

Abstract
The secreted Chlamydia protease CPAF cleaves a defined set of mammalian and Chlamydia proteins in vitro. As a result, this protease has been proposed to modulate a range of bacterial and host cellular functions. However, it has recently come into question the extent to which many of its identified substrates constitute bona fide targets of proteolysis in infected host cell rather than artifacts of post lysis degradation. Here we clarify the role played by CPAF in cellular models of infection by analyzing Chlamydia trachomatis mutants deficient for CPAF activity. Using reverse genetic approaches, we identified two C. trachomatis strains possessing nonsense, loss-of-function mutations in cpa (CT858), and a third strain containing a mutation in Type II secretion (T2S) machinery that inhibited CPAF activity by blocking zymogen secretion and subsequent proteolytic maturation into the active hydrolase. HeLa cells infected with T2S− or CPAF− C. trachomatis mutants lacked detectable in vitro CPAF proteolytic activity, and were not defective for cellular traits that have been previously attributed to CPAF activity, including resistance to staurosporine-induced apoptosis, Golgi fragmentation, altered NFκB-dependent gene expression, and resistance to reinfection. However, CPAF-deficient mutants did display impaired generation of infectious elementary bodies (EBs), indicating an important role for this protease in the full replicative potential of C. trachomatis. In addition, we provide compelling evidence in live cells that CPAF-mediated protein processing of at least two host protein targets, vimentin filaments and the nuclear envelope protein Lamin-associated protein 1 (LAP1), occurs rapidly after the loss of the inclusion membrane integrity, but before loss of plasma membrane permeability and cell lysis. CPAF-dependent processing of host proteins correlates with a loss of inclusion membrane integrity, and so we propose that CPAF plays a role late in infection, possibly during the stages leading to the dismantling of the infected cell prior to the release of EBs during cell lysis.
INTRODUCTION
The obligate, intracellular bacterial pathogen Chlamydia trachomatis primarily infects epithelial cells of the urogenital tract and the conjunctiva, leading to sexually transmitted diseases and conjunctivitis [1]. Disease is often associated with immune damage resulting from chronic inflammation due to repeated and recurring infections. In women, genital tract infections can result in severe sequelae such as pelvic inflammatory disease, ectopic pregnancies and infertility [2]. Similarly, recurrent and untreated C. trachomatis conjunctival infections can lead to trachoma, a prominent cause of blindness worldwide [3].
C. trachomatis infection begins with the attachment and entry of elementary bodies (EB), the infectious form of Chlamydia, into epithelial cells. Upon entry, the EB form transitions into the replicative reticulate body (RB) form, and establishes a parasitophorous vacuole (“inclusion”) that avoids fusion with lysosomal compartments and ensuing degradation of its contents [1]. At mid-to-late stages of infection, RB replication becomes asynchronous to generate more RBs and intermediary forms that transition back to the EB form. By the end of the cycle, bacteria within the inclusion are released to infect neighboring cells. The process of exit from infected cells can occur through a lytic phase, where there is a programmed dismantling of the inclusion membrane that precedes loss of plasma membrane integrity, and a non lytic, “extrusion” exit mechanism that leads to the exocytosis of the entire inclusion or inclusion fragments [4].
To modulate host cellular functions, Chlamydia employs a type III secretion (T3S) system to translocate “effector” proteins that mediate cell invasion, re-routing of lipid transport, and manipulation of signaling pathways important in immunity [5]. In addition, some chlamydial virulence proteins contain “classical” signal peptides and presumably use the Sec secretory system to cross the bacterial cytoplasmic membrane [6]. How these signal peptide-containing proteins are further translocated from the bacterial periplasmic space to the inclusion lumen and eventually across the inclusion membrane is unclear. Potential delivery pathways include outer membrane vesicles [7,8] and the Type II secretion (T2S) machinery, which in gram-negative bacteria is required to secrete a subset of folded proteins across the outer membrane [9]. One prominent example of a protein that may follow this secretion pathway is the chlamydial protease-like activity factor (CPAF), a serine protease [10]. Late in infection, CPAF localizes to the inclusion lumen and the host cell cytoplasm as assessed by immunofluorecence microscopy and subcellular fractionation [11,12].
CPAF was the first Chlamydia virulence factor for which a biochemical activity (protease) and a target (transcription factors) had been established [11]. CPAF-mediated degradation of the transcription factors RFX-5 and USF-1 were linked to the loss of expression of MHC Class I and II - a cellular phenotype that had been observed in infected cells [13]. CPAF has also been reported to cleave the pro-apoptotic factors Bim and Puma [10,14], p65/RelA, a transcription factor required for NFκB signaling [15], intermediate filaments vimentin, and keratins 8 and 18, [16,17], the adherence junction protein nectin1 [18], the MHC-like lipid presentation protein CD1d [19], the pro-inflammatory mediator HMGB1 [20], the mitotic cell cycle regulator CyclinB1 [21], securin [22,23], the Golgi tethering factor Golgin 84 [23], and PARP – a mediator of DNA-damage during apoptosis [24]. Because of CPAF’s apparent preference for proteins important in host immunity and signaling, it has been proposed that CPAF-mediated proteolysis represents a core strategy employed by Chlamydia to modify host-signaling pathways and usurp the cellular machinery for its own benefit [25,26]. Additional functions of CPAF may include death of the infected host cell as ectopic over expression of CPAF in mammalian cells initiates a host cell death pathway that mimics the necrotic cell death observed at the end of the Chlamydia life cycle [24]. In addition, Chlamydia proteins have also been reported to be targets of CPAF-mediated processing including early T3S effectors, which we postulated plays a role in protection from re-infection [27], and the abundant outer membrane protein OmcB [28], which may represent a mechanism to generate soluble form(s) of OmcB that can access the host cell cytoplasm [29].
As more CPAF targets began to emerge, correlations were made between the degradation of these factors and cellular phenotypes of Chlamydia-infected cells. This list includes, but is not limited to, Golgi fragmentation, inclusion expansion, resistance to re-infection, modulation of apoptosis and pro-inflammatory signaling pathways, disruption of the cell cycle, cell junction defects, and centrosomal abnormalities (reviewed in [25]). Because molecular genetic tools to specifically inactivate chlamydial genes were not available at the time, many of these correlations could not be formally tested in CPAF-deficient mutants. The development of small molecule and peptide inhibitors that blocked CPAF activity in vitro and in vivo promised to be helpful in assessing some of these questions [27]. Indeed, cell-permeable inhibitors blocked CPAF activity in cell culture infection models, decreased the replicative potential of Chlamydia [23,27,28] and often reversed cellular phenotypes of infected cells that have been associated with CPAF. However, these CPAF inhibitors also varied in their effect in cells and displayed varied levels of toxicity [23,27].
CPAF is fairly selective in its target specificity and broad degradation of proteins is not observed when crude cell lysates are treated with recombinant CPAF [27]. However, the significance of CPAF-dependent proteolysis in vivo has recently come into question since post-cell lysis sample preparation could expose putative substrates under examination to degradation by nonspecific proteases that are commonly released upon cellular lysis [30]. As such, the extent to which CPAF cleaves reported substrates in intact infected cells remains unclear [30]. Definitive experimental evidence for the in vivo target specificity of CPAF and the consequences of its proteolytic activity has been hampered by the difficulty in identifying CPAF substrate recognition sites that can be mutated, the redundancies in the function of host cell targets, and the lack of a system to generate defined mutations in Chlamydia. Furthermore, while cell permeable inhibitors of CPAF demonstrated a role in chlamydial pathogenesis, off-target effects are difficult to anticipate or control and thus can limit their usefulness as compared to genetic knockout approaches [23,27,28].
In this study, we sought to clarify the role played by CPAF by performing a phenotypic analysis of C. trachomatis mutants that are either defective for CPAF secretion or have loss-of-function mutations in cpa. In this manner, we confirm that CPAF is required for the efficient generation of Chlamydia infectious progeny. We also report that many of the cellular phenotypes of Chlamydia-infected cells that had been previously ascribed to CPAF, are not CPAF-dependent. However, it is important to note that this does not mean that all identified CPAF substrates are not targeted for proteolysis in vivo. We provide compelling evidence for CPAF-mediated processing of vimentin as well as a new CPAF substrate we identified in vitro, the nuclear envelope protein Lamin Associated protein-1 (LAP1). These proteolytic events occurred in intact live cells late in infection soon after loss of inclusion membrane integrity, suggesting that the bulk of active CPAF is sequestered within the inclusion lumen.
MATERIALS AND METHODS
Reagents
Reagents were obtained from the following sources: mouse anti-GM130 (BD Biosciences), rabbit anti-Chlamydia MOMP (K. Fields, U. of Kentucky), mouse anti-CT813 (G. Zhong, U. of Texas Health and Science Center), mouse anti-vimentin (Invitrogen, clone V6630), rabbit anti-GAPDH (Abcam), mouse anti-EGFP monoclonal antibody (Clontech), rabbit anti-LAP1 antibodies (William Dauer, U. of Michigan), rabbit anti-RpoB/C (M. Tan, UC Irvine), rabbit anti-CPAF (27), mouse anti-phosphotyrosine (Cell signaling), Alexa Fluor 555-conjugated anti-mouse, Alexa Fluor 488-conjugated anti-rabbit, Hoescht 33258 (Invitrogen), FluorSave Reagent (Calbiochem), Staurosporine (Cell Signaling), IL-1β (Biolegend), Cyclohexamide (Sigma-Aldrich), Rifampicin (Sigma-Aldrich), Trimethoprim (Sigma-Aldrich), Slow Fade Gold Antifade Reagent (Invitrogen), JetPrime (Polyplus transfection). Enhanced green fluorescent protein (EGFP)-tagged rat vimentin was provided by Ronald Liem (Columbia University, NY), and tandem dimer Tomato (tdTomato) was provided by Marc Caron (Duke University, NC). Full-length lamin-associated protein 1 (LAP1) was PCR-amplified from MGC Human sequence-verified cDNA clone 3458117 (Thermo Fisher) and inserted into pLEGFP-C1 (Clontech) downstream of EGFP to express an EGFP-LAP1 fusion protein.
Cell Culture and Chlamydia Infections
HeLa cells (CCL-2: ATCC) and Vero cells (CCL-81: ATCC) were maintained in DMEM HG supplemented with 10% FBS (CellGro Mediatech). C. trachomatis LGV-L2 434/Bu and C. trachomatis mutant strains were propagated by infecting HeLa cells with elementary bodies (EBs) that had been stored in sucrose-phosphate-glutamate (SPG) buffer (0.25 M sucrose, 10 mM sodium phosphate, 5 mM L-glutamic acid, pH7.0) and purified on Omnipaque (GE Healthcare) density gradients [31]. EBs were added to HeLa cells at the indicated multiplicities of infection (MOIs), and infections were synchronized by centrifugation at 2500 × g for 30 min at 10°C. Rifampin, spectinomycin, and trimethoprim resistant C. trachomatis LGV-L2 variants were generated as previously described [32]. The mutations leading to antibiotic resistance in these strains were determined by whole genome sequencing: H471Y in CTL0567 (rpoB), G1197 in CTL_r01/CTL_r02 (16S rRNA copies 1 and 2), and G408R in CTL0369 lead to RifR, SpcR, and TmpR, respectively.
Plaque assays
Plaque assays were performed as previously described [33]. Briefly, monolayers of Vero cells grown in a 6-well plate were infected with a concentration of ~100 inclusion-forming units (IFUs) per well. Cells were incubated for 2 hours at 37 °C and 5% CO2, allowing for bacterial internalization. The growth medium in infected cell monolayers was replaced with 6 mL of a DMEM/agarose overlay/well (DMEM HG, 10% FBS, 50 μg/mL Gentamicin, 500 ng/mL cyclohexamide, 1X nonessential amino acids (Gibco), 0.5% SeaKem LE agarose (Lonza)) allowed to solidify for 10 minutes, and dried in a sterile cabinet without lids for 15 minutes. Cells were incubated for 10 to 14 days until plaques were observed. Plaques were isolated using a pipette tip and transferred to Vero cell monolayers for amplification.
Identification and whole genome sequencing of CPAF-deficient LGV-L2 strains
Identification of cpa mutants
LGV-L2 strains containing the null alleles C127T (Q43*) and G882A (W294*) in cpa (CTL0233) were initially identified by whole genome sequencing of pools of chemically-mutagenized and plaque-purified C. trachomatis LVG-L2 434/Bu strains (Bastidas R. J. and Valdivia R. H. unpublished results). Strain CTL2-M532 harboring the cpaC127T allele and strain CTL2-M169 harboring the cpaG882A allele were identified from among two independent pools of mutants and the mutations in the cpa locus (CTL0233) were confirmed by Sanger sequencing.
Genomic sequencing
Strains were harvested from infected Vero cells grown in a 6-well cell culture plate by hypotonic lysis of host cells with 800 μl of dH2O per well (for 20 minutes) followed by addition of 200 μl of 5X SPG buffer. Lysates were sonicated (2 × 10 seconds in ice water) and bacterial cells spun down at 14,000 rpm, for 15 minutes at 4 °C. Bacterial pellets were pooled and resuspended in 1X DNAse I buffer (New England Biolabs, Ipswich, MA, USA). Depletion of host DNA was achieved by treating cell suspensions with 4 units of DNAse I (New England Biolabs) for 1 hour at 37 °C. Bacterial pellets were washed with PBS buffer and total DNA isolated with a DNA isolation kit (DNeasy tissue and blood kit, Qiagen) following the manufacturers instructions. M169 (1 μg) and M532 enriched DNA (1 μg) were each sheared with an Adaptive Focused Acoustics S220 instrument (Covaris). DNA sequencing libraries were prepared with a library construction kit (TruSeq DNA Sample Preparation Kit v2, Illumina, Inc. San Diego, CA, USA) according to the manufacturer’s instructions. Libraries were sequenced in a MiSeq DNA Sequencing Platform (Illumina) at the Duke University IGSP DNA Sequencing Core facility. Genome assembly and single nucleotide variant (SNV) identification was performed with Geneious Software version 6 (Biomatters - http://www.geneious.com). The C. trachomatis LGV L2 434/Bu genome (GenBank no. NC_010287) was used as reference sequence. All non-synonymous single nucleotide variants (SNVs) identified in M169 and M532 (Table 1 and Table S1) were verified by Sanger dideoxy DNA sequencing.
Generation of M169 RST recombinant strains
Recombinant LGV-L2 strains were generated as previously described [33]. Briefly, confluent Vero cells grown on a 24-well plate were co-infected with M169 (RifR) and a SpcR mapping strain at a MOI ratio of 3:3. Recombinant progeny were selected from among plaques that formed in the presence of 200 ng/mL Rif and 200 μg/mL Spc. Plaque-purified recombinants were further expanded in Vero cells and genotyped to assess the segregation of mutations present in the CTL2-M169 parental strain. RifR and SpcR M169 recombinants harboring the cpaG882A allele were further backcrossed to a trimethoprim resistant (TmpR) mapping strain as described above and progeny selected from plaques that formed in the presence of Spc and 150 μg/mL Tmp. These second-generation, plaque-purified recombinants were further expanded in Vero cells and a M169-derived recombinant strain harboring the cpaG882A allele (M169 RST17 (CPAF−)) was identified by genotyping with SNV specific primers. A co-isogenic recombinant strain (RST5 (CPAF+)) that shares the same background SNVs as RST17 (CPAF−) and that inherited a wild type cpa allele was also identified. The genomes of both RST recombinant strains were re-sequenced and the relevant SNVs identified (Table 2 and in Table S2). A list of C. trachomatis strains described in this study can be found in Table S3.
Western blot analyses
HeLa cells were grown in 6-well plates to confluency and infected with C. trachomatis LGV-L2 RifR parent or its derived mutant strains at an MOI of 1. At the indicated hours post infection, cells were washed with 1X PBS (Invitrogen), lysed with 1% SDS buffer (1% SDS, 150 mM NaCl, 50 mM Tris-HCl pH 7.5) heated in boiling water bath immediately before addition to cells. Lysates were incubated at 65 °C for 10 minutes to solubilize and denature proteins and sonicated 2×10 seconds to shear DNA. Protein concentrations were determined by the DC protein assay (Bio-Rad). Equal amounts of lysate were loaded into SDS-PAGE 4–15% gradient gels (Bio-Rad), transferred to 0.45 μM nitrocellulose membranes using a Trans-Blot SD Semi-Dry Electrophoretic Transfer Cell (Bio-Rad), blocked in Odyssey blocking buffer (LI-COR) and incubated in primary antibodies, followed by incubation with goat anti-rabbit IRDye 680LT (LI-COR) or goat anti-mouse IRDye 800CW (LI-COR). Membranes were imaged with the LI-COR Odyssey infrared imaging system.
In Vitro CPAF cleavage assays
HeLa cells grown in 6-well plates were mock-infected or infected with the C. trachomatis LGV-L2 RifR strain or with the indicated mutant strains at an MOI of 1. At 40 hours post infection (hpi) crude protein extracts were prepared by lysing infected cells in RIPA buffer (50 mM Tris pH 7.5, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP-40) supplemented with a protease inhibitor cocktail (Roche). Purified recombinant CPAF (40 μg) [34] or crude protein extracts were prepared as described above and incubated with 20 μg recombinant GST-CT695 for 1hr at 37 °C, and cleavage was assessed by colloidal blue Commassie Blue staining (Invitrogen). Figures were compiled and intensities adjusted for display using Photoshop CS6.
To test the effectiveness of 1% SDS buffer in preventing post-lysis degradation by CPAF, the activity of recombinant CPAF in this denaturing buffer was assessed. Crude protein extracts were prepared from HeLa cells by rinsing monolayers with PBS, adding 1% SDS buffer pre-warmed to 100 °C, transferring to a microfuge tube, heating at 65 °C for 10 minutes, and then clarifying the lysate by centrifugation (10,000 × g, 15 minutes, room temperature). For the in vitro CPAF cleavage assays, 200 μg of total HeLa protein extract with or without 100 μM of the CPAF-inhibitory peptide Pep2 [34] was mixed with 0.5, 2.5, or 5 μg recombinant CPAF in a final volume of 100 μl. The reactions were assembled at room temperature, incubated at 37 °C for 20 minutes, and then inactivated with SDS-PAGE sample buffer and incubation at 65 °C for 10 minutes. As a positive control, reactions were also performed under non-denaturing conditions. Crude protein extracts were generated from HeLa cells by rinsing monolayers with PBS, adding ice-cold TNEX buffer (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, complete protease inhibitor cocktail (Roche)), incubating at 4°C for 10 minutes and then clarifying the lysate by centrifugation (10,000 × g, 15 minutes, 4 °C). Reactions were assembled on ice, incubated at 37 °C for 20 minutes, and subsequently processed as described for the reactions in 1% SDS buffer. CPAF activity was determined by monitoring vimentin cleavage via western blot analysis.
In vitro cleavage assays were used to demonstrate that CPAF cleaves EGFP-LAP1. HeLa cells transfected with the EGFP-LAP1 construct were harvested in TNEX buffer, and crude protein extracts were generated as described above. For the reactions, 100 μg of transfected or non-transfected extract was incubated with either recombinant CPAF or TNEX protein extracts prepared from mock-infected or LGV-L2-infected (44 hours post-infection) HeLa cells. The reactions were assembled on ice, incubated at 37 °C for 30 minutes, and then inactivated with SDS-PAGE sample buffer and incubation at 65 °C for 10 minutes. Cleavage was assessed by western blot analysis with mouse anti-EGFP) and rabbit anti-LAP1 antibodies.
IFU burst assays
Vero cells were seeded onto 96-well plates (density of 15,000 cells/well) and infected with each of the strains analyzed (six biological replicates per each timepoint) at a MOI = ~0.6. To determine the input inclusion forming units (IFUs), a set of infected wells were fixed with 100% methanol (EMD Millipore) for 10 minutes on ice and stained with a polyclonal anti-LGV-L2 sera followed by Alexafluor-conjugated secondary antibodies and cells samples mounted for fluorescence microscopy using the FluorSave reagent. Images were acquired in a Zeiss Axioskop 2 upright epifluorescence microscope and the number of inclusions in at least 5 different fields per replicate were counted. Output IFUs at 30 hpi and 48 hpi were determined by lysing infected cells as described in [33] and infecting Vero cells with serial dilutions of harvested cell lysates. After 40–42 hpi, cells were fixed, immunostained, and the number of inclusions determined as described above. To determine the infectious progeny generated per input bacteria (“IFU burst”), the total number of output IFUs was divided by the total number of input IFUs.
Immunofluorescence microscopy
For routine indirect immunofluorescence, HeLa cells were grown on glass coverslips and infected at the indicated MOIs. Cells were fixed with 3% formaldehyde/0.025% glutaraldehyde or 4% paraformaldehyde or methanol in phosphate-buffered saline (PBS) for 20 minutes and permeabilized with 0.1–0.2% Triton X-100 for 10 minutes. After blocking with 5% bovine serum albumin (BSA) in PBS for 20 minutes, cells were stained with specific antibodies followed by Alexa-conjugated secondary antibodies at room temperature at 20 minutes. Host and bacterial DNA were stained with 1 μg/mL Hoechst. Coverslips were mounted with FluorSave, or Slow Fade.
Golgi Fragmentation Analysis
HeLa cells were seeded onto coverslips to visualize the Golgi apparatus in cell infected with RifR or M532 (CPAF-) C. trachomatis strains. A set of infected cells were treated with Z-WEHD-fmk (75 μM, Enzo Life Sciences) for 9h after infection as a positive control for blocking Golgi fragmentation. At 24h after infection, cells were fixed with 4% paraformaldehyde for 20 minutes at room temperature, permeabilized, and blocked with BSA-PBS. Bacteria, the Golgi apparatus, and DNA were detected by incubating cells with rabbit anti-Chlamydia MOMP and mouse anti-GM130 antibodies for 20 minutes followed by incubation with fluorescently conjugated secondary antibodies and Hoescht for 20 minutes at room temperature. Cells were mounted with FluorSave and allowed to cure at room temperature overnight before imaging. Images were acquired with a Zeiss 780 scanning confocal microscope with the same settings for each sample with a 100× objective and processed using ImageJ. Confocal images of specific samples were used to quantify Golgi fragmentation. The number and area of Golgi elements in 18 cells at minimum per condition were counted after a applying a threshold calculated with the Otsu algorithm for each image and using the Analyse Particles function in ImageJ software excluding particles smaller than 0.1 μm. Three independent experiments were performed and imaged to analyze fragmentation. Figures were compiled and intensities adjusted for display using Adobe Photoshop CS6. All raw data is available upon request.
Detergent Extraction Assays
For immunofluorescence assays after detergent extraction, HeLa cells were seeded in 96-well plates, with three biological replicates per condition (+Triton X-100 and −Triton X-100), and infected at an MOI of 1 with the indicated strains. At 44 hpi, 52 hpi, or 60 hpi cells in the untreated condition were washed twice with ice cold PBS and fixed with 3% formaldehyde/0.025% glutaraldehyde at room temperature for 20 minutes prior to permeabilization and blocking with BSA-PBS. For live cell samples extracted with Triton X-100, infected cells were first washed twice with ice cold PBS before incubation with pre-chilled 0.5% Triton X-100 in PBS supplemented with 100 μM anti-CPAF peptide [34] on ice for 5 minutes. Cells were then fixed, blocked with BSA-PBS and immunostained with mouse monoclonal anti-vimentin antibodies, followed by fluorophore conjugated secondary anti-mouse antibodies. Samples were mounted with Slow Fade Gold Antifade reagent and images from at least 5 fields for each replicate were acquired with a Zeiss Axioskop 2 upright epifluorescence microscope using Axiovision v3.0 software. The number of infected cells with altered vimentin staining was calculated for each replicate in 0.5% Triton X-100 treated and untreated samples. The average percentage of infected cells with altered vimentin (6–8% of total cells) staining in the untreated samples was subtracted from the treated control for each replicate. Figures were compiled and intensities adjusted for display using Photoshop CS6. Two-way ANOVA with Bonferroni’s post test was performed using GraphPad Prism for Windows, GraphPad Software, San Diego California USA.
NF-κB Reporter Assays
The NF-κB luciferase HeLa reporter cell line used in this study was generated by stably transducing HeLa cells with an NF-κB-Luciferase reporter system (SABiosciences) following the manufacturer’s instructions. NF-κB activity was assayed by infecting reporter cells in triplicate for each condition in 96-well plates with the indicated strains at an MOI of 10, 30, or 50. Experiments were performed in duplicate. At the time of infection, cells were treated with 10 ng/mL of IL-1β (BioLegend). After 24 hours, cells were lysed and luciferase activity measured using the britelite plus reagent (PerkinElmer) according to manufacturer’s instructions and luminescence values determined using an EnSpire 2300 Multilabel reader (PerkinElmer). Luminescence values obtained for each sample were normalized to the IL-1β treated, mock-infected control. Data was analyzed and figures generated using GraphPad Prism (GraphPad Software, San Diego California USA).
Apoptosis Induction Assay
HeLa cells grown on glass coverslips were infected with a MOI of 0.5 with the indicated strains (two biological replicates and two technical replicates per condition). Six hours prior to fixation cells were treated with 2μM staurosporine (Cell Signaling). At 24 hpi, 36 hpi, and 48 hpi infected cells were fixed with methanol and stained with anti-LGV-L2 antibodies followed by Hoechst and Alexa-conjugated secondary antibodies. The number of infected or mock-infected cells with condensed nuclei was counted using a Zeiss Axioskop 2 upright epifluorescence microscope with Axiovision v3.0 software for 200 infected cells in each condition. Data was analyzed and figures generated using GraphPad Prism.
Assessment of secondary infections
HeLa cells were seeded on coverslips in 24-well plates (50,000 cells/well) and incubated overnight. For primary infections, cells were infected with purified EBs from either C. trachomatis L2 434/Bu or its mutant RifR derivatives, as indicated at an MOI of 1. At 29 hpi, secondary infections were performed using a C. trachomatis L2 434/Bu strain transformed with the GFP-expressing plasmid pGFP-SW2 [35]. After 1h, cells were fixed (3% formaldehyde-0.025% glutaraldehyde, 20 min, RT) and immunostained with mouse anti-phospho tyrosine monoclonal antibodies without permeabilization and counter-stained with anti-mouse Alexafluor 555-conjugated secondary antibodies (Invitrogen). Coverslips were mounted in Slow Fade Gold Antifade media and images were acquired using a Zeiss Axioskop 2 upright epifluorescence microscope with Axiovision v3.0 software. Phosphotyrosine-containing foci were counted in 35 cells for each of two biological replicates, in two independent experiments performed by two independent observers. One-way ANOVA with Bonferroni’s multiple comparison test was performed using GraphPad Prism.
EGFP-Vimentin and LAP1-EGFP transfection and live cell microscopy
Cells were seeded onto #1.5 glass-bottom plates, infected with RifR L2 434/Bu, M532 (CPAF−), M169 (CPAF−), RSTE4 (TS2−) strains, and co-transfected with either EGFP-Vimentin or EGFP-LAP1 and tdTomato using the lipid-based JetPrime reagent 4h after infection. Cells were imaged every seven minutes from 54–76hpi after infection at 37°C under 5% CO2 using a motorized Zeiss Axio Observer Z1 widefield fluorescence microscope equipped with a 40× air objective. Fifteen stage positions at minimum were recorded with 6–23 instances of inclusion rupture in transfected cells observed for each condition. Only cells expressing moderate levels of the fluorescent reporters were included in the analysis. Images were viewed with Metamorph to manually assess inclusion rupture by tdTomato diffusion into the inclusion lumen and the structured or diffuse nature of the EGFP signal. Images were deconvolved using Huygens Essential, and processed with ImageJ and Photoshop for presentation.
RESULTS AND DISCUSSION
A Chlamydia mutant defective for Type II secretion accumulates unprocessed CPAF zymogen
C. trachomatis remains poorly characterized because of its history of intractability to routine molecular genetic manipulation. Recently we developed a combinatorial approach to rapidly generate a comprehensive library of genetically defined mutants [33]. Chemical mutagenesis coupled with whole-genome sequencing (WGS) was used to generate Chlamydia mutants with distinct phenotypes and to map the underlying genetic lesions. As a result, we identified a C. trachomatis LGV-L2 variant (RSTE4 (T2S-)) bearing a point mutation in the Type II secretion ATPase GspE [33]. This mutant is attenuated for growth and accumulates insoluble glycogen granules within inclusions, presumably as a result of the impaired secretion of glycogen processing enzymes [33]. Given the known role of T2S in the export of folded hydrolases from the bacterial periplasm [9], we hypothesized that this secretion system is also responsible for the export of CPAF. Indeed, immunofluorescence analysis with anti-CPAF antibodies of HeLa cells infected with C. trachomatis RSTE4 (T2S−) mutants revealed that all CPAF was exclusively associated with bacteria within the inclusion and no immunoreactive material was present in the host cytoplasm (Fig. 1A–B). An immunoblot analysis of total protein lysates from cells infected with RSTE4 (T2S−) further showed that the processed 35kDa and 29kDa bands of mature CPAF [36], which are prominent in cells infected with wild type C. trachomatis, are absent. Instead, a higher molecular weight 70kDa band consistent with the size of unprocessed CPAF zymogen [36] was detected, albeit at lower abundance (Fig. 1C). Lysates from cells infected with an unrelated C. trachomatis slow-growing mutant (SPQ6-2 (TrxB*))[33] also showed the processed forms of CPAF, indicating that the accumulation of unprocessed CPAF in cells infected with the T2S− mutants is not due to a slower replication rate. Consistent with this observation, crude lysates from cells infected with a gspE mutant lacked in vitro CPAF activity against a purified substrate (recombinant GST-CT695) (Fig 1D). Overall these results suggest that CPAF is a substrate of T2S and that secretion is coupled to its processing into an active form.
Figure 1. Identification of C. trachomatis strains deficient in CPAF secretion or expression.

AB). CPAF fails to accumulate in the cytoplasm of cells infected with a T2S-deficient mutant and is not detectable in a CPAF-truncation mutant (M169). The subcellular localization of CPAF in HeLa cells infected with the indicated C. trachomatis mutant strains was assessed by indirect immunofluorescence with polyclonal anti-CPAF antibodies (green), DNA with Hoescht (blue), and the inclusion membrane with anti-CT813 antibodies (red). Immunoreactive anti-CPAF material could not be detected in cells infected with a CPAF-deficient strain, or in the cytoplasm of cells infected with a T2S-deficient strain. The relative distribution of CPAF at 32 hpi in infected cells (n=100) is shown in panel B. C) T2S-deficient mutants fail to process CPAF into its active form. Immunoblot analysis of total protein lysates from HeLa cells infected with the indicated strains at 48 hpi. Strains containing mutations in cpa that do not express CPAF and T2S-deficient strains exhibit accumulation of a larger molecular weight band consistent with the size of unprocessed CPAF (proCPAF). The C terminal fragment and N terminal fragment of CPAF are indicated by “CPAFc” and “CPAFn”, respectively. D) Lysates from cells infected with CPAF or T2S-deficient C. trachomatis mutants do not process CPAF substrates in vitro. Purified recombinant CPAF (rCPAF) or lysates from HeLa cells that were mock infected or infected with the indicated strain were harvested at 40 hpi and incubated with recombinant GST-CT695, an in vitro CPAF substrate. Cleavage products were monitored by SDS PAGE and staining with Coomassie Blue. FL: full length GST-CT695; C: cleavage product. E) M169-derived recombinant C. trachomatis strains retaining the cpa mutation do not express CPAF. Immunoblot analysis of total protein lysates of HeLa cells infected with RST17 (CPAF-) or its nearly co-isogenic recombinant sibling RST5 (CPAF+) for 48 hours. CPAF expression is restored in RST5 (CPAF+) bacteria but still absent in the RST17 (CPAF−) strain. The genotypes of these strains are shown in Tables 2 and Table S2.
Identification of Chlamydia strains with loss-of-function mutations in CPAF
The finding that RSTE4 (T2S−) mutants did not possess CPAF activity suggested that CPAF is not absolutely essential for bacterial viability. This is consistent with previous observations made with CPAF-inhibitors, where 5–10 fold differences in the yield of infectious units were reported [23,27,28]. We subsequently generated a bank of chemically mutagenized C. trachomatis LGV-L2 strains and screened for mutants that consistently formed small plaques on Vero cell monolayers overlaid with soft agar. We isolated ~1000 small plaque forming mutants, which were amplified in Vero cells, and their DNA isolated and sequenced (Bastidas and Valdivia, unpublished data). Among this collection of mutants, we identified two strains (M169 and M532) with nonsense mutations in cpa (Table 1 and supplementary Table S1). We determined that a representative CPAF truncation mutant (M169) had no detectable CPAF protein by immunofluorescence microscopy (Fig 1A–B) or western blots (Fig 1C). Furthermore, as observed for the T2S-deficient Chlamydia mutant, lysates of HeLa cells infected with the strains M169 and M532 lacked in vitro protease activity (Fig 1D).
CPAF mutants are defective for the generation of infectious elementary bodies
We next sequenced the genome of the CPAF-deficient isolate M169 and identified 19 additional non-parental SNVs (Table S1). To segregate the cpa mutation from these other mutations, we co-infected Vero cells with M169, which had been generated in a RifR background, and a wild type SpcR LGV-L2 strain. Progeny from this co-infection were amplified in the presence of rifampicin and spectinomycin to isolate recombinant strains as previously described [33]. In this manner, we generated SpcRRifR recombinants where only four mutations were inherited from the parental M169 strain. These strains were further “crossed” to a TmpR LGV-L2 strain by new co-infections to further decrease the number of extraneous mutations. As a result two strains were produced, RST5 (CPAF+) and RST17 (CPAF−) (Table 2), where the only differences between their genotypes in addition to a nonsense mutation in cpa (CTL0233) in RST17 (CPAF−) is the presence of an L to F substitution in a non-conserved residue of FtsH (CTL0213) in RST5 (CPAF+) and a S to F substitution at residue 555 in CTL0884 in RST17 (CPAF−) that was present in the SpcR strain used for crosses.
Next, we determined if RST5 (CPAF+) displayed any differences in growth potential as compared to its nearly co-isogenic partner RST17 (CPAF−). Cells infected with the cpa mutant RST17 (CPAF−) consistently displayed a ~3 fold decrease in EB yields per input EB as compared to RST5 (CPAF+) (Fig 2). These findings support a role for CPAF in the generation of fully infectious forms of C. trachomatis.
Figure 2. C. trachomatis mutants deficient in CPAF expression are impaired for the generation of infectious progeny.

Vero cells were infected with the indicated strains at an MOI=0.3 and infectious progeny released per input IFU was calculated at 30hpi and 48hpi. RST17 (CPAF−) mutants produce a 3-fold lower yield of infectious units compared with RST5 (CPAF+) bacteria at 48hpi. Three independent experiments, SEM, two-way ANOVA with Bonferroni post-hoc, ***p<0.001.
HeLa cells infected with CPAF-deficient Chlamydia mutants display Golgi fragmentation, activation of NFκB, and resistance to apoptosis and re-infection
Because the expected consequences of the degradation of previously identified CPAF targets correlated with cellular phenotypes observed in C. trachomatis infected cells, and because these phenotypes were reversed by CPAF inhibitors, it has been widely assumed that CPAF may be required for these cellular disruptions [23,25,27,28]. We tested this premise through the use of T2S− and CPAF− C. trachomatis mutants. We monitored the following cellular phenotypes associated with Chlamydia infections:
Golgi fragmentation
C. trachomatis infection leads to fragmentation of the Golgi apparatus into mini-stacks [37], a phenotype that can be recapitulated by expression of active CPAF in infected cells [23,37]. Golgi fragmentation in infected cells correlates with the cleavage of golgin-84 [37]. The levels of Golgi fragmentation in cells infected with wild type LGV-L2 or M532 (CPAF−) were assessed by quantifying the number of GM130-positive Golgi structures, using previously described methods [23,37]. No significant differences between the wild type and the cpa mutant were observed, as both strains exhibited Golgi fragmentation (Fig 3A–B). As a control, we treated infected cells with the caspase-1 inhibitor z-WEHD-fmk, which has been reported to block CPAF activity, and to inhibit golgin-84 cleavage and Golgi fragmentation [23]. Indeed, z-WEHD-fmk partially rescued Golgi fragmentation in both wild type and the cpa mutant (Fig 3C). Golgin-84 was not cleaved in cells infected with cpa mutants (data not shown). These experiments suggest that CPAF is not essential for the Golgi fragmentation phenotype observed in Chlamydia infected cells.
Figure 3. CPAF is not required for many of the cellular phenotypes associated with C. trachomatis infection.

A–C) C. trachomatis-induced Golgi fragmentation occurs in a z-WEHD-fmk-dependent manner in the absence of CPAF. HeLa cells were infected with the indicated strains for 24hpi and processed for immunofluorescence to visualize the Golgi apparatus (GM130, red), C. trachomatis, (MOMP, green) and DNA (Hoescht, blue). No difference was observed in Golgi fragmentation as assessed visually and by post-acquisition processing to assess size and number of Golgi elements between wild-type and a CPAF-deficient strain infections (panels A and B). In the same experiment, cells were treated with 75μM z-WEHD-fmk from 9hpi, which inhibited Golgi fragmentation in cells infected with wild-type and a CPAF-deficient strain (panels A and C). Three independent experiments, SEM. D) HeLa cells stably expressing an NFκB luciferase reporter were infected with the indicated strains at an MOI of 10, 30, or 50 for 24 hours and were simultaneously treated with 10 ng/ml IL-1β. Luciferase activity was measured in cell lysates and normalized to the treated mock-infected control. CPAF-deficient strains and RifR (WT) strains show an MOI dependent decrease in luciferase activity. E) Infected or mock-infected HeLa cells were treated with 2 μM staurosporine for 6 hours prior to the indicated time points and the percentage of infected cells with condensed nuclei determined. F) HeLa cells were either infected with the indicated strain or left uninfected for 29 hours prior to being infected at an MOI=25 with GFP-expressing LGV-L2 for 1 hour. The number of phospho-tyrosine foci per cell was determined for 35 cells. HeLa cells infected with the CPAF-deficient strain (M169) remain protected from superinfection. **: p < 0.01, SEM, n=3.
Expression NF-κB-dependent genes late in infection
The RelA/p65 subunit of the NF-κB transcription factor has been identified as a CPAF target [38]. RelA/p65 is required for the expression of inflammatory genes during Chlamydia infections and Chlamydia infected cells are impaired in their ability to express NF-κB-dependent genes upon stimulation with IL-1β [38]. These cleavage events and loss of responsiveness to IL-1β can be recapitulated in a cell line expressing active CPAF [38]. To formally test the role of CPAF in this process, we infected HeLa cells stably expressing a luciferase reporter under the control of an NFκB-responsive promoter with increasing MOIs of RST5 (CPAF+), RST17 (CPAF−), and M532 (CPAF−) for 24h in the presence of 10 ng/mL of IL-1β (Fig 3D). Mock infected cells displayed a robust activation of the luciferase reporter upon treatment with IL-1β. The expression of the NF-κB-dependent reporter was inhibited by infection in a dose-dependent manner. However this inhibition was independent of the presence of CPAF (Fig. 3D)
Staurosporine-induced programmed cell death
Chlamydia infected cells are remarkably resistant to extrinsic and intrinsic pro-apoptotic stimuli [39,40]. Because BH3-domain pro-apoptotic proteins have been described as targets of CPAF-mediated degradation, it has been inferred that CPAF activity is required for host cell resistance to cell death, especially late in infection [21,41,42]. We formally tested this premise by infecting HeLa cells with wild type or CPAF-deficient mutants. At timepoints of 18, 30 and 42h post infection, staurosporine was added for 6h to induce host cell death [43]. Unlike mock infected control cells, where staurosporine treatment led to nuclear condensation in greater than 70% of cells, neither wild type nor CPAF-deficient mutants showed apparent signs of nuclear condensation or cell death (Fig. 3E). These findings imply that CPAF is likely not essential for the anti-apoptosis state of Chlamydia-infected cells.
Protection from reinfection
We recently reported that early effector proteins like Tarp and CT694 are targets of CPAF-mediated degradation [27]. We postulated that one instance where such effectors would encounter CPAF is when effectors are delivered by an EB attempting to infect a cell that has an established inclusion. Consistent with this hypothesis, pre-infected cells are partially protected from reinfection [27]. Because this protection from re-infection is reversed by treating infected cells with a CPAF-specific inhibitory peptide, we postulated that CPAF may play a central role in niche protection [27]. To formally test this premise, we pre-infected cells with wild type or M169 (CPAF−) mutants for 30h, followed by a second round of infection with GFP-expressing LGV-L2 for 1h. Cells were fixed and immunostained with anti-phospho-tyrosine (Tyr) antibodies, which prominently labels Tarp that is translocated at EB entry sites. As previously reported, pre-infection decreased the number of phosphor-Tyr positive EBs [27]. However, these data suggest that protection from re-infection is not dependent on CPAF, since strain M169 (CPAF−) was not less susceptible to reinfection (Fig. 3F).
CPAF mediates cleavage of intermediate filaments late in infection
CPAF cleaves the head domain at the amino terminus of intermediate filaments in vitro [34,44,45]. Given the possibility that post lysis proteolysis may mask the extent to which truly CPAF-dependent processing occurs in live cells [30,46], we reassessed our protein harvesting protocols to minimize post-lysis proteolysis. In particular, because of the intrinsic liability of its parent thioester to long term storage that likely explains batch to batch variability on the effectiveness of some commercial batches of lactacystin (data not shown), we shifted to performing lactacystin-free protein extractions either in hot 1% SDS at pH 7.0 or in normal sample buffer supplemented with a CPAF-inhibitory peptide (termed Pep2, which is cell permeable and inhibits CPAF reversibly with a 50 nM KI) [27]. Under either of these lysis conditions, the activity of recombinant CPAF activity is completely inhibited (Fig. 4A). We therefore performed a time course of infection with C. trachomatis from 12–56h and harvested total protein under these conditions. An immunoblot analysis of total protein lysates indicated that a vimentin cleavage product is detected as compared to the CPAF− control strain, and this degradation occurred at a later timepoint during infection. We also observed that the vimentin degradation occurred to a lesser extent than previously described [45]. This difference could be attributed to the degree to which the vimentin is proteolyzed intracellularly versus postlysis or to other factor such as the degree to which vimentin fragments are extracted in 1% SDS. To exemplify the dynamic range of CPAF-mediated cleavage of vimentin occurred, the example shown in Fig 4B highlights an experiment where the least amount of vimentin proteolytic processing was observed. In parallel, we monitored the release of lactate dehydrogenase (LDH) from wells with infected cells and did not observe significant differences in LDH release suggesting that cells remained intact during the course of infection (Fig. 4C). These findings are consistent with vimentin and other CPAF targets (see below) being processed late during the course of infection.
Figure 4. Evidence for CPAF mediated processing of vimentin in intact cells.
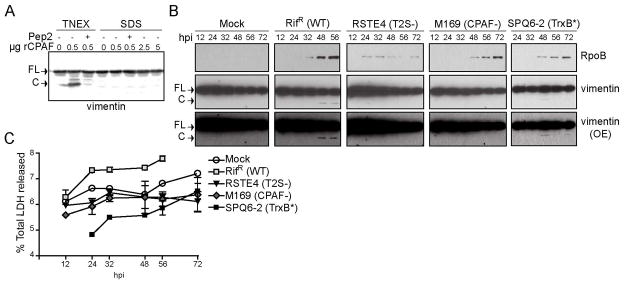
A) CPAF is not active in 1% SDS buffer. HeLa cell lysates prepared under denaturing conditions in 1% SDS buffer were incubated with the 0.5, 2.5, and 5 μg recombinant CPAF (rCPAF) for 20 minutes at 37°C. As a positive control, rCPAF was also incubated with HeLa lysates prepared under non-denaturing conditions in TNEX buffer. Where indicated, 100 μM CPAF inhibitor peptide (CIP) was included as a control. CPAF activity was assessed by monitoring the generation of vimentin cleavage products by immunoblot analysis. B) Proteolytic processing of vimentin at later stages of infection is dependent on CPAF. The bottom panel represents an increased exposure (OE) of the middle panel. FL: full-length vimentin; C: cleavage product C) LDH release by cells infected with wild type and CPAF-deficient C. trachomatis strains. The supernatants of infected cells were collected at the indicated time points and the amount of LDH release was measured and compared to total LDH levels. Error bars represent standard deviation, n=3.
We previously observed that extraction of live cells with 0.5% Triton X-100 on ice for 5 minutes preferentially extracted filaments proximal to the inclusion, but not those at the periphery within the same cell or within adjacent cells [45]. These findings were interpreted as evidence of spatially constrained alteration of the cytoskeletal properties of intermediate filaments by CPAF. Although these detergent extractions were performed in the presence of lactacystin, the effectiveness of this inhibitor is somewhat difficult to control since the active form omuralide contains a beta lactone which is labile over longer periods of time in aqueous buffers. To address potential issues arising from CPAF proteolysis during the detergent extraction process, we repeated these experiments in the presence of the CPAF inhibitory peptide Pep2 [34]. HeLa cells were infected with C. trachomatis for 44, 52 and 60h, chilled on ice and extracted with 0.5% Triton X-100 with excess amounts of the Pep2. Under these conditions, no solubilization of filaments adjacent to the inclusion for the majority of infected cells was observed (Fig. 5A lower left panel). These results indicate that either processing does not occur in all infected cells or that the extent of processing in the averaged infected cell does not lead to gross alterations in the physical properties of filaments.
Figure 5. The filament forming properties of vimentin are altered in a CPAF-dependent manner at late stages of infection.
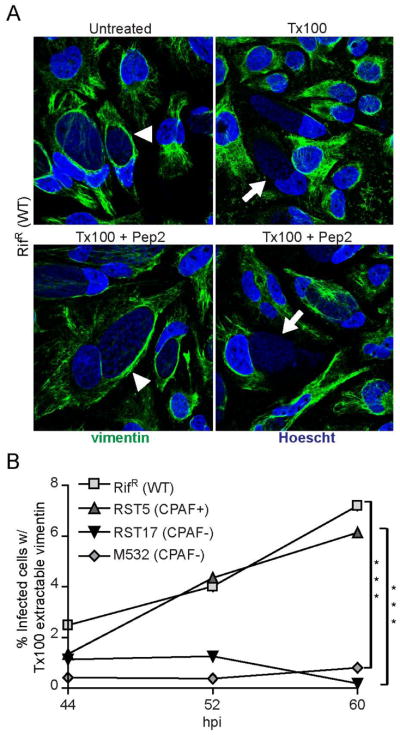
A) A subset of infected cells contain vimentin filaments that are sensitive to detergent extraction. Top panel: Immunofluorescence images of HeLa cells infected with RifR (WT) at 52 hpi. Infected cells were either untreated or treated with 0.5% Tx100 for 5 minutes on ice, prior to fixation and immunostaining for vimentin localization. Vimentin cage forms around the inclusion of infected cells (arrowhead), which are sensitive to Tx100 extraction (arrow). Bottom panel: Infected HeLa cells were treated with 0.5 % Tx100 in the presence of 100 μM CPAF-inhibitory peptide (Pep2). Note the presence of inclusions with both detergent resistant (arrowhead) and detergent sensitive (arrow) vimentin filaments. Blue: Hoescht; Green: vimentin. B) The proportion of inclusions with detergent extractable vimentin filaments increases with infection times in a CPAF-dependent manner. Cells were infected with RifR (WT) or CPAF-mutant strains and the percentage of infected cells with Tx100 extractable vimentin (Figure 5A, bottom right) was quantified at 44 hpi, 52 hpi, and 60 hpi. All extractions were done in the presence of 100 μM Pep2. Cells with extractable intermediate filaments were not apparent in HeLa cells infected CPAF-deficient mutants. ***:p<0.001, SEM, n=3.
Nonetheless, in a significant subset of infected cells, the vimentin filament network was completely extractable with Triton X-100 even when CPAF activity was carefully inhibited during the extraction process (Fig 5A lower right panel). The proportion of these cells increased to ~7% of all cells as infection progressed to >60h. To determine if CPAF played a role in this processing, we infected HeLa cells with RST5 (CPAF+) or the CPAF-deficient mutants RST17 and M532 and recorded the number of cells with Tx100-extractable vimentin filaments in the presence of Pep2 (Fig. 5B). The presence of these “detergent-sensitive” soluble vimentin filaments in infected cells was completely dependent on CPAF. These results strongly suggest that at late stages of infection a subset of infected cells display CPAF-mediated degradation of one of its well-described substrates.
Given the observation that brief extractions (<5 min) of live cells with cold Triton X-100 in the absence of robust inhibition of CPAF lead to the preferential solubilization of filaments at the inclusion periphery post-lysis, we infer that the bulk of active CPAF resides within inclusion lumen where it may access chlamydial substrates, and then host substrates in a live cell when the integrity of the inclusion membrane is compromised. To formally test this premise, we performed live cell microcopy in HeLa cells co-expressing EGFP-vimentin and tdTomato during infection with either wild type (RifR) or a CPAF-deficient mutant (M532). tdTomato is a red fluorescent protein that localizes to the host cell cytoplasm and is excluded from the inclusion lumen. Upon loss of inclusion membrane integrity, tdTomato becomes evenly distributed throughout the infected cell. Similarly, loss of plasma membrane integrity can be monitored as the overall loss of cell-associated red fluorescence, when cytoplasmic proteins rapidly diffuse into the extracellular media. In this manner, we determined that on average, plasma membrane integrity is not compromised until >30 min after the loss of inclusion membrane integrity (data not shown). Time-lapse events captured by fluorescence microscopy showed that in cells infected with wild type Chlamydia, loss of inclusion membrane integrity at late stages of infection preceded the rapid (<7 min) processing of vimentin filaments (Fig. 7A and Movie S1). In contrast, in CPAF-deficient mutants, the vimentin network remained unaltered upon loss of inclusion membrane integrity or even after loss of plasma membrane integrity.
Figure 7. Cleavage of vimentin and LAP1 in live infected cells occurs after inclusion rupture and is dependent on CPAF and T2S.

A) CPAF-dependent modification of vimentin filaments occurs immediately after inclusion rupture. HeLa cells were infected with the indicated strains and transfected with N-terminally EGFP-tagged vimentin and tdTomato vectors and imaged using widefield deconvolution live-cell microscopy every seven minutes after 54hpi for 14 hours (Movie S1). Inclusion rupture was assessed by the influx of dTomato signal into the inclusion lumen. Images acquired 7 min before and after inclusion rupture are shown. In cells infected with RifR (WT) LGV-L2, the filamentous EGFP-vimentin signal became diffuse immediately after inclusion rupture. EGFP-vimentin remained in a filamentous form after loss of inclusion integrity in cells infected with CPAF or T2S-deficient strains (arrows). B) CPAF-dependent processing of LAP1 occurs after inclusion rupture. HeLa cells were transfected with EGFP-LAP1 and tdTomato expression vectors and infected and imaged as in (A) (Movie S2). Loss of EGFP-LAP1 localization to the nuclear membrane occurs rapidly following inclusion rupture in cells infected with RifR (WT) but not in cells infected with CPAF or T2S-deficient strains.
CPAF mediates cleavage of the nuclear envelope protein LAP1
As part of the characterization of the molecular basis of developmental transitions and the metabolic properties of the EB and RB forms of C. trachomatis, we previously performed a large-scale, label-free quantitative proteomic analysis of LGV-L2 strain EB and RB forms and also used these methods to analyze the membranes of the pathogen-containing inclusion vacuole [31]. During this analysis, in addition to chlamydial proteins expressed and processed, we observed other proteins from the host cell were degraded only in infected cells. One of these proteins, is the human laminin-associated protein-1 (LAP1)/Torsin 1A-interacting protein. LAP1 is a Type II membrane protein and a major constituent of the mammalian nuclear envelope, where it forms multimeric assemblies that are suspended in the inner nuclear membrane and are specifically associated with B-type lamins, where they serve to link nuclear lamins to the dystonia ER ATPase Torsin1A [46]. Mass spectrometry-based mapping of LAP1 cleavage sites indicated that the CPAF cleavage site most likely localized to domains of the protein within the nucleoplasm (unpublished data). Importantly, LAP1 cleavage was also observed in cell lysates of Chlamydia infected cells that had been harvested under conditions that inhibited all CPAF activity. Consistent with these findings, we determined that endogenous LAP1 and an EGFP-LAP1 fluorescent construct were also cleaved in vitro by recombinant CPAF (Fig 6A) and this cleavage was blocked by Pep2 (not shown). As with vimentin, LAP1 was cleaved late during infection and these cleavage events were not observed in HeLa cells infected with the CPAF-deficient mutant M169 or T2S-defective mutants (Fig. 6B).
Figure 6. CPAF cleaves LAP1 during infection.
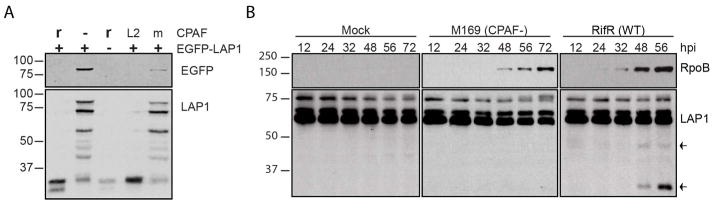
A.) CPAF cleaves EGFP-LAP1. HeLa lysates transfected (+) or not (-) with EGFP-LAP1 were incubated with (r) or without (-) recombinant CPAF or with lysates from L2-infected HeLa cells (L2) or mock-infected (m) HeLa cells. Blots were probed with anti-EGFP (EGFP) or anti-LAP1 (LAP1). B.) LAP1 cleavage during infection is CPAF-dependent. HeLa cells infected with either the CPAF-strain or RifR (WT) strain or mock-infected were harvested in 1% SDS buffer at the indicated times post-infection. Blots were probed with anti-LAP1 and anti-rpoB. Arrows indicate LAP1 cleavage products.
Based on our observations with vimentin processing in live cells, we predicted that LAP1 cleavage would also occur upon loss of inclusion membrane integrity late in infection, where CPAF is released into the host cytosol. We formally tested this prediction by infecting cells that had been co-transfected with tdTomato and EGFP-LAP1 with either RST17 (CPAF−) or RST5 (CPAF+) Chlamydia strains. As previously described [46], EGFP-LAP1 prominently labeled the nuclear envelope (Fig 7B). This localization pattern was not altered in infected cells. Time lapse microscopy of live infected cells revealed that upon loss of inclusion membrane integrity, as assessed by the influx of tdTomato into the inclusion lumen, the localization of EGFP-LAP1 rapidly changed from the nuclear envelope to the cytoplasm, which most likely resulted from the cleavage of the EGFP moiety from the portion of the protein anchored to the nuclear membrane (Fig 7B and Movie S2). As with vimentin cleavage, these proteolytic events occurred within cells in which the integrity of the plasma membrane was not compromised and were dependent on CPAF.
CONCLUSIONS AND PERSPECTIVES
In the last two years there has been remarkable advances in the development of systems for genetic analysis in C. trachomatis. This includes the generation and mapping of chemically generated mutants as well as the stable delivery of recombinant DNA into C. trachomatis [33,35,47,48]. We had previously identified and characterized a mutant defective in T2S [33], which we now report is also defective for CPAF secretion (Fig 1A). In addition, we identified two C. trachomatis LGV-L2 strains with nonsense mutations in cpa (Fig 1, Table 1). Equipped with these mutants we can now clarify the role that CPAF plays in the various unique cellular phenotypes that have been attributed to Chlamydia infections. Overall, our findings suggest that CPAF is not essential for blocking apoptosis, Golgi fragmentation, resistance to re-infection, or inhibition of pro-inflammatory signaling pathways. However, while CPAF does not appear necessary for these cellular processes to occur, we cannot exclude the possibility that this protease plays an axillary role or whether there are redundant factors contributing to these cellular phenotypes.
Tan and colleagues brought to the forefront the issue that the unique challenges of inhibiting CPAF’s proteolytic activity post lysis can confound the degree of proteolytic activity that occurs within live infected cells [30]. Indeed, the level of true CPAF-mediated proteolysis that occurs within infected cells has come into doubt with an understandable impulse to label most proteolytic events observed to date as artifacts [48]. Our findings indicate that it is premature to assume that traditional targets of CPAF are not cleaved during infection. We presented evidence that the bulk of CPAF activity resides within the inclusion lumen and that CPAF rapidly cleaves at least one of its described targets when the inclusion membrane is compromised during late stages of infection (Fig 7 and Movie S1). Our results also indicate that the new CPAF substrate we identified, LAP1, is similarly processed upon loss of inclusion membrane integrity. This could represent a pathogenic strategy to destroy key components in the cytoplasm or subcellular structures (e.g. nuclear envelope for LAP1) prior to bacterial exit from a lysing cell or a transient response to mechanical or immune stress on the integrity of the inclusion. Such a strategy may help degrade or modify factors that constitute danger signals to be sensed by immune cells, or modify physical barriers that limit bacterial dispersal. Indeed, a role for chlamydial proteases in the dissolution of the infected host cell late in infection was first reported by Gerald Stoke almost 40 year ago [49]. CPAF mutants display a small (~2.5 fold) decrease in the generation of IFUs (Fig 2), suggesting that CPAF also performs functions important for replication before the late exit stages. These functions could include the processing and turnover of bacterial proteins within the inclusion, as has been suggested for the outer membrane protein OmcB [28].
In addition, CPAF released from infected cells may play a prominent role in tissue colonization or dissemination in infected animals. Indeed, experimentally infected animals and human patients develop a strong humoral response to CPAF, and immunization with recombinant CPAF is protective against infections [50,51], suggesting that this protein may have extracellular roles in tissue infections. It should also be emphasized that our findings are restricted to the role of CPAF in strain LGV-L2. We do not know if CPAF plays different roles in urogenital and ocular C. trachomatis serovars.
At this stage, there are many unresolved questions as to the function of CPAF, yet these mutant strains offer a distinct advantage to deconstruct the function of CPAF, especially in light of very recent advanced in transformation and genetic manipulation of Chlamydia [35]. In this manner, we are equipped to address several issues such as what are the cytosolic substrates of CPAF and what are their roles in supporting pathogenesis? Is CPAF activity in the host cytosol regulated? Could there be additional structural motifs that are only present in a subset of ‘true’ CPAF substrates that are cleaved in the cytoplasm of cells with intact inclusions? In such a scenario, CPAF in the cytoplasm could acts as a molecular “scalpel” to specifically modify host cellular processes and as a “hammer” when inclusion membrane integrity is compromised and the bulk of active CPAF is released in to the cytoplasm. Preliminary analyses of the sequence specificity of CPAF (D. McCafferty, unpublished results) indicates a capacity for this enzyme to conduct endoproteolysis at multiple sites, as well as the participation of CPAF in complexes with other proteases. Therefore, CPAF may exhibit additional interactions with host proteins, mediated by intrinsic substrate specificity or by release into subcellular compartments, that may lead to the proteolytic activation of host hydrolases (or removal of inhibitory factors) to promote lytic egress as has been observed in viral and parasitic infections. Future work with Chlamydia expressing modified forms of CPAF should help address some of the outstanding questions remaining as to the function of this unusual protease.
Supplementary Material
Video S1. Vimentin cleavage in live cells occurs after inclusion rupture and requires CPAF and T2S. HeLa cells were infected with the indicated strains and transfected with N-terminally EGFP-tagged vimentin and tdTomato vectors and imaged using widefield deconvolution live-cell microscopy every seven minutes after 54hpi for 14 hours. Images of representative cells from each condition ten frames prior to and following inclusion rupture were selected and processed into aligned and synchronous video which plays at 2 frames/sec. The frame immediately following loss of inclusion membrane integrity is annotated. Inclusion rupture was assessed by the influx of tdTomato signal into the inclusion lumen and occurs prior to 01:17:00. In cells infected with RifR (WT) LGV-L2, the filamentous EGFP-vimentin signal (cyan arrowheads) became diffuse immediately after inclusion rupture. EGFP-vimentin remained in a filamentous form after loss of inclusion integrity (magenta arrowheads) in cells infected with CPAF or T2S-deficient strains. For the cell shown, loss of plasma membrane integrity occurs at 01:31:00 for WT and CPAF-strains and at 01:45:00 for the T2S-deficient strain. Elapsed time is displayed as hr:min:sec.
Video S1. LAP1 cleavage in live cells occurs after inclusion rupture and requires CPAF and T2S. HeLa cells were infected with the indicated strains and transfected with N-terminally EGFP-tagged LAP1 and tdTomato vectors and imaged using widefield deconvolution live-cell microscopy every seven minutes after 54hpi for 14 hours. Images of representative cells from each condition ten frames prior to and following inclusion rupture were selected and processed into aligned and synchronous video which plays at 2 frames/sec. The frame immediately following loss of inclusion membrane integrity is annotated. Inclusion rupture was assessed by the influx of tdTomato signal into the inclusion lumen and occurs prior to 01:17:00. Loss of EGFP-LAP1 localization to the nuclear membrane (cyan arrowheads) occurs rapidly following inclusion rupture in cells infected with RifR (WT) but not in cells infected with CPAF or T2S-deficient strains (magenta arrowheads). Elapsed time is displayed as hr:min:sec.
Acknowledgments
We thank K. Fields, M. Tan, G. Zhong, W. Dauer, R. Liem, M. Caron, and K. Maksimchuk for antibodies and reagents, and the Duke IGSP DNA Sequencing Core for their technical support. We are also grateful to A. Olive and S. Grieshaber for their helpful comments on the manuscript, and members of the Valdivia laboratory for their support during the design and implementation of this work. The work described here was supported by funds from the NIAID/NIH (AI100759 to RHV, and AI107951 to DGM) and the Burroughs Wellcome Program in Infectious Diseases (RHV).
Footnotes
The authors have no conflicts of interest to disclose.
References
- 1.Belland R, Ojcius DM, Byrne GI. Chlamydia. Nature reviews Microbiology. 2004;2:530–531. doi: 10.1038/nrmicro931. [DOI] [PubMed] [Google Scholar]
- 2.Darville T, Hiltke TJ. Pathogenesis of genital tract disease due to Chlamydia trachomatis. The Journal of infectious diseases. 2010;201(Suppl 2):S114–125. doi: 10.1086/652397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu VH, Harding-Esch EM, Burton MJ, Bailey RL, Kadimpeul J, Mabey DCW. Epidemiology and control of trachoma: systematic review. Tropical Medicine & International Health. 2010:673–691. doi: 10.1111/j.1365-3156.2010.02521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hybiske K, Stephens RS. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc Natl Acad Sci U S A. 2007;104:11430–11435. doi: 10.1073/pnas.0703218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valdivia RH. Chlamydia effector proteins and new insights into chlamydial cellular microbiology. Curr Opin Microbiol. 2008;11:53–59. doi: 10.1016/j.mib.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Chen D, Lei L, Lu C, Flores R, DeLisa MP, et al. Secretion of the chlamydial virulence factor CPAF requires the Sec-dependent pathway. Microbiology. 2010;156:3031–3040. doi: 10.1099/mic.0.040527-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giles DK, Whittimore JD, LaRue RW, Raulston JE, Wyrick PB. Ultrastructural analysis of chlamydial antigen-containing vesicles everting from the Chlamydia trachomatis inclusion. Microbes and infection / Institut Pasteur. 2006;8:1579–1591. doi: 10.1016/j.micinf.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 8.Giles DK, Wyrick PB. Trafficking of chlamydial antigens to the endoplasmic reticulum of infected epithelial cells. Microbes and infection / Institut Pasteur. 2008;10:1494–1503. doi: 10.1016/j.micinf.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sandkvist M. Type II secretion and pathogenesis. Infect Immun. 2001;69:3523–3535. doi: 10.1128/IAI.69.6.3523-3535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhong G, Fan P, Ji H, Dong F, Huang Y. Identification of a chlamydial protease-like activity factor responsible for the degradation of host transcription factors. J Exp Med. 2001;193:935–942. doi: 10.1084/jem.193.8.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhong G, Fan P, Ji H, Dong F, Huang Y. Identification of a chlamydial protease-like activity factor responsible for the degradation of host transcription factors. The Journal of experimental medicine. 2001;193:935–942. doi: 10.1084/jem.193.8.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaw AC, Vandahl BB, Larsen MR, Roepstorff P, Gevaert K, et al. Characterization of a secreted Chlamydia protease. Cellular microbiology. 2002;4:411–424. doi: 10.1046/j.1462-5822.2002.00200.x. [DOI] [PubMed] [Google Scholar]
- 13.Zhong G, Liu L, Fan T, Fan P, Ji H. Degradation of transcription factor RFX5 during the inhibition of both constitutive and interferon gamma-inducible major histocompatibility complex class I expression in chlamydia-infected cells. The Journal of experimental medicine. 2000;191:1525–1534. doi: 10.1084/jem.191.9.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pirbhai M, Dong F, Zhong Y, Pan KZ, Zhong G. The secreted protease factor CPAF is responsible for degrading pro-apoptotic BH3-only proteins in Chlamydia trachomatis-infected cells. J Biol Chem. 2006;281:31495–31501. doi: 10.1074/jbc.M602796200. [DOI] [PubMed] [Google Scholar]
- 15.Christian JG, Vier J, Paschen SA, Hacker G. Cleavage of the NF-{kappa}B-family protein p65/RelA by the chlamydial protease chlamydial protease-like activity factor (CPAF) impairs pro-inflammatory signalling in cells Infected with chlamydiae. J Biol Chem. 2010 doi: 10.1074/jbc.M110.152280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar Y, Valdivia RH. Actin and intermediate filaments stabilize the Chlamydia trachomatis vacuole by forming dynamic structural scaffolds. Cell Host Microbe. 2008;4:159–169. doi: 10.1016/j.chom.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong F, Su H, Huang Y, Zhong Y, Zhong G. Cleavage of host keratin 8 by a Chlamydia-secreted protease. Infect Immun. 2004;72:3863–3868. doi: 10.1128/IAI.72.7.3863-3868.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun J, Kintner J, Schoborg RV. The host adherens junction molecule nectin-1 is downregulated in Chlamydia trachomatis-infected genital epithelial cells. Microbiology. 2008;154:1290–1299. doi: 10.1099/mic.0.2007/015164-0. [DOI] [PubMed] [Google Scholar]
- 19.Kawana K, Quayle AJ, Ficarra M, Ibana JA, Shen L, et al. CD1d degradation in Chlamydia trachomatis-infected epithelial cells is the result of both cellular and chlamydial proteasomal activity. J Biol Chem. 2007;282:7368–7375. doi: 10.1074/jbc.M610754200. [DOI] [PubMed] [Google Scholar]
- 20.Yu H, Schwarzer K, Forster M, Kniemeyer O, Forsbach-Birk V, et al. Role of high-mobility group box 1 protein and poly(ADP-ribose) polymerase 1 degradation in Chlamydia trachomatis-induced cytopathicity. Infect Immun. 2010;78:3288–3297. doi: 10.1128/IAI.01404-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paschen SA, Christian JG, Vier J, Schmidt F, Walch A, et al. Cytopathicity of Chlamydia is largely reproduced by expression of a single chlamydial protease. The Journal of cell biology. 2008;182:117–127. doi: 10.1083/jcb.200804023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown HM, Knowlton AE, Grieshaber SS. Chlamydial infection induces host cytokinesis failure at abscission. Cellular microbiology. 2012;14:1554–1567. doi: 10.1111/j.1462-5822.2012.01820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christian JG, Heymann J, Paschen SA, Vier J, Schauenburg L, et al. Targeting of a chlamydial protease impedes intracellular bacterial growth. PLoS pathogens. 2011;7:e1002283. doi: 10.1371/journal.ppat.1002283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paschen SA, Christian JG, Vier J, Schmidt F, Walch A, et al. Cytopathicity of Chlamydia is largely reproduced by expression of a single chlamydial protease. J Cell Biol. 2008;182:117–127. doi: 10.1083/jcb.200804023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhong G. Chlamydia trachomatis secretion of proteases for manipulating host signaling pathways. Frontiers in microbiology. 2011;2:14. doi: 10.3389/fmicb.2011.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhong G. Killing me softly: chlamydial use of proteolysis for evading host defenses. Trends in microbiology. 2009;17:467–474. doi: 10.1016/j.tim.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jorgensen I, Bednar MM, Amin V, Davis BK, Ting JP, et al. The Chlamydia protease CPAF regulates host and bacterial proteins to maintain pathogen vacuole integrity and promote virulence. Cell host & microbe. 2011;10:21–32. doi: 10.1016/j.chom.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hou S, Lei L, Yang Z, Qi M, Liu Q, et al. Chlamydia trachomatis outer membrane complex protein B (OmcB) is processed by the protease CPAF. Journal of bacteriology. 2013;195:951–957. doi: 10.1128/JB.02087-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qi M, Gong S, Lei L, Liu Q, Zhong G. A Chlamydia trachomatis OmcB C-terminal fragment is released into the host cell cytoplasm and is immunogenic in humans. Infection and immunity. 2011;79:2193–2203. doi: 10.1128/IAI.00003-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen AL, Johnson KA, Lee JK, Sutterlin C, Tan M. CPAF: a Chlamydial protease in search of an authentic substrate. PLoS pathogens. 2012;8:e1002842. doi: 10.1371/journal.ppat.1002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saka HA, Thompson JW, Chen YS, Kumar Y, Dubois LG, et al. Quantitative proteomics reveals metabolic and pathogenic properties of Chlamydia trachomatis developmental forms. Molecular microbiology. 2011;82:1185–1203. doi: 10.1111/j.1365-2958.2011.07877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nguyen BD, Valdivia RH. Forward genetic approaches in Chlamydia trachomatis. Journal of visualized experiments : JoVE. 2013:e50636. doi: 10.3791/50636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nguyen BD, Valdivia RH. Virulence determinants in the obligate intracellular pathogen Chlamydia trachomatis revealed by forward genetic approaches. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:1263–1268. doi: 10.1073/pnas.1117884109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bednar MM, Jorgensen I, Valdivia RH, McCafferty DG. Chlamydia protease-like activity factor (CPAF): characterization of proteolysis activity in vitro and development of a nanomolar affinity CPAF zymogen-derived inhibitor. Biochemistry. 2011;50:7441–7443. doi: 10.1021/bi201098r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Kahane S, Cutcliffe LT, Skilton RJ, Lambden PR, et al. Development of a transformation system for Chlamydia trachomatis: restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS pathogens. 2011;7:e1002258. doi: 10.1371/journal.ppat.1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dong F, Pirbhai M, Zhong Y, Zhong G. Cleavage-dependent activation of a chlamydia-secreted protease. Molecular microbiology. 2004;52:1487–1494. doi: 10.1111/j.1365-2958.2004.04072.x. [DOI] [PubMed] [Google Scholar]
- 37.Heuer D, Rejman Lipinski A, Machuy N, Karlas A, Wehrens A, et al. Chlamydia causes fragmentation of the Golgi compartment to ensure reproduction. Nature. 2009;457:731–735. doi: 10.1038/nature07578. [DOI] [PubMed] [Google Scholar]
- 38.Christian J, Vier J, Paschen SA, Hacker G. Cleavage of the NF-kappaB family protein p65/RelA by the chlamydial protease-like activity factor (CPAF) impairs proinflammatory signaling in cells infected with Chlamydiae. The Journal of biological chemistry. 2010;285:41320–41327. doi: 10.1074/jbc.M110.152280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Byrne GI, Ojcius DM. Chlamydia and apoptosis: life and death decisions of an intracellular pathogen. Nature reviews Microbiology. 2004;2:802–808. doi: 10.1038/nrmicro1007. [DOI] [PubMed] [Google Scholar]
- 40.Sharma M, Rudel T. Apoptosis resistance in Chlamydia-infected cells: a fate worse than death? FEMS immunology and medical microbiology. 2009;55:154–161. doi: 10.1111/j.1574-695X.2008.00515.x. [DOI] [PubMed] [Google Scholar]
- 41.Fischer SF, Vier J, Kirschnek S, Klos A, Hess S, et al. Chlamydia inhibit host cell apoptosis by degradation of proapoptotic BH3-only proteins. The Journal of experimental medicine. 2004;200:905–916. doi: 10.1084/jem.20040402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pirbhai M, Dong F, Zhong Y, Pan KZ, Zhong G. The secreted protease factor CPAF is responsible for degrading pro-apoptotic BH3-only proteins in Chlamydia trachomatis-infected cells. The Journal of biological chemistry. 2006;281:31495–31501. doi: 10.1074/jbc.M602796200. [DOI] [PubMed] [Google Scholar]
- 43.Fan T, Lu H, Hu H, Shi L, McClarty GA, et al. Inhibition of apoptosis in chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. The Journal of experimental medicine. 1998;187:487–496. doi: 10.1084/jem.187.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dong F, Su H, Huang Y, Zhong Y, Zhong G. Cleavage of host keratin 8 by a Chlamydia-secreted protease. Infection and immunity. 2004;72:3863–3868. doi: 10.1128/IAI.72.7.3863-3868.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar Y, Valdivia RH. Actin and intermediate filaments stabilize the Chlamydia trachomatis vacuole by forming dynamic structural scaffolds. Cell host & microbe. 2008;4:159–169. doi: 10.1016/j.chom.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goodchild RE, Dauer WT. The AAA+ protein torsinA interacts with a conserved domain present in LAP1 and a novel ER protein. The Journal of cell biology. 2005;168:855–862. doi: 10.1083/jcb.200411026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kari L, Goheen MM, Randall LB, Taylor LD, Carlson JH, et al. Generation of targeted Chlamydia trachomatis null mutants. Proc Natl Acad Sci U S A. 2011;108:7189–7193. doi: 10.1073/pnas.1102229108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Conrad T, Yang Z, Ojcius D, Zhong G. A path forward for the chlamydial virulence factor CPAF. Microbes and infection / Institut Pasteur. 2013 doi: 10.1016/j.micinf.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stokes GV. Formation and destruction of internal membranes in L cells infected with Chlamydia psittaci. Infection and immunity. 1973;7:173–177. doi: 10.1128/iai.7.2.173-177.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murthy AK, Guentzel MN, Zhong G, Arulanandam BP. Chlamydial protease-like activity factor--insights into immunity and vaccine development. Journal of reproductive immunology. 2009;83:179–184. doi: 10.1016/j.jri.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murthy AK, Li W, Guentzel MN, Zhong G, Arulanandam BP. Vaccination with the defined chlamydial secreted protein CPAF induces robust protection against female infertility following repeated genital chlamydial challenge. Vaccine. 2011;29:2519–2522. doi: 10.1016/j.vaccine.2011.01.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Vimentin cleavage in live cells occurs after inclusion rupture and requires CPAF and T2S. HeLa cells were infected with the indicated strains and transfected with N-terminally EGFP-tagged vimentin and tdTomato vectors and imaged using widefield deconvolution live-cell microscopy every seven minutes after 54hpi for 14 hours. Images of representative cells from each condition ten frames prior to and following inclusion rupture were selected and processed into aligned and synchronous video which plays at 2 frames/sec. The frame immediately following loss of inclusion membrane integrity is annotated. Inclusion rupture was assessed by the influx of tdTomato signal into the inclusion lumen and occurs prior to 01:17:00. In cells infected with RifR (WT) LGV-L2, the filamentous EGFP-vimentin signal (cyan arrowheads) became diffuse immediately after inclusion rupture. EGFP-vimentin remained in a filamentous form after loss of inclusion integrity (magenta arrowheads) in cells infected with CPAF or T2S-deficient strains. For the cell shown, loss of plasma membrane integrity occurs at 01:31:00 for WT and CPAF-strains and at 01:45:00 for the T2S-deficient strain. Elapsed time is displayed as hr:min:sec.
Video S1. LAP1 cleavage in live cells occurs after inclusion rupture and requires CPAF and T2S. HeLa cells were infected with the indicated strains and transfected with N-terminally EGFP-tagged LAP1 and tdTomato vectors and imaged using widefield deconvolution live-cell microscopy every seven minutes after 54hpi for 14 hours. Images of representative cells from each condition ten frames prior to and following inclusion rupture were selected and processed into aligned and synchronous video which plays at 2 frames/sec. The frame immediately following loss of inclusion membrane integrity is annotated. Inclusion rupture was assessed by the influx of tdTomato signal into the inclusion lumen and occurs prior to 01:17:00. Loss of EGFP-LAP1 localization to the nuclear membrane (cyan arrowheads) occurs rapidly following inclusion rupture in cells infected with RifR (WT) but not in cells infected with CPAF or T2S-deficient strains (magenta arrowheads). Elapsed time is displayed as hr:min:sec.