-κB, A549 cells
Introduction
The chemokine CXCL8 (interleukin [IL]-8) plays a crucial role in lung inflammation by recruiting neutrophils to the sites of injury or infection.1 Elevated CXCL8 levels are typical in airway diseases such as chronic obstructive pulmonary disease, cystic fibrosis, and severe asthma, and are believed to be an important factor in the pathogenesis of such disorders.2–4 In addition, CXCL8 also has mitogenic, motogenic, and angiogenic properties and may contribute to cancer development.5 Thus, elucidating the mechanisms of CXCL8 regulation is of considerable therapeutic interest.
Activation of CXCL8 production is known to be mediated via several signal pathways, including different mitogen-activated protein kinases (MAPKs) and the nuclear factor kappa B (NF-κB) transcription factor. The MAPKs are members of a family of serine-threonine kinases, to which many externally activated signaling pathways converge.6 Both extracellular signal-regulated kinase-1 and -2 (ERK1/2), the c-jun-N-terminal kinases (JNK1/2), and the p38 MAPKs seem to influence CXCL8 synthesis, the contribution of each depending on the cell type and stimulant.7–13 The central element of the classical NF-κB pathway is a homo- or heterodimer, most typically consisting of the p65/p50 dimer, which within resting cells is inactivated in the cytosol due to binding to IκB. Upon stimulation, IκB is phosphorylated, ubiquitinated, and degraded, thus allowing p65/p50 to translocate to the nucleus and bind to κB-sites in promoters of target genes.14 NF-κB signaling is regarded as indispensable for inducible CXCL8 regulation. The effect of NF-κB activation alone on the CXCL8 response is, however, normally marginal. Therefore, a combined activation of NF-κB and at least one of the main MAPK cascades are required to stimulate any considerable increase in CXCL8 production. Maximal CXCL8 responses are believed to require a combined activation of NF-κB along with all the three main MAPK cascades.15 In particular, post-transcriptional stabilization of CXCL8 messenger (m)RNA by p38 has been suggested to be essential to achieve high levels of CXCL8 release, as CXCL8 mRNA otherwise is highly unstable and degrades rapidly in the absence of p38 activity.15 The MAPK and NF-κB cascades are usually regarded as separate pathways. However, it has also been reported that MAPK activation may be localized upstream to the activation of NF-κB, possibly depending on both the stimulator and cell type.12,16–19
A large range of toxic agents are known to interact with the lung epithelial cell layer, triggering innate immune responses and the release of proinflammatory cytokines, including CXCL8.20 However, the ability to stimulate CXCL8 production varies considerably. Proinflammatory cytokines, such as IL-1 and tumor necrosis factor (TNF)-α, may upregulate CXCL8 release by 100-fold, while other agents cause more moderate 5–10-fold increases.15 An important question is which mechanisms determine the magnitude and persistence of the observed CXCL8 responses. We have previously reported varying potentials to induce CXCL8 responses in lung cells by different toxic agents.21–26 Of note, the phorbol ester, 12-O-tetradecanoylphorbol-13-acetate (TPA), induced a strong CXCL8 response in A549 cells that markedly exceeded the effect of sodium fluoride (NaF),9 and with a different time course of response. TPA, a classical promoting agent in studies of cancer development, is a rapid and strong activator of protein kinase C (PKC), and also a potent MAPK activator.27 TPA usually seems to exert its cellular effects via ERK1/2 activation,18,19 although it has also been reported that TPA may activate p38.28 Fluoride exposure in the form of hydrogen fluoride is involved in airway disease, as reflected by an association with occupational asthma in epidemiologic studies,29 and with respiratory inflammation in experimental human clinical studies.30,31 To investigate how these effects might be elicited, mechanistic studies of fluoride responses are of essence. In addition to our previous in vitro studies on fluoride exposure in lung epithelial cells,8,9,26,32–34 several mechanistic studies on fluoride exposure have emerged the recent years, including studies on the role of MAPK in COX-2 upregulation in lung epithelial cells,35 on oxidative stress and lipoxygenase activity in monocytes/macrophages,36,37 and on MAPK and oxidative stress in microglia cells.38 NaF has been reported to mediate its effect via a persistent G-protein activation and/or phosphatase inhibition.39,40 In A549 cells, NaF induced a weak, delayed, but persistent PKC activation, in contrast to TPA that elicited a strong, rapid, but transient effect. Upon PKC inhibition, the stimulatory effect of TPA on CXCL8 release was nearly abolished, whereas the response of NaF on CXCL8 release was only partially reduced.9 Furthermore, NaF elicited a persistent activation/phosphorylation of MAPKs, involving both EGF receptor and Src-dependent processes in A549 cells. The NaF effect was partially reduced by the inhibition of the MAPKs ERK1/2, p38, and JNK1/2, respectively, whereas the combined inhibition of different MAPKs (p38/JNK, p38/ERK1/2, and ERK1/2/JNK1/2) completely abolished the response.8,9,33 In the present study, we have compared the CXCL8 responses induced by TPA and NaF with respect to the involvement of MAPK and NF-κB pathways, with emphasis on the magnitude and duration of the response.
Materials and methods
Reagents
Nutrition mixture F12 HAM Kaighn’s modification culture medium was obtained from Sigma-Aldrich Co. (St Louis, MO, USA). Fetal bovine serum (FBS) was from EuroClone SpA (Milano, Italy). Ampicillin and fungizone were purchased from Bristol-Myers Squibb (New York, NY, USA) and penicillin/streptomycin from BioWhittaker® (VWR International, Radnor, PA, USA). NaF was obtained from Honeywell International, Inc. (Morristown, NJ, USA). TPA, SB202190, PD98059, SP600125, curcumin, MG132, and BAY11-7082 were purchased from Calbiochem-Novabiochem Corporation (San Diego, CA, USA); phenylmethylsulfonyl fluoride and pyrrolidine dithiocarbamate (PDTC) were from Sigma-Aldrich Co. JNK2 SMARTpool small interfering (si)RNA reagent against JNK2 and nonspecific control pool (negative control) were from Upstate Biotechnology, Inc. (Lake Placid, NY, USA). HiPerFect transfection reagent was from Qiagen NV (Venlo, the Netherlands). SiRNA against p65 and nonsense siRNA were from Cell Signaling Technology, Inc. (Danvers, MA, USA). Cytokine ELISA assay for human CXCL8 (IL-8) (Cyto-Set® CHC1304) was supplied by Thermo Fisher Scientific (Waltham, MA, USA). Acrylamide/Bis-acrylamide mix (30%), TEMED, and Bio-Rad DC™ Protein Assay were from Bio-Rad Laboratories Inc. (Hercules, CA, USA). The Compartment Protein Extraction Kit was from BioChain Institute, Inc. (Newark, CA, USA). The Absolutely RNA™ reverse transcription (RT) polymerase chain reaction (PCR) Miniprep kit was from Stratagene California (La Jolla, CA, USA). The predesigned TaqMan® Gene Expression Assays, TaqMan Universal PCR Master Mix, and the High-Capacity cDNA Archive Kit were purchased from Thermo Fisher Scientific. SuperSignal® West Dura chemoluminiscence system was obtained from Thermo Fisher Scientific, and the stripping solution (Mild Antibody Stripping Solution®) was obtained from Chemicon International, Inc. (Billerica, MA, USA). All other chemicals were purchased from commercial sources at the highest purity available.
Antibodies
Specific antibodies against phospho-p38 (Thr180/Tyr182), p38, phospho-JNK1/2 (Thr183/Tyr185), JNK 1/2, IκBα, phospho-IκBα (Ser32), p65, phopho-p65 (Ser536), glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and histon H1 were obtained from Cell Signaling Technology, Inc. Antibodies against phospho-ERK1/2 (Tyr-204) were obtained from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). The antibody against JNK2 was from Upstate Biotechnology, Inc., and the antibody against β-actin was obtained from Sigma-Aldrich Co.
Cell cultures and exposure
A549 cells, a human epithelial lung cell line, from the American Type Culture Collection (Manassas, VA, USA) were cultured in F12 HAM Kaighn’s modification medium, supplemented with ampicillin (0.1 mg/mL), penicillin (0.1 mg/mL), streptomycin (0.1 mg/mL), fungizone (0.25 μg/mL), and 10% heat-inactivated FBS. The cells (passage number 79–100) were plated in 35 mm six-well culture dishes (2×104 cells/well) and grown to near confluency at 37°C in a humidified atmosphere of 5% CO2 in air. The cells were exposed to varying concentrations of NaF (1.0–3.75 mM) and TPA (1–100 nM) for the indicated periods of time. When used, pharmacological inhibitors were added 1 hour prior to NaF or TPA exposure and kept in the culture medium during the whole incubation period. The concentrations of MAPK inhibitors PD98059 (25 μM), SB202190 (10 μM), and SP600125 (20 μM) were based on previous studies of concentration– response relationships for the respective inhibitors.9 For the NF-κB inhibitors, different concentrations were examined: curcumin (0–100 μM); MG132 (0–10 μM); BAY11-7082 (0–10 μM); or PDTC (0–75 μM).
Separation of nuclei and cytosol
For the extraction of nuclei and cytosol, we used a Compartment Protein Extraction Kit from BioChain Institute, Inc. The cells were lysed, and cytosolic and nuclei proteins were isolated according to producer’s recommendations. Protein concentrations in the lysate fractions were measured by Bio-Rad DC Protein Assay. Then, 6 μg of protein were loaded on a 10% Acrylamide/Bis gel. The amounts of p65 in the cytosolic and nuclei fractions were analyzed by Western blotting.
Transfection with siRNA against JNK and p65
A549 cells at a density of 200,000 cells/well were transfected with 10 nM siRNA against JNK2 and p65, using HiPerfect transfection reagent (18 μL/well) according to the procedure by Qiagen NV using the fast-forward protocol.41 The effectiveness of gene silencing was analyzed by Western blotting at 48 hours, 72 hours, or 96 hours by measuring the JNK2 and p65 protein levels in relation to β-actin. A negative siRNA control, SMARTpool (Upstate Biotechnology, Inc.), was used for the JNK experiments and SignalSilence® Control siRNA for the p65 experiments. Cells transfected with siRNA against JNK2 and p65 or negative control siRNA were exposed to NaF and TPA for 20 hours, 48 hours after transfection, and CXCL8 levels were measured by ELISA.
Measurements of CXCL8
CXCL8 protein levels in the cell medium were determined by ELISA assay. The growth medium was harvested and centrifuged at 250× g to remove cells. The final supernatants were stored at −70°C. CXCL8 levels were determined according to the manufacturer’s guidelines. Absorbance was measured and quantified using a plate reader (Sunrise™; Tecan Trading AG, Maennedorf, Switzerland) complete with software (Magellan version 1.10), and color intensity was converted to nanograms of CXCL8 using appropriate standards.
Measurements of CXCL8 mRNA
CXCL8 mRNA levels were determined by real-time PCR. Total mRNA was isolated from cells according to the supplier’s recommendations using the Absolutely RNA™RTPCR Miniprep kit, and reverse transcribed to cDNA on a PCR System 2400 (PerkinElmer) using a High-Capacity cDNA Archive Kit (Applied Biosystems; Thermo Fisher Scientific). Real-time PCR was performed using the Applied Biosystems 7500 Real-Time PCR System, with predesigned TaqMan Gene Expression Assays (18S, Hs99999901_s1 and CXCL8, Hs00174103_m1) and TaqMan Universal PCR Master Mix. For these analyses, 1 μg of total RNA was reverse transcribed to complementary (c)DNA using a High-Capacity cDNA Archive Kit. The cDNAs were diluted 1:20 in a solution of nuclease-free water, TaqMan Universal Master Mix, primers, and probe before performing the real-time PCR. The expression of each gene of interest (GOI) in each sample was normalized against housekeeping genes (HKG), and expressed as the fold change compared to the untreated control, as calculated by the ΔΔCt-method:
| (1) |
| (2) |
| (3) |
Immunoblotting
Total and phosphorylated protein levels were detected by Western blotting. Cells were resuspended in ice-cold lysis buffer (20 mM Tris-HCL; pH=7.5; 150 mM NaCl; 1 mM EDTA; 1 mM EGTA; 2.4 mM Na-pyrophosphate; 1.0 mM orthovanadate; 1 mM NaF; 21 mM leupeptin; 1.5 mM aprotinin; 15 mM pepstatin A; and 1% Triton™-X), sonicated for 5×1 second, and centrifuged for 8,000× g for 10 minutes. Protein determination was done in the supernatant by the Bio-Rad DC Protein Assay. Proteins (12.5 μg/well) from whole-cell lysates were separated by 10% SDS-PAGE and blotted onto nitrocellulose membranes. To ensure that the protein levels of each well were equal, Ponceau staining was used for loading control. The membranes were then probed with antibodies against the respective phosphorylated MAPKs (p-ERK1/2, p-JNK1/2, or p-p38) prior to incubation with horseradish peroxidase-conjugated secondary antibodies. The blots were developed using the SuperSignal West Dura chemoluminiscence system according to the manufacturer’s instructions. Finally, the membranes were stripped by incubation for 15 minutes at room temperature with Mild Antibody Stripping Solution and reprobed for the total amount of the respective kinases (ERK2, JNK2, p38) and/or β-actin. Using a similar procedure, the membranes were probed with antibodies against different NF-κB components (IκBα, p-IκBα, p65, p-p65) and reprobed with histon H1 and GAPDH.
Statistical analysis
Statistical calculations were performed by Student’s t-test or analysis of variance with post-tests for multiple comparisons, as indicated in the figure legends. Significance was assigned to a P-value ≤0.05.
Results
CXCL8 responses to fluoride and TPA in A549 cells
A549 cells were exposed to NaF (0–5 mM) and TPA (0–100 nM) for 20 hours. NaF significantly increased CXCL8 levels at 2.5 mM, with a maximal response (~6-fold increase) at 3.75 mM, and a subsequent reduction at 5 mM (Figure 1A). In accordance with previous results obtained by propidium iodide and Hoechst 33342 staining,32 NaF appeared to have a marginal effect on cell viability at 3.75 mM, whereas a substantial reduction in viability was observed at 5 mM, as judged visually by microscopy (a decrease in cell density and an increase in rounded/floating cells – not quantified). In comparison, TPA was much more potent, with responses in the nM range, and with a 20-fold increase in CXCL8 release from the cells at 100 nM, as measured after 20 hours’ exposure (Figure 1B). No change in viability was observed (not quantified). The time–course relationships for CXCL8 mRNA expression were also examined. NaF (3.75 mM) only slowly upregulated CXCL8 mRNA levels (~2-fold at 1 hour; ~12-fold at 2 hours), reaching a maximal level (~65-fold) at 6–10 hours. In contrast, TPA triggered a much more rapid and marked mRNA response for CXCL8, with a half-maximal response (45–50-fold) at ~1 hour, a peak at 2 hours (~90-fold), and then a progressive decline up to 10 hours (Figure 2). The cumulative mRNA response for NaF and TPA seemed to be of approximately the same magnitude for up to 10 hours.
Figure 1.
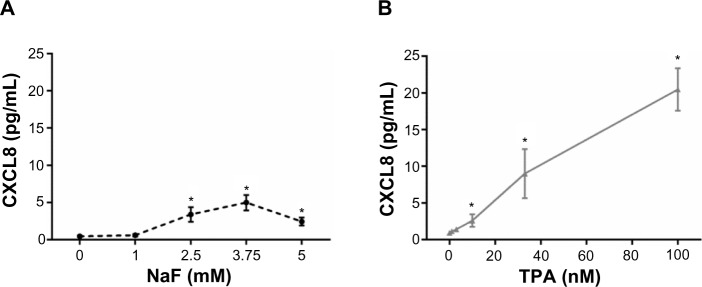
Concentration-dependent release of CXCL8.
Notes: CXCL8 after exposure to (A) NaF and (B) TPA.Aa549 cells were exposed to NaF (0–5 mM) and TPA (0–100 nM) for 20 hours, and assessed for the release of CXCL8 by ELISA. The data represent the mean ± SEM of three independent experiments. *Significant increase of CXCL8 release; P≤0.05. One-way ANOVA with Dunnett’s multiple comparisons test. The statistics were performed on log-transformed data.
Abbreviations: CXCL8, interleukin-8; NaF, sodium fluoride; TPA, 12-O-tetradecanoylphorbol-13-acetate; ELISA, enzyme-linked immunosorbent assay; SEM, standard error of the mean; ANOVA, analysis of variance.
Figure 2.
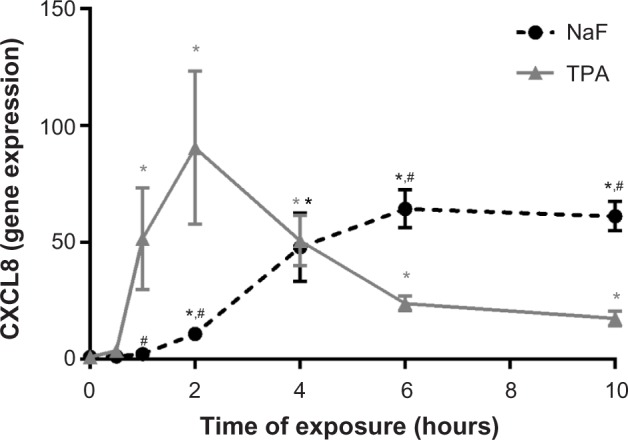
Time-dependent increase in CXCL8 mRNA expression in A549 cells upon exposure to NaF and TPA.
Notes: A549 cells were exposed to 3.75 mM of NaF and 100 nM of TPA for up to 10 hours, and assessed for CXCL8 mRNA by real-time PCR. The data represent the mean ± SEM of four independent experiments. *Significant increase (P<0.05) in CXCL8 mRNA. #Significantly different (P<0.05) from the CXCL8 increase in TPA-treated cells. Two-way ANOVA with Šidák’s multiple comparisons test. The statistics were performed on log-transformed data.
Abbreviations: CXCL8, interleukin-8; NaF, sodium fluoride; TPA, 12-O-tetradecanoylphorbol-13-acetate; mRNA, messenger RNA; PCR, polymerase chain reaction; SEM, standard error of the mean; ANOVA, analysis of variance.
Importance of time of exposure for the CXCL8 responses
To assess whether short-term or prolonged signaling was required for the CXCL8 responses triggered by NaF and TPA, the culture medium was removed from the A549 cell cultures after incubation for 0.5 hour, 1 hour, 3 hours, 4 hours, 6 hours, 8 hours, and 10 hours. The cells were subsequently washed (three times) and resupplemented with fresh medium up to the time point for CXCL8 analysis at 20 hours. These experiments showed only a slight increase in CXCL8 responses with medium shift after 0.5–3 hours for NaF exposure. Further, after medium shift at 4–6 hours, the response seemed to be approximately half of the response in the cells cultured for 20 hours without medium replacement (Figure 3A), suggesting that prolonged signaling was required for the NaF-induced response. For TPA exposure, on the other hand, the media shift as early as 30 minutes did not reduce the maximal CXCL8 release; it was even higher when TPA was removed from the cells (Figure 3B), suggesting that the early signaling was sufficient.
Figure 3.
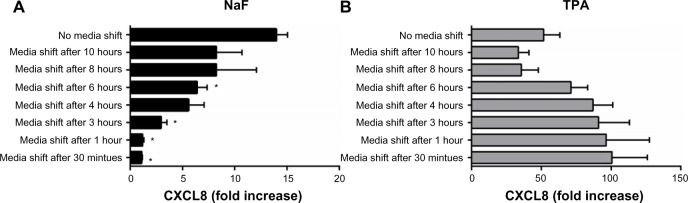
Effect of short-term exposure on NaF- and TPA-induced CXCL8 responses.
Notes: (A) NaF- and (B) TPA-induced CXCL8 responses. A549 cells were exposed to 3.75 mM of NaF or 100 nM of TPA for 0.5 hour, 1 hour, 3 hours, 4 hours, 6 hours, 8 hours, 10 hours, and 20 hours. The culture medium was replaced with medium without NaF/TPA at the respective time points, and further cultured up to 20 hours. The CXCL8 releases were measured by ELISA 20 hours after exposure start. The data represent the mean ± SEM of three independent experiments. *Significantly different from no media shift, P<0.05 (one-way ANOVA with Dunnetts multiple comparison test).
Abbreviations: NaF, sodium fluoride; CXCL8, interleukin-8; TPA, 12-O-tetradecanoylphorbol-13-acetate; ELISA, enzyme-linked immunosorbent assay; SEM, standard error of the mean; ANOVA, analysis of variance.
Involvement of MAPK in NaF- versus TPA-induced CXCL8 release
We have previously shown that NaF induced a time- and concentration-dependent increase in the activation/phosphorylation of the MAPKs ERK1/2, p38, and JNK, with a persistent increase in the period from 1–4 hours in A549 cells.8,9,33 In the present study, we compared the effects of NaF (3.75 mM) and TPA (100 nM) in the same experiments on the duration of MAPK phosphorylation upon exposure up to 4 hours for NaF and TPA, as assessed by Western analysis (Figure 4). TPA strongly enhanced ERK1/2 phosphorylation, with a maximum response already noted after 15–30 minutes. After 2 hours, the TPA-induced ERK1/2 phosphorylation was diminished, but it remained elevated compared to the control at least 4 hours after exposure (Figure 4Ab). NaF induced a much weaker, although sustained, ERK1/2 phosphorylation, with relatively similar levels from 15 minutes to 4 hours (~2.5%–5% of the maximal value for TPA) (Figure 4Aa). Thus, the ERK1/2 phosphorylation after TPA exposure was ~25-fold higher than after NaF exposure. In contrast, NaF exposure led to a stronger induction of p38 phosphorylation, with a distinct and sustained 3-fold increase lasting from 1–4 hours after exposure (Figure 4Ba), whereas TPA only weakly and transiently affected p38 phosphorylation, peaking at 30 minutes (~1.7-fold increase), returning to control levels after 1 hour (Figure 4Bb). Furthermore, NaF induced a very strong JNK1/2 phosphorylation (~20-fold) after 1 hour, which sharply declined at 2 hours (Figure 4Ca). TPA induced a low-to-moderate and transient JNK1/2 response, peaking at 15 minutes, returning to background levels after 1 hour (Figure 4Cb).
Figure 4.
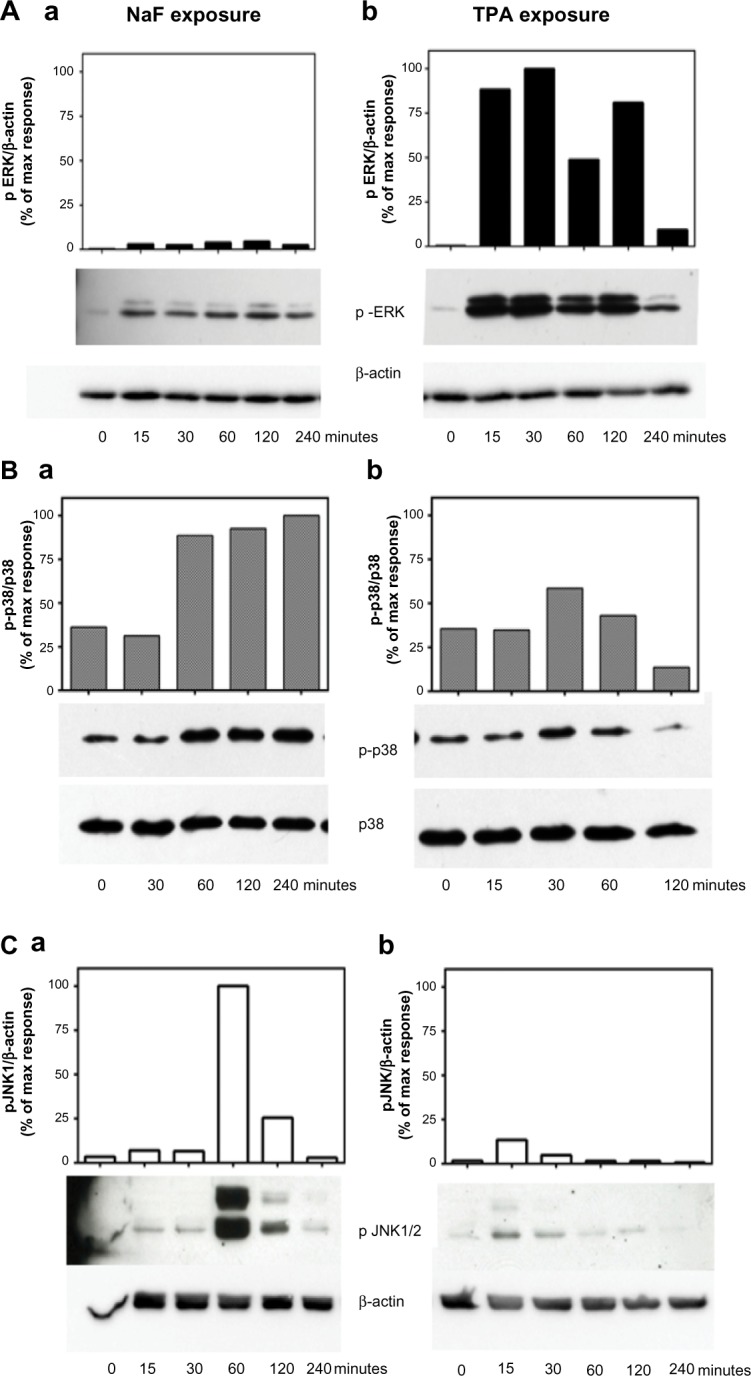
Time-dependent changes of MAPK ERK1/2, p38, and JNK1/2 phosphorylation upon NaF and TPA exposure.
Notes: (A) MAPK ERK1/2, (B) p38, and (C) JNK1/2. a549 cells were exposed to (Aa, Ba, and Ca) NaF (3.75 mM) and (Ab, Bb, and Cb) TPA (100 nM) for the time periods indicated. The MAPK phosphorylations were assessed by Western analysis. The phosphorylated ERK1/2 was related to β-actin. Phosphorylated p38 was related to total p38, whereas phosphorylated JnK1/2 was related to β-actin. A typical experiment is shown by Western blot and subsequent optical quantification. Values are expressed in percentage of the maximal phosphorylation observed. The experiment is representative of 3–5 experiments.
Abbreviations: NaF, sodium fluoride; ERK, extracellular signal-regulated kinase; TPA, 12-O-tetradecanoylphorbol-13-acetate; max, maximum; JNK, c-jun-N-terminal kinase; MAPK, mitogen-activated protein kinase.
Selective pharmacological MAPK inhibitors were then used to assess the role of the three MAPK cascades in TPA-induced CXCL8 responses in A549 cells. Figure 5A shows that pretreatment with the MEK/ERK1/2 inhibitor PD98059 (25 μM) reduced the responses substantially. The p38 inhibitor SB202190 (10 μM) affected CXCL8 release from TPA-exposed cells marginally, whereas the JNK1/2 inhibitor SP600125 (20 μM) seemed to slightly reduce the response, but this was not statistically significant (Figure 5A). Similarly, JNK2 silencing with siRNA only resulted in a slight, statistically nonsignificant reduction in TPA-induced CXCL8 (Figure 5B). The effect of siRNA on the JNK2 levels is shown in Figure 5C. Of note, the level of inhibition of TPA-induced CXCL8 by the three MAPK inhibitors appeared to reflect their activation level by TPA (Figure 4). Thus, the failure of SB202190 and SP600125 (and siJNK2) to elicit a statistically significant suppression of TPA-induced CXCL8 was likely due to a too low activation of p38 and JNK in TPA-exposed cells. In Table 1, the percentage inhibition obtained by PD98059, SB202190, and SP9000125 in TPA-exposed cells are compared to similar experiments in NaF-exposed cells, based upon our previously published study.8
Figure 5.

Involvement of MAPKs in TPA-induced CXCL8 release.
Notes: (A) A549 cells were pretreated for 1 hour with the p38 inhibitor SB202190 (10 μM), the ERK1/2 inhibitor PD98059 (25 μM), and the JNK1/2-inhibitor SP600125 (20 μM) prior to exposure with 100 nM of TPA for 20 hours, and assessed for CXCL8 release by ELISA. (B) The cells were transfected with siRNA against JNK2 and nonspecific siRNA for 48 hours, exposed for 100 nM of TPA for 20 hours, and assessed for CXCL8 release. The values are presented in percentage of the TPA response, and represent the mean ± SEM of three independent experiments. *Significant reduction as analyzed by multiple t-tests (P≤0.05). (C) JNK1/2 levels after exposure to siRNA against JNK2 as shown by a typical Western blot. The JNK1/2 levels were related to β-actin.
Abbreviations: CXCL8, interleukin-8; TPA, 12-O-tetradecanoylphorbol-13-acetate; siRNA, small interfering RNA; JNK, c-jun-N-terminal kinase; MAPKs, mitogen-activated protein kinases; ERK, extracellular signal-regulated kinase; ELISA, enzyme-linked immunosorbent assay; SEM, standard error of the mean.
Table 1.
The relative effect of MAPK inhibitors on NaF- and TPA-induced CXCL8 release
| Inhibitor | % inhibition of CXCL8 release
|
|
|---|---|---|
| NaF | TPA | |
| PD98059 | 43.6 | 55.4 |
| SB202190 | 41.0 | 12.4ns |
| SP600125 | 58.5 | 32.6ns |
Notes: The table compares the percent inhibition of CXCL8 release by the p38 inhibitor SB202190 (10 μM), the ERK1/2 inhibitor PD98059 (25 μM), and the JNK1/2-inhibitor SP600125 (20 μM) in A549 cells exposed to 3.75 mM of NaF8 or 100 nM of TPA for 20 hours. nsNot statistically significant. Data from Refsnes M, Skuland T, Schwarze PE, Øvrevik J, Låg M. Fluoride-induced IL-8 release in human epithelial lung cells: relationship to EGF-receptor-, SRC- and MAP-kinase activation. Toxicol Appl Pharmacol. 2008;227(1):56–67.8
Abbreviations: MaPK, mitogen-activated protein kinase; NaF, sodium fluoride; TPA, 12-O-tetradecanoylphorbol-13-acetate; CXCL8, interleukin-8; ERK, extracellular signal-regulated kinase; JNK, c-jun-N-terminal kinase.
Involvement of NF-κB activation in fluoride- versus TPA-induced CXCL8 release
IκBα degradation and p65 phosphorylation and translocation
In Figure 6A, the time courses for fluoride- and TPA-induced IκBα degradation are presented. The IκBα levels were only slightly reduced during the first hour of NaF (3.75 mM) exposure, but more strongly diminished (50%–60%) at 2–4 hours (Figure 6Aa). After TPA (100 nM) exposure, the IκBα levels were abruptly, but transiently, reduced (~90% reduction) after 30 minutes, already returning to initial levels after 2 hours’ exposure (Figure 6Ab). Similarly, TPA induced a more marked increase in IκBα phosphorylation than did NaF in the period up to 1–2 hours, with the increase in IκBα phosphorylation preceding the decline in IκBα levels (data not shown). NaF elicited a gradual phosphorylation of p65, increasing from 30 minutes after exposure and reaching a sustained and maximal response (~2-fold) from 1–4 hours (Figure 6Ba). In contrast, TPA induced a rapid phosphorylation of p65, already peaking after 15–30 minutes (~3-fold increase), and gradually returning to background levels from 1–2 hours after exposure (Figure 6Bb). The time course for p65 translocation from the cytosol to the nuclei after NaF and TPA exposure is presented in Figure 6C. Upon NaF exposure, no translocations were observed at 15 minutes and 30 minutes, whereas the highest level of translocation was observed at 2 hours (2.2-fold compared to untreated controls), followed by a reduction at 4 hours (Figure 6Ca). TPA exposure resulted in a rapid and more marked translocation to the nuclei, with ~4-fold increase already observed after 30 minutes, and with a persistent response (>3-fold) up to 4 hours (Figure 6Cb).
Figure 6.

Time-dependent changes of NaF- and TPA-induced IκBα degradation, p65 phosphorylation, and p65 translocation.
Notes: (A) NaF- and TPA-induced IκBα degradation; (B) p65 phosphorylation; and (C) p65 translocation. A549 cells were exposed to 3.75 mM of NaF (Aa, Ba, Ca) and to 100 nM of TPA (Ab, Bb, Cb) for the time periods indicated. The IκBα, β-actin, p65, and p-p65 levels were measured in cell lysates, and translocated p65 in nuclei. The data are shown by typical Western blots (out of 3–5 separate experiments) and by optical quantification of the protein bands. (A) IκBα levels were related to β-actin; (B) p65 phosphorylation levels in lysates were related to p65; and (C) the p65 levels in the nuclei were related to histon H1 (a stable marker in nuclei). The levels of the cytosolic marker (GAPDH) in the nuclear samples are also shown. The blot is typical for three experiments.
Abbreviations: NaF, sodium fluoride; TPA, 12-O-tetradecanoylphorbol-13-acetate; max, maximum; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Effect of NF-κB inhibitors on the NaF- and TPA-induced CXCL8 responses
The degradation of IκBα may be prevented by different proteosomal inhibitors,42–46 and this has been demonstrated to affect the cytokine responses to different agents including TPA.46 In initial experiments, we pretreated the cells with curcumin, a well-known proteosomal inhibitor,44–46 and they were exposed to NaF or TPA. Curcumin substantially reduced the fluoride- and TPA-induced IκBα degradation (Figure 7A) and abolished the p65 phosphorylation, as assessed in cell lysates (Figure 7B) and p65 translocation to the nuclei (Figure 7C) after exposure to both agents. Furthermore, curcumin almost completely abolished the CXCL8 response to both NaF and TPA (Figure 8D). However, since curcumin has been reported to inhibit MAPKs and other signaling systems,46 we examined the concentration-dependent effects of other well-established proteosomal inhibitors, such as BAY11-7082, MG132, and PDTC, known to inhibit IκBα degradation in a range of studies.43,47 For NaF, small, but not significant reductions in CXCL8 release were observed for BAY11-7082, MG132 and PDTC (Figure 8Aa, Ba and Ca). For TPA, PDTC showed a minor, nonsignificant reduction (Figure 8Cb), whereas BAY11-7082 and MG132 did not exert any reduction (Figure 8Ab and Bb). No apparent signs of cytotoxicity were observed for the inhibitors. We also assessed the potential role of NF-κB in fluoride- and TPA-induced responses by using siRNA against p65. The NaF- and TPA-induced CXCL8 releases were partially suppressed (~50% and ~40% reduction, respectively) by siRNA, although inhibition of the TPA-induced response did not reach statistical significance (Figure 9A). Of note, p65 protein levels were almost completely knocked down in by siRNA transfection, as assessed by Western blotting (Figure 9B).
Figure 7.

Effect of curcumin on NaF- and TPA-induced changes in NF-κB in A549 cells.
Notes: (A) Effects of curcumin (50 μM, 75 μM, and 100 μM) on IκBα degradation in lysates; (B) p65 phosphorylation in lysates; and (C) p65 translocation to nuclei. The cells were pretreated with curcumin at the indicated concentrations for 1 hour, and further exposed to NaF (3.75 mM) (Aa, Ba, and Ca) and TPA (100 nM) (Ab, Bb, and Cb) for 2 hours and 30 minutes, respectively. IκBα levels in lysates were related to β-actin, phosphorylation of p65 in lysates was related to total p65, and translocation of p65 was related to histon in the nuclei. The data are shown by typical Western blots, and also presented by optical quantification of the blots, given in relation to the responses (100%) to NaF and TPA at 2 hours and 30 minutes. The experiments in (A) and (C) are typical for four experiments and in (B) for two experiments.
Abbreviations: NaF, sodium fluoride; TPA, 12-O-tetradecanoylphorbol-13-acetate; max, maximum; NF-κB, nuclear factor kappa B.
Figure 8.

Concentration–response relationships for different NF-κB inhibitors versus fluoride- and TPA-induced CXCL8 release.
Notes: A549 cells were pretreated with different NF-κB inhibitors for 1 hour, and then further exposed to 3.75 mM of NaF (Aa, Ba, Ca, and Da) and 100 nM of TPA (Ab, Bb, Cb, and Db) for 20 hours. (A) BAYII-7082 (2.5–10 μM); (B) MG132 (2.5–10 μM); (C) PDTC (25–75 μM); and (D) curcumin (50–100 μM). The CXCL8 released to the medium was measured by ELISA. The data represent the mean ± SEM from 3–4 experiments.
Abbreviations: NaF, sodium fluoride; CXCL8, interleukin-8; TPA, 12-O-tetradecanoylphorbol-13-acetate; PDTC, pyrrolidine dithiocarbamate; NF-κB, nuclear factor kappa B; ELISA, enzyme-linked immunosorbent assay; SEM, standard error of the mean.
Figure 9.

Effects of siRNA against p65 on NaF- and TPA-induced CXCL8 release.
Notes: (A) A549 cells were pretreated for 24 hours with siRNA against p65 (si p65), and then further exposed to NaF (3.75 mM) and TPA (100 nM) for 20 hours. The cells were also transfected with nonsense siRNA, si NT. The CXCL8 released to the medium was measured by ELISA. (B) The p65 levels are presented after exposure with siRNA against p65 (si p65) and a negative control (si NT). The p65 levels are related to β-actin levels. The data represent the mean ± SEM from 3–4 experiments. *Significant reduction as analyzed by student’s t-test (P≤0.05). The statistics were performed on log-transformed data.
Abbreviations: CXCL8, interleukin-8; siRNA, small interfering RNA; NaF, sodium fluoride; TPA, 12-O-tetradecanoylphorbol-13-acetate; ELISA, enzyme-linked immunosorbent assay; SEM, standard error of the mean; si p65; small interference RNA against p65; si NT; non-targeting small interference RNA.
Effect of MAPK inhibitors on the NF-κB-system
To examine whether ERK1/2, JNK1/2, and p38 exerted their effect via NF-κB-dependent or independent pathways, we examined the ability of selective MAPK inhibitors to interfere with NF-κB. Subsequent to NaF exposure, neither of the MAPK inhibitors affected the IκBα degradation (Figure 10Aa). Whereas the NaF-induced p65 phosphorylation was partially reduced by the p38 inhibitor, SB202190, the ERK inhibitor, PD98059, and JNK inhibitor, SP600125, did not exert any effects (Figure 10Ba). In contrast, the NaF-induced p65 translocation was partially reduced by all the MAPK inhibitors (Figure 10Ca). With respect to TPA exposure, the inhibitors of MAPK appeared to be without effects on NF-κB signaling (Figure 10Ab, 10Bb, and 10Cb).
Figure 10.

Relationship between MAPK and NF-κB activity after fluoride and TPA exposure.
Notes: Effect of MAPK inhibitors on (A) IκBα degradation, (B) p65 phosphorylation, and (C) p65 translocation. A549 cells were pretreated for 1 hour with the p38 inhibitor SB202190 (10 μM), the ERK1/2 inhibitor PD98059 (25 μM), and the JNK1/2 inhibitor SP202190 (20 μM) prior to 2 hours of exposure with 3.75 mM of NaF (Aa, Ba, and Ca) or 30 minutes of exposure with 100 nM of TPA (Ab, Bb, and Cb). Typical blots are shown, and quantification data representing the mean ± SEM of 3–6 independent experiments. The data are presented in percentages of the control values. The IκBα levels in lysates were related to β-actin, p65 phosphorylation in lysates was related to total p65, and p65 in nuclei was related to histon H1.
Abbreviations: NaF, sodium fluoride; TPA, 12-O-tetradecanoylphorbol-13-acetate; PD, PD98059; SB, SB202190; SP, sP6001254; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor kappa B; ERK, extracellular signal-regulated kinase; JNK, c-jun-N-terminal kinase; SEM, standard error of the mean.
Discussion
The present study compared the CXCL8 response to NaF with the phorbol ester, TPA – two compounds that show no structural similarity, but exhibit different activation patterns of intracellular signaling (slow/rapid; persistent/transient; weak/strong). Previously, we have reported on the differential activation of PKC by NaF and TPA in A549 cells,9 whereas the present study shows the patterns of MAPK and NF-κB activation in relation to CXCL8 induction after exposure to these stimulants.
NaF exposure elicited a moderate but sustained phosphorylation of p38, ERK1/2, and the p65 subunit of NF-κB, as well as a strong but transient phosphorylation of JNK. This was accompanied by a gradual, but sustained increase in CXCL8 mRNA levels and a low-to-moderate increase in CXCL8 release from A549 cells. Consistent with this gradual, sustained activation of intracellular signaling pathways and CXCL8 expression, our experiments – with the removal of NaF by washing and shift of medium – indicated that a long-term/persistent response was required for triggering the NaF-induced CXCL8 response. Furthermore, all the three MAPK pathways and NF-κB signaling seem involved in CXCL8 regulation in NaF-exposed A549 cells. This corroborates and extends previous results showing that a combined inhibition of different MAPKs was necessary to completely abrogate CXCL8 responses in NaF-exposed A549 cells.8
In contrast to NaF, TPA exposure resulted in a rapid, strong, but relatively transient increase in phosphorylation of ERK1/2, with only marginal effects on p38 and JNK, as compared to NaF. Moreover, TPA also elicited a more transient phosphorylation of p65, although p65 nuclear translocation appeared to persist longer than in NaF-exposed cells. In line with the effects on ERK and p65 phosphorylation, TPA also resulted in a rapid, transient increase in CXCL8 mRNA levels, accompanied by a strong increase in CXCL8 protein release. Contrary to the effects observed in NaF-exposed cells, the effects of TPA seemed to be predominately mediated through ERK1/2 and NF-κB signaling, as inhibitors of p38 and JNK1/2 had little impact on the TPA-induced CXCL8 response. This also contradicts earlier suggestions that JNK1/2 signaling is indispensable for inducible CXCL8 regulation.15 Furthermore, the wash-out experiment showed that a short-term exposure to TPA was sufficient to trigger a CXCL8 response, suggesting that these early transient signaling events appear to be central and perhaps sufficient to elicit the full TPA response. Notably, the wash-out experiment also indicated that CXCL8 responses were reduced upon prolonged TPA exposure, as compared to shorter/more acute exposure times. The mechanism for this remains obscure, but it may be related to time-dependent downregulation/desensitization of cellular signaling by TPA. In support of this, we have previously observed that pretreatment with TPA markedly lowered the CXCL8 response to repeated TPA exposures.9
The general concept of CXCL8 regulation is that combined activation of all the MAPKs, in addition to NF-κB, is required to achieve a maximal activation of the CXCL8 response.15 Interestingly, the present study shows that TPA, via a rapid and strong activation of ERK in combination with NF-κB, induces a strong upregulation of CXCL8 production at 20 hours, by far surpassing the CXCL8 response induced by the combined, moderate, and sustained activation of the different MAPKs in NaF-exposed A549 cells. Although it cannot be excluded that activation of additional MAPKs (p38 and JNK) would have enhanced the TPA-induced CXCL8 response even further, our results show that strong activation of ERK1/2 alone, in combination with NF-κB signaling, may be sufficient for a marked CXCL8 response. Of note, inhibition of ERK1/2 by PD98059 resulted in a partial and almost identical reduction of CXCL8 induction after both NaF and TPA exposure, while p65 knockdown by siRNA omitted a slightly stronger relative suppression of NaF-induced CXCL8 release. Thus, the greater CXCL8 release induced by TPA compared to NaF may not solely be due to its stronger effects on ERK1/2- and NF-κB-dependent mechanisms. An interesting observation is that even though TPA induced a considerably stronger CXCL8 release than did NaF when analyzed at 20 hours, the cumulative CXCL8 mRNA responses in the period up to 10 hours appeared to be relatively similar. The reason for this apparent discrepancy is unclear, but it may possibly be related to effects at the translational level.
Although TPA induced a strong increase in CXCL8 release (20-fold) compared to NaF (6-fold), it should also be noted that the level of CXCL8 induction by TPA is considerably lower than the more than 100-fold increase in CXCL8 synthesis, which has been reported after exposure with cytokines such as IL-1 and TNF-α.15 Such massive increases in CXCL8 are believed to require additional mRNA stabilization by p38.15 In our present study, p38 was only marginally affected by TPA exposure, and in line with the notion that p38 activity is required to stabilize CXCL8 mRNA, TPA-induced CXCL8 mRNA levels dropped rapidly after 2 hours’ exposure, which is also when TPA-induced phosphorylation of ERK1/2 and p65 returned to basal levels. In contrast, the sustained increase in CXCL8 mRNA levels in NaF-exposed A549 cells appeared to be accompanied by comparably sustained increases in p38 and NF-κB phosphorylation levels, lasting at least up to 4 hours after exposure.
NF-κB is considered to play an important role in CXCL8 regulation.15 Thus, regulation of the CXCL8 promoter activity has been shown to involve a strong cooperation between a high-affinity NF-κB site and an activating protein-1 (AP-1) recognition sequence.15,48 The AP-1 recognition sequence binds proteins like c-jun and c-fos that are activated by MAPKs. In accordance with this, the attenuation of NF-κB signaling by use of curcumin or siRNA transfection against p65 suppressed CXCL8 responses induced by both NaF and TPA. However, despite that, silencing of p65 by siRNA resulted in an almost complete downregulation of p65 protein levels; this only led to a partial (50%) reduction of NaF-induced CXCL8 and a 40% (and not statistically significant) reduction of TPA-induced CXL8 responses in the A549 cells. This contrasts the common assumption that NF-κB signaling is a prerequisite for inducible CXCL8 regulation.15 Curcumin treatment, on the other hand, nearly completely abrogated the CXCL8 responses induced by both TPA and NaF. Notably, curcumin, which inhibited IκBα degradation, p65 phosphorylation, and translocation in our present study, is also known to suppress AP-1 and other signaling pathways.44–46 Thus, the inhibitory effects of curcumin on NaF- and TPA-induced CXCL8 responses are likely not restricted to NF-κB inhibition, but they may involve suppression of additional signaling pathways such as AP-1. Somewhat surprisingly, a range of chemical inhibitors of IκBα degradation commonly used to assess NF-κB signaling (MG132, BAY11-7082, and PDTC) mostly failed to reduce the CXCL8 responses to TPA or NaF treatment, though nonsignificant reductions in the response to NaF were observed. Although we did not verify that these inhibitors actually prevented IκB degradation, it seems unlikely that they all simultaneously failed to function properly at every concentration tested. A more likely explanation is that the lack of convincing inhibitory effects is related to unspecific properties of the inhibitors. Almost all of these inhibitors have been reported to possess AP-1-stimulating activity, which may counteract the effects of reduced NF-κB signaling and mask the expected inhibition of the CXCL8 responses.43,47 Thus, these classical inhibitors of IκBα degradation seem less suited for studies on the role of NF-κB in cytokine regulation.
In addition to activating the AP-1 site of the CXCL8 promoter, MAPKs may also be localized upstream to NF-κB. Thus, different MAPK inhibitors have been reported to interfere with the activation of NF-κB upon exposure to different agents. Both ERK1/2 and JNK1/2 have been reported as upstream activators of NF-κB in IL-1- and TNF-α-exposed lung epithelial cells, including A549 cells.12,16,17 ERK1/2 has previously been reported to regulate NF-κB activation in TPA-exposed A549 and U937 cells.18,19 In contrast to this, our current findings did not reveal any effects of either ERK1/2, p38, or JNK1/2 inhibition on TPA-induced NF-κB signaling in the A549 cells. The reason for this discrepancy from previous studies remains unclear. However, in NaF-exposed A549 cells, inhibition of all the three MAPKs partially reduced NF-κB translocation. Moreover, p38 inhibition also reduced p65 phosphorylation at serine 536 in NaF-exposed cells, which is important to CXCL8 expression as it promotes coupling of p65 to the basal transcriptional machinery.49–51 This observation is consistent with reports of p38 regulating p65 Ser536-phosphorylation at the CXCL8 promoter in pneumococci-stimulated human bronchial epithelial cells.50 Notably, p38 has often been associated with post-transcriptional effects on mRNA stabilization, but it may also function as an early responder to different toxicants, and to be an upstream regulator of the TACE (TGF-α converting enzyme)/transforming growth factor-α/epidermal growth factor receptor pathway, which has been proposed as a central, convergent pathway in innate immunity.52,53
This study is restricted to molecular mechanisms regulating CXCL8 responses to NaF and TPA in A549 cells. Fluoride-induced IL-6 release appears to follow similar time course and concentration–response relationships as CXCL8, suggesting that comparable mechanisms may be involved in the regulation of these cytokines.26 Although we have not assessed the regulatory mechanism for fluoride- and TPA-induced IL-6, we have previously found that the p38 inhibitor, SB202190, elicited the identical inhibition of IL-6 and CXCL8, while the ERK1/2 inhibitor, PD98059, was somewhat less efficient in inhibiting IL-6 than CXCL8, from stone particle-exposed A549 cells.54 Despite that, the A549 cell line is one of the best described lung cell models and is commonly used for toxicity testing and pharmacological studies, the generalization of the results obtained from immortalized cell lines should be done with great caution. Thus, one cannot exclude the possibility that NaF and TPA may exert their effects on CXCL8 through different mechanisms in normal lung cells. However, we have previously shown by use of other exposure agents (crystalline silica) that the role of MAPKs in the regulation of CXCL8 release from A549 cells reSEMbles their role in the regulation of MIP-2 release from primary rat type 2 cells.55
Overall, the potential of different toxicants and other activators to induce CXCL8 responses seems to depend upon the respective signals elicited and the magnitude and duration of the signaling. Our results suggest that the stronger release of CXCL8 to TPA than to NaF after 20 hours’ exposure in lung epithelial cells (A549 cells) seems ascribed to the early and strong upregulation of the ERK1/2 and NF-κB activation induced by TPA. For NaF, both the magnitude and persistency of the signaling seem important, involving the combined activation of ERK1/2, p38, JNK, and NF-κB pathways. Our findings also underscore that a strong, rapid, and relatively transient activation of ERK1/2 in combination with NF-kB may be sufficient to induce a strong induction of CXCL8, which may exceed the effects of a more moderate ERK1/2 activation in combination with the activation of p38, JNK, and NF-κB.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Mukaida N. Pathophysiological roles of interleukin-8/CXCL8 in pulmonary diseases. Am J Physiol Lung Cell Mol Physiol. 2003;284(4):L566–L577. doi: 10.1152/ajplung.00233.2002. [DOI] [PubMed] [Google Scholar]
- 2.Lezcano-Meza D, Teran LM. Occupational asthma and interleukin-8. Clin Exp Allergy. 1999;29(10):1301–1303. doi: 10.1046/j.1365-2222.1999.00723.x. [DOI] [PubMed] [Google Scholar]
- 3.Barnes PJ. New molecular targets for the treatment of neutrophilic diseases. J Allergy Clin Immunol. 2007;119(5):1055–1062. doi: 10.1016/j.jaci.2007.01.015. quiz 1063–1064. [DOI] [PubMed] [Google Scholar]
- 4.Burgel PR, Nadel JA. Epidermal growth factor receptor-mediated innate immune responses and their roles in airway diseases. Eur Respir J. 2008;32(4):1068–1081. doi: 10.1183/09031936.00172007. [DOI] [PubMed] [Google Scholar]
- 5.Xie K. Interleukin-8 and human cancer biology. Cytokine Growth Factor Rev. 2001;12(4):375–391. doi: 10.1016/s1359-6101(01)00016-8. [DOI] [PubMed] [Google Scholar]
- 6.Puddicombe SM, Davies DE. The role of MAP kinases in intracellular signal transduction in bronchial epithelium. Clin Exp Allergy. 2000;30(1):7–11. doi: 10.1046/j.1365-2222.2000.00709.x. [DOI] [PubMed] [Google Scholar]
- 7.Hashimoto S, Gon Y, Takeshita I, et al. Diesel exhaust particles activate p38 MAP kinase to produce interleukin 8 and RANTES by human bronchial epithelial cells and N-acetylcysteine attenuates p38 MAP kinase activation. Am J Respir Crit Care Med. 2000;161(1):280–285. doi: 10.1164/ajrccm.161.1.9904110. [DOI] [PubMed] [Google Scholar]
- 8.Refsnes M, Skuland T, Schwarze PE, Øvrevik J, Låg M. Fluoride-induced IL-8 release in human epithelial lung cells: relationship to EGF-receptor-, SRC- and MAP-kinase activation. Toxicol Appl Pharmacol. 2008;227(1):56–67. doi: 10.1016/j.taap.2007.09.022. [DOI] [PubMed] [Google Scholar]
- 9.Refsnes M, Thrane EV, Låg M, Thoresen GH, Schwarze PE. Mechanisms in fluoride-induced interleukin-8 synthesis in human lung epithelial cells. Toxicology. 2001;167(2):145–158. doi: 10.1016/s0300-483x(01)00448-6. [DOI] [PubMed] [Google Scholar]
- 10.Furuichi S, Hashimoto S, Gon Y, Matsumoto K, Horie T. p38 mitogen-activated protein kinase and c-Jun-NH2-terminal kinase regulate interleukin-8 and RANTES production in hyperosmolarity stimulated human bronchial epithelial cells. Respirology. 2002;7(3):193–200. doi: 10.1046/j.1440-1843.2002.00401.x. [DOI] [PubMed] [Google Scholar]
- 11.Jung YD, Fan F, McConkey DJ, et al. Role of P38 MAPK, AP-1, and NF-kappaB in interleukin-1beta-induced IL-8 expression in human vascular smooth muscle cells. Cytokine. 2002;18(4):206–213. doi: 10.1006/cyto.2002.1034. [DOI] [PubMed] [Google Scholar]
- 12.Li J, Kartha S, Iasvovskaia S, et al. Regulation of human airway epithelial cell IL-8 expression by MAP kinases. Am J Physiol Lung Cell Mol Physiol. 2002;283(4):L690–L699. doi: 10.1152/ajplung.00060.2002. [DOI] [PubMed] [Google Scholar]
- 13.Li LF, Ouyang B, Choukroun G, et al. Stretch-induced IL-8 depends on c-Jun NH2-terminal and nuclear factor-kappaB-inducing kinases. Am J Physiol Lung Cell Mol Physiol. 2003;285(2):L464–L475. doi: 10.1152/ajplung.00031.2003. [DOI] [PubMed] [Google Scholar]
- 14.Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3(3):221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 15.Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M. Multiple control of interleukin-8 gene expression. J Leukoc Biol. 2002;72(5):847–855. [PubMed] [Google Scholar]
- 16.Hu J, Nakano H, Sakurai H, Colburn NH. Insufficient p65 phosphorylation at S536 specifically contributes to the lack of NF-kappaB activation and transformation in resistant JB6 cells. Carcinogenesis. 2004;25(10):1991–2003. doi: 10.1093/carcin/bgh198. [DOI] [PubMed] [Google Scholar]
- 17.Lin FS, Lin CC, Chien CS, Luo SF, Yang CM. Involvement of p42/p44 MAPK, JNK, and NF-kappaB in IL-1beta-induced ICAM-1 expression in human pulmonary epithelial cells. J Cell Physiol. 2005;202(2):464–473. doi: 10.1002/jcp.20142. [DOI] [PubMed] [Google Scholar]
- 18.Chang MS, Chen BC, Yu MT, Sheu JR, Chen TF, Lin CH. Phorbol 12-myristate 13-acetate upregulates cyclooxygenase-2 expression in human pulmonary epithelial cells via Ras, Raf-1, ERK, and NF-kappaB, but not p38 MAPK, pathways. Cell Signal. 2005;17(3):299–310. doi: 10.1016/j.cellsig.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 19.Jang BC, Jung TY, Paik JH, et al. Tetradecanoyl phorbol acetate induces expression of Toll-like receptor 2 in U937 cells: involvement of PKC, ERK, and NF-kappaB. Biochem Biophys Res Commun. 2005;328(1):70–77. doi: 10.1016/j.bbrc.2004.12.144. [DOI] [PubMed] [Google Scholar]
- 20.Parker D, Prince A. Innate immunity in the respiratory epithelium. Am J Respir Cell Mol Biol. 2011;45(2):189–201. doi: 10.1165/rcmb.2011-0011RT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hetland RB, Refsnes M, Myran T, Johansen BV, Uthus N, Schwarze PE. Mineral and/or metal content as critical determinants of particle-induced release of IL-6 and IL-8 from A549 cells. J Toxicol Environ Health A. 2000;60(1):47–65. doi: 10.1080/009841000156583. [DOI] [PubMed] [Google Scholar]
- 22.Totlandsdal AI, Cassee FR, Schwarze P, Refsnes M, Låg M. Diesel exhaust particles induce CYP1A1 and pro-inflammatory responses via differential pathways in human bronchial epithelial cells. Part Fibre Toxicol. 2010;7:41. doi: 10.1186/1743-8977-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Øvrevik J, Refsnes M, Namork E, et al. Mechanisms of silica-induced IL-8 release from A549 cells: initial kinase-activation does not require EGFR activation or particle uptake. Toxicology. 2006;227(1–2):105–116. doi: 10.1016/j.tox.2006.07.029. [DOI] [PubMed] [Google Scholar]
- 24.Låg M, Rodionov D, Ovrevik J, Bakke O, Schwarze PE, Refsnes M. Cadmium-induced inflammatory responses in cells relevant for lung toxicity: Expression and release of cytokines in fibroblasts, epithelial cells and macrophages. Toxicol Lett. 2010;193(3):252–260. doi: 10.1016/j.toxlet.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 25.Kocbach A, Namork E, Schwarze PE. Pro-inflammatory potential of wood smoke and traffic-derived particles in a monocytic cell line. Toxicology. 2008;247(2–3):123–132. doi: 10.1016/j.tox.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 26.Refsnes M, Becher R, Lâg M, Skuland T, Schwarze PE. Fluoride-induced interleukin-6 and interleukin-8 synthesis in human epithelial lung cells. Hum Exp Toxicol. 1999;18(11):645–652. doi: 10.1191/096032799678839572. [DOI] [PubMed] [Google Scholar]
- 27.Verma AK, Hsieh JT, Pong RC. Mechanisms involved in ornithine decarboxylase induction by 12-O-tetradecanoylphorbol-13-acetate, a potent mouse skin tumor promoter and an activator of protein kinase C. Adv Exp Med Biol. 1988;250:273–290. doi: 10.1007/978-1-4684-5637-0_25. [DOI] [PubMed] [Google Scholar]
- 28.Kim JH, Na HK, Pak YK, et al. Roles of ERK and p38 mitogen-activated protein kinases in phorbol ester-induced NF-kappaB activation and COX-2 expression in human breast epithelial cells. Chem Biol Interact. 2008;171(2):133–141. doi: 10.1016/j.cbi.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 29.Søyseth V, Kongerud J. Prevalence of respiratory disorders among aluminium potroom workers in relation to exposure to fluoride. Br J Ind Med. 1992;49(2):125–130. doi: 10.1136/oem.49.2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lund K, Refsnes M, Sandstrøm T, et al. Increased CD3 positive cells in bronchoalveolar lavage fluid after hydrogen fluoride inhalation. Scand J Work Environ Health. 1999;25(4):326–334. doi: 10.5271/sjweh.442. [DOI] [PubMed] [Google Scholar]
- 31.Lund K, Refsnes M, Ramis I, et al. Human exposure to hydrogen fluoride induces acute neutrophilic, eicosanoid, and antioxidant changes in nasal lavage fluid. Inhal Toxicol. 2002;14(2):119–132. doi: 10.1080/089583701753403944. [DOI] [PubMed] [Google Scholar]
- 32.Refsnes M, Schwarze PE, Holme JA, Låg M. Fluoride-induced apoptosis in human epithelial lung cells (A549 cells): role of different G protein-linked signal systems. Hum Exp Toxicol. 2003;22(3):111–123. doi: 10.1191/0960327103ht322oa. [DOI] [PubMed] [Google Scholar]
- 33.Thrane EV, Refsnes M, Thoresen GH, Låg M, Schwarze PE. Fluoride-induced apoptosis in epithelial lung cells involves activation of MAP kinases p38 and possibly JNK. Toxicol Sci. 2001;61(1):83–91. doi: 10.1093/toxsci/61.1.83. [DOI] [PubMed] [Google Scholar]
- 34.Refsnes M, Kersten H, Schwarze PE, Lag M. Involvement of protein kinase C in fluoride-induced apoptosis in different types of lung cells. Ann N Y Acad Sci. 2002;973:218–220. doi: 10.1111/j.1749-6632.2002.tb04636.x. [DOI] [PubMed] [Google Scholar]
- 35.Ridley W, Matsuoka M. Fluoride-induced cyclooxygenase-2 expression and prostaglandin E2 production in A549 human pulmonary epithelial cells. Toxicol Lett. 2009;188(3):180–185. doi: 10.1016/j.toxlet.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 36.Gutowska I, Baranowska-Bosiacka I, Baśkiewicz M, et al. Fluoride as a pro-inflammatory factor and inhibitor of ATP bioavailability in differentiated human THP1 monocytic cells. Toxicol Lett. 2010;196(2):74–79. doi: 10.1016/j.toxlet.2010.03.1167. [DOI] [PubMed] [Google Scholar]
- 37.Gutowska I, Baranowska-Bosiacka I, Safranow K, et al. Fluoride in low concentration modifies expression and activity of 15 lipoxygenase in human PBMC differentiated monocyte/macrophage. Toxicology. 2012;295(1–3):23–30. doi: 10.1016/j.tox.2012.02.014. [DOI] [PubMed] [Google Scholar]
- 38.Yan L, Liu S, Wang C, et al. JNK and NADPH oxidase involved in fluoride-induced oxidative stress in BV-2 microglia cells. Mediators Inflamm. 2013;2013:895975. doi: 10.1155/2013/895975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sternweis PC, Gilman AG. Aluminum: a requirement for activation of the regulatory component of adenylate cyclase by fluoride. Proc Natl Acad Sci U S A. 1982;79(16):4888–4891. doi: 10.1073/pnas.79.16.4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wergedal JE, Lau KH. Human bone cells contain a fluoride sensitive acid phosphatase: evidence that this enzyme functions at neutral pH as a phosphotyrosyl protein phosphatase. Clin Biochem. 1992;25(1):47–53. doi: 10.1016/0009-9120(92)80045-i. [DOI] [PubMed] [Google Scholar]
- 41.QIAGEN . HiPerFect Transfection Reagent Handbook. Venlo: QIAGEN; 2010. [Accessed November 5, 2014]. Available from: http://www.qiagen.com/nz/resources/resourcedetail?id=c475824e-3da5-40b6-9870-1752ffb015a6&lang=en. [Google Scholar]
- 42.Wu HM, Wen HC, Lin WW. Proteasome inhibitors stimulate interleukin-8 expression via Ras and apoptosis signal-regulating kinase-dependent extracellular signal-related kinase and c-Jun N-terminal kinase activation. Am J Respir Cell Mol Biol. 2002;27(2):234–243. doi: 10.1165/ajrcmb.27.2.4792. [DOI] [PubMed] [Google Scholar]
- 43.Joshi-Barve S, Barve SS, Butt W, Klein J, McClain CJ. Inhibition of proteasome function leads to NF-kappaB-independent IL-8 expression in human hepatocytes. Hepatology. 2003;38(5):1178–1187. doi: 10.1053/jhep.2003.50470. [DOI] [PubMed] [Google Scholar]
- 44.Jobin C, Bradham CA, Russo MP, et al. Curcumin blocks cytokine-mediated NF-kappa B activation and proinflammatory gene expression by inhibiting inhibitory factor I-kappa B kinase activity. J Immunol. 1999;163(6):3474–3483. [PubMed] [Google Scholar]
- 45.Pan MH, Lin-Shiau SY, Lin JK. Comparative studies on the suppression of nitric oxide synthase by curcumin and its hydrogenated metabolites through down-regulation of IkappaB kinase and NFkappaB activation in macrophages. Biochem Pharmacol. 2000;60(11):1665–1676. doi: 10.1016/s0006-2952(00)00489-5. [DOI] [PubMed] [Google Scholar]
- 46.Lin JK. Molecular targets of curcumin. Adv Exp Med Biol. 2007;595:227–243. doi: 10.1007/978-0-387-46401-5_10. [DOI] [PubMed] [Google Scholar]
- 47.Wu HM, Chi KH, Lin WW. Proteasome inhibitors stimulate activator protein-1 pathway via reactive oxygen species production. FEBS Lett. 2002;526(1–3):101–105. doi: 10.1016/s0014-5793(02)03151-4. [DOI] [PubMed] [Google Scholar]
- 48.Henriquet C, Gougat C, Combes A, Lazennec G, Mathieu M. Differential regulation of RANTES and IL-8 expression in lung adenocarcinoma cells. Lung Cancer. 2007;56(2):167–174. doi: 10.1016/j.lungcan.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buss H, Dörrie A, Schmitz ML, Hoffmann E, Resch K, Kracht M. Constitutive and interleukin-1-inducible phosphorylation of p65 NF-{kappa}B at serine 536 is mediated by multiple protein kinases including I{kappa}B kinase (IKK)-{alpha}, IKK{beta}, IKK{epsilon}, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J Biol Chem. 2004;279(53):55633–55643. doi: 10.1074/jbc.M409825200. [DOI] [PubMed] [Google Scholar]
- 50.Schmeck B, Zahlten J, Moog K, et al. Streptococcus pneumoniae-induced p38 MAPK-dependent phosphorylation of RelA at the interleukin-8 promotor. J Biol Chem. 2004;279(51):53241–53247. doi: 10.1074/jbc.M313702200. [DOI] [PubMed] [Google Scholar]
- 51.Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W. IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J Biol Chem. 1999;274(43):30353–30356. doi: 10.1074/jbc.274.43.30353. [DOI] [PubMed] [Google Scholar]
- 52.Brill A, Chauhan AK, Canault M, Walsh MT, Bergmeier W, Wagner DD. Oxidative stress activates ADAM17/TACE and induces its target receptor shedding in platelets in a p38-dependent fashion. Cardiovasc Res. 2009;84(1):137–144. doi: 10.1093/cvr/cvp176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Skuland T, Ovrevik J, Låg M, Schwarze P, Refsnes M. Silica nanoparticles induce cytokine responses in lung epithelial cells through activation of a p38/TACE/TGF-α/EGFR-pathway and NF-κΒ signalling. Toxicol Appl Pharmacol. 2014;279(1):76–86. doi: 10.1016/j.taap.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 54.Øvrevik J, Låg M, Hetland RB, Schwarze PE, Refsnes M. Stone particle-induced interleukin-6 and -8 release involves activation of MAP kinases and tyrosine kinases. Ann Occup Hyg. 2002;46(Suppl 1):390–392. [Google Scholar]
- 55.Øvrevik J, Låg M, Schwarze P, Refsnes M. p38 and Src-ERK1/2 pathways regulate crystalline silica-induced chemokine release in pulmonary epithelial cells. Toxicol Sci. 2004;81(2):480–490. doi: 10.1093/toxsci/kfh214. [DOI] [PubMed] [Google Scholar]