

Abstract
Ribonuclease A (RNase A) is a small globular enzyme that lyses RNA. The remarkable solution stability of its structure and enzymatic activity has led to its investigation to develop a new class of drugs for cancer chemotherapeutics. However, the successful clinical application of RNase A has been reported to be limited by insufficient stability and loss of enzymatic activity when it was coupled with a biomaterial carrier for drug delivery. The objective of this study was to characterize the structural stability and enzymatic activity of RNase A when it was adsorbed on different surface chemistries (represented by fused silica glass, high-density polyethylene, and poly(methyl-methacrylate)). Changes in protein structure were measured by circular dichroism, amino acid labeling with mass spectrometry, and in vitro assays of its enzymatic activity. Our results indicated that the process of adsorption caused RNase A to undergo a substantial degree of unfolding with significant differences in its adsorbed structure on each material surface. Adsorption caused RNase A to lose about 60% of its native-state enzymatic activity independent of the material on which it was adsorbed. These results indicate that the native-state structure of RNase A is greatly altered when it is adsorbed on a wide range of surface chemistries, especially at the catalytic site. Therefore, drug delivery systems must focus on retaining the native structure of RNase A in order to maintain a high level of enzymatic activity for applications such as antitumor chemotherapy.
I. Introduction
Ribonucleases, such as ribonuclease A (RNase A), which catalyzes the breakdown of the phosphodiester backbone of ribonucleic acid (RNA) into smaller components, are being investigated as potential chemotherapy agents.1−3 RNase A has been shown to have a cytotoxic effect that is specific for many malignant tumor cells from in vitro experiments. Its effectiveness on tumor cells is believed to be due, in part, to this enzyme’s exceptional stability even under harsh environmental conditions.4,5 The aqueous solution stability of RNase A is recognized to be a result of its compact globular structure (14 kDa with four disulfide bonds) and its hydrophilicity.6−8
Unfortunately, the successful clinical application of RNase A has been limited by factors including its short half-life in vivo due to rapid glomerular filtration and inactivation by antibodies.9,10 Attempts have been made to increase the in vivo residence time and delivery concentration by coupling it to material surfaces of various drug delivery platforms.9,11−13 However, studies have indicated that this often causes loss of native-state enzyme activity due to adsorption-induced changes in the structure and/or stearic hindrance of the active site.12−16 These findings indicate that greater understanding is needed regarding how interactions with material surfaces influence the adsorbed structure and enzymatic activity of RNase A in order to support the therapeutic use of this enzyme in antitumor drug delivery applications.
The effect of different material surfaces and adsorption conditions on the structure and enzymatic activity of adsorbed RNase A is not very well understood. Previous studies on the adsorption behavior of proteins have shown that the adsorbed orientation and adsorption-induced changes in protein conformation and enzymatic activity are a result of the combination of a protein’s internal stability relative to the ability of protein–surface and protein–protein interactions (PPI) on the surface to perturb the protein’s structure.17−19 Although many previous studies have been published that relate adsorption-induced loss in protein structure to loss of bioactivity, most of these studies were done using techniques like circular dichroism spectropolarimetry (CD), which, though useful, are only scalar indicators of the molecular processes underlying the involved processes.10,19−23 Alternatively, techniques like amino acid labeling and mass spectrometry (AAL/MS), though localized, can be used to identify the shifts in the solvent exposure of the residues within the tertiary structure of adsorbed protein.12,17,23−29 Additionally, these types of techniques are especially relevant in applications that require molecular-scale understanding of the processes underlying the loss in the bioactivity of a protein, despite retaining its near-native secondary structure.29
The objective of this study was, therefore, to investigate how different adsorption conditions would influence the structure and enzymatic activity of adsorbed RNase A. Toward this purpose, we have used an AAL/MS technique along with CD to quantitatively investigate the effects of adsorption on bovine pancreatic ribonuclease A (RNase A) when it is adsorbed on fused silica glass (glass), high-density polyethylene (HDPE), and poly(methyl-methacrylate) (PMMA) to further explore how surface chemistry influences the relationship between adsorbed conformation and enzymatic activity. The combined use of these techniques provides insight into the protein’s adsorbed orientation, adsorption-induced changes in protein secondary and tertiary structures, and adsorption-induced effects on the solvent accessibility of RNase A’s catalytic site.
II. Materials and Methods
II.a. Material Surface Preparation and Characterization
The selected material surfaces include fused silica glass (glass), high-density polyethylene (HDPE), and poly(methyl methacrylate) (PMMA). Custom cut glass (0.375″ (L) × 0.0625″ (W) × 1.625″ (H), Chemglass Life Sciences) was procured and cleaned at 50 °C by immersion in piranha solution (7:3 v/v H2SO4(EMD Chemicals, SX 1244)/H2O2) for at least 30 min followed by basic wash (1:1:5 v/v NH4OH (BDH Chemicals, BDH3016)/H2O2/H2O), and this procedure was repeated twice. Standard safety procedures were followed during the handling, storage, and disposal of these wash solutions. HDPE and PMMA surfaces were spin-coated onto the silicon wafer (6″ diameter, University Wafer) from dodecalin (0.5% (w/w) at 1500 rpm for 60 s) and chloroform solutions (1.5% (w/w) at 1000 rpm for 60 s), respectively. All chemicals including the polymers of HDPE (Mw =125 000 Da, Sigma 181900) and PMMA (Mw = 350 000 Da, Sigma 445746) and the solvents such as dodecalin (Sigma 294772) and chloroform (EMD Chemicals, CX 1054) were used as supplied by the manufacturer. Prior to conducting the adsorption studies, all of the substrates were rinsed in absolute ethanol followed by nanopure water and then dried under nitrogen gas.
Characterization of the material surfaces was performed to determine the static air–water contact angle, surface composition, film thickness, and surface roughness of the substrates. For each of the surfaces, the static air–water contact angle was analyzed using a contact angle goniometer (Krüss, DSA-20E). The surface composition was verified via X-ray photoelectron spectroscopy (NESCA/BIO, University of Washington), and the average RMS surface roughness was analyzed using atomic force microscopy (Asylum Research, MFP-3D) over an area of 5 × 5 μm2. The thickness of the polymer films was characterized using variable-angle spectroscopic ellipsometry (Sopra Inc., GES-5). See section S.1.b in the Supporting Information for further details regarding the material characterization procedures followed in this research.
II.b. Protein Adsorption and Equilibration
The adsorption of RNAse A (Sigma R6513) on the material surfaces was carried out using previously described methods (see S.1.a in the Supporting Information).19 Briefly, 10 mM potassium phosphate buffer solution (PPB; pH 7.4) was prepared by mixing appropriate amounts of 1 M monobasic potassium phosphate (Sigma, P8708) or 1 M dibasic potassium phosphate (Sigma, P8508), following which the buffer concentration was verified by titrating against 0.065 M potassium hydrogen phthalate.
Protein adsorption was conducted in 10 mM PPB under protein concentrations of 0.03 and 1.00 mg/mL for 2 h in order to vary the surface coverage of adsorbed protein on each surface, following which the material surfaces were gently rinsed under a steady gentle flow (12 mL/min) of PPB for 5 min to remove weakly adsorbed protein. The surfaces with the adsorbed layer of protein were then immersed in PPB for 15 h to allow the adsorbed protein layers to structurally equilibrate on the surface at room temperature (≈25 °C). Control studies were conducted to ensure that RNase A itself did not undergo a significant change in structure and/or activity during this frame in PPB solution due to simple aging. The effect of adsorption time from protein solution and equilibration time in PPB for different surfaces on the surface coverage and structure of the protein when adsorbed from a given solution concentration is provided in sections S.2.a and S.2.b of the Supporting Information. From these studies, it was determined that the designated times of 2 h for initial adsorption followed by 15 h of relaxation under PPB were sufficient for system equilibration for each of our treatment conditions.
II.c. Characterization of Secondary Structure
The secondary structures of RNase A both in solution and on each surface were determined using previously described CD spectropolarimetry methods (see section S.1.d in the Supporting Information).30 Briefly, the CD spectra of RNase A in solution was obtained at room temperature using a Jasco J-810 spectropolarimeter in a 0.1 mm path-length quartz cuvette (Starna) from 190 to 300 nm in 10 mM PPB solution (pH 7.4). The structure of the adsorbed RNase A was determined under similar conditions but using a custom-made cuvette that was designed to hold four sets of the adsorbed surfaces perpendicular to the CD beam, which enhances the signal-to-noise ratio. The amount of protein on a given surface (Qads) was determined from eq 1 using the absorbance at 205 nm (A205)24
| 1 |
where ε205 represents the molar extinction coefficient at 205 nm (units of M–1 cm–1), which was determined from the calibration plot of standardized solution of RNase A at different concentrations. The solution concentrations (Csoln) of RNase A were standardized using the E (1%) of 7.0 (g/100 mL)−1 cm–1 at 280 nm method provided by the supplier.
II.d. Characterizing Orientation and Tertiary Structure of RNase A Using Amino Acid Labeling/Mass Spectrometry (AAL/MS)
AAL/MS uses amino acid-specific, nonreversible chemical labeling to probe the adsorbed orientation and adsorption-induced changes in the tertiary structure of proteins.24 This method is based on the principle that only solvent-accessible amino acids can undergo chemical labeling. Mass spectrometry is then used to identify whether the targeted amino acids are labeled. Amino acid residues that are found to be labeled in solution but unlabeled following adsorption indicate regions of blockage by the surface (i.e., indicative of adsorbed orientation) or by neighboring proteins (i.e., indicative of protein–protein interactions). Alternatively, amino acids that are unlabeled in solution but become labeled following adsorption are indicative of sites in the protein that underwent adsorption-induced tertiary unfolding that exposed otherwise unavailable residues.
Application of AAL/MS to multiple different amino acid types that are distributed throughout a protein enables a fairly comprehensive picture to be generated regarding the primary distribution of sites in the protein that are tightly adsorbed to the surface (or blocked by neighboring proteins) and sites undergoing adsorption-induced tertiary unfolding.
II.d.1. Batch Labeling of Target Amino Acids
Arg, Lys, Asp, Glu, Tyr, and His in RNAse A were individually labeled under common reactive conditions to facilitate direct comparison of the labeling profiles from each of these amino acids using previously developed methods (see section S.1.e.2 in the Supporting Information).31−34 For consistency between treatments, the reaction between the labeling agent and its targeted amino acid was carried out at 5× the overall molar concentration of the targeted amino acid type contained within the protein in the dark at 25 °C for 3 h in PPB. The solution pH was maintained at 7.4 by adding required amounts of 1 M monobasic potassium phosphate (Sigma, P8708) or 1 M dibasic potassium phosphate (Sigma, P8508), following which the buffer concentration was verified by titrating against 0.065 M potassium hydrogen phthalate.
II.d.2. Analysis by Mass Spectrometry
Proteolytic digestion of modified and unmodified RNAse A from in-solution and adsorbed states was done using sequence-grade porcine trypsin (Promega) after being chemically labeled as described in section S.1.f of the Supporting Information. Trypsin-digested peptides were subsequently analyzed using an ultra performance liquid chromatography system (UPLC, Waters) coupled with a quadruple time-of-flight mass spectrometer (Q-TOF MS, Waters) with electrospray ionization in both ESI+-MS and ESI+-MS/MS (SetMass without fragmentation) mode operated by Masslynx software (v4.1). The intensities obtained from mass matching were subsequently used to quantify the extent of solvent exposure for the targeted residues.
II.d.3. Correlating Mass Spectra to Configuration of Adsorbed RNase A
It has been previously shown that the peak intensities of the mass spectra of trypsin digests of a protein following chemical labeling are directly related to the extent of the solvent exposure of the targeted amino acid.26 These previous methods, however, were developed only for application to individual types of amino acid residues within a protein. Our group has developed a method to target multiple amino acids within a protein24 by (i) using an unlabeled peptide sequence of the protein to normalize the absolute extent of labeling from the peptide intensities of each of the batch experiments to a common reference state35 and (ii) calculating a relative ratio of the normalized extent of modification for each targeted amino acid residue of the protein in its adsorbed state to that in its solution state, which we refer to as the residue profile. These combined methods are subsequently used to probe the adsorbed configuration of a protein on a surface.
Accordingly, the normalized extents of modification (%) for a target residue in its solution (Isoln) and adsorbed (Iads) states were subsequently estimated from a given mass spectrum by calculating the ratio of the weighted intensity of peptide fragments containing the labeled target amino acid to the total weighted intensities of all peptide fragments, as shown in section S.1.h of the Supporting Information. Following which, the net intensity parameter of amino acid labeling in the protein’s adsorbed state (Iads) was determined by dividing it by its net intensity parameter in solution to obtain the overall relative degree of labeling in the amino acid’s adsorbed versus solution state. If the weighted intensity for a given residue in solution (Isoln) or in its adsorbed state (Iads) was found to be less than 0.10 (which was considered to be the limit of detection), then a low ceiling threshold value of 0.10 was designated for the respective intensity value instead of zero in order to avoid the mathematical problems of dividing by zero or taking the log(0) in eq 1.24 Similarly, the maximum values that could be expected for Isoln and Iads was 1.0, which corresponds to the condition when all of the peptide fragments containing the target residues were labeled.24 The base 10 logarithm of Iads/Isoln was then taken to provide the residue profile value for each targeted amino acid, as indicated in eq 1. A given residue’s profile could then be used to represent an averaged response of the ensemble of configurations that the adsorbed protein adopts on a given surface. A positive shift in the profile of a given residue indicates that on average it has more solvent exposure after being adsorbed compared to when it is in solution, whereas a negative shift in its profile indicates that, on average, it has lower solvent exposure in its adsorbed state compared to that in solution.
| 2 |
Thus, the expected range of Iads/Isoln values is from 0.1 to 10 (using 0.1 and 1.0 as the minimum and maximum intensity values, respectively).24 If the extent of labeling within the adsorbed and native states of the protein was similar, then the Iads/Isoln value would be close to 1.0. Modification of residues within the solution provided a measure of the variability in Isoln of about 0.25 (95% confidence interval (CI) about the mean). Since reacting conditions between different modifications in the adsorbed and solution states were kept identical, similar variability in labeling was also expected in the adsorbed state of the protein. Therefore, we considered Iads/Isoln values beyond the range of 0.75 (i.e., 1 – CI) to 1.25 (i.e., 1 + CI) as representing a significant change in solvent accessibility. However, among the residues showing significant change in solvent accessibility, residues with Iads/Isoln > 5.0 and < 0.2 indicate a 5-fold shift in their state of solvation, with a corresponding log-ratio p-value36 < 0.0001.36,37 These metrics are listed in Table 1.
Table 1. Metrics To Determine the Configuration of an Adsorbed Protein Based on Its Labeling Profilea.
| Iads/Isoln | profile = log[Iads/Isoln] | solvent exposure of residues | physical meaning |
|---|---|---|---|
| ≥5.0 | ≥0.70 | more than the native state | accessibility increased by tertiary unfolding |
| 1.25–5.0 | 0.10 to 0.70 | ||
| 0.75–1.25 | –0.12 to 0.10 | similar to the native state | native structure |
| 0.20–0.75 | –0.12 to–0.70 | less than the native state | accessibility decreased by surface or protein–protein effects |
| ≤0.20 | ≤−0.70 |
Iads/Isoln values between 0.75 and 1.25 are considered not to be significantly different than that of the native solution-state structure. Iads/Isoln values that are 5× higher or lower than the native solution-state condition are designated as undergoing a high level of change.
The labeling agents for the reacting conditions used in the current study did not significantly affect the secondary structure of RNase A in either the solution or the adsorbed state, as determined by CD (see section section S.2.c in the Supporting Information). Thus, the profile shifts can be considered to be solely mediated by the shift in solvent exposure of the residue as a result of the adsorption process. The resulting profile values for the combined set of targeted amino acids for RNase A were determined accordingly and mapped onto the native structure of the protein for visualization, which was represented by the Protein Data Bank’s38 tertiary structure model of RNase A, 6RSA,39 with UCSF Chimera used as the visualization software.
II.e. Characterization of Enzymatic Activity
A spectrophotometric assay was used to measure the enzymatic activity of RNase A to complement the CD and AAL/MS data. Taken together, these combined methods enable correlations to be examined between adsorbed orientation and conformation with adsorption-induced changes in RNase A’s enzymatic activity. These enzymatic activity studies were also carried out in CD cuvettes.19 Briefly, ribonucleic acid, which is the substrate for RNase A, was prepared in PPB to a final concentration of 20 mg/mL (Baker’s yeast, Sigma R6750) and exposed to RNase A both in solution and following RNase A adsorption. An initial calibration plot for solution-state enzymatic activity was obtained for a working range of 0.1–30 μg of RNAse A (based on the equivalently adsorbed amount of protein on different surfaces) by monitoring the absorbance at 300 nm (ΔA300) at pH 7.4. A time period of 10 min was found to be sufficient for complete catalysis. The amount of adsorbed protein was quantified by the layer’s absorbance at 205 nm (A205), both before and after the bioactivity assays were performed to ensure that they did not cause a measurable amount of the protein to be desorbed from the adsorbent surface. The specific activities of the adsorbed proteins were then calculated by normalizing the ΔA300 absorbance values by the total amount of protein adsorbed on the surface (Qads × area of adsorbent surface). The relative enzymatic activities (%) of the adsorbed RNase A enzymes were then determined by normalizing the measured adsorbed-state-specific activity by the solution-state-specific activity.
II.f. Statistical Analysis
The mean and 95% confidence interval (CI) for each measurement were calculated for each set of experimental data collected. Statistical differences were determined using a one-tailed Student’s t test, with values of p ≤ 0.05 considered to be statistically significant.
III. Results and Discussion
III.a. Surface Characterization
Table 2 presents the results analyzed by the characterization techniques applied to the surfaces used in this study. All of the measured values reported in Table 2 fall within the expected range.
Table 2. Surface Characterizationa.
| surface | C (%) | S (%) | N (%) | O (%) | roughness (nm) | contact angle (deg) | thickness (nm) |
|---|---|---|---|---|---|---|---|
| glassc | 25.4 (2.3) | b | 0.6 (0.5) | 49.2 (2.2) | <10.0 | 23 (4) | NA |
| PMMA | 75.6 (1.3) | b | b | 23.7 (1.4) | <1.5 | 63 (3) | 90 (10) |
| HDPE | 96.3 (2.7) | b | b | 3.4 (2.6) | <8.0 | 97 (5) | 100 (10) |
Surface composition, static contact angle, film thickness, and surface roughness analyses for each surface. Mean (±95% CI); n = 3.
Indicates a negligible value.
Fused glass slide also contains Zn (0.7 ± 0.3%), Al (0.9 ± 0.4%), and Si (22.0 ± 1.0%). The presence of extra carbon composition is believed to be originating from surface contamination due to the exposure of samples to air after the cleaning procedure. These are the typical adventitious and unavoidable hydrocarbon impurities that adsorb spontaneously from ambient air onto the glass surfaces;40 NA refers to the thickness of the custom cut glass described in the Materials and Methods.
III.b. Role of Adsorbed Configuration of RNase A on the Enzymatic Activity
Unlike many proteins, RNase A is a very hydrophilic molecule. It is composed of more than 70% polar and charged amino acids and therefore it was expected to interact more strongly with the glass and PMMA than the HDPE surface via hydrogen bonding and/or electrostatic effects. The influence of adsorption conditions on the secondary structure, surface coverage, average distance between protein, and relative enzymatic activity of the adsorbed RNase A on each of our three surfaces for each solution concentration are presented in Table 3.
Table 3. Secondary Structure Content (%), Surface Coverage, Avg. Distance Between Proteins, and Relative Enzymatic Activity (%) for Adsorbed RNase A from Two Different Protein Solution Concentrations (0.03 and 1.00 mg/mL) on (a) Glass, (b) HDPE, and (c) PMMA (n = 3; Average ± 95% CI Values)a.
| surface | solution conc. (mg/mL) | surface coverage (μg/cm2) | avg. distance between proteins (nm)b | helices (%) | sheets (%) | relative enzymatic activity (%) |
|---|---|---|---|---|---|---|
| glass | 0.03 | 0.08 ± 0.01 | 5.8 | 5 ± 2 | 52 ± 8 | 38 ± 8 |
| 1.00 | 0.16 ± 0.03 | 4.1 | 19 ± 4 | 26 ± 5 | 39 ± 9 | |
| HDPE | 0.03 | 0.10 ± 0.01 | 5.2 | 18 ± 2 | 25 ± 3 | 43 ± 6 |
| 1.00 | 0.17 ± 0.03 | 4.0 | 9 ± 2 | 29 ± 5 | 35 ± 8 | |
| PMMA | 0.03 | 0.08 ± 0.02 | 5.8 | 8 ± 2 | 31 ± 3 | 33 ± 5 |
| 1.00 | 0.16 ± 0.03 | 4.1 | 18 ± 3 | 24 ± 4 | 45 ± 9 |
For comparison, the helical and β-sheet content of RNaseA in solution were found to be 20% (±3%) and 42% (±4%), respectively. The theoretical full surface coverage of RNase A for adsorption in side-on and end-on orientations is τside (0.21 μg/cm2) and τend (0.28 μg/cm2), respectively.41.
Average distance between the centers of adsorbed RNase A assuming monolayer coverage with the enzymes arranged in an evenly spaced hexagonal array.42,43 For comparison sake, per the Protein Data Bank (PDB) structure of RNase A (PDB ID: 6RSA(39)), the long and short axis dimensions of RNase A are approximately 4.2 and 2.8 nm, respectively.
III.b.1.a. Role of Surface Coverage and Surface Chemistry on the Secondary Structural Content of Adsorbed RNase A
As shown in Table 3, adsorption of RNase A to each surface for 2 h of exposure in the protein solutions followed by 15 h of equilibration in buffer resulted in a significant shift in its secondary structure for each surface and for each solution concentration. These results reflect combined influences of protein–surface interactions, protein–protein interactions (PPI), and internal protein stability effects.19 These time frames (i.e., 2 h adsorption, 15 h relaxation) were selected to represent equilibrated conditions where the amount and structure of the adsorbed protein were found to stabilize and undergo no further noticeable changes (see S.2.a and S.2.b in the Supporting Information). In addition, control studies were conducted to measure the secondary structure of RNase A in solution over time frames of at least 24 h and showed no significant change in either the secondary structure or enzymatic activity during this time, thus supporting that the changes in the structure of RNase A on the materials surfaces were due to interactions of the protein with the surface rather than being simply an aging phenomenon of the protein itself.
When RNase A was adsorbed from a 1.00 mg/mL solution concentration followed by 15 h of equilibration under pure PPB (i.e., protein-free PPB solution), the resulting surface coverage of the adsorbed protein on each surface was within 25% of a saturated, close-packed monolayer with side-on protein orientation. In contrast, when adsorbed from a 0.03 mg/mL solution and equilibrated, the resulting surface coverage of the RNase A was about half of that obtained when it was adsorbed from the 1.00 mg/mL solution. These results show that different degrees of surface coverage for RNase A were obtained in our studies by varying the protein solution concentration from which it is adsorbed. As intended, the higher solution concentration resulted in higher surface coverage, which can subsequently be associated with a greater degree of PPI effects on the surface.
As can be seen from the data presented in Table 3, when the coverage on the surfaces was low (e.g., 0.08 μg/cm2, nearly 3× less than the closed-packed side-on arrangement of 0.21 μg/cm2), in which case the effects of PPI can be expected to be relatively low, the protein–surface interactions induced about 70% and 85% loss in the native helical content of adsorbed RNase A on the PMMA and glass surfaces, respectively. We hypothesize that these responses are indicative of the surface destabilizing the helical structures of RNase A by competing with the hydrogen bonding that stabilizes the helices of the native-state structure. However, at high surface coverages (0.16 μg/cm2, close to the close-packed side-on arrangement), where PPI effects can be expected to be substantially greater, these effects apparently tend to inhibit surface-induced unfolding and result in the native-state helical content being largely preserved with less than 1% loss in the native structure.
In contrast to the trends observed for PPI effects on the native-state helical content of RNase A, changes in the surface coverage of RNase A on the HDPE and PMMA surfaces had minimal influence on the β-sheet content of the protein, with a general decrease in β-sheet structures, ranging from 24 to 31%, with an average of about 28% β-sheet structure (or a 29% loss in the native-state percent of β-sheet). However, on the silica glass surfaces, when PPI effects were minimized, it was observed that there was a large increase in the β-sheet content of the adsorbed RNase A (i.e., 24% gain in β-sheet), suggesting that the glass surface has a particularly strong tendency to act as a planar template for the alignment of the polypeptide chain segments as the protein unfolds, presumably mediated mainly by electrostatic effects.44 However, this trend was not observed at the higher protein surface coverage on glass, with PPI effects apparently inhibiting the ability of the protein to unfold and spread out on the surface. Thus, while it is relatively easy to predict that the competing influence of hydrogen-bondable groups in glass and PMMA for the hydrogen bonds that stabilize the helical secondary structure of the protein would induce a loss in helicity, its effect on the on the β-sheet structure of RNase A is less predictable. The hydrogen-bonding groups of the surface can either compete for the hydrogen bonds that stabilize the β-sheet structure, leading to a reduction in β-sheet content, or serve as a template to form new β-sheet-like structure by attracting and aligning peptide segments along the surface. On the basis of this understanding, we consider that the change in helical structure provides a more sensitive and straightforward indicator of the degree of adsorption-induced disruption of the native-state secondary structure of a protein.
In direct contrast to the stabilizing effect of PPIs on the helical content of adsorbed RNase A on PMMA and glass, it is apparent that PPI on the hydrophobic HDPE surface had a destabilizing effect on the helical structure: adsorption to HDPE induced more than 50% loss in the native-state percent helicity at higher surface coverage (0.17 μg/cm2) compared to less than 1% loss at low surface coverage (0.10 μg/cm2). As an explanation for these interesting results, we propose that, in the absence of PPI effects, the adsorption of RNase A to HDPE results in the replacement of the hydrophobic interactions between the side chains of the amino acid residues making up the helices and the β-sheet in the native-state structure with hydrophobic interactions with the HDPE surface. This process thus could result in unfolding the tertiary structure while maintaining the stability of the helical secondary structure of the protein. We further propose that the presence of high PPI effects disrupts this process in RNaseA, leading to the separation and destabilization of the helical and β-sheet structures while inhibiting the helices from being restabilized by the hydrophobic surface. Obviously, these specific types of molecular-level interactions are speculative at this time. We hope to provide support for these hypothesized molecular-level events through molecular simulation studies in the near future.
III.b.1.b. Impact of a Loss in Secondary Structure on the Enzyme Activity of Adsorbed RNase A
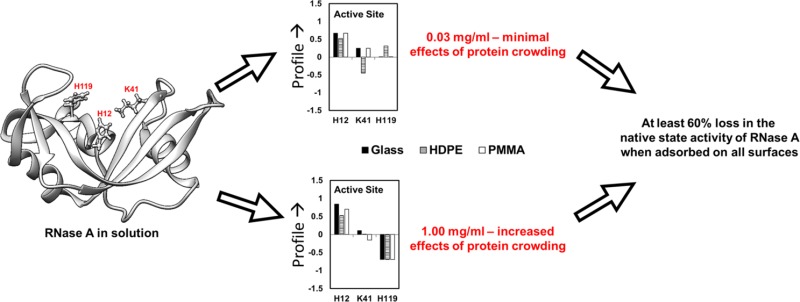
The key element in the current study is the activity of the adsorbed RNase A and the factors influencing its activity. In many studies, the extent of helical unfolding has often been associated with the loss in activity for other proteins. Additionally, at least one of the three residues involved in catalysis are within the helical conformation of the protein structure (Figure 1).45 The native-state structure of the RNase A resembles a kidney shape, with the active site residues (H12, K41, and H119) laying in the concave cleft (Figure 1).45
Figure 1.
Ribbon diagram of the three-dimensional structure of ribonuclease A.45 The three residues most important for catalysis, His12, His119, and Lys41, are marked in red.
As can be inferred from the results presented in Table 3, the loss in helix is not a clear indicator of the loss in enzymatic activity, especially since the relative enzymatic activity of RNase A was unchanged for a wide range of helical unfolding. Even when the helical content within the protein was equivalent to that of its native-state structure (i.e., when RNase A was adsorbed under minimal PPI conditions on the HDPE surface and when adsorbed under high PPI conditions on the glass and PMMA surfaces), there were no significant differences (p > 0.05) in its activity when compared to that of the unfolded states of RNase A under conditions exhibiting a large degree of unfolding. Thus, it is evident that the reduction in helical content of RNase A does not correlate well with the loss in the native-state activity. As a result, more sensitive assays that would provide molecular (or domain) level insights on the tertiary structure and orientation of the adsorbed RNase A might serve as a better indicator of how the adsorbed configuration of the protein might affect its enzymatic activity level.
III.b.2.a. AAL/MS Technique To Identify the Orientation and Tertiary Structural Shift in Adsorbed Protein
In our study, the AAL/MS technique was used to identify the areas within the protein that underwent orientation and tertiary structural shift by estimating the changes in the absolute extent of modification (%) in the adsorbed states of the protein relative to that in its solution state, or profile, using eq 2. The extent of modification (%) was assumed to be directly proportional to the solvent exposure of the target residues, as the labeling conditions used in the current study were not found to significantly affect the structure of protein structure in solution or the adsorbed state (see S.2.c. in the Supporting Information). Additionally, sequence coverage of 100% was obtained with the tryptic digests of in-solution and adsorbed RNase A. In our experiment, a total of 34 residues that were distributed throughout the protein were labeled in solution, of which H12, K41, and H119, form the catalytic site in RNase A and are utilized by these enzymes to cleave phosphodiester bonds in RNA. Residues H12 and H119 acts as an acid or a base to both accept and donate electrons, whereas K41 stabilizes the transition state of the catalytic reaction.45,46
III.b.2.b. Active Sites and Solvent Accessibility of Amino Acid Residues in the Solution Phase
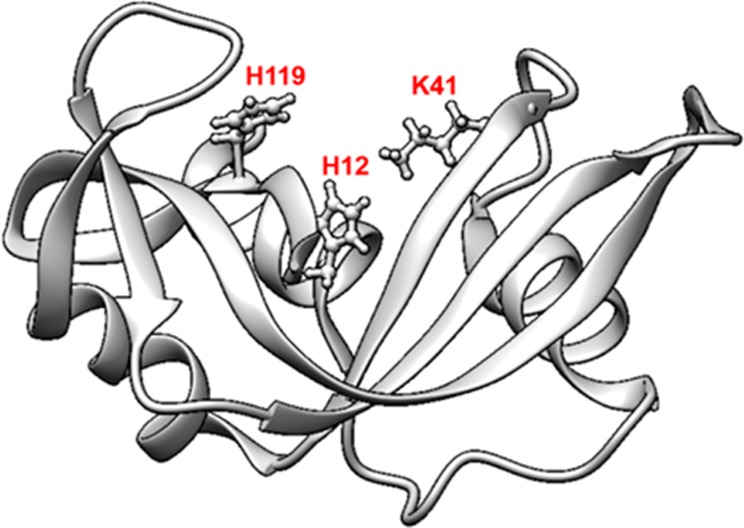
Figure 2 illustrates these effects in a space-filled model of RNase A with targeted amino acid residues color-coded by their degree of solvent exposure, as determined by the side-chain modification experiment. On the basis of these results, two of the catalytic residues (K41 and H119) were solvent-exposed, whereas the third (H12) was buried in the solution-state structure.
Figure 2.
Space-filled model of RNase A with amino acid residues color coded by their solvent accessibility, as determined from targeted amino acid labeling in solution. Color coding: charged amino acid residues (Asp, Glu, Lys, Arg, His) with high solvent accessibility (green) and moderate solvent accessibility (blue), tyrosine residues with high solvent accessibility (orange) and low solvent accessibility (black). Nontargeted amino acid residues are color coded in light gray. Figure illustrated using UCSF Chimera. The arrows point out the location of the three key amino acid residues that provide the catalytic function of the enzyme (H12, K41, H119).
On the basis of the absolute extent of modification (%) for a target residue in its solution state, Isoln, charged residues such as Arg (R10, R33, R39, and R85), Lys (K1, K7, K31, K37, K41, K61, K66, K91, K98, and K104), His (H105, H119), Asp (D10, D38, and D53), and Glu (E2, E9, E49, and E86) at physiological pH were found to be solvent-exposed. However, some of the charged residues were found to be less solvent-exposed or buried (D14, D121, D83, H12, and H48). In contrast to charged residues, most Tyr amino acids were found to be less solvent-exposed or buried inside the protein structure (Y25, Y73, Y97, and Y115), whereas Y92 and Y76, which are located on the outer surface of the protein, were found to be solvent-exposed (see section S.2.e in the Supporting Information for raw data).
III.b.2.c. Active Sites and Solvent Accessibility of Amino Acid Residues in the Adsorbed Phase
The resulting profiles for each of the targeted amino acids were determined and are shown in Figures 3 and 4 for each of the three different surfaces when adsorbed from 0.03 and 1.00 mg/mL protein solutions, respectively. The data presented in Figure 3 (0.03 mg/mL results) were separated according to the classification shown in Table 1 (i.e., surface type and solution concentration), and the resulting residues belonging to each group are presented in Table 4. Similarly, Figure 4 presents the profile values for RNase A adsorbed from 1.0 mg/mL solution, with the division of residues according to Table 1 categories presented in Table 5.
Figure 3.

Labeling profile of the targeted residues in RNase A on glass, PMMA, and HDPE surfaces when adsorbed from 0.03 mg/mL protein solution. The residues within the active site of RNase A are shown separately in the right-hand plot to more clearly show their response. The profiles within about ±0.1 of zero are not significantly different from those in the solution state (n = 3). Residues showing no difference in their solvation between the solution and adsorbed states have profile values equal to 0 (e.g., Y115 for all three surfaces).
Figure 4.

Labeling profile of the targeted residues in RNase A on glass, PMMA, and HDPE surfaces when adsorbed from 1.00 mg/mL protein solutions. The residues within the active site of RNase A are shown separately in the right-hand plot to more clearly show their response. Profiles within about ±0.1 of zero are not significantly different from those in the solution state (n = 3). Residues showing no difference in their solvation between the solution and adsorbed states have profile values equal to 0 (e.g., R85 for the glass and HDPE surfaces).
Table 4. Labeling Profile of RNase A on Each Surface When Adsorbed from 0.03 mg/mL Solutiona.
| surface | profile ≤ −0.72 | –0.72 < profile <−0.12 | –0.12 ≤ profile ≤ 0.1 | 0.1 < profile < 0.7 | profile ≥ 0.7 |
|---|---|---|---|---|---|
| glass | K1, R39, R85, E111 | K7, R10, K31, D38, D53, K98 | E9, R33, K37, E49, K61, Y76, K91, Y115, H119, D121 | E2, H12, D14, K41, K66, D83, E86, Y92, K104, H105 | Y25, H48, Y73, Y97 |
| HDPE | D53, K61, K66, Y76, K91, K98 | K104, R39, K41, E49, Y73, Y92 | K1, E9, D38, R85, Y115 | E2, K7, R10, H12, H15, Y25, K31, R33, K37, D83, E86, Y97, E111, H119 | D14, H48, D121 |
| PMMA | E111 | D38, R39, K66, Y76, K91, Y92, K98 | K1, E9, R10, D53, K61, R85, Y97, Y115, H119, D121 | E2, K7, H12, D14, Y25, K31, R33, K37, K41, E49, Y73, D83, E86, K104, H105 | H48 |
His12, Lys41, and His119 are the main catalytic residues.
Table 5. Labeling Profile of RNase A on Each Surface When Adsorbed from 1.00 mg/mL Solutiona.
| surface | profile ≤ −0.72 | –0.72 < profile < −0.12 | –0.12 ≤ profile ≤ 0.1 | 0.1 < profile < 0.7 | profile ≥ 0.7 |
|---|---|---|---|---|---|
| glass | K1, K7, E9, D38, K61, K66, Y76, K91,K104, H105,H119 | K31, R39, K41, D53, Y73, R85, E86 | E2, R10, D14, R33, K37, H48, E49, D83, Y92, K98, E111, Y115 | H12, Y25, Y97, D121 | |
| HDPE | K1, K7, K31, K61, K66, K91, K98 | K37, D38, Y76, K104, H105, H119 | R39, K41, D53, R85, Y92, E111 | E2, E9, R10, H12, Y25, R33, E49, Y73, D83, E86 | D14, H48, Y97, Y115, D121 |
| PMMA | K1, K66, K31, R85, K7 | R33, K37, D38, R39, K41, D53, K61, Y76, K91, Y92, E111, H105, H119 | Y25, Y73, K98, K104, Y115 | E2, E9, R10, E49, D83, E86, Y97, D121 | H12, D14, H48 |
His12, Lys41, and His119 are the main catalytic residues.
As can be seen from Figure 3 and Figure 4, the labeling profiles reveal substantial differences in residue solvent accessibility among each surface when adsorbed in both 0.03 and 1.00 mg/mL solutions. These data also reflect the combined influences of protein–surface interactions, PPI, and internal protein stability effects upon the adsorption configuration of adsorbed RNase A. As noted in Table 1, a positive profile value for a designated amino acid is indicative of an adsorption-induced increase in the solvent accessibility of its side group, which implies that, on average, a tertiary unfolding event has taken place in that location of the protein. In contrast, a negative shift in the profile indicates that adsorption has reduced solvent accessibility of the designated amino acid’s side chain, which implies that this part of the protein has been sterically blocked by either the surface (i.e., orientation effect) or a neighboring protein (i.e., protein–protein effect).33,47 In order to provide a graphical understanding of the locations of the amino acid residues in RNase A that underwent adsorption-induced changes in their solvated state, Figures 5–7 present images of the native-state structure of RNase A with the residues color coded by their respective profile values from Tables 4 and 5.
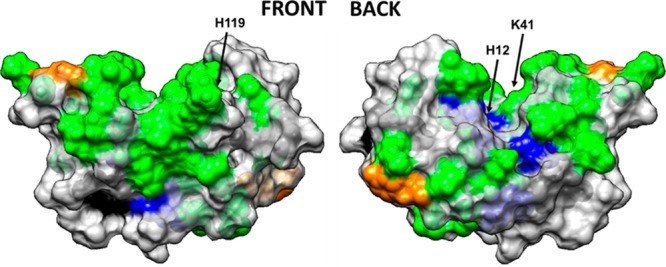
Figure 5.
Solvation profile of residues in RNase A adsorbed from (A) 0.03 and (B) 1.00 mg/mL on the glass surface. Residue color code: yellow (− −), orange (−), dark gray (native state), green (+), blue (++), and light gray (nontargeted). The arrows point to the location of the three key amino acid residues that provide the catalytic function of the enzyme (H12, K41, and H119).
Figure 7.
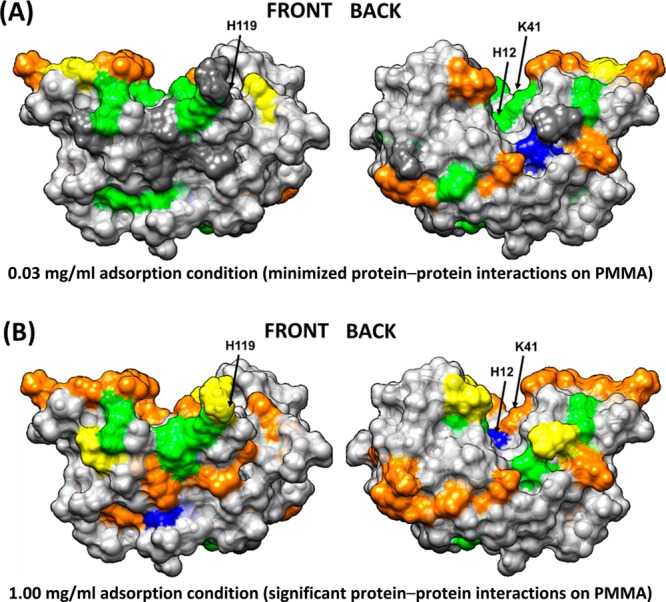
Solvation profiles of residues in RNase A adsorbed in (A) 0.03 and (B) 1.00 mg/mL on the PMMA surface. Residue color code: yellow (− −), orange (−), dark gray (native state), green (+), blue (++), and light gray (nontargeted). The arrows point to the location of the three key amino acid residues that provide the catalytic function of the enzyme (H12, K41, and H119).
The data presented in Tables 4 and 5 for the amino acids with negative profiles (i.e., loss in solvent accessibility) are visually depicted as yellow and orange in Figures 5–7 on the protein’s native-state structure. The loss in solvent accessibility of the amino acid residues displaying a negative profile can be caused by close contact with either the adsorbent surface or neighboring adsorbed proteins (i.e., from PPI effects). The regions of the RNase A that underwent a high degree of adsorption-induced tertiary unfolding, as evidenced by increased solvent accessibility following adsorption, are green and blue in Figures 5–7.
III.b.2.d. Orientation and Configuration of Adsorbed RNase A
As is evident from Figures 3 and 5a and Table 4, when adsorbed from the low solution concentration (i.e., low PPI effects), the adsorbed RNase A predominantly interacts with glass along the positively charged patch of amino acid residues, as shown by the residues with greatly reduced solvent accessibility in Table 4. These interactions are likely due to the electrostatic interactions between the negatively charged glass surface (determined by streaming potential technique) and the protein. However, when adsorbed from high protein solution concentration (Figures 4 and 5b and Table 5), the resulting increased PPI effects appear to have interfered with the ability of electrostatic interactions to orient RNase A on the surface, with the negative profiles being not as strong and shifted toward different positions that are more evenly distributed around the protein’s surface, presumably due to closer contact with neighboring proteins and different orientations adopted by the protein when approaching surface saturation.
The strongly hydrophobic HDPE surface, which does not have hydrogen-bonding capability, has the potential to primarily interact with the hydrophobic side-chain functional groups of amino acids within RNase A. As shown in Figure 6A, when adsorbed from 0.03 mg/mL with minimal PPI effects, the amino acids showing the lowest solvent accessibility are all positioned along what we refer to as the back surface of the protein, with no apparent areas of lost solvent accessibility on the front surface (Figure 3 and Table 4). These results provide evidence that RNase A adsorbs on HDPE with its back face oriented toward the surface. When adsorbed from 1.00 mg/mL (Figure 6B), with higher PPI effects, a similar loss in solvent accessibility is indicated on the back surface, as with the low PPI effect case, but with a few additional areas on the front surface showing substantial loss in solvent accessibility due to either altered orientation or blockage from neighboring proteins (Figure 4 and Table 5).
Figure 6.
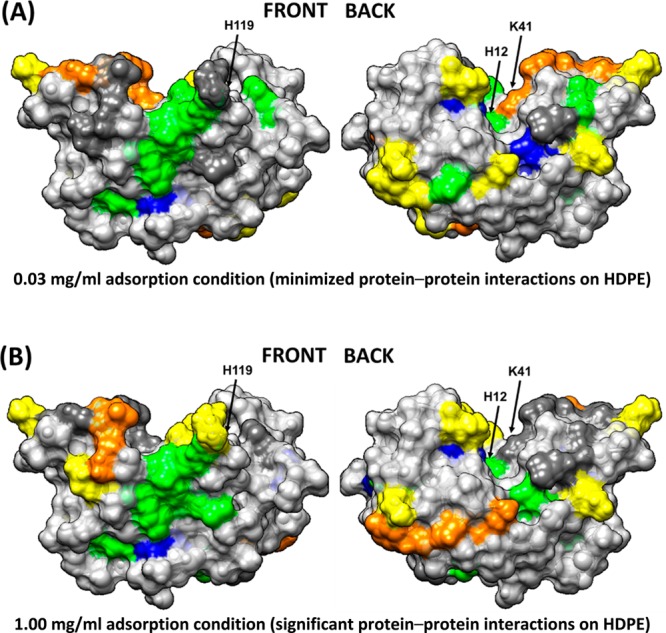
Solvation profile of residues in RNase A adsorbed in (A) 0.03 and (B) 1.00 mg/mL on the HDPE surface. Residue color code: yellow (− −), orange (−), dark gray (native state), green (+), blue (++), and light gray (nontargeted). The arrows point to the location of the three key amino acid residues that provide the catalytic function of the enzyme (H12, K41, and H119).
Comparing the results of a RNase A adsorbed on the neutral and moderately hydrophobic PMMA surface from 0.03 mg/mL, Figure 7A shows areas of loss in solvation along the back surface very similar to that on the HDPE surface but with a lower degree of solvent accessibility loss compared to that on the HDPE surface (i.e., orange instead of yellow color coding in Figure 7A), which we assume to reflect the weaker degree of hydrophobic interactions on PMMA (Figure 3 and Table 4). Adsorption to PMMA under 1.00 mg/mL conditions, with a greater degree of PPI effects, showed a loss in solvent accessibility similar to the 0.03 mg/mL condition but with a few additional areas of loss in solvent accessibility, due to either altered adsorbed orientation or blocking by neighboring adsorbed RNase A (Figure 4 and Table 5).
III.b.2.e. Tertiary Unfolding of an Adsorbed RNase A
As shown in Figures 5–7, there are many similarities in the amino acid residues of RNase A that underwent increased solvent exposure, indicative of tertiary unfolding on each of our three model surfaces, with relatively minor differences indicated between these three surfaces (Figures 3–7 and Tables 4 and 5). We interpret these results to reflect regions in the RNase A structure that are less stable and prone to adsorption-induced unfolding. One point that is particularly relevant is that RNase A shows a substantial degree of increased solvent accessibility deep within its bioactive cleft for each surface and solution concentration, as indicated by the solvent exposure of H12, which is not solvent accessible in solution (see Tables 4 and 5 and Figures 2 and 5–7). These results suggest the disturbance of the structure of its binding site. This similarity may be responsible for the nearly equivalent loss in enzymatic activity that we measured for each adsorbed condition, which is addressed in the following section.
III.c. Molecular Mechanism Underlying the Enzymatic Activity of Adsorbed RNase A
The adsorption processes not only altered RNase A’s native-state structure but also substantially reduced its enzymatic activity. As shown in Table 3, RNase A lost at least 60% of its solution-state activity on each of our three surfaces, which represent a broad range of surface chemistries, with no significant difference in the loss of activity for any of the applied adsorption conditions. The one common feature shown in Figures 5–7 for each of the adsorption conditions, which may explain these results, is a substantial increase in solvent accessibility of the H12 residue that is buried deep within the bioactive site pocket of RNase A, thus indicating that adsorption caused a substantial degree of tertiary unfolding to occur in this region of the protein. As shown in Figure 1, H12 is one of the three key residues responsible for this enzyme’s catalytic function. On the basis of these results, we propose that RNase A is susceptible to adsorption-induced unfolding of its binding site when adsorbed to a broad range of surface chemistries and that this unfolding behavior causes substantial loss in its enzymatic activity.
The presented studies investigated the influence of adsorption on the structure and enzymatic activity of RNase A and are thus primarily relevant to protein-based drug delivery systems where proteins are adsorbed onto or within some sort of larger carrier particles for the purpose of delivering higher payloads of a protein to a target. In such approaches, the outer surface of the carrier particles are often tethered with stealth molecules, such as poly(ethylene glycol) (PEG), to enhance their residence time in the bloodstream. Targeting molecules such as antibodies are then also typically linked to the drug delivery particles to selectively bind and concentrate the drug-bearing particles to their intended delivery site. As shown from the presented fundamental studies, adsorption of an enzyme to a material support can lead to a reduction in the enzyme’s activity by either structural unfolding or steric blocking of the enzyme’s active site. Other simpler approaches for the delivery of protein-based pharmaceutical agents have been taken such as the direct PEGylation of otherwise free enzymes to slow their clearance from the blood through mechanisms such as inhibiting their detection by phagocytic cells. However, this strategy can also reduce enzymatic activity by sterically blocking the binding of its intended substrate or receptor.12,29,48 The key element in either of these strategies is to design the enzyme delivery system in such a manner as to preserve its activity so that it can perform its intended function once it is delivered to its intended target. This process requires a residue-level understanding of both the solvent accessibility and structural integrity of the enzyme’s active site along with the development and application of methods to make these types of assessments, such as the presented method of AAL/MS.
IV. Conclusions
RNase A is known to be structurally robust in solution. However, our results demonstrate that it undergoes dramatic changes in both its structure and enzymatic activity during adsorption on biomaterial surfaces with a broad range of surface chemistries and solution conditions. Using a complementary array of experimental techniques, which included CD and AAL/MS, we quantitatively demonstrated that the orientation and adsorption-induced changes in the secondary and tertiary structures of adsorbed RNase A are unique for each surface type and degree of PPI effects occurring in the adsorbed layer of protein. However, the effect of adsorption on the enzymatic activity of RNase A was not significantly different for any of the applied conditions, with about a 60% loss in enzymatic activity occurring irrespective of the type of adsorbent surface or degree of protein–protein interactions on the surface. Our results indicate that the similar loss in enzymatic activity observed with RNase A, despite undergoing varying extent of structural unfolding, is most likely due to the localized structural unfolding of the catalytic site. Therefore, drug delivery systems must focus on retaining the native structure of RNase A’s catalytic site in order to maintain a high level of enzymatic activity for applications such as antitumor chemotherapy.
Acknowledgments
This project received support from the Defense Threat Reduction Agency, Joint Science and Technology Office for Chemical and Biological Defense (grant no. HDTRA1-10-1-0028). The facilities used in this research were also supported by NIH grant nos. 5P20RR021949 and 8P20GM103444. We also would like to thank Megan Grobman, Dr. Lara Gamble, and Dr. David Castner of NESAC/BIO at the University of Washington for assistance with surface characterization with XPS under the funding support by NIBIB (grant no. EB002027).
Supporting Information Available
(i) Experimental methodology and (ii) results for the different techniques used in the current study to determine the surface coverage, adsorbed configuration, and bioactivity of adsorbed protein. The experimental methodology contains descriptions of the adsorption procedure, XPS technique, ellipsometry technique, CD spectroscopy, and AAL/MS technique. The Results and Discussion section contains descriptions of the (a) surface coverage and (b) helix content of the protein when adsorbed from different solution concentrations and surface chemistries, (c) the effect of labeling on the structure of the protein in solution and in the adsorbed state, (d) surface coverage following trypsin treatment, and (e) raw data on the extent of modification in solution and profile of the target residues in adsorbed RNase A. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Rybak S. M.; Newton D. L. Natural and engineered cytotoxic ribonucleases: therapeutic potential. Exp. Cell Res. 1999, 253, 325–335. [DOI] [PubMed] [Google Scholar]
- Arnold U.; Ulbrich-Hofmann R. Natural and engineered ribonucleases as potential cancer therapeutics. Biotechnol. Lett. 2006, 28, 1615–1622. [DOI] [PubMed] [Google Scholar]
- Ilinskaya O. N.; Makarov A. A. Why ribonucleases induce tumor cell death. Mol. Biol. 2005, 39, 1–10. [Google Scholar]
- Leland P. A.; Raines R. T. Cancer chemotherapy—ribonucleases to the rescue. Chem. Biol. 2001, 8, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klink T. A.; Raines R. T. Conformational stability is a determinant of ribonuclease A cytotoxicity. J. Biol. Chem. 2000, 275, 17463–17467. [DOI] [PubMed] [Google Scholar]
- Poklar N.; Petrovčič N.; Oblak M.; Vesnaver G. Thermodynamic stability of ribonuclease A in alkylurea solutions and preferential solvation changes accompanying its thermal denaturation: a calorimetric and spectroscopic study. Protein Sci. 1999, 8, 832–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend M. W.; Deluca P. P. Stability of ribonuclease A in solution and the freeze-dried state. J. Pharm. Sci. 1990, 79, 1083–1086. [DOI] [PubMed] [Google Scholar]
- Klink T. A.; Woycechowsky K. J.; Taylor K. M.; Raines R. T. Contribution of disulfide bonds to the conformational stability and catalytic activity of ribonuclease A. Eur. J. Biochem. 2000, 267, 566–572. [DOI] [PubMed] [Google Scholar]
- Daly S. M.; Przybycien T. M.; Tilton R. D. Adsorption of poly(ethylene glycol)-modified ribonuclease A to a poly(lactide-co-glycolide) surface. Biotechnol. Bioeng. 2005, 90, 856–868. [DOI] [PubMed] [Google Scholar]
- Li C.; Lin Q.; Wang J.; Shen L.; Ma G.; Su Z.; Hu T. Preparation, structural analysis and bioactivity of ribonuclease A–albumin conjugate: tetra-conjugation or PEG as the linker. J. Biotechnol. 2012, 162, 283–288. [DOI] [PubMed] [Google Scholar]
- Michaelis M.; Cinatl J.; Pouckova P.; Langer K.; Kreuter J.; Matousek J. Coupling of the antitumoral enzyme bovine seminal ribonuclease to polyethylene glycol chains increases its systemic efficacy in mice. Anticancer Drugs 2002, 13, 149–54. [DOI] [PubMed] [Google Scholar]
- Mathé C.; Devineau S.; Aude J.-C.; Lagniel G.; Chédin S.; Legros V.; Mathon M.-H.; Renault J.-P.; Pin S.; Boulard Y.; Labarre J. Structural determinants for protein adsorption/non-adsorption to silica surface. PLoS One 2013, 8, e81346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai S. S.; Hammouda B.; Hong K.; Pozzo D. C.; Przybycien T. M.; Tilton R. D. The conformation of the Poly(ethylene glycol) chain in mono-PEGylated lysozyme and mono-PEGylated human growth hormone. Bioconjugate Chem. 2011, 22, 2317–2323. [DOI] [PubMed] [Google Scholar]
- Lee C. S.; Belfort G. Changing activity of ribonuclease A during adsorption: a molecular explanation. Proc. Natl. Acad. Sci. U.S.A. 1989, 86, 8392–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi C.; Fong C. C.; Zhang Q.; Lee S. T.; Yang M. The structure and function of ribonuclease A upon interacting with carbon nanotubes. Nanotechnology 2008, 19, 0957–4484. [DOI] [PubMed] [Google Scholar]
- Smith B. D.; Soellner M. B.; Raines R. T. Synthetic surfaces for ribonuclease adsorption. Langmuir 2005, 21, 187–90. [DOI] [PubMed] [Google Scholar]
- Kastantin M.; Langdon B. B.; Schwartz D. K. A bottom-up approach to understanding protein layer formation at solid–liquid interfaces. Adv. Colloid Interface Sci. 2014, 207, 240–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W. K.; McGuire J.; Bothwell M. K. Concentration effects on adsorption of bacteriophage T4 lysozyme stability variants to silica. J. Colloid Interface Sci. 2002, 252, 473–6. [DOI] [PubMed] [Google Scholar]
- Wei Y.; Thyparambil A. A.; Latour R. A. Quantification of the influence of protein–protein interactions on adsorbed protein structure and bioactivity. Colloids Surf., B 2013, 110, 363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaraman B.; Latour R. A. The relationship between platelet adhesion on surfaces and the structure versus the amount of adsorbed fibrinogen. Biomaterials 2010, 31, 832–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Somorjai G. A. Molecular packing of lysozyme, fibrinogen, and bovine serum albumin on hydrophilic and hydrophobic surfaces studied by infrared-visible sum frequency generation and fluorescence microscopy. J. Am. Chem. Soc. 2003, 125, 3150–3158. [DOI] [PubMed] [Google Scholar]
- Hu Y.-J.; Ou-Yang Y.; Dai C.-M.; Liu Y.; Xiao X.-H. Site-selective binding of human serum albumin by palmatine: spectroscopic approach. Biomacromolecules 2009, 11, 106–112. [DOI] [PubMed] [Google Scholar]
- Rabe M.; Verdes D.; Seeger S. Understanding protein adsorption phenomena at solid surfaces. Adv. Colloid Interface Sci. 2011, 162, 87–106. [DOI] [PubMed] [Google Scholar]
- Thyparambil A. A.; Wei Y.; Wu Y.; Latour R. A. Determination of orientation and adsorption-induced changes in the tertiary structure of proteins on material surfaces by chemical modification and peptide mapping. Acta Biomater. 2014, 10, 2404–2414. [DOI] [PubMed] [Google Scholar]
- Engel M. F. M.; Visser A.; van Mierlo C. P. M. Conformation and orientation of a protein folding intermediate trapped by adsorption. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 11316–11321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J. S.; Bowden E. F. Determination of the orientation of adsorbed cytochrome c on carboxyalkanethiol self-assembled monolayers by in situ differential modification. J. Am. Chem. Soc. 2006, 128, 6813–6822. [DOI] [PubMed] [Google Scholar]
- Ovod V.; Scott E. A.; Flake M. M.; Parker S. R.; Bateman R. J.; Elbert D. L. Exposure of the lysine in the gamma chain dodecapeptide of human fibrinogen is not enhanced by adsorption to poly(ethylene terephthalate) as measured by biotinylation and mass spectrometry. J. Biomed. Mater. Res., Part A 2012, 100A, 622–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Q.; Hu T.; Yu J. Molecular insight into the steric shielding effect of PEG on the conjugated staphylokinase: biochemical characterization and molecular dynamics simulation. PLoS One 2013, 8, e68559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaraman B.; Fears K. P.; Latour R. A. Investigation of the effects of surface chemistry and solution concentration on the conformation of adsorbed proteins using an improved circular dichroism method. Langmuir 2009, 25, 3050–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner A.; Amon S.; Rizzi A.; Lindner W. Use of the arginine-specific butane dione/phenylboronic acid tag for analysis of peptides and protein digests using matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 1321–1330. [DOI] [PubMed] [Google Scholar]
- Suckau D.; Mak M.; Przybylski M. Protein surface topology-probing by selective chemical modification and mass-spectrometric peptide-mapping. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 5630–5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fears K. P.; Sivaraman B.; Powell G. L.; Wu Y.; Latour R. A. Probing the conformation and orientation of adsorbed enzymes using side-chain modification. Langmuir 2009, 25, 9319–9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J. The beginning of a beautiful friendship: cross-linking/mass spectrometry and modelling of proteins and multi-protein complexes. J. Struct. Biol. 2011, 173, 530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza V. L.; Vachet R. W. Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom Rev. 2009, 28, 785–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman D. G.; Bland J. M. How to obtain the confidence interval from a P value. BMJ 2011, 343, d2090. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Hsiao T.-H.; Suresh U.; Chen H.-I.; Wu X.; Wolf S. E.; Chen Y. A novel significance score for gene selection and ranking. Bioinformatics 2012, 30, 801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borah B.; Chen C. W.; Egan W.; Miller M.; Wlodawer A.; Cohen J. S. Nuclear magnetic resonance and neutron diffraction studies of the complex of ribonuclease A with uridine vanadate, a transition-state analog. Biochemistry 1985, 24, 2058–2067. [DOI] [PubMed] [Google Scholar]
- Serra J.; González P.; Liste S.; Serra C.; Chiussi S.; León B.; Pérez-Amor M.; Ylänen H. O.; Hupa M. FTIR and XPS studies of bioactive silica based glasses. J. Non-Cryst. Solids 2003, 332, 20–27. [Google Scholar]
- Anand G.; Sharma S.; Dutta A. K.; Kumar S. K.; Belfort G. Conformational transitions of adsorbed proteins on surfaces of varying polarity. Langmuir 2010, 26, 10803–10811. [DOI] [PubMed] [Google Scholar]
- Ostuni E.; Grzybowski B. A.; Mrksich M.; Roberts C. S.; Whitesides G. M. Adsorption of proteins to hydrophobic sites on mixed self-assembled monolayers. Langmuir 2003, 19, 1861–1872. [Google Scholar]
- Konradi R.; Textor M.; Reimhult E. Using complementary acoustic and optical techniques for quantitative monitoring of biomolecular adsorption at interfaces. Biosensors 2012, 2, 341–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fears K. P.; Latour R. A. Assessing the influence of adsorbed-state conformation on the bioactivity of adsorbed enzyme layers. Langmuir 2009, 25, 13926–13933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raines R. T. Ribonuclease A. Chem. Rev. 1998, 98, 1045–1066. [DOI] [PubMed] [Google Scholar]
- Chatani E.; Hayashi R. Functional and structural roles of constituent amino acid residues of bovine pancreatic ribonuclease A. J. Biosci. Bioeng. 2001, 92, 98–107. [DOI] [PubMed] [Google Scholar]
- Lins L.; Thomas A.; Brasseur R. Analysis of accessible surface of residues in proteins. Protein Sci. 2003, 12, 1406–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X.; Li D.; Yu J.; Ma G.; Su Z.; Hu T. Phenyl linker-induced dense PEG conformation improves the efficacy of C-terminally monoPEGylated staphylokinase. Biomacromolecules 2013, 14, 331–341. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.