

Abstract
Peroxiredoxins (Prxs) make up an ancient family of enzymes that are the predominant peroxidases for nearly all organisms and play essential roles in reducing hydrogen peroxide, organic hydroperoxides, and peroxynitrite. Even between distantly related organisms, the core protein fold and key catalytic residues related to its cysteine-based catalytic mechanism have been retained. Given that these enzymes appeared early in biology, Prxs have experienced more than 1 billion years of optimization for specific ecological niches. Although their basic enzymatic function remains the same, Prxs have diversified and are involved in roles such as protecting DNA against mutation, defending pathogens against host immune responses, suppressing tumor formation, and—for eukaryotes—helping regulate peroxide signaling via hyperoxidation of their catalytic Cys residues. Here, we review the current understanding of the physiological roles of Prxs by analyzing knockout and knockdown studies from ∼25 different species. We also review what is known about the structural basis for the sensitivity of some eukaryotic Prxs to inactivation by hyperoxidation. In considering the physiological relevance of hyperoxidation, we explore the distribution across species of sulfiredoxin (Srx), the enzyme responsible for rescuing hyperoxidized Prxs. We unexpectedly find that among eukaryotes appearing to have a “sensitive” Prx isoform, some do not contain Srx. Also, as Prxs are suggested to be promising targets for drug design, we discuss the rationale behind recently proposed strategies for their selective inhibition.
Introduction to Peroxiredoxins and Scope of This Review
Peroxiredoxins (Prxs) are nature’s dominant peroxidases. From archaea to humans, they are widely expressed and possess the same catalytic components.3 Prxs serve to protect cells from oxidative stress and prevent damage to DNA, lipids, and other proteins by reducing hydroperoxides and peroxynitrite.4 With catalytic rates of ∼107 M–1 s–1 and an abundance that implies that they account for the reduction of more than 90% of cytosolic peroxide, they are crucial for regulating intracellular peroxide levels in most organisms.5 Cells encounter peroxides in a variety of ways—as a byproduct of cellular processes, as a consequence of environmental conditions, or even as a result of deliberate attacks by other cells6—and Prxs have been finely tuned to address the needs of their respective organisms. Given the ubiquity of Prxs, it is presumed that they make up an ancient enzyme family that arose at the time of the great Oxidation Event, some 2.4 billion years ago, to aid cells in coping with increased oxygen levels and to facilitate aerobic metabolism.7 Because of their retention over the millennia, with no major alterations in the protein fold or catalytic mechanism, Prxs can be seen as being integral to the existence of life on Earth.
We have come to understand that Prxs serve a function much more complex than simply purging cells of a toxic molecule. This is in part due to the discovery that peroxide not only creates oxidative stress and participates in stress-related signaling, such as activating the bacterial transcription regulator OxyR,8 but also in eukaryotes is an integral part of normal, “non-oxidative-stress-related”a cell regulation events.1 Such non-stress-related peroxide signaling is now known to be an important factor involved in cell proliferation, angiogenesis, senescence, and apoptosis.6,9,10 Non-oxidative-stress-related peroxide signaling occurs, for instance, as a part of insulin-stimulated activation of NADPH-oxidases (NOXs),11 or adrenocorticotropic hormone (ACTH)-stimulated activation of a cytochrome P450 that contributes to peroxide buildup (Figure 1). The peroxide bolus produced by such enzymes becomes a chemical signal that leads to changes in protein activities through the reversible oxidation of protein residues, like an active site cysteine of protein tyrosine phosphatases.4 Other phospho-regulatory enzymes, such as kinases CAMKII, PKA, and PKG, are oxidant-sensing and also can be regulated by hydrogen peroxide (recently reviewed by Burgoyne et al.11). Thus, a complex interplay exists between Prxs and transcription factors, phosphatases, kinases, and any cellular molecule capable of being modified by peroxide (Figure 1). The important influence of Prxs in cell homeostasis is supported by the observations that Prxs are overexpressed in some human breast,12 lung,13 and thymic14 cancers, and that when the most abundant Prx isoform is knocked out in mice the animals develop malignant tumors and hemolytic anemia and die prematurely.15
Figure 1.

Examples of non-stress-related peroxide signaling. The white panel (left) shows a general scheme of growth factor-triggered peroxide signaling.6 Binding of growth factor to receptors (green) leads to the activation of oxidases (orange) and the production of superoxide that is subsequently converted to peroxide. Certain aquaporins (dark red) facilitate the entry of peroxide into the cell137 where kinases (light purple), phosphatases (dark purple), and transcription factors6,100 (dark blue) can be oxidatively activated or deactivated.137 Active Prxs (cyan toroid) degrade peroxides but also can be inactivated by hyperoxidation (dark toroid); Srx (light red) reactivates hyperoxidized Prxs. The magenta and purple panels convey other examples of peroxide signaling highlighted in the text. In LPA-mediated signaling104 (magenta, bottom), binding of LPA to its receptor (green) activates NADPH oxidase (NOX, orange), and through endocytosis, a “redoxosome” is formed. Superoxide/peroxide accumulates in the redoxosome, and it serves as a hub for modifying regulatory factors. In murine adrenal corticosteroid production108 (purple, top right), binding of ACTH to its receptor (green) leads to the activation of the cAMP-PKA pathway (the transcription factor cAMP response element-binding protein is denoted with an asterisk) and then phosphorylation and activation of steroidogenic acute regulatory protein (StAR); StAR makes cholesterol available for CYP11A1- and CYP11B1-catalyzed conversion via 11-deoxycorticosterone (DOC) to corticosterone (CS) and also produces superoxide from which superoxide dismutase (SOD) produces peroxide. The peroxide increasingly inactivates PrxIII and after further buildup initiates a negative feedback loop by activating p38, which in turn suppresses the synthesis of StAR.
There have been a number of recent reviews of Prxs that have highlighted structure–function relations,1,5 enzymology,16,17 and their roles in signaling.18 Here, we seek to complement these reviews by organizing current knowledge of the physiological roles of Prxs, exploring how evolution has optimized Prx dynamics and thermodynamics to modulate their sensitivity to hyperoxidation, assessing the distribution of its partner enzyme sulfiredoxin (Srx),19 and describing how drug design might take advantage of the conformational changes that Prxs undergo.20−22 At present, there are ∼120 Prx structures in the Protein Data Bank (PDB) and more than 15000 annotated Prx genes,23 so a wealth of data is available.
Peroxidase Function of Prxs
Catalytic Cycle
Prxs have been classified into subgroups on the basis of functional site sequence similarity.3 These are Prx1, Prx5, Prx6, Tpx, AhpE, and PrxQ [proposed recently24 to replace the uninformative name of BCP (bacterioferritin comigratory protein) that has been used for some members of this group]. These subgroups have variations in their oligomerization, conformation, and some secondary structure elements, and most organisms possess multiple isoforms5 (for example, humans contain four Prx1 subtypes, one Prx5 subtype, and one Prx6 subtype, whereas Escherichia coli has one Prx1, one Tpx, and one PrxQ). For all Prxs, however, catalysis is facilitated by a peroxidatic Cys (CP) contained within a universally conserved Pxxx(T/S)xxC active site motif3 (Figure 2). The active site lowers the CP side chain pKa from ∼8.4 to ∼6 or even lower so that it is kept predominantly in a nucleophilic, thiolate state.25−27
Figure 2.

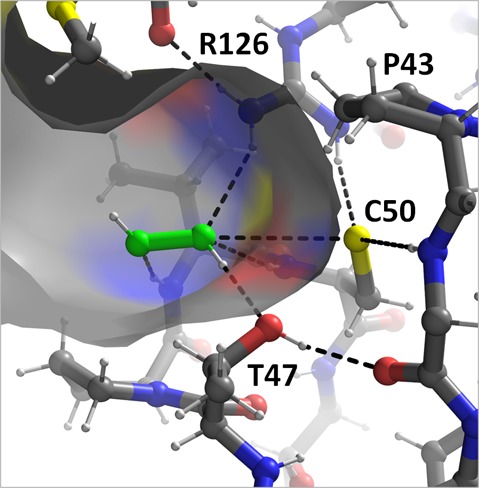
Catalysis by peroxiredoxins. (A) Michaelis complex of peroxide (green) bound to the FF active site of ApTpx (PDB entry 3a2v) with atom coloring (gray carbons, white hydrogens, yellow sulfurs, red oxygens, and blue nitrogens) showing key hydrogen bonds (dashed lines). (B) The normal Prx catalytic cycle (black) is shown along with the hyperoxidation shunt (gray). To illustrate the change in conformation necessary for Prx catalysis, the center shows a morph between FF and LU conformations for the Prx1 subfamily member StAhpC; the CP- and CR-containing chains are colored white and dark gray, respectively, and the C-terminal region beyond CR is not shown. (C) An organic peroxide and peroxynitrous acid are shown bound to the active site in ways that mimic the interactions made by peroxide in panel A. “BB” refers to a backbone NH hydrogen bond donor. The placement of the hydrophobic collar seen in some organic peroxide selective Prxs is noted by orange circles. (D) Chemical structures of some other molecules recently reported to react with Prxs (see the text).
The conformation of the enzyme that possesses a substrate-ready active site pocket (Figure 2A) is termed “fully folded” (FF). In the catalytic cycle (Figure 2B), the peroxide substrate binds to the FF active site where it is attacked by the nucleophilic CP in an SN2-type reaction to form Cys-sulfenic acid (CP-SOH) and water or alcohol. Subsequently, the active site locally unfolds, an event that for some Prxs involves the rearrangement of as many as ∼35 residues20 (Figure 2B, center). As discussed later in more detail, a second peroxide can react with CP-SOH to hyperoxidize the enzyme to a dead-end CP-SO2–. Some organisms, mainly eukaryotes, contain Srx, which converts the hyperoxidized form back to CP-SOH in an ATP-dependent reaction.28 For a minority of Prxs, termed “1-Cys” Prxs, the CP-SOH form is reduced directly by an intracellular reductant such as glutathione or ascorbate.29 The majority of Prxs, called “2-Cys” Prxs, have a second resolving Cys (CR) that forms a disulfide bond with CP.5 Depending on the Prx, the CR may be contained within the same chain or, for some oligomeric Prxs, in the chain of another subunit. The formation of the CP–CR disulfide requires the active site to locally unfold, i.e., adopting a “locally unfolded” (LU) conformation, that often involves substantial rearrangements of both the CP and CR regions (Figure 2B).5
To complete the catalytic cycle, the disulfide is commonly reduced by thioredoxin (Trx), or a thioredoxin-like protein,30 and the Prx is returned to the FF conformation. Recently, the first structure of a Prx–Trx complex was obtained, showing one Trx on each side of a Prx dimer trapped in a mixed disulfide with CR.31 However, given that this particular yeast Prx possesses an unconventional N-terminal CR, it is unclear how representative the details of this interaction may be for Prxs in general.
Reactivity toward Various Substrates
Structural work has greatly elucidated the features important for substrate interactions, with a peroxide-bound complex of Aeropyrum pernix thiol peroxidase32 providing a view of a true Prx Michaelis complex (Figure 2A). Other ligands bound at the active sites of Prx crystal structures include molecules such as oxidized dithiothreitol (DTT),24,33 benzoate,34 acetate,32 formate,35 and glycerol,32 with the oxygens of these molecules mimicking those of a peroxide. Analysis of these complexes led to a proposal that the roughly 105-fold rate increase of the enzyme over that of free cysteine is largely due to an extensive set of hydrogen bonds that stabilize the transition state of the reaction,33 and this was supported by recently determined experimental thermodynamic activation energies as well as quantum mechanics/molecular mechanics simulations.36,37
Interestingly, though Prxs share a universal catalytic cycle and active site, some are observed to have relatively broad substrate specificity, while others are more selective.38 For instance, Salmonella typhimurium alkyl hydroperoxide reductase C (StAhpC) is ∼100-fold more reactive with hydrogen peroxide than with organic peroxides primarily because of differences in Km.39 In contrast, human PrxV26 and E. coli thiol peroxidase (EcTpx)38 are ∼100- and ∼200-fold, respectively, more reactive with organic peroxides. The preference of some Prxs for organic peroxides has been attributed to a “hydrophobic collar” of apolar side chains around their active site that can make favorable hydrophobic interactions with the hydrocarbon part of the substrate (Figure 2C). Such a conserved hydrophobic collar was first observed in the Tpx subfamily,38 but other Prxs that efficiently reduce organic peroxides, such as human PrxV,33 also possess analogous collars. One commonality among various hydrophobic collars is that a dimer partner is frequently seen to contribute a bulky hydrophobic side chain to the collar across the dimer interface.24,31,33,38 The significance of this interaction is not fully understood but may be related to a positive cooperativity seen for one Prx when consuming organic peroxides.31 Another possible contributor to substrate specificity proposed for a PrxQ from Xanthomonas campestris is for an extended β-strand to fold down and cap the active site after binding an organic peroxide.35
The ability of Prxs to reduce peroxynitrite is also well-established.40 AhpCs from the genera Salmonella, Mycobacterium, and Helicobacter were shown to efficiently reduce peroxynitrite,41 as were Prxs from other organisms such as Trypanosoma cruzi tryparedoxin peroxidase42 and human PrxV.26,43 Experiments indicate that the CP thiolate reacts with peroxynitrous acid44 (i.e., the protonated form that is readily formed at physiological pHs45), and this is consistent with the protonated form being better able to mimic peroxide binding in the Prx active site (Figure 2C). Additionally, lowering the pH from 7.8 to 7.4 (and increasing the fraction of peroxynitrous acid present) increased the rate of peroxynitrite reduction by human PrxV26 from ∼107 to ∼108 M–1 s–1.
Hypochlorous acid (HOCl) is among the reactive oxygen species released extracellularly by neutrophils to overwhelm pathogen redox systems,46 and HOCl can also lead to generation of chloramines via spontaneous HOCl-amino reactions47,48 (Figure 2D). HOCl and chloramines readily oxidize thiol groups, and recent studies indicate Prxs are targets of these chemical species.47,48 Human PrxIII did become oxidized when cells were treated with micromolar levels (thought to be representative of in vivo concentrations) of NH2Cl and HOCl, but reported rates are similar to that of free thiols, suggesting the reaction is not substantially facilitated by the enzyme.47,48 Given the prevalence of glutathione and other cellular thiols, Prxs are not thought to be major sinks for HOCl or chloramines.47 Nevertheless, it can be seen that a major evolutionary advantage conferred by Prxs is the ability to eliminate many forms of peroxide and apparently even some other reactive species.
Knockdown and Knockout Studies as Probes of the Physiological Roles of Prxs
Prxs influence a variety of cellular processes, and one approach to discern their various physiological roles is to observe the phenotypes that arise when cells or whole organisms are made deficient in terms of these enzymes. Summarized here are the results of extensive knockdown studies in cells from humans and in other organisms (Table S1 of the Supporting Information) and of knockout studies for vertebrates (Table 1), other eukaryotes (Table S2 of the Supporting Information), and prokaryotes (Table S3 of the Supporting Information).
Table 1. Summary of Prx Knockout Studies in Vertebrates.
| organism/enzyme | ref | brief phenotypic observations |
|---|---|---|
| Mus musculus-PrxI | (66) | malignant cancers, hemolytic anemia, premature death |
| M. musculus-PrxI | (58) | increased DNA oxidation, increased c-Myc activation in embryonic fibroblasts |
| M. musculus-PrxI | (65) | increased susceptibility to Ras-induced breast cancer |
| M. musculus-PrxII | (59) | increased protein oxidation in red blood cells, hemolytic anemia |
| M. musculus-PrxII | (67) | increased plaque formation, predisposition to develop atherosclerosis |
| M. musculus-PrxII | (69) | enlarged thymus, increased T cell proliferation |
| M. musculus-PrxII | (68) | increased splenocytes, bone marrow differentiation |
| M. musculus-PrxII | (71) | increased p21 and p53 levels, increased cellular senescence |
| M. musculus-PrxII | (60) | increased protein cysteine oxidation in red blood cell fractions |
| M. musculus-PrxIII | (61) | increased lung damage from inflammation, increased DNA damage |
| M. musculus-PrxIII | (62) | increased fat mass, increased protein carbonylation in adipose tissue |
| M. musculus-PrxIII | (72) | reduced litter size, increased oxidative stress in placenta tissue |
| M. musculus-PrxIII | (73) | increased macrophage apoptosis by lipopolysaccharide treatment |
| M. musculus-PrxIV | (74) | testicular atrophy, reduced sperm viability in oxidative stress |
| M. musculus-PrxVI | (134) | increased lung damage, decreased animal survival due to hyperoxia |
| M. musculus-PrxVI | (131) | increased ischemic reperfusion injury, increased cardiomyocyte apoptosis |
| M. musculus-PrxVI | (135) | decreased lung surfactant degradation |
| M. musculus-PrxVI | (64) | increased LDL oxidation by macrophages, increased plasma lipid H2O2 levels |
| M. musculus-PrxVI | (136) | increased UPR, increased apoptosis in lens epithelial and aging cells |
Prx Deficiency in Eukaryotes
Humans have six Prx isoforms, which are localized in discrete parts of the cell. PrxI, PrxII, and PrxVI are primarily cytosolic. PrxIII is mitochondrial. PrxIV is in the endoplasmic reticulum and PrxV is in the cytosol as well as the mitochondria and peroxisomes.4 The effects of Prx knockdowns have been characterized in at least one cell line for each isoform (Table S1 of the Supporting Information). One commonality of these studies is an increase in the level of oxidative damage to cellular components such as increases in levels of protein carbonylation49 and DNA oxidation.50 These effects are typically accompanied by reduced rates of growth and survival and an increased rate of apoptotic cell death, especially under conditions of oxidative stress.49,51−56 It is perhaps not surprising, therefore, that Prx deficiency also contributes to cellular degeneration and decreases the viability of cancer cells. For example, PrxI was designated as a tumor suppressor upon the discovery that a histone deacetylase exerted its antitumor properties by increasing the level of PrxI expression in cancerous esophageal cells.57 Additionally, knockdowns of PrxII51 and PrxVI55 in breast cancer cells were found to inhibit metastases.
Further elucidating the protective role of Prxs in mammals are knockout analyses conducted on the homologous mouse enzymes (Table 1). As was seen in the human cell knockdowns, Prx knockout mice show increased levels of oxidative damage to proteins, lipids, and DNA that detrimentally affect a host of cellular processes and often result in abnormal cellular regulation and growth.58−64 Mouse PrxI knockouts exhibit the most severe phenotype in which c-Myc levels increase,58 Akt kinase levels are elevated in fibroblasts and mammary epithelial cells,65 and death occurs by 9 months because of the development of malignant tumors.66 PrxII-knockout animals showed an increased level of atherosclerosis,67 an increased number of splenocytes, bone marrow differentiation, and peripheral blood mononuclear cells,68 an enlarged thymus, an increased rate of T-cell proliferation,69,70 and elevation of p21 and p53 levels and increased cell senescence.71 PrxIII-null mice exhibited alterations in fat metabolism, with increased fat mass, downregulation of adiponectin, impaired glucose tolerance and insulin resistance,62 and a reduced litter size and general sensitivity to oxidative stress as observed in placenta,72 macrophage,73 and lung cells.61 PrxIV was also found to influence reproductive success, as PrxIV-knockout mice displayed testicular atrophy and reduced sperm viability under conditions of oxidative stress.74
Prxs are further seen to be important for the viability of less complex eukaryotes (Tables 1 and 2 of the Supporting Information). Caenorhabditis elegans Prx knockdowns show a 70% reduction in brood size, and individual growth is retarded.75,76 Also, studies of Prx-deficient disease-causing eukaryotes have implicated Prxs as pathogenicity factors for a number of organisms, with Schistosoma showing decreased rates of survival and larval size,77−79Trypanosoma brucei exhibiting a 16-fold increase in sensitivity to peroxide-induced death,80 and Leishmania infantum having decreased infectivity in mice.81 In addition, Tpx1 knockouts of Plasmodia have increased sensitivity to paraquat and nitroprusside,82 produce 60% fewer gametes, exhibit delayed gaetocytemia,83 grow fewer sporozoites in mosquitoes, and are less effective at infecting mice.84 Thus, these results are consistent with Prxs being crucial components of pathogen redox defenses.
Several studies have utilized fungal model organisms to analyze the effects of Prx knockouts (Table S2 of the Supporting Information). In Saccharomyces cerevisiae, which has multiple Prx and glutathione peroxidase (Gpx) isoforms, the knockout of individual Prxs resulted in increased sensitivity to reactive oxygen and nitrogen species as well as an increased number of DNA mutations.85 Not surprisingly, these effects were magnified when all Prx isoforms were knocked out,85 and dual Prx/Gpx-null strains exhibited a ∼50% shorter life span.86 The less extensively studied Neurospora crassa showed altered circadian periods and phases when a Prx was knocked out and peroxide-dependent transcriptional responses were lost.87 Alterations to circadian rhythms were also seen for Arabidopsis thaliana, the only plant for which a Prx deficiency has been well-characterized.87 Interestingly, Prx knockdowns in this model plant impacted several plant-specific processes, such as an increased level of foliar ascorbate oxidation,88 altered gene expression in the chloroplast, and reduction of photosystem II and cytochrome b6 content89 (Table S1 of the Supporting Information).
These studies demonstrate that Prxs in eukaryotes are essential to normal function, as their absence results in damage to cell components and promotes deterioration of cell cycle regulation; the latter especially emphasizes that a vital role is played by Prxs in non-oxidative-stress-related peroxide signaling. An interesting observation that arises from the different effects seen in the knockout or knockdown of single Prxs is that, despite their high level of sequence similarity and shared peroxidase functionality, Prx isoforms do not have fully overlapping functions. This is illustrated especially well for humans and mice, for which the deficiency in each isoform resulted in distinct, deleterious phenotypes (Tables 1 and Table S1 of the Supporting Information). One obvious contributor to this lack of compensation is the discrete tissue expression profiles and cellular locations of eukaryotic Prx isoforms.53 Besides the restrictions imposed by localization, the cytosol, nucleus, ER, and mitochondria all have distinct redox environments90,91 (for a recent review, see ref (92)), and therefore, Prx isoforms have been specifically tuned for optimal function in only certain cellular compartments.
Prx Deficiency in Bacteria
Unlike their eukaryotic counterparts, bacteria are not known to utilize non-oxidative-stress-related peroxide signaling. Thus, the lack of an evolutionary pressure to allow for the localized buildup of peroxide constitutes a major difference in the functional optimization of bacterial Prxs. As a consequence, many bacterial Prxs have evolved to be highly “robust” against inactivation by hyperoxidation, even at millimolar peroxide concentrations.93 The advantage of this robustness is especially apparent for pathogenic bacteria as Prxs are utilized to defend against the reactive oxygen species employed by attacking macrophages.94 Investigations into the role of bacterial Prxs, therefore, have been largely focused on disease-causing species (Table S3 of the Supporting Information).
The most extreme dependence on Prxs so far observed for a bacterial species is that of Helicobacter, for which knockouts displayed no growth under microaerobic conditions,95 were more susceptible to killing by macrophages, and nearly lost their ability to colonize mouse stomachs.96 Likewise, for Staphylococcus aureus(97)and Mycobacterium bovis,98 Prx-deficient strains were shown to have reduced infectivity. In general, minimal effects of some Prx knockouts may be due to compensation by other redox defense enzymes. Some support for this is found in that more adverse phenotypes are observed for Vibrio parahemolyticus(99) and Brucella abortus(94) when two enzymes are knocked out at once. As discussed above, substrate specificity may influence the essentiality of a certain isoform or set of isoforms, and for Prxs specific for organic peroxides, like E. coli Tpx, it is important to note that the impact of the loss of its activity may be underestimated by challenges with H2O2 alone.
Prx Hyperoxidation
Potential Physiological Value of Prx Hyperoxidation
As noted above, the CP-SOH state of a Prx can react with a second peroxide and become hyperoxidized to a Cys-sulfinate (CP-SO2–), which inactivates the enzyme’s peroxidase function (Figure 2B). Prokaryotic Prxs typically are rather resistant to hyperoxidation, requiring millimolar concentrations of substrate, and have been termed robust isoforms.100 In contrast, many eukaryotic Prxs are quite readily hyperoxidized even though this makes them worse peroxidases. For example, human PrxII is converted almost entirely to the hyperoxidized state in the presence of only 40 μM peroxide (with no reducing agent present), with a kSOH → kSO2 rate on the order of ∼1.0 × 103 M–1 s–1 or higher.101,102 Such isoforms are termed “sensitive”, because even at low peroxide levels they are sensitive to being inactivated through hyperoxidation.100 To facilitate comparisons of sensitivity between Prxs, the quantity Chyp1% was recently introduced as a normalized way to quantify this property;93Chyp1% defines the peroxide concentration at which 1% of Prx molecules become hyperoxidized during each turnover. Using this terminology, it is apparent that human PrxI (Chyp1% = 62 μM), human PrxII (Chyp1% ∼ 1.5 μM), and human PrxIII (Chyp1% ∼ 18 μM) are much more sensitive than StAhpC (Chyp1% = ∼10000 μM).93,102
When Prx hyperoxidation was first discovered, its physiological relevance was questioned, as in vivo peroxide concentrations in healthy cells are thought to rarely exceed 1–15 μM.103 It has since been hypothesized that peroxide levels may locally reach concentrations at which hyperoxidation can occur,4 such as in the vicinity of peroxide-producing enzymes such as NOXs (Figure 1). Recently, the growth factor lysophosphatidic acid (LPA) was shown to stimulate cellular internalization of NOX components into early endosomes, termed “redoxosomes”, to serve as hubs for oxidative regulation104 (Figure 1). Strong support for the existence of local peroxide buildup is an elegant study proving that protein tyrosine phosphatases, which are not highly reactive with peroxide, actually do become oxidized in vivo.105 Further, Prx hyperoxidation is observed in vivo in a variety of organisms and has been discussed as a marker of ancient circadian rhythms,87,106 though the meaning or relevance of this latter observation is not yet clear.
In terms of what evolutionary advantages could be conferred to the many eukaryotes that contain sensitive Prxs, there is as of yet no final consensus. One explanation, termed the “floodgate hypothesis”, proposes that Prx hyperoxidation is important for enabling non-stress-related peroxide signaling in eukaryotes.100 In this model, low peroxide concentrations are reduced efficiently, but when levels spike locally because of the purposeful production of H2O2 by enzymes such as NOX during signaling events,6 Prxs are inactivated to allow the H2O2 to build up sufficiently in a local area to oxidize downstream target proteins (Figure 1). The dysregulation of this signaling pathway provides an explanation for how knockouts of sensitive isoforms in mammals (PrxI–IV) could result in the development of cancers,66 increased cell senescence,71 and malformed tissue and organs72,74 (Table 1 and Table S1 of the Supporting Information). As noted above, the downstream targets that have been most extensively studied are the protein tyrosine phosphatases that become inactivated through the oxidation of a catalytic Cys residue (reviewed by Frijhoff et al.107). Nevertheless, the best documented example of such a floodgate-style function of a Prx is in fact the role of PrxIII in the negative feedback control of mammalian corticosteroid production (Figure 1). As this process occurs in adrenal gland mitochondria as a circadian cycle, an ACTH-activated cytochrome P450 produces H2O2 as a byproduct of making corticosteroids, and the inactivation of PrxIII allows peroxide to build up sufficiently to lead to p38 activation and a shutting down of the synthesis of the steroidogenic acute regulatory protein108 (Figure 1).
Additional proposals that have been put forth for the possible benefits of Prx hyperoxidation include their serving as chaperones,81,109,110 regulating senescence through protein–protein interactions with p38MAPKα,111 and peroxide exposure dosimeters.2 Also, most recently, Day et al.112 showed that under extreme oxidative conditions the inactivation of Prxs can serve to preserve the Trx pool for use by more essential cellular systems.2 In that study, the survival of Schizosaccharomyces pombe was greatly diminished when its single Prx was not inactivated by millimolar levels of peroxide.112 The authors showed the Prx inactivation allowed the reduced Trx pool to be retained for use by Trx-dependent repair enzymes such as methionine sulfoxide reductase.112 Though S. pombe in nature would not normally encounter such high peroxide levels, these results provide a valuable insight into the importance of maintaining a reduced Trx pool. In relation to this, it was proposed that the eukaryotic pathogen Schistosoma might possess both sensitive and robust isoforms because it allows for the switching between reduction sources; because the latter enzyme is preferentially reduced by the glutathione/glutathione reductase system,113 the organism does not exclusively rely on Trx when enduring a peroxide burst from a macrophage. It is also noteworthy that although Schistosoma do not possess catalase, peroxide disproportionation by catalases, present in most cells, is in principle an alternative approach by which cells can prevent the depletion of their reduced Trx.2
Structural Features Influencing the Sensitivity of Prx to Hyperoxidation
So what are the structural features that give rise to sensitivity to hyperoxidation? It was discovered that many Prx1 subfamily sensitive Prxs contain two motifs that pack against the FF active site, a “GGLG” and a C-terminal extension with a “YF”, which are not present in most robust isoforms100 (Figure 3). By inhibiting the local unfolding of the active site, these motifs serve to rigidify and stabilize the FF active site and make the enzyme more susceptible to hyperoxidation.100 This mode of action and the greater importance of the C-terminal YF motif to sensitivity were proven shortly thereafter by a study showing that C-terminal swapping between sensitive and robust isoforms from the eukaryotic parasite Schistosoma resulted in variants with reversed sensitivity.113 Likewise, a truncation of the C-terminal YF motif in human PrxIV greatly diminished the enzyme’s sensitivity.114 On the basis of such results, it has sometimes been generalized that only eukaryotes possess sensitive isoforms and that sensitive and robust Prxs can be reliably distinguished by the presence or absence of the GGLG and YF motifs, but these are both oversimplifications.
Figure 3.
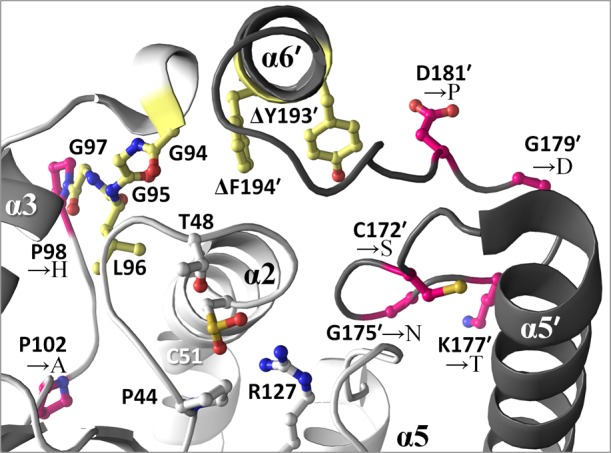
Studies probing the structural basis for Prx hyperoxidation. The active site and C-terminal region are shown for HsPrxII (PDB entry 1qmv), with the GGLG and YF regions colored yellow. Sites where mutations have been introduced as a means to explore the impact on hyperoxidation for PrxI subfamily enzymes are colored pink.101 Elimination of the YF motif by C-terminal truncation (indicated by Δ) has also been conducted.114
With regard to the first point, some prokaryotes do possess sensitive Prxs. A number of bacterial Prx isoforms have the GG(L/V/I)G and YF (or YL or FL) motifs, and some have been shown to be sensitive,110,115,116 although they appear to be used for antioxidant defense rather than regulating peroxide signaling. Examples of this are two cyanobacterial species, Anabaena and Synechocystis, that both have sensitive Prxs.115Anabaena expresses its sensitive isoform abundantly and utilizes an Srx to rescue any hyperoxidized forms, while Synechocystis (which has no Srx) expresses its moderately sensitive Prx only at low levels to mop up endogenous peroxide and rapidly produces catalase to defend against higher peroxide levels.115 Similarly, the bacterium Vibrio vulnificus was shown to possess both a sensitive and a robust Prx,116 with trace amounts of peroxide inducing the expression of the sensitive isoform, whereas only high levels of peroxide induced the robust isoform, suggesting that the two Prxs are utilized for discrete levels of oxidative stress.
With regard to the second point, a recent study of human PrxII and PrxIII explored through mutagenesis the importance of secondary features associated with the two regions101 (Figure 3). Both PrxII and PrxIII contain the GGLG and YF motifs, but nevertheless, PrxIII is ∼10-fold more robust. Swapping the identities of nearby residues between these two isoforms generated more robust PrxII variants and also more sensitive PrxIII variants, although again it was the presence of the C-terminal YF positions that was most critical to promoting sensitivity.101 This proves that positions other than the GGLG and YF motifs can also contribute to sensitivity or robustness. This is especially exemplified by E. coli Tpx, which is a fairly sensitive Prx (Chyp1% of 156 μM for cumene hydroperoxide93) even though it does not contain either motif and is actually in a different Prx subfamily. Also, Perkins et al.20 showed that even conservative mutations such as CR → Ser or Ala, commonly used to study the properties of Prxs, can actually perturb the C-terminal packing sufficiently to shift the FF ↔ LU equilibrium toward LU and make the enzyme less sensitive. Such modulations of sensitivity have been recently shown to occur physiologically, as the C-terminal lysine acetylation of human PrxI117 and N-terminal acetylation of human PrxII118 led the enzymes to become robust. Further, nitration of human PrxII Tyr193 (in the YF motif), detected in Alzheimer patient brains, converted the enzyme to being robust and may play a role in the development of the disease.119 Thus, a small alteration to even one residue can potentially reduce the fraction of the active FF population by orders of magnitude and thereby inhibit hyperoxidation.
These complexities reinforce the point that various Prxs have been optimized to suit diverse needs, and although trends do exist, caution must be employed when attempting to draw firm conclusions about Prx sensitivity solely from a sequence fingerprint. In general, enzymatic characterization is necessary to be certain, and there remains much to learn about the occurrence and roles of sensitive versus robust Prxs.
Distribution of Sulfiredoxin among Eukaryotes
Sulfiredoxin (Srx) catalyzes the ATP-driven rescue of CP-SO2– back to CP-SOH19 and is present in many eukaryotes and a few cyanobacteria.115 Upon its discovery,120 Srx provided an explanation for how eukaryotes could allow sensitive Prxs to be hyperoxidized without being wastefully irreversibly inactivated. A crystal structure of a Prx–Srx complex140 revealed that the two enzymes embrace with the locally unfolded Prx C-terminus wrapping around the backside of Srx, and the Prx CP being placed into the Srx Gly-Cys-His-Arg (GCHR) active site pocket near the bound ATP (Figure 4).28
Figure 4.
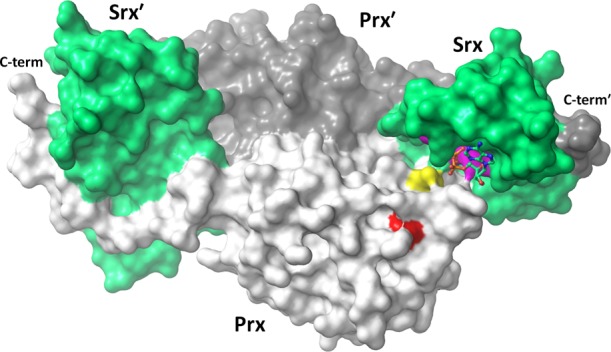
Prx–Srx embrace. Shown is a crystal structure of a human PrxI dimer (light and dark gray) in complex with two Srx chains (green, PDB entry 3hy2). Highlighted are the Prx CP (yellow), the GGLG motif (red), the Srx active site (purple), and its bound ATP (sticks). The Prx C-terminal YF motif is disordered and not shown.
Srx appears to be remarkably important for organisms that express it. Knockouts of Srx cope poorly with oxidative stress,121 with cells showing dramatically increased levels of Prx hyperoxidation, apoptosis, and mitochondrial membrane potential collapse.122 Conversely, the overexpression of Srx has been observed to influence cell proliferation and pro-cancerous activity, including altering the states of p21, p23, and p53.123 In yeast, the overexpression of Srx was shown to increase the replicative life span by 20%.124 We expect that these phenotypes are largely due to altered Prx regulation, but Srx has also been reported to possess deglutathionylation activity.125 Two recent reviews provide further details about the structure, function, and physiology of Srx.125,126 Here, assuming that the presence of Srx in an organism would suggest a signaling-related physiological role for Prx hyperoxidation, we have investigated the distribution of Srx in nature to seek insight into the occurrence, and evolutionary roots, of peroxide signaling pathways.
To perform an updated analysis of the distribution of Srx, we used BLAST127 to retrieve 335 Srx sequences from the nonredundant protein database. Only sequences containing the “GCHR” Srx active site fingerprint121 were included as a way to filter out proteins such as the functionally unrelated bacterial chromosomal partition protein B (ParB), which is a known homologue.128 An evolutionary tree (Figure 5) reveals that Srx is present and clusters distinctly in animals, fungi, plants, and some protists, and as reported in a 2005 Srx evolution study,128 some cyanobacteria are the only prokaryotes to contain an srx gene. This apparent wide distribution of Srx among eukaryotes implies a relatively ancient existence of functional Prx hyperoxidation.
Figure 5.

Relatedness tree for Srx sequences. An unrooted phylogenetic tree of 335 Srx sequences is shown. Select organisms or groups of organisms are noted. Sequences were retrieved from the nonredundant protein database by BLAST127 on January 31, 2014, with an expect threshold of 100 using the human Srx1 sequence, and additional searches using distantly related Srx sequences did not identify further homologues. Sequences were aligned with MUSCLE,138 and evolutionary distances were calculated using PhyML.139
As a next step, we analyzed the 220 available sequenced eukaryotic genomes and surprisingly found that only 56% of them contained an srx gene: fungi and protists quite commonly lack Srx, and while most animals and plants contain Srx, a few animal exceptions seem to exist (Table 2). For example, Xenopus apparently does not have Srx, and subsequent searches for an amphibian srx gene did not yield any examples. Also, especially noteworthy is the fact that many organisms causing human disease, some of which had been mentioned in the 2005 study,128 do not possess Srx (Table 2). These include Entamoeba, apicomplexans (such as Plasmodia species and Toxoplasma gondii), the Diplomonad Giardia lamblia, the parabasalid Trichomonas vaginalis, euglenozoa (Trypanosoma and Leishmania species), the nematodes Loa loa (eye worm) and Brugia malayi (causes elephantitis), and the flatworm Schistosoma mansoni.
Table 2. Presence of Sulfiredoxin in Eukaryotesa.
| animals | fungi | protists | plants |
|---|---|---|---|
| vertebrates | ascomycetes | choanoflagellates (0/1) | eudicots (11/11) |
| mammals (24/24) | saccharomycetes (25/25) | amoebozoa | monocots (6/6) |
| birds (9/9) | sordariomycetes (0/9) | dictyostelium (3/3) | ferns (1/1) |
| reptiles (1/2)b | leotiomycetes (0/2) | entamoeba (0/2) | mosses (1/1) |
| amphibians (0/2)c | eurotiomycetes (0/15) | acanthamoeba (1/1) | green algae (6/8) |
| fish (6/6) | dothideomycetes (0/3) | alveolates | red algae (0/3) |
| lancelets (1/1) | pezizomycetes (0/1) | apicomplexans (0/16) | |
| ascidians (1/1) | schizosaccharomycetes (1/1) | ciliates (0/2) | |
| echinoderms (1/1) | basidiomycetes (3/11) | stramenopiles | |
| arthropods | microsporidians (0/4) | diatoms (0/2) | |
| insects (19/20)d | oomycetes (0/1) | ||
| mites/ticks (1/1) | eustigmatophytes (0/1) | ||
| nematodes (1/5) | cryptomonads (0/1) | ||
| flatworms (0/1) | haptophyta (0/1) | ||
| cnidarians (1/2) | euglenozoa (0/7) | ||
| placozoans (0/1) | heterolobosea (1/1) | ||
| poriferans (0/1) | parabasalids (0/1) | ||
| diplomonads (0/1) |
Across 220 organisms with sequenced genomes, the fractions of the total found to possess an Srx are given in parentheses. Groups containing any members with an Srx-encoding gene are highlighted in bold.
Searches of the Anolis carolinensis genome did not yield an Srx sequence, but that of Ophiophagus hannah (king cobra) did.
Frogs from the genus Xenopus. Additional searches yielded no amphibian Srx-possessing representatives.
The mosquito Anopheles gambiae had no Srx, but two other mosquitos, Aedes aegypti and Culex quinquefasciatus, possessed an Srx gene.
The fact that Srx is present in a diverse range of eukaryotes yet is apparently absent from certain groups seems to be an important observation. For those eukaryotes lacking Srx, some possibilities for how they differ are that the Prx repair function is performed by a different enzyme, that hyperoxidized Prxs are not rescued, and/or that non-stress-related peroxide signaling is either not as important or not similarly regulated by Prx hyperoxidation. In Sc. mansoni, which does not possess Srx but does have a sensitive Prx isoform,113 it has been shown that Prxs that become hyperoxidized are not repaired.77 Whether they use peroxide in non-stress-related signaling is unknown. Like Schistosoma, many of the eukaryotes that do not contain Srx do have at least one Prx isoform that contains the GGLG and YF motifs (Table S4 of the Supporting Information). As discussed earlier, the presence of the GGLG and YF motifs does not necessarily prove that a Prx is sensitive, but as is seen for the Schistosoma enzyme, some may indeed be sensitive.
From these analyses, the additional question of why organisms that seem to lack the ability to rescue hyperoxidized Prxs would retain sensitive isoforms arises. Perhaps some of these organisms, such as was seen for Vibrio,116 minimize waste by tightly regulating their sensitive Prxs to be expressed only at basal levels of peroxide. A further consideration is that because of cellular compartmentalization, even organisms that do contain Srx may not necessarily efficiently rescue all hyperoxidized Prxs. This is illustrated by a recent study showing that in human fibrosarcoma cells, when ER-localized human PrxIV hyperoxidation is induced through ER stress-generating agents, no rescue was observed, leading the authors to conclude that no ER-localized Srx exists.129 We propose that the distribution pattern of Srx in eukaryotes holds important clues about the physiological roles of facile Prx hyperoxidation and that it is worthy of further study.
Efficacy of Targeting Prxs for Drug Design
From the wealth of studies summarized above, we can conclude that Prxs play prominent roles in protecting DNA and other cellular components from oxidative damage, as well as influencing cell signaling, regulation, and proliferation in multicellular eukaryotes. So what rationale is there for the development of Prx-based therapeutics? A particularly interesting development for mammalian Prxs is the recent proposal that certain isoforms, especially PrxV and PrxVI, are danger signals associated with ischemic brain injury.130,131 These enzymes are released poststroke by necrotic brain cells and are specifically detected by toll-like receptors of infiltrating macrophages, stimulating inflammatory cytokine production and promoting ischemic brain damage.130 Antibodies against these Prxs were able to attenuate injury, providing evidence that implicates them as viable targets for future stroke therapeutics.130 Also, given that some cancers overexpressing Prxs are resistant to radiation or other therapies,12−14 it is tempting to envision that inhibiting human Prxs could have therapeutic value in some circumstances. For Prxs from pathogens, however, the case that they are drug targets seems very clear as Prx deficiencies in both prokaryotic and eukaryotic pathogens are linked to viability and infectivity.
The oft-noted challenge with regard to Prxs as drug targets is that the Prx active site is highly conserved, making it very challenging to make selective inhibitors targeting the active site. As an idea for designing inhibitors that would not target the active site, Perkins et al.20 proposed that the delicately balanced FF ↔ LU equilibrium could be shifted by a small molecule to stabilize a single conformation (either the FF or the LU), thereby preventing the structural changes required for Prx catalysis. Surface regions of the protein that are involved in the FF ↔ LU transition are rather divergent in sequence and structure and can therefore be targeted. One such example is the C-terminal region of the Prx1 subfamily. If the LU form were stabilized, it would directly result in the loss of peroxidase activity. Alternatively, if the FF form were stabilized, and the CP was blocked from resolving with the CR, this would directly enhance activity but would indirectly lead to inhibition by promoting hyperoxidation.20 Because most pathogens do not possess an Srx to rescue the hyperoxidized form (e.g., Table 2), these Prxs would be permanently inactivated. Further, the affinity of such an inhibitor could perhaps even be tuned so that it would dissociate and go on to inactivate other Prxs, thereby leading to an increased potency beyond a 1:1 ratio. Structures of many pathogenic Prxs are available (for detailed reviews, see refs (5) and (132))—including bacterial isoforms StAhpC, HpAhpC, Haemophilus influenza Tpx, MtAhpC, and MtTpx and eukaryotic isoforms132Plasmodium yoelii PrxI, Plasmodium vivax 2-Cys, and Plasmodium falciparum Trx-Px2—so rational drug design techniques such as virtual ligand screening133 could be applied to identify leads. These approaches for Prx-targeted therapeutics warrant investigation, because two decades of Prx research can now be used for guidance, and if the effort is successful, it could provide novel antibiotics for some of the most virulent modern diseases.
Glossary
Abbreviations
- Prx
peroxiredoxin
- Srx
sulfiredoxin
- NOX
NADPH oxidase
- ACTH
adrenocorticotropic hormone
- PDB
Protein Data Bank
- CP
peroxidatic cysteine
- CR
resolving cysteine
- FF
fully folded
- LU
locally unfolded
- DTT
dithiothreitol
- HOCl
hypochlorous acid
- Gpx
glutathione peroxidase
- LPA
lysophosphatidic acid
- ParB
chromosomal partitioning protein B.
Supporting Information Available
Summary of Prx knockdown studies (Table S1), summary of Prx knockout studies in various eukaryotes (Table S2), summary of Prx knockout studies in prokaryotes (Table S3), and a list of representative eukaryotes that lack Srx but have Prxs with GGLG/YF motifs (Table S4). This material is available free of charge via the Internet at http://pubs.acs.org.
This study was supported in part by National Institutes of Health Grant R01 GM050389 to L.B.P. and P.A.K.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Footnotes
Supplementary Material
References
- Hall A.; Karplus P. A.; Poole L. B. (2009) Typical 2-Cys peroxiredoxins: Structures, mechanisms and functions. FEBS J. 276, 2469–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus P. A.; Poole L. B. (2012) Peroxiredoxins as Molecular Triage Agents, Sacrificing Themselves to Enhance Cell Survival During a Peroxide Attack. Mol. Cell 45, 275–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson K. J.; Knutson S. T.; Soito L.; Klomsiri C.; Poole L. B.; Fetrow J. S. (2010) Analysis of the peroxiredoxin family: Using active-site structure and sequence information for global classification and residue analysis. Proteins: Struct., Funct., Bioinf. 79, 947–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee S. G.; Woo H. A.; Kil I. S.; Bae S. H. (2012) Peroxiredoxin Functions as a Peroxidase and a Regulator and Sensor of Local Peroxides. J. Biol. Chem. 287, 4403–4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A.; Nelson K.; Poole L. B.; Karplus P. A. (2011) Structure-based Insights into the Catalytic Power and Conformational Dexterity of Peroxiredoxins. Antioxid. Redox Signaling 15, 795–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio M.; Trinei M.; Migliaccio E.; Pelicci P. G. (2007) Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals?. Nat. Rev. Mol. Cell Biol. 8, 722–728. [DOI] [PubMed] [Google Scholar]
- Copley S. D.; Novak W. R. P.; Babbitt P. C. (2004) Divergence of Function in the Thioredoxin Fold Suprafamily: Evidence for Evolution of Peroxiredoxins from a Thioredoxin-like Ancestor. Biochemistry 43, 13981–13995. [DOI] [PubMed] [Google Scholar]
- Zheng M.; Åslund F.; Storz G. (1998) Activation of the OxyR Transcription Factor by Reversible Disulfide Bond Formation. Science 279, 1718–1722. [DOI] [PubMed] [Google Scholar]
- Frey R. S.; Ushio-Fukai M.; Malik A. B. (2009) NADPH Oxidase-Dependent Signaling in Endothelial Cells: Role in Physiology and Pathophysiology. Antioxid. Redox Signaling 11, 791–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antico Arciuch V. G.; Elguero M. E.; Poderoso J. J.; Carreras M. C. (2011) Mitochondrial Regulation of Cell Cycle and Proliferation. Antioxid. Redox Signaling 16, 1150–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne J. R.; Oka S.; Ale-Agha N.; Eaton P. (2013) Hydrogen Peroxide Sensing and Signaling by Protein Kinases in the Cardiovascular System. Antioxid. Redox Signaling 18, 1042–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dy N.; Sj A.; Ra L.; Sw K.; Ia P.; Hz C. (2000) Overexpression of peroxiredoxin in human breast cancer. Anticancer Res. 21, 2085–2090. [PubMed] [Google Scholar]
- Chang J. W.; Jeon H. B.; Lee J. H.; Yoo J. S.; Chun J. S.; Kim J. H.; Yoo Y. J. (2001) Augmented Expression of Peroxiredoxin I in Lung Cancer. Biochem. Biophys. Res. Commun. 289, 507–512. [DOI] [PubMed] [Google Scholar]
- Nonn L.; Berggren M.; Powis G. (2003) Increased Expression of Mitochondrial Peroxiredoxin-3 (Thioredoxin Peroxidase-2) Protects Cancer Cells Against Hypoxia and Drug-Induced Hydrogen Peroxide-Dependent Apoptosis. Mol. Cancer Res. 1, 682–689. [PubMed] [Google Scholar]
- Neumann C. A.; Krause D. S.; Carman C. V.; Das S.; Dubey D. P.; Abraham J. L.; Bronson R. T.; Fujiwara Y.; Orkin S. H.; Van Etten R. A. (2003) Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature 424, 561–565. [DOI] [PubMed] [Google Scholar]
- Rhee S. G.; Chae H. Z.; Kim K. (2005) Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radical Biol. Med. 38, 1543–1552. [DOI] [PubMed] [Google Scholar]
- Barranco-Medina S.; Lázaro J.-J.; Dietz K.-J. (2009) The oligomeric conformation of peroxiredoxins links redox state to function. FEBS Lett. 583, 1809–1816. [DOI] [PubMed] [Google Scholar]
- Rhee S. G.; Yang K.-S.; Kang S. W.; Woo H. A.; Chang T.-S. (2005) Controlled Elimination of Intracellular H2O2: Regulation of Peroxiredoxin, Catalase, and Glutathione Peroxidase via Post-translational Modification. Antioxid. Redox Signaling 7, 619–626. [DOI] [PubMed] [Google Scholar]
- Rhee S. G.; Jeong W.; Chang T.-S.; Woo H. A. (2007) Sulfiredoxin, the cysteine sulfinic acid reductase specific to 2-Cys peroxiredoxin: Its discovery, mechanism of action, and biological significance. Kidney Int. 72, S3–S8. [DOI] [PubMed] [Google Scholar]
- Perkins A.; Nelson K. J.; Williams J. R.; Parsonage D.; Poole L. B.; Karplus P. A. (2013) The Sensitive Balance between the Fully Folded and Locally Unfolded Conformations of a Model Peroxiredoxin. Biochemistry 52, 8708–8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cioli D.; Valle C.; Angelucci F.; Miele A. E. (2008) Will new antischistosomal drugs finally emerge?. Trends Parasitol. 24, 379–382. [DOI] [PubMed] [Google Scholar]
- Piñeyro M. D.; Parodi-Talice A.; Arcari T.; Robello C. (2008) Peroxiredoxins from Trypanosoma cruzi: Virulence factors and drug targets for treatment of Chagas disease?. Gene 408, 45–50. [DOI] [PubMed] [Google Scholar]
- Maglott D.; Ostell J.; Pruitt K. D.; Tatusova T. (2005) Entrez Gene: Gene-centered information at NCBI. Nucleic Acids Res. 33, D54–D58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins A.; Gretes M. C.; Nelson K. J.; Poole L. B.; Karplus P. A. (2012) Mapping the Active Site Helix-to-Strand Conversion of CxxxxC Peroxiredoxin Q Enzymes. Biochemistry 51, 7638–7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson K. J.; Parsonage D.; Hall A.; Karplus P. A.; Poole L. B. (2008) Cysteine pKa Values for the Bacterial Peroxiredoxin AhpC. Biochemistry 47, 12860–12868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo M.; Clippe A.; Manta B.; Ferrer-Sueta G.; Smeets A.; Declercq J.-P.; Knoops B.; Radi R. (2007) Pre-steady state kinetic characterization of human peroxiredoxin 5: Taking advantage of Trp84 fluorescence increase upon oxidation. Arch. Biochem. Biophys. 467, 95–106. [DOI] [PubMed] [Google Scholar]
- Ferrer-Sueta G.; Manta B.; Botti H.; Radi R.; Trujillo M.; Denicola A. (2011) Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chem. Res. Toxicol. 24, 434–450. [DOI] [PubMed] [Google Scholar]
- Jeong W.; Park S. J.; Chang T.-S.; Lee D.-Y.; Rhee S. G. (2006) Molecular Mechanism of the Reduction of Cysteine Sulfinic Acid of Peroxiredoxin to Cysteine by Mammalian Sulfiredoxin. J. Biol. Chem. 281, 14400–14407. [DOI] [PubMed] [Google Scholar]
- Monteiro G.; Horta B. B.; Pimenta D. C.; Augusto O.; Netto L. E. S. (2007) Reduction of 1-Cys peroxiredoxins by ascorbate changes the thiol-specific antioxidant paradigm, revealing another function of vitamin C. Proc. Natl. Acad. Sci. U.S.A. 104, 4886–4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jönsson T. J.; Ellis H. R.; Poole L. B. (2007) Cysteine Reactivity and Thiol–Disulfide Interchange Pathways in AhpF and AhpC of the Bacterial Alkyl Hydroperoxide Reductase System. Biochemistry 46, 5709–5721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian F.-M.; Yu J.; Ma X.-X.; Yu X.-J.; Chen Y.; Zhou C.-Z. (2012) Structural Snapshots of Yeast Alkyl Hydroperoxide Reductase Ahp1 Peroxiredoxin Reveal a Novel Two-cysteine Mechanism of Electron Transfer to Eliminate Reactive Oxygen Species. J. Biol. Chem. 287, 17077–17087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T.; Kado Y.; Yamaguchi T.; Matsumura H.; Ishikawa K.; Inoue T. (2010) Crystal Structure of Peroxiredoxin from Aeropyrum pernix K1 Complexed with Its Substrate, Hydrogen Peroxide. J. Biochem. 147, 109–115. [DOI] [PubMed] [Google Scholar]
- Hall A.; Parsonage D.; Poole L. B.; Karplus P. A. (2010) Structural Evidence that Peroxiredoxin Catalytic Power Is Based on Transition-State Stabilization. J. Mol. Biol. 402, 194–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrard C.; Capron A.; Marchand C.; Clippe A.; Wattiez R.; Soumillion P.; Knoops B.; Declercq J.-P. (2004) Crystal Structure of a Dimeric Oxidized form of Human Peroxiredoxin 5. J. Mol. Biol. 337, 1079–1090. [DOI] [PubMed] [Google Scholar]
- Liao S.-J.; Yang C.-Y.; Chin K.-H.; Wang A. H.-J.; Chou S.-H. (2009) Insights into the Alkyl Peroxide Reduction Pathway of Xanthomonas campestris Bacterioferritin Comigratory Protein from the Trapped Intermediate–Ligand Complex Structures. J. Mol. Biol. 390, 951–966. [DOI] [PubMed] [Google Scholar]
- Portillo-Ledesma S.; Sardi F.; Manta B.; Tourn M. V.; Clippe A.; Knoops B.; Alvarez B.; Coitino E. L.; Ferrer-Sueta G. (2014) Deconstructing the catalytic efficiency of peroxiredoxin-5 peroxidatic cysteine. Biochemistry 53, 6113–6125. [DOI] [PubMed] [Google Scholar]
- Zeida A.; Reyes A. M.; Lebrero M. C. G.; Radi R.; Trujillo M.; Estrin D. A. (2014) The extraordinary catalytic ability of peroxiredoxins: A combined experimental and QM/MM study on the fast thiol oxidation step. Chem. Commun. 50, 10070–10073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A.; Sankaran B.; Poole L. B.; Karplus P. A. (2009) Structural Changes Common to Catalysis in the Tpx Peroxiredoxin Subfamily. J. Mol. Biol. 393, 867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsonage D.; Karplus P. A.; Poole L. B. (2008) Substrate specificity and redox potential of AhpC, a bacterial peroxiredoxin. Proc. Natl. Acad. Sci. U.S.A. 105, 8209–8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo M., Ferrer-Sueta G., Thomson L., Flohé L., and Radi R. (2007) in Peroxiredoxin Systems (Flohé L., and Harris J. R., Eds.) Vol. 44, pp 83–113, Springer, Dordrecht, The Netherlands. [DOI] [PubMed] [Google Scholar]
- Bryk R.; Griffin P.; Nathan C. (2000) Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature 407, 211–215. [DOI] [PubMed] [Google Scholar]
- Piacenza L.; Peluffo G.; Alvarez M. N.; Kelly J. M.; Wilkinson S. R.; Radi R. (2008) Peroxiredoxins play a major role in protecting Trypanosoma cruzi against macrophage- and endogenously-derived peroxynitrite. Biochem. J. 410, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubuisson M.; Vander Stricht D.; Clippe A.; Etienne F.; Nauser T.; Kissner R.; Koppenol W. H.; Rees J.-F.; Knoops B. (2004) Human peroxiredoxin 5 is a peroxynitrite reductase. FEBS Lett. 571, 161–165. [DOI] [PubMed] [Google Scholar]
- Trujillo M.; Radi R. (2002) Peroxynitrite Reaction with the Reduced and the Oxidized Forms of Lipoic Acid: New Insights into the Reaction of Peroxynitrite with Thiols. Arch. Biochem. Biophys. 397, 91–98. [DOI] [PubMed] [Google Scholar]
- Beckman J. S.; Beckman T. W.; Chen J.; Marshall P. A.; Freeman B. A. (1990) Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 87, 1620–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton M. B.; Kettle A. J.; Winterbourn C. C. (1998) Inside the Neutrophil Phagosome: Oxidants, Myeloperoxidase, and Bacterial Killing. Blood 92, 3007–3017. [PubMed] [Google Scholar]
- Stacey M. M.; Peskin A. V.; Vissers M. C.; Winterbourn C. C. (2009) Chloramines and hypochlorous acid oxidize erythrocyte peroxiredoxin 2. Free Radical Biol. Med. 47, 1468–1476. [DOI] [PubMed] [Google Scholar]
- Stacey M. M.; Vissers M. C.; Winterbourn C. C. (2012) Oxidation of 2-Cys Peroxiredoxins in Human Endothelial Cells by Hydrogen Peroxide, Hypochlorous Acid, and Chloramines. Antioxid. Redox Signaling 17, 411–421. [DOI] [PubMed] [Google Scholar]
- De Simoni S.; Goemaere J.; Knoops B. (2008) Silencing of peroxiredoxin 3 and peroxiredoxin 5 reveals the role of mitochondrial peroxiredoxins in the protection of human neuroblastoma SH-SY5Y cells toward MPP+. Neurosci. Lett. 433, 219–224. [DOI] [PubMed] [Google Scholar]
- Kropotov A.; Serikov V.; Suh J.; Smirnova A.; Bashkirov V.; Zhivotovsky B.; Tomilin N. (2006) Constitutive expression of the human peroxiredoxin V gene contributes to protection of the genome from oxidative DNA lesions and to suppression of transcription of noncoding DNA. FEBS J. 273, 2607–2617. [DOI] [PubMed] [Google Scholar]
- Stresing V.; Baltziskueta E.; Rubio N.; Blanco J.; Arriba M.; Valls J.; Janier M.; Clézardin P.; Sanz-Pamplona R.; Nieva C.; Marro M.; Dmitri P.; Sierra A. (2013) Peroxiredoxin 2 specifically regulates the oxidative and metabolic stress response of human metastatic breast cancer cells in lungs. Oncogene 32, 724–735. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S. S.; Leung K. S.; Hicks M. J.; Hastings P. J.; Youssoufian H.; Plon S. E. (2006) Defective mitochondrial peroxiredoxin-3 results in sensitivity to oxidative stress in Fanconi anemia. J. Cell Biol. 175, 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavender T. J.; Bulleid N. J. (2010) Peroxiredoxin IV protects cells from oxidative stress by removing H2O2 produced during disulphide formation. J. Cell Sci. 123, 2672–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavender T. J.; Sheppard A. M.; Bulleid N. J. (2008) Peroxiredoxin IV is an endoplasmic reticulum-localized enzyme forming oligomeric complexes in human cells. Biochem. J. 411, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang X.-Z.; Li D.-Q.; Hou Y.-F.; Wu J.; Lu J.-S.; Di G.-H.; Jin W.; Ou Z.-L.; Shen Z.-Z.; Shao Z.-M. (2007) Identification of the functional role of peroxiredoxin 6 in the progression of breast cancer. Breast Cancer Res. 9, R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. Y.; Chun E.; Lee K.-Y. (2011) Phospholipase A2 of peroxiredoxin 6 has a critical role in tumor necrosis factor-induced apoptosis. Cell Death Differ. 18, 1573–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino I.; Matsubara H.; Hanari N.; Mori M.; Nishimori T.; Yoneyama Y.; Akutsu Y.; Sakata H.; Matsushita K.; Seki N.; Ochiai T. (2005) Histone Deacetylase Inhibitor FK228 Activates Tumor Suppressor Prdx1 with Apoptosis Induction in Esophageal Cancer Cells. Clin. Cancer Res. 11, 7945–7952. [DOI] [PubMed] [Google Scholar]
- Egler R. A.; Fernandes E.; Rothermund K.; Sereika S.; de Souza-Pinto N.; Jaruga P.; Dizdaroglu M.; Prochownik E. V. (2005) Regulation of reactive oxygen species, DNA damage, and c-Myc function by peroxiredoxin 1. Oncogene 24, 8038–8050. [DOI] [PubMed] [Google Scholar]
- Lee T.-H.; Kim S.-U.; Yu S.-L.; Kim S. H.; Park D. S.; Moon H.-B.; Dho S. H.; Kwon K.-S.; Kwon H. J.; Han Y.-H.; Jeong S.; Kang S. W.; Shin H.-S.; Lee K.-K.; Rhee S. G.; Yu D.-Y. (2003) Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood 101, 5033–5038. [DOI] [PubMed] [Google Scholar]
- Yang H.-Y.; Kwon J.; Choi H.-I.; Park S. H.; Yang U.; Park H.-R.; Ren L.; Chung K.-J.; Kim Y. U.; Park B.-J.; Jeong S.-H.; Lee T.-H. (2012) In-depth analysis of cysteine oxidation by the RBC proteome: Advantage of peroxiredoxin II knockout mice. Proteomics 12, 101–112. [DOI] [PubMed] [Google Scholar]
- Li L.; Shoji W.; Takano H.; Nishimura N.; Aoki Y.; Takahashi R.; Goto S.; Kaifu T.; Takai T.; Obinata M. (2007) Increased susceptibility of MER5 (peroxiredoxin III) knockout mice to LPS-induced oxidative stress. Biochem. Biophys. Res. Commun. 355, 715–721. [DOI] [PubMed] [Google Scholar]
- Huh J. Y.; Kim Y.; Jeong J.; Park J.; Kim I.; Huh K. H.; Kim Y. S.; Woo H. A.; Rhee S. G.; Lee K.-J.; Ha H. (2012) Peroxiredoxin 3 Is a Key Molecule Regulating Adipocyte Oxidative Stress, Mitochondrial Biogenesis, and Adipokine Expression. Antioxid. Redox Signaling 16, 229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manevich Y.; Fisher A. B. (2005) Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radical Biol. Med. 38, 1422–1432. [DOI] [PubMed] [Google Scholar]
- Wang X.; Phelan S. A.; Petros C.; Taylor E. F.; Ledinski G.; Jürgens G.; Forsman-Semb K.; Paigen B. (2004) Peroxiredoxin 6 deficiency and atherosclerosis susceptibility in mice: Significance of genetic background for assessing atherosclerosis. Atherosclerosis 177, 61–70. [DOI] [PubMed] [Google Scholar]
- Cao J.; Schulte J.; Knight A.; Leslie N. R.; Zagozdzon A.; Bronson R.; Manevich Y.; Beeson C.; Neumann C. A. (2009) Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J. 28, 1505–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann C. A.; Krause D. S.; Carman C. V.; Das S.; Dubey D. P.; Abraham J. L.; Bronson R. T.; Fujiwara Y.; Orkin S. H.; Van Etten R. A. (2003) Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature 424, 561–565. [DOI] [PubMed] [Google Scholar]
- Park J.-G.; Yoo J.-Y.; Jeong S.-J.; Choi J.-H.; Lee M.-R.; Lee M.-N.; Lee J. H.; Kim H. C.; Jo H.; Yu D.-Y.; Kang S. W.; Rhee S. G.; Lee M.-H.; Oh G. T. (2011) Peroxiredoxin 2 Deficiency Exacerbates Atherosclerosis in Apolipoprotein E-Deficient Mice. Circ. Res. 109, 739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon E.-Y.; Noh Y.-W.; Han Y.-H.; Kim S.-U.; Kim J.-M.; Yu D.-Y.; Lim J.-S. (2006) T lymphocytes and dendritic cells are activated by the deletion of peroxiredoxin II (Prx II) gene. Immunol. Lett. 102, 184–190. [DOI] [PubMed] [Google Scholar]
- Moon E.-Y.; Han Y. H.; Lee D.-S.; Han Y.-M.; Yu D.-Y. (2004) Reactive oxygen species induced by the deletion of peroxiredoxin II (PrxII) increases the number of thymocytes resulting in the enlargement of PrxII-null thymus. Eur. J. Immunol. 34, 2119–2128. [DOI] [PubMed] [Google Scholar]
- Michalek R. D.; Crump K. E.; Weant A. E.; Hiltbold E. M.; Juneau D. G.; Moon E.-Y.; Yu D.-Y.; Poole L. B.; Grayson J. M. (2012) Peroxiredoxin II Regulates Effector and Secondary Memory CD8+ T Cell Responses. J. Virol. 86, 13629–13641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y.-H.; Kim H.-S.; Kim J.-M.; Kim S.-K.; Yu D.-Y.; Moon E.-Y. (2005) Inhibitory role of peroxiredoxin II (Prx II) on cellular senescence. FEBS Lett. 579, 4897–4902. [DOI] [PubMed] [Google Scholar]
- Li L.; Shoji W.; Oshima H.; Obinata M.; Fukumoto M.; Kanno N. (2008) Crucial role of peroxiredoxin III in placental antioxidant defense of mice. FEBS Lett. 582, 2431–2434. [DOI] [PubMed] [Google Scholar]
- Li L.; Kaifu T.; Obinata M.; Takai T. (2009) Peroxiredoxin III-deficiency Sensitizes Macrophages to Oxidative Stress. J. Biochem. 145, 425–427. [DOI] [PubMed] [Google Scholar]
- Iuchi Y.; Okada F.; Tsunoda S.; Kibe N.; Shirasawa N.; Ikawa M.; Okabe M.; Ikeda Y.; Fujii J. (2009) Peroxiredoxin 4 knockout results in elevated spermatogenic cell death via oxidative stress. Biochem. J. 419, 149. [DOI] [PubMed] [Google Scholar]
- Isermann K.; Liebau E.; Roeder T.; Bruchhaus I. (2004) A Peroxiredoxin Specifically Expressed in Two Types of Pharyngeal Neurons is Required for Normal Growth and Egg Production in Caenorhabditis elegans. J. Mol. Biol. 338, 745–755. [DOI] [PubMed] [Google Scholar]
- Ranjan M.; Gruber J.; Ng L. F.; Halliwell B. (2013) Repression of the mitochondrial peroxiredoxin antioxidant system does not shorten life span but causes reduced fitness in Caenorhabditis elegans. Free Radical Biol. Med. 63, 381–389. [DOI] [PubMed] [Google Scholar]
- Sayed A. A.; Cook S. K.; Williams D. L. (2006) Redox Balance Mechanisms in Schistosoma mansoni Rely on Peroxiredoxins and Albumin and Implicate Peroxiredoxins as Novel Drug Targets. J. Biol. Chem. 281, 17001–17010. [DOI] [PubMed] [Google Scholar]
- de Moraes Mourão M.; Dinguirard N.; Franco G. R.; Yoshino T. P. (2009) Phenotypic Screen of Early-Developing Larvae of the Blood Fluke, Schistosoma mansoni, using RNA Interference. PLoS Neglected Trop. Dis. 3, e502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai T.; Osada Y.; Ohta N.; Kanazawa T. (2009) Peroxiredoxin-1 from Schistosoma japonicum functions as a scavenger against hydrogen peroxide but not nitric oxide. Mol. Biochem. Parasitol. 164, 26–31. [DOI] [PubMed] [Google Scholar]
- Wilkinson S. R.; Horn D.; Prathalingam S. R.; Kelly J. M. (2003) RNA Interference Identifies Two Hydroperoxide Metabolizing Enzymes That Are Essential to the Bloodstream Form of the African Trypanosome. J. Biol. Chem. 278, 31640–31646. [DOI] [PubMed] [Google Scholar]
- Castro H.; Teixeira F.; Romao S.; Santos M.; Cruz T.; Flórido M.; Appelberg R.; Oliveira P.; Ferreira-da-Silva F.; Tomás A. M. (2011) Leishmania Mitochondrial Peroxiredoxin Plays a Crucial Peroxidase-Unrelated Role during Infection: Insight into Its Novel Chaperone Activity. PLoS Pathog. 7, e1002325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komaki-Yasuda K.; Kawazu S.; Kano S. (2003) Disruption of the Plasmodium falciparum 2-Cys peroxiredoxin gene renders parasites hypersensitive to reactive oxygen and nitrogen species. FEBS Lett. 547, 140–144. [DOI] [PubMed] [Google Scholar]
- Yano K.; Komaki-Yasuda K.; Tsuboi T.; Torii M.; Kano S.; Kawazu S. (2006) 2-Cys Peroxiredoxin TPx-1 is involved in gametocyte development in Plasmodium berghei. Mol. Biochem. Parasitol. 148, 44–51. [DOI] [PubMed] [Google Scholar]
- Yano K.; Otsuki H.; Arai M.; Komaki-Yasuda K.; Tsuboi T.; Torii M.; Kano S.; Kawazu S.-I. (2008) Disruption of the Plasmodium berghei 2-Cys peroxiredoxin TPx-1 gene hinders the sporozoite development in the vector mosquito. Mol. Biochem. Parasitol. 159, 142–145. [DOI] [PubMed] [Google Scholar]
- Wong C.-M.; Siu K.-L.; Jin D.-Y. (2004) Peroxiredoxin-null yeast cells are hypersensitive to oxidative stress and are genomically unstable. J. Biol. Chem. 279, 23207–23213. [DOI] [PubMed] [Google Scholar]
- Fomenko D. E.; Koc A.; Agisheva N.; Jacobsen M.; Kaya A.; Malinouski M.; Rutherford J. C.; Siu K.-L.; Jin D.-Y.; Winge D. R.; Gladyshev V. N. (2011) Thiol peroxidases mediate specific genome-wide regulation of gene expression in response to hydrogen peroxide. Proc. Natl. Acad. Sci. U.S.A. 108, 2729–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. S.; Green E. W.; Zhao Y.; van Ooijen G.; Olmedo M.; Qin X.; Xu Y.; Pan M.; Valekunja U. K.; Feeney K. A.; Maywood E. S.; Hastings M. H.; Baliga N. S.; Merrow M.; Millar A. J.; Johnson C. H.; Kyriacou C. P.; O’Neill J. S.; Reddy A. B. (2012) Peroxiredoxins are conserved markers of circadian rhythms. Nature 485, 459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baier M.; Noctor G.; Foyer C. H.; Dietz K.-J. (2000) Antisense Suppression of 2-Cysteine Peroxiredoxin in Arabidopsis Specifically Enhances the Activities and Expression of Enzymes Associated with Ascorbate Metabolism But Not Glutathione Metabolism. Plant Physiol. 124, 823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkemeyer P.; Laxa M.; Collin V.; Li W.; Finkemeier I.; Schöttler M. A.; Holtkamp V.; Tognetti V. B.; Issakidis-Bourguet E.; Kandlbinder A.; Weis E.; Miginiac-Maslow M.; Dietz K.-J. (2006) Peroxiredoxin Q of Arabidopsis thaliana is attached to the thylakoids and functions in context of photosynthesis. Plant J. 45, 968–981. [DOI] [PubMed] [Google Scholar]
- Schafer F. Q.; Buettner G. R. (2001) Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radical Biol. Med. 30, 1191–1212. [DOI] [PubMed] [Google Scholar]
- Go Y.-M.; Jones D. P. (1780) Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta 2008, 1273–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banach-Latapy A.; He T.; Dardalhon M.; Vernis L.; Chanet R.; Huang M.-E. (2013) Redox-sensitive YFP sensors for monitoring dynamic compartment-specific glutathione redox state. Free Radical Biol. Med. 65, 436–445. [DOI] [PubMed] [Google Scholar]
- Nelson K. J.; Parsonage D.; Karplus P. A.; Poole L. B. (2013) Evaluating peroxiredoxin sensitivity toward inactivation by peroxide substrates. Methods Enzymol. 527, 21–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele K. H.; Baumgartner J. E.; Valderas M. W.; Roop R. M. (2010) Comparative Study of the Roles of AhpC and KatE as Respiratory Antioxidants in Brucella abortus 2308. J. Bacteriol. 192, 4912–4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker L. M. S.; Raudonikiene A.; Hoffman P. S.; Poole L. B. (2001) Essential Thioredoxin-Dependent Peroxiredoxin System from Helicobacter pylori: Genetic and Kinetic Characterization. J. Bacteriol. 183, 1961–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olczak A. A.; Richard W. Seyler J.; Olson J. W.; Maier R. J. (2003) Association of Helicobacter pylori Antioxidant Activities with Host Colonization Proficiency. Infect. Immun. 71, 580–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove K.; Coutts G.; Jonsson I.-M.; Tarkowski A.; Kokai-Kun J. F.; Mond J. J.; Foster S. J. (2007) Catalase (KatA) and Alkyl Hydroperoxide Reductase (AhpC) Have Compensatory Roles in Peroxide Stress Resistance and Are Required for Survival, Persistence, and Nasal Colonization in Staphylococcus aureus. J. Bacteriol. 189, 1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson T.; Lisle G. W.; de Marcinkeviciene J. A.; Blanchardand J. S.; Collins D. M. (1998) Antisense RNA to ahpC, an oxidative stress defence gene involved in isoniazid resistance, indicates that AhpC of Mycobacterium bovis has virulence properties. Microbiology 144, 2687–2695. [DOI] [PubMed] [Google Scholar]
- Wang H.-W.; Chung C.-H.; Ma T.-Y.; Wong H. (2013) Roles of Alkyl Hydroperoxide Reductase Subunit C (AhpC) in Viable but Nonculturable Vibrio parahaemolyticus. Appl. Environ. Microbiol. 79, 3734–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood Z. A.; Poole L. B.; Karplus P. A. (2003) Peroxiredoxin Evolution and the Regulation of Hydrogen Peroxide Signaling. Science 300, 650–653. [DOI] [PubMed] [Google Scholar]
- Haynes A. C.; Qian J.; Reisz J. A.; Furdui C. M.; Lowther W. T. (2013) Molecular Basis for the Resistance of Human Mitochondrial 2-Cys Peroxiredoxin 3 to Hyperoxidation. J. Biol. Chem. 288, 29714–29723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peskin A. V.; Dickerhof N.; Poynton R. A.; Paton L. N.; Pace P. E.; Hampton M. B.; Winterbourn C. C. (2013) Hyperoxidation of Peroxiredoxins 2 and 3. J. Biol. Chem. 288, 14170–14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder E.; Eaton P. (2008) Hydrogen peroxide as an endogenous mediator and exogenous tool in cardiovascular research: Issues and considerations. Curr. Opin. Pharmacol. 8, 153–159. [DOI] [PubMed] [Google Scholar]
- Klomsiri C.; Rogers L. C.; Soito L.; McCauley A. K.; King S. B.; Nelson K. J.; Poole L. B.; Daniel L. W. (2014) Endosomal H2O2 production leads to localized cysteine sulfenic acid formation on proteins during lysophosphatidic acid-mediated cell signaling. Free Radical Biol. Med. 71, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque A.; Andersen J. N.; Salmeen A.; Barford D.; Tonks N. K. (2011) Conformation-Sensing Antibodies Stabilize the Oxidized Form of PTP1B and Inhibit Its Phosphatase Activity. Cell 147, 185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill J. S.; Reddy A. B. (2011) Circadian clocks in human red blood cells. Nature 469, 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frijhoff J.; Dagnell M.; Godfrey R.; Östman A. (2014) Regulation of Protein Tyrosine Phosphatase Oxidation in Cell Adhesion and Migration. Antioxid. Redox Signaling 20, 1994–2010. [DOI] [PubMed] [Google Scholar]
- Kil I. S.; Lee S. K.; Ryu K. W.; Woo H. A.; Hu M.-C.; Bae S. H.; Rhee S. G. (2012) Feedback Control of Adrenal Steroidogenesis via H2O2-Dependent, Reversible Inactivation of Peroxiredoxin III in Mitochondria. Mol. Cell 46, 584–594. [DOI] [PubMed] [Google Scholar]
- Angelucci F.; Saccoccia F.; Ardini M.; Boumis G.; Brunori M.; Di Leandro L.; Ippoliti R.; Miele A. E.; Natoli G.; Scotti S.; Bellelli A. (2013) Switching between the Alternative Structures and Functions of a 2-Cys Peroxiredoxin, by Site-Directed Mutagenesis. J. Mol. Biol. 425, 4556–4568. [DOI] [PubMed] [Google Scholar]
- Chuang M.-H.; Wu M.-S.; Lo W.-L.; Lin J.-T.; Wong C.-H.; Chiou S.-H. (2006) The antioxidant protein alkylhydroperoxide reductase of Helicobacter pylori switches from a peroxide reductase to a molecular chaperone function. Proc. Natl. Acad. Sci. U.S.A. 103, 2552–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner-Ivey B.; Manevich Y.; Schulte J.; Kistner-Griffin E.; Jezierska-Drutel A.; Liu Y.; Neumann C. A. (2013) Role for Prdx1 as a specific sensor in redox-regulated senescence in breast cancer. Oncogene 32, 5302–5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day A. M.; Brown J. D.; Taylor S. R.; Rand J. D.; Morgan B. A.; Veal E. A. (2012) Inactivation of a Peroxiredoxin by Hydrogen Peroxide Is Critical for Thioredoxin-Mediated Repair of Oxidized Proteins and Cell Survival. Mol. Cell 45, 398–408. [DOI] [PubMed] [Google Scholar]
- Sayed A. A.; Williams D. L. (2004) Biochemical Characterization of 2-Cys Peroxiredoxins from Schistosoma mansoni. J. Biol. Chem. 279, 26159–26166. [DOI] [PubMed] [Google Scholar]
- Wang X.; Wang L.; Wang X.; Sun F.; Wang C. (2012) Structural insights into the peroxidase activity and inactivation of human peroxiredoxin 4. Biochem. J. 441, 113–118. [DOI] [PubMed] [Google Scholar]
- Pascual M. B.; Mata-Cabana A.; Florencio F. J.; Lindahl M.; Cejudo F. J. (2010) Overoxidation of 2-Cys Peroxiredoxin in Prokaryotes Cyanobacterial 2-Cys Peroxiredoxins Sensitive to Oxidative Stress. J. Biol. Chem. 285, 34485–34492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang Y.-J.; Oh M. H.; Choi S. H. (2012) Distinct Characteristics of Two 2-Cys Peroxiredoxins of Vibrio vulnificus Suggesting Differential Roles in Detoxifying Oxidative Stress. J. Biol. Chem. 287, 42516–42524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmigiani R. B.; Xu W. S.; Venta-Perez G.; Erdjument-Bromage H.; Yaneva M.; Tempst P.; Marks P. A. (2008) HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc. Natl. Acad. Sci. U.S.A. 105, 9633–9638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J. H.; Lim J. C.; Lee D.-Y.; Kim K. S.; Piszczek G.; Nam H. W.; Kim Y. S.; Ahn T.; Yun C.-H.; Kim K.; Chock P. B.; Chae H. Z. (2009) Novel Protective Mechanism against Irreversible Hyperoxidation of Peroxiredoxin Nα-terminal Acetylation of Human Peroxiredoxin II. J. Biol. Chem. 284, 13455–13465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall L. M.; Manta B.; Hugo M.; Gil M.; Batthyany C.; Trujillo M.; Poole L. B.; Denicola A. (2014) Nitration transforms a sensitive peroxiredoxin 2 into a more active and robust peroxidase. J. Biol. Chem. 289, 15536–15543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biteau B.; Labarre J.; Toledano M. B. (2003) ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 425, 980–984. [DOI] [PubMed] [Google Scholar]
- Liu X. P.; Liu X. Y.; Zhang J.; Xia Z. L.; Liu X.; Qin H. J.; Wang D. W. (2006) Molecular and functional characterization of sulfiredoxin homologs from higher plants. Cell Res. 16, 287–296. [DOI] [PubMed] [Google Scholar]
- Baek J. Y.; Han S. H.; Sung S. H.; Lee H. E.; Kim Y.; Noh Y. H.; Bae S. H.; Rhee S. G.; Chang T.-S. (2012) Sulfiredoxin Protein Is Critical for Redox Balance and Survival of Cells Exposed to Low Steady-state Levels of H2O2. J. Biol. Chem. 287, 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei K.; Townsend D. M.; Tew K. D. (2008) Protein cysteine sulfinic acid reductase (sulfiredoxin) as a regulator of cell proliferation and drug response. Oncogene 27, 4877–4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molin M.; Yang J.; Hanzén S.; Toledano M. B.; Labarre J.; Nyström T. (2011) Life Span Extension and H2O2 Resistance Elicited by Caloric Restriction Require the Peroxiredoxin Tsa1 in Saccharomyces cerevisiae. Mol. Cell 43, 823–833. [DOI] [PubMed] [Google Scholar]
- Lowther W. T.; Haynes A. C. (2010) Reduction of Cysteine Sulfinic Acid in Eukaryotic, Typical 2-Cys Peroxiredoxins by Sulfiredoxin. Antioxid. Redox Signaling 15, 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong W.; Bae S. H.; Toledano M. B.; Rhee S. G. (2012) Role of sulfiredoxin as a regulator of peroxiredoxin function and regulation of its expression. Free Radical Biol. Med. 53, 447–456. [DOI] [PubMed] [Google Scholar]
- Altschul S. F.; Madden T. L.; Schäffer A. A.; Zhang J.; Zhang Z.; Miller W.; Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu M. K.; Koonin E. V. (2005) Evolution of Eukaryotic Cysteine Sulfinic Acid Reductase, Sulfiredoxin (Srx), from Bacterial Chromosome Partitioning Protein ParB. Cell Cycle 4, 947–952. [DOI] [PubMed] [Google Scholar]
- Cao Z.; Subramaniam S.; Bulleid N. J. (2014) Lack of an Efficient Endoplasmic Reticulum-localized Recycling System Protects Peroxiredoxin IV from Hyperoxidation. J. Biol. Chem. 289, 5490–5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shichita T.; Hasegawa E.; Kimura A.; Morita R.; Sakaguchi R.; Takada I.; Sekiya T.; Ooboshi H.; Kitazono T.; Yanagawa T.; Ishii T.; Takahashi H.; Mori S.; Nishibori M.; Kuroda K.; Akira S.; Miyake K.; Yoshimura A. (2012) Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat. Med. 18, 911–917. [DOI] [PubMed] [Google Scholar]
- Nagy N.; Malik G.; Fisher A. B.; Das D. K. (2006) Targeted disruption of peroxiredoxin 6 gene renders the heart vulnerable to ischemia-reperfusion injury. Am. J. Physiol. 291, H2636–H2640. [DOI] [PubMed] [Google Scholar]
- Gretes M. C.; Poole L. B.; Karplus P. A. (2012) Peroxiredoxins in Parasites. Antioxid. Redox Signaling 17, 608–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M.; Coleman R. G.; Fraser J. S.; Shoichet B. K. (2014) Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery. Nat. Chem. 6, 575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Feinstein S. I.; Manevich Y.; Ho Y.-S.; Fisher A. B. (2004) Lung injury and mortality with hyperoxia are increased in peroxiredoxin 6 gene-targeted mice. Free Radical Biol. Med. 37, 1736–1743. [DOI] [PubMed] [Google Scholar]
- Fisher A. B.; Dodia C.; Feinstein S. I.; Ho Y.-S. (2005) Altered lung phospholipid metabolism in mice with targeted deletion of lysosomal-type phospholipase A2. J. Lipid Res. 46, 1248–1256. [DOI] [PubMed] [Google Scholar]
- Fatma N.; Singh P.; Chhunchha B.; Kubo E.; Shinohara T.; Bhargavan B.; Singh D. P. (2011) Deficiency of Prdx6 in lens epithelial cells induces ER stress response-mediated impaired homeostasis and apoptosis. Am. J. Physiol. 301, C954–C967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sies H. (2014) Role of Metabolic H2O2 Generation: Redox Signalling and Oxidative Stress. J. Biol. Chem. 289, 8735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. (2004) MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S.; Lethiec F.; Duroux P.; Gascuel O. (2005) PHYML Online: A web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 33, W557–W559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jönsson T. J.; Johnson L. C.; Lowther W. T. (2009) Protein Engineering of the Quaternary Sulfiredoxin Peroxiredoxin Enzyme Substrate Complex Reveals the Molecular Basis for Cysteine Sulfinic Acid Phosphorylation. J. Biol. Chem. 284, 33305–33310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.