

Conspectus
Lipoxygenase enzymes insert oxygen in a polyunsaturated lipid, yielding a hydroperoxide product. When the acyl chain is arachidonate, with three cis-pentadiene units, 12 positionally and stereochemically different products might result. The plant lipids, linoleate and linolenate, have, respectively, four and eight potential oxygen insertion sites. The puzzle of how specificity is achieved in these reactions grows as more and more protein structures confirm the conservation of a lipoxygenase protein fold in plants, animals, and bacteria. Lipoxygenases are large enough (60–100 kDa) that they provide a protein shell completely surrounding an active site cavity that has the shape of a long acyl chain and contains a catalytic metal (usually iron).
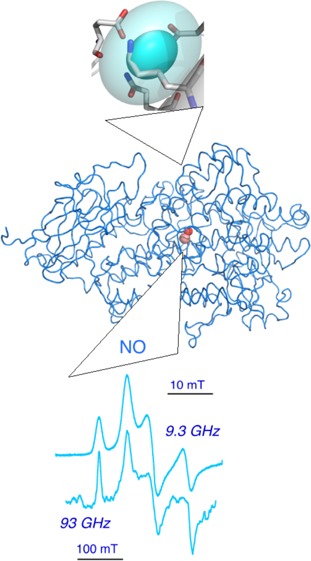
This Account summarizes electron paramagnetic resonance (EPR) spectroscopic, and other, experiments designed to bridge the gap between lipid–lipoxygenase interactions in solution and crystal structures. Experiments with spin-labeled lipids give a picture of bound lipids tethered to protein by an acyl chain, but with a polar end emerging from the cavity to solvent exposure, where the headgroup is highly flexible. The location of a spin on the polar end of a lysolecithin was determined by pulsed, dipolar EPR measurements, by representing the protein structure as a five-point grid of spin-labels with coordinates derived from 10 distance determinations between spin pairs. Distances from the lipid spin to each grid site completed a six-point representation of the enzyme with a bound lipid. Insight into the dynamics that allow substrate/product to enter/exit the cavity was obtained with a different set of spin-labeled protein mutants. Once substrate enters the cavity, the rate-limiting step of catalysis involves redox cycling at the metal center. Here, a mononuclear iron cycles between ferric and ferrous (high-spin) forms. Two helices provide pairs of side-chain ligands to the iron, resulting in characteristic EPR signals. Quantitative comparison of EPR spectra of plant and bacterial lipoxygenases has suggested conservation of a unique geometry of lipoxygenase iron centers. High frequency (94 GHz) EPR is consistent with a similar metal center in a manganese version of lipoxygenase. Overall, established and emerging EPR experiments have been developed and applied to the lipoxygenase family of enzymes to elucidate changes in the solution structures that are related to function.
1. Introduction
The best-characterized group of lipoxygenases is the isoform set in soybeans. There are crystal structures of four of these.1−4 Overall, the enzymes have two domains, the larger of which is the lipoxygenase fold. This domain is mostly helical, composed of ∼20 helices and several sheets, in plant, animal and bacterial lipoxygenases (Conspectus figure). The structures of the soybean set enclose a cavity that wraps around the water (hydroxyl) ligand of a redox-active iron. In the apoenzyme, the cavity contains several well-ordered solvent molecules. Side chains of seven conserved residues line the substrate cavity in the structures of the soybean isoforms, and these are largely nonpolar (Figure 1, gray side chains).4 The cavity has two lobes; several configurations of linoleic acid can be modeled into the lobe containing catalytic iron5 (Figure 1, magenta carbon chain). The other lobe contains an arginine of debated function. Four residues with side chains that coordinate nonheme iron are at equivalent sequence positions in known lipoxygenase structures: three histidines and a carboxyl, provided by the C-terminus, an unusual ligand to a protein-bound metal; a fifth side chain coordinating iron is most often asparagine (Figure 1, cyan carbons), but in some cases it is another histidine. Water (or hydroxide) is a sixth metal ligand.
Figure 1.

Representations of conserved residues lining the substrate cavity in four isoforms of soybean lipoxygenase (gray, bold letters are residue numbers of SBL1 and smaller numbers refer to residue positions in soybean lipoxygenase isoforms LOX-3, VLXB, and VLXD.) Protein side chain ligands to iron (carbon in cyan) are shown, together with a linoleic acid models (purple). A conserved Arg (gray) occurs in a subcavity extension. Catalytic iron is colored salmon and the water O ligand, dark gray. Adapted with permission from Figure 5A, ref (4), 2006 John Wiley & Sons.
One arrangement of linoleic acid in the soybean cavity5 is presented in Figure 1, but models having the polar and methyl ends in reversed orientation can be made also. The reverse placement idea5 has guided experiments on lipoxygenases for many years, especially because the product of the enzymatic reaction with linoleic acid would be the 13S hydroperoxide for the orientation shown in Figure 1, but it would be 9S if the substrate orientation were reversed (Figure 2). Lipid substrates for lipoxygenase contain at least one cis,cis-1,4-pentadiene unit. Linoleate has one of these units, and arachidonate has three (Figure 2). The rate-limiting step of the enzymatic reaction is a proton-coupled electron transfer involving a bis-allylic C–H bond of substrate and the catalytic iron.6 A proton transfers to the −OH ligand of metal and an electron reduces ferric iron. Hydrogen removal and oxygen insertion, (n ± 2) carbons away, are usually antarafacial (Figure 2). A case of suprafacial oxygenation has been found more recently in a natural lipoxygenase.7 One electron transfers to/from the metal center in the catalytic cycle are illustrated in the bottom line of Figure 2. The ferrous and ferric ions are high spin (four and five unpaired electrons, respectively) in lipoxygenases. With this background in mind, experiments were designed to ask: where is the polar end of a lipid when it is bound to lipoxygenase, and how does it get there? Do the spectra of the metal center account for all of the metal in the “EPR-visible” states, and is there a unique spectral signature for lipoxygenases from different species?
Figure 2.

Substrates (linoleate and arachidonate) and product hydroperoxide (HPODE) of the lipoxygenase catalytic reaction and one-electron redox reactions at the iron atom are depicted.
2. Lipid Binding to Lipoxygenase
The relatively large size of soybean lipoxygenase-1 (SBL1) (∼100 kDa) compared to the size of lipids means that detecting a difference in rotational correlation time of a lipid in solution and one bound tightly to the enzyme is suitable for EPR studies with spin-labeled lipids.
Doxy-stearates (stearic acid substituted with a 1-oxyl-2,2,4,4-tetramethyloxazolidine ring, DSAs)8 and a headgroup spin-labeled lysolecithin (lyso-oleoylphosphatidylTEMPOcholine, LOPTC)9 (Figure 3) provided an overview of the affinity of these lipids for lipoxygenase. The doxyl lipids have a free radical ring placed at carbons 5, 8, 10, 12, and 16 of the 18-carbon stearate chain. EPR determinations of protein-bound and free DSAs showed that the affinity of these lipids for the enzyme depends on the length of the methyl end of the hydrocarbon chain, and increases with ΔΔG of −190 cal/methylene.8 (Note that since the entire DSA length has 18 carbons, in each case, there is also an unfavorable contribution to ΔΔG from exposure to solvent of the remaining methylenes on the polar end of the chain.) The depth of penetration into the hydrophobic cavity is limited by the bulk of the doxl ring of these stearate analogues. This conclusion has some analogy to understanding synthetic inhibitors of lipoxygenase that have long hydrocarbon chains variously substituted with aromatic rings.10 To characterize motions of the polar end of a lipid bound to lipoxygenase, a spin-labeled lyso-lecithin (LOPTC) was also examined by EPR.9 In the absence of protein, this lipid forms small micelles in water with polar ends extending outward. Close packing of the head groups gives rise to rapid spin exchange, and a single broad EPR signal. With lipoxygenase present, monomeric lipid is bound to the protein and the EPR signals indicate that, although the lipid is tethered to the protein by an acyl chain, the polar end is highly flexible, consistent with the polar end extending into solvent.11
Figure 3.

Spin-labeled lipids used to study interactions with lipoxygenase are (A) a series of stearic acid derivatives with a doxy ring placed at different carbon atoms and (B) a lyso-oleoylphosphatidyl choline with a spin-label replacing one choline methyl group.
3. The EPR Structure of a Lipid–Lipoxygenase Complex
Pulsed dipolar EPR spectroscopy (PDS) techniques are a subset of time-domain EPR based on pulse sequences that select for dipolar spin–spin components of relaxation.12 Dipolar contributions to relaxation depend on the distance between spin pairs and on the orientation of the interspin vector in the magnetic field. Spin pairs are either introduced into a macromolecule by mutation and, generally, chemical modification, or include a natural spin, for instance an iron–sulfur center.13 Dipolar interaction between nitroxide spin-labels is the most extensively studied, but nitroxide-to-metal dipolar relaxation provides an emerging alternative.14,15
To determine the location of the polar end of a lipid substrate analogue bound to the SBL1 structure, collaboration on PDS experiments was initiated with Peter Borbat and Jack Freed at the ACERT EPR Center (Cornell).9
The experimental objective was to determine distances from the polar end of a paramagnetic lipid to nitroxide spins placed on selected sites of lipoxygenase, and then by triangulation place the lipid spin. The lipids examined included primarily LOPTC but also 8-DSA (Figure 3). The internal cavity of SBL1 in the crystal structures is not open, but it nears the surface in two places that are distant from each other. A geometrical solution to locating the spin of a small molecule then requires a minimum of four grid sites that adequately represent the volume of the macromolecule. We chose five sites near the surface of the SBL1 structure and introduced spins by mutating natural side chains to cysteine and reaction with a methanethiosulfonate reagent that substitutes a spin-label moiety on sulfur, creating the amino acid R1 (Figure 4A).
Figure 4.

Lipid polar headgroup located by pulsed dipolar EPR spectroscopy with (A) a grid of five lipoxygenase side chains, mutated in pairs by spin-labeling. The spin-labeled residue numbers are, proceeding clockwise from the uppermost, F270R1, L480R1, A619R1, and F782R1, and the one in the middle is A569R1. The chemical structure shown is the spin-label side chain of R1 that has five rotatable bonds. (B) Calculation of one-dimensional probability distributions, P(r), based on time-domain decays (see Supporting Information in ref (9)). Pairs of residue numbers are indicated, for example, as 480:782 for double-labeled pair L480R1 and F782R1. Structural inset (B, upper right) gives the 1 and 2σ volume distribution solved for the spin of a bound LOPTC (cyan ovals). Pulsed EPR data and analysis were by Peter Borbat (Cornell). Adapted with permission from parts of Figures 2, 3, and 6 of ref (9), 2012, Elsevier.
Final determination of a spin location based on sparse grid sites spread over a macromolecule requires having approximate coordinates of at least one site. Criteria for introducing R1 to selected sites in SBL1 included that the sites be near the surface but in a niche that was likely to limit side-chain motion (few side chain rotamers). The relatively large size of SBL1 was an advantage here because most of the surface is distant from the active site (Figure 4A). Computer programs are available that fit rotamers of commonly used nitroxide spin-labels into locations in a protein structure.16−18 Availability of similar crystal structures of four isoforms of lipoxygenase in soybeans allowed limiting the initial calculated side chain torsion angle (χ1 and χ2) choices to ones based on native residue angles. Additional angles χ3 to χ5, derived from calculation, completed the set (Figure 4A right inset). The SBL1 grid sites chosen had 1 (F270R1, A619R1, and F782R1), 2 (L480R1) and 10 (A569R1) solutions with acceptable clash criteria. Coordinates calculated for F782R1, primarily, were chosen to align a grid of spins, from PDS, with the protein structure coordinates.
Distances between the grid sites depicted in Figure 4A were determined with the PDS pulse-sequence variations double electron–electron resonance (DEER) and double quantum coherence (DQC) applied at 17.3 GHz. Spin labels are composed of three molecular entities, differing in nitrogen nuclear spin states, that overlap in one part of the EPR spectrum but are resolved in other parts. Thus, a hard pulse to the overlap part and detection in a resolved region can be used to separate pump from detection responses. Examples of time-domain decays for three doubly labeled samples involving residue 480R1 (left side of Figure 4B) illustrate the distance-dependent dipolar oscillations. After data processing (subtraction of singly spin-labeled background and normalization), the depth of the initial decays indicated that each pair was almost fully labeled. Decays were also collected after LOPTC (or 8-DSA) was added to singly labeled SBL1 (examples: right side of Figure 4B). In these cases, the depth of the initial decay indicated partial occupancy of the protein site by the lipid. Titrations by DEER, and enzyme inhibition determination, indicated a partially occupied lipid-binding site under conditions of the experiments (0.8 equiv of LOPTC, 0.2 mM SBL1). Transformation of time-domain data to a distance probability distribution12,19 gives a one-dimensional representation of the range of R1 rotamers that have been trapped by freezing. The distance distributions (Figure 4B) between spin pairs attached to the protein were more narrowly defined than for the LOPTC to protein spin pairs. This observation is consistent with evidence for motion deduced from solution EPR spectra of the respective spins.9
Assembling the 15 distance probability distributions for pairs of protein–protein and protein–lipid spins into a representation of the volume, uncertainty, and alignment of LOPTC with the SBL1 structure was accomplished by statistical analyses (Supporting Information for ref (9)), similar to distance-geometry approaches used in NMR. The result is a polyhedron representing the 1 and 2σ uncertainty of the LOPTC spin, drawn simply as an ellipsoid (Figure 4B, right upper structure figure). The major radius in the ellipsoid representation is ∼6 Å at 2σ, with the width being attributed primarily to motion of the LOPTC headgroup. This solution overlaps SBL1 side chains of residues E236, K260, Q264, and Q544. The last three residues are in helices 2 and 11 of the protein structure and E236 is in a loop preceding helix 2.9
4. Entrance to Lipoxygenase Substrate Cavity
The headgroup location of LOPTC, just outside helices 2 and 11, is consistent with suggestions of an entrance between these helices, made possible by side chain displacements.2 As the pH optimum for activity is 8–9 for SBL1, and the pH of crystals is 7 or less, singly spin-labeled helix 2 mutants were examined20 in solution at pH values 7–9 to determine points of flexibility in helix 2. Replacing natural side chains with the spin-label R1 group was possible at almost all of the 21 residues, without major adverse effects on enzyme activity. The study revealed reversible changes in side chain and/or backbone flexibility with pH. The most flexible residues are those in the center of SBL1 helix 2 assigned as a π-helix in the crystal structure. The solution structure of the π-helical segment changes from having side chains with nanosecond motion that correlates with solvent exposure in the crystal structure (pH 7) to a state in which these residues have uniform motion of intermediate rate. Additional changes in motion are observed when a lipid is added at pH 9. The α-helical portions of helix 2 have little pH sensitivity under these conditions. The structure of the π-helix and changes in EPR spectra of that segment are illustrated in Figure 5. The similarity of line shapes from these residues, at pH 9, suggests a correlated backbone flexibility that might result, for example, from hydrogen bonds breaking and reforming in nanoseconds in this π-helix.20
Figure 5.
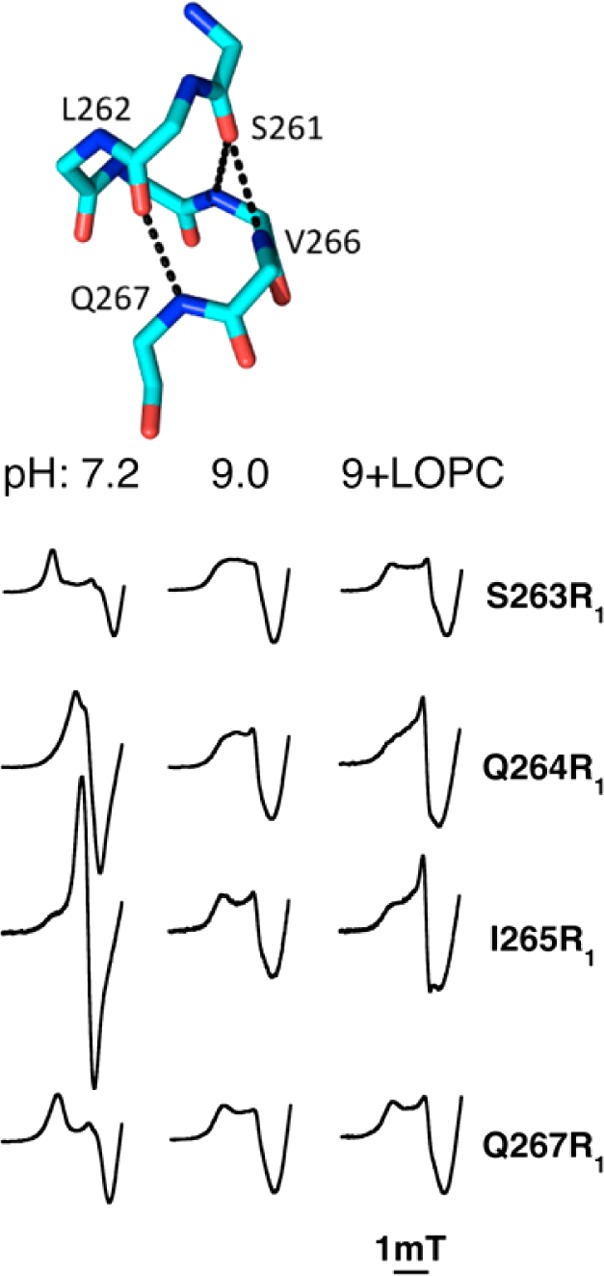
A π-helix in SBL1 helix 2 (upper) has a change in nanosecond fluctuations in the presence of a substrate analogue and upon a pH switch from 7 to 9, while few changes (not shown) occur in neighboring α-helix residues. Mutant lipoxygenases have spin-label R1 groups substituted for natural side chains at single sites in helix 2. The EPR spectra (X-band, low field portion of spectrum) of selected R1 substitutions at π-helix residues 263–265 and 267 are shown below. Adapted with permission from ref (20). Copyright 2014 American Chemical Society.
Relaxation of spin-labeled side chains by the lipoxygenase ferrous metal was also examined in this study. The natural side chain ultimate atoms have distances from iron of ∼10–25 Å, meaning that the metal should cause differential dipolar relaxation, depending on the nitroxide spin to iron distances. Other factors, including two symmetries of the iron center and spin-label exposure to solvent also contributed to rates of relaxation (at 60 K). By examining all 21 residues in helix 2, again, the π-helical segment stood out as having most variable relaxation rates with solvent pH and lipid binding conditions.20 It is suggested that the flexible π-helical region of lipoxygenase helix 2 is the site at which substrate gains access to the substrate cavity.
5. EPR of Metal Centers in Lipoxygenases
Evidence that both ferric and ferrous oxidation states of iron are involved in the lipoxygenase catalytic cycle came initially from observation of a high spin iron-like EPR signal after the enzyme turned over a single equivalent of substrate, or was exposed to an equivalent of its own product, a lipid allylic hydroperoxide.21,22 The resting enzyme did not give a signal at conventional EPR frequencies, suggesting it is ferrous, now confirmed by other types of spectroscopy.23
Quantitative accounting for EPR spectra of metals with more than one unpaired electron, for example, in the high-spin ferric iron (five unpaired electrons) of lipoxygenase, is made complex by distributions in zero field splitting energies (D and E). Yang and Gaffney initiated calculations to account quantitatively for multiple signals from the ferric centers in the non-heme iron proteins phenylalanine hydroxylase and diferric transferrin.24 Collaboration with Silverstone (Johns Hopkins) led to computation of energy levels, in cases where D and the EPR frequency are of the same magnitude.25,26 This type of calculation has many applications, one of which leads to assigning the EPR spectrum of manganese lipoxygenase at 9.4 and 94 GHz,27 described in subsection 5.2 below.
5.1. Iron Lipoxygenases
Initial EPR studies of ferric lipoxygenase revealed overlapping spectral components, the relative amounts of which varied with sample preparation details.28 In the reaction scheme (Figure 2), there should be no free radicals present when iron is ferric, although ferrous enzyme intermediates have a bound substrate or hydroperoxide radical. But, in the standard method of converting resting ferrous lipoxygenase to a ferric form, by adding one equivalent of lipid hydroperoxide, one unpaired electron is missing in the spin count. An unassigned radical derivative of lipid hydroperoxide likely reacts with oxygen, when oxygen is in excess, and free radical chain reactions are quenched.
Practically, oxidation of dilute (∼2 μM) resting lipoxygenase with a single equivalent of hydroperoxide under aerobic conditions converts all of the iron to ferric, with an EPR signal dominated by one or two components, after lipid byproducts are removed during sample concentration. The formation of ferric enzyme is substoichiometric when carried out with higher protein to oxygen concentrations.21,29
Discovery of a bacterial lipoxygenase from Pseudomonas aeruginosa (PaLOX), that occurs naturally with a lipid chain bound in the active site, provided an opportunity for comparison of EPR spectra of iron in that cavity with iron in a lipid-free cavity in ferric SBL1, in collaboration with Manresa and others at Barcelona.29 Somewhat surprisingly, the ferric form of both proteins gave EPR spectra that were nearly identical qualitatively and quantitatively. An experimental spectrum of ferric PaLOX appears in Figure 6A. Assignment of the EPR spectra included a distribution of zero field energies as the major line broadening mechanism,29 as reported earlier for SBL1 EPR (Figure 6B).28 In Figure 6B, features of the major component are labeled 1, and the minor component, 2. Applying a distribution function to the calculated subspectra results in less broadening of the lowest field 1a feature than the upfield 1b feature, and thus an experimental spectrum having almost a single low field peak visible. The feature 1c is from a transition in an upper pair of energy levels and has a g-value almost invariant with zero-field energies, so it becomes more prominent when a distribution in these energies is significant. (A g-value is (spectrometer frequency/resonance field position).) A second component, 2, contributes slightly to the experimental spectrum (Figure 6A), but can be the major component for samples in some buffers. For example, peaks 1 and 2 contribute almost equally to spectra of samples prepared in tris buffers.5 Recent high-resolution crystal structures do show nonwater solvent molecules in lipoxygenase cavities.30 Note that peaks 2b and 1c overlap at g = 5.86, which is also a g-value of heme iron, if any is present as an impurity. Similarly, two symmetries are seen in ferrous-nitric oxide lipoxygenase EPR spectra. In this case, the apparent line width is governed more by relaxation than by distribution in zero field energies and thus is narrower at 94 GHz than at 9.431 (Conspectus figure; EPR spectra adapted with permission from Figure 15 of reference (31), 2009, Springer).
Figure 6.

Origin of lipoxygenase high-spin ferric, multicomponent EPR signals: (A) an experimental example from a bacterial lipoxygenase (PaLOX) and (B) corresponding components of calculated spectra. The distinguishing features labeled in the calculated 9.4 GHz spectra are at g-values 7.3 (1a), 5.86 (1c), ∼4.7 (1b), 6.3 (2a), and 5.86 (2b). Each of the calculated spectra labeled “1” represents the same number of spins and that labeled “2” has half that amount. A simulated spectrum is based on a distribution of amplitudes of the subspectra “1”. A simulation of the experimental spectrum of the lipoxygenase from Pseudomonas aeruginosa appears in ref (29).
5.2. High Frequency EPR of Manganese Lipoxygenase
Manganese, instead of iron, is the metal in a fungal lipoxygenase discovered by Oliw and Su (Uppsala).7 This enzyme is also unusual in forming bis-allylic hydroperoxides and then catalyzing rearrangement of them to allylic hydroperoxide products characteristic of iron lipoxygenase reactions.32 Sequence and mutagenesis results suggest protein side chain ligands to metal and their arrangements are likely the same as those in iron lipoxygenases. Progress toward a crystal structure of manganese lipoxygenase is reported33 and completion should establish structural the similarities, and differences, of manganese and iron lipoxygenases.
In collaboration with the Oliw group, we asked whether EPR provides evidence for a collection of N- and O-side chain ligands to metal that are similar to the iron cases.27 Oliw had shown that the Mn2+ state (five unpaired electrons) of the resting enzyme was observable by EPR and that addition of substrate abolished this signal (as expected for the even number of unpaired electrons in Mn3+), thus revealing that maganese lipoxygenase cycles between 2+ and 3+ states, as do iron versions of the enzyme.34 However, the Mn2+ EPR spectrum also confirmed that the value of the zero field splitting (∼0.08 cm–1) was of the same magnitude as the EPR measurement frequency, 9.4 GHz (or ∼0.32 cm–1). At a 10-fold higher EPR frequency (94 GHz), a spectrum that could be analyzed more simply was obtained (Figure 7C, D). The result, in comparison with other manganese containing proteins, suggested that manganese lipoxygenase has three N- and one or more O- metal ligands.27 Even higher EPR frequencies are being used elesewhere to characterize manganese superoxide dismutases, enzymes with metal–ligand composition similar to lipoxygenases, but different geometry.35
Figure 7.

Transitions contributing to EPR absorption line shapes of manganese lipoxygenase at two frequencies: (A) calculations at 9.4 GHZ (X) of intensities for different angular orientations and pairs of energy levels (dots) and (B) line shapes resulting from these transitions; (C) an experimental 94 GHz (W) EPR spectrum of manganese lipoxygenase and (D) the same calculation of line shapes shown in (B) but at the higher EPR frequency 94 GHz. Colors show the pair of energy levels involved in significant transitions (level “1” is lowest in energy): 1 → 2 (red), 1 → 3 (gray), 1 → 4 (orange), 2 → 3 (black), 2 → 4 (green), 3 → 4 (light brown), 3 → 5 (light blue), 4 → 5 (purple) and 5 → 6 (dark green). Calculation parameters included zero field energies D = 0.1 cm–1 and E/D = 0.06. The inset in (C) compares the calculated (lower) and experimental (upper) hyperfine details in the central transition of manganese lipoxygenase. The straight red lines in (C) indicate the same magnetic field range in the inset and the full spectrum. (A) and (B) are adapted with permission from Figure 9 of ref (31) 2009, Springer; and (C) and (D) are adapted with permission from ref (27) 2001, Springer.
Computation, to first order, of signals for high spin manganous (five unpaired electrons) EPR is essentially the same as for high spin ferric electron centers, except six manganese nuclear hyperfine splittings are superimposed and energies are generally smaller. Figure 7 illustrates how some of the complexity of X-band manganous spectra is resolved at a higher EPR frequency.
Odd numbers of electrons are most suitable for EPR. There are six configurations for the five unpaired electrons of manganous or ferric ions, and there are 15 possible transitions between the six energy levels corresponding to these configurations. The probability that a transition between two levels will be observed at a particular frequency, field-scan range, and molecular orientation (θ, ϕ) in the magnetic field is given by an intensity factor25 (the product of the square of the transition dipole and sin θ). Taking the zero field term D = 0.1 cm–1 as a first approximation to manganous lipoxygenase zero field splitting, 9 of the 15 possible pairs of energy levels contribute significantly to spectra taken at the conventional 9.4 GHz EPR frequency (Figure 7A). In this kind of diagram, each dot represents a unique molecular orientation in the field.25 Most intense features in the EPR spectrum (Figure 7B) will occur where the density of dots and their intensities are highest in Figure 7A. At the higher 94 GHz EPR frequency, a spectrum arising from the same magnetic parameters will be simplified to a nested set of five transitions. These predictions are born out in the experimental spectrum of manganese lipoxygenase (Figure 7C) and the simulation (Figure 7D). The middle transitions in the spectra cover a narrow enough field range that Mn nuclear hyperfine transitions are resolved. Characteristic details of the hyperfine pattern refined the estimate of zero field splitting parameters for manganese lipoxygenase to approximately D = 0.08 cm–1 and E/D = 0.06.27
6. Summary and Outlook
Variations in lipoxygenase function are broader than forming a lipid hydroperoxide with unique stereochemistry. For instance, the human epidermal lipoxygenase eLOX3 appears to have a perfectly normal iron-binding site, from sequence comparison with functional lipoxygenases.36 Nevertheless, it is devoid of activity with normal substrates for lipoxygenases. Instead, it functions catalytically as a hydroperoxide isomerase, converting preformed hydroperoxides to epoxyalcohols. Similarly, manganese lipoxygenase isomerizes and rearranges hydroperoxides, in addition to having a lipoxygenase function.37 These conversions of lipid hydroperoxides to epoxyalcohols proceed by an oxygen rebound reaction in which both O atoms of the cleaved hydroperoxide are retained in the product. Steric factors and/or the metal redox potential likely influence the balance of lipoxygenase and isomerase reactions.37 This Account covers EPR approaches to examine selected aspects of lipoxygenase structure in solution or frozen solution. Working with solutions allows flexibility in conditions, for instance, those leading to changes in structure with pH.20 A quite obvious extension is to versions of the enzyme that form products oxidized at different positional sites. More challenging is to extend these ideas to differentiate details of lipid and hydroperoxide reactivity by the enzyme. Here some ingenuity in synthesis of paramagnetic substrate/product analogues will be needed.
Acknowledgments
The author is deeply indebted to Ph.D. students whose dissertations and papers, over 1986–2014, made this review possible. They are Dimitrios V. Mavrophilipos (initiated studies of SBL1), An-Suei Yang (computed EPR spectra of iron centers with distributions of zero-field splitting), Brendan C. Maguire (cloned lipoxygenase from soybeans), Jeffrey C. Boyington (solved lipoxygenase crystal structure with L. M. Amzel), Kutbuddin S. Doctor (simulated high- and low-frequency iron EPR), Fayi Wu (constructed no-cysteine SBL1 and determined affinity for doxyl stearates), and, currently, Miles D. Bradshaw (finding location of entry to the active site in SBL1). Earlier, Shaw T. Chen and Gary L. Willingham set the stage (membrane studies). The author’s work has been supported by The National Institutes of Health grants, presently by GM065268 to B.J.G.
Biography
Betty J. Gaffney received her B.S. and Ph.D. degrees at Stanford, studying asymmetric synthesis with Harry S. Mosher. As a postdoc, she worked on protein-modification reagents (dia- and paramagnetic) with Koji Nakanishi at Tohoku University, Sendai, Japan,38 and then developed her interest in biological lipids and EPR with Harden M. McConnell at Stanford.39 She was a Professor of Chemistry at Johns Hopkins University (1974–1996), and since then is Professor of Biological Science at Florida State University.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Boyington J. C.; Gaffney B. J.; Amzel L. M. The 3-Dimensional Structure of an Arachidonic-Acid 15-Lipoxygenase. Science 1993, 260, 1482–1486. [DOI] [PubMed] [Google Scholar]
- Minor W.; Steczko J.; Stec B.; Otwinowski Z.; Bolin J. T.; Walter R.; Axelrod B. Crystal structure of soybean lipoxygenase L-1 at 1.4 Å resolution. Biochemistry 1996, 35, 10687–10701. [DOI] [PubMed] [Google Scholar]
- Skrzypczak-Jankun E.; Borbulevych O. Y.; Zavodszky M. I.; Baranski M. R.; Padmanabhan K.; Petricek V.; Jankun J. Effect of crystal freezing and small-molecule binding on internal cavity size in a large protein: X-ray and docking studies of lipoxygenase at ambient and low temperature at 2.0 Å resolution. Acta Crystallogr., Sect. D 2006, 62, 766–775. [DOI] [PubMed] [Google Scholar]
- Youn B.; Sellhorn G. E.; Mirchel R. J.; Gaffney B. J.; Grimes H. D.; Kang C. Crystal structures of vegetative soybean lipoxygenase VLX-B and VLX-D, and comparisons with seed isoforms LOX-1 and LOX-3. Proteins 2006, 65, 1008–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffa G.; Imber A. N.; Maguire B. C.; Laxmikanthan G.; Schneider C.; Gaffney B. J.; Brash A. R. On the relationships of substrate orientation, hydrogen abstraction, and product stereochemistry in single and double dioxygenations by soybean lipoxygenase-1 and its Ala542Gly mutant. J. Biol. Chem. 2005, 280, 38756–38766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S.; Sharma S. C.; Scouras A. D.; Soudackov A. V.; Carr C. A.; Hammes-Schiffer S.; Alber T.; Klinman J. P. Extremely elevated room-temperature kinetic isotope effects quantify the critical role of barrier width in enzymatic C–H activation. J. Am. Chem. Soc. 2014, 136, 8157–8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C.; Oliw E. H. Manganese lipoxygenase – Purification and characterization. J. Biol. Chem. 1998, 273, 13072–13079. [DOI] [PubMed] [Google Scholar]
- Wu F.; Gaffney B. J. Dynamic behavior of fatty acid spin labels within a binding site of soybean lipoxygenase-1. Biochemistry 2006, 45, 12510–12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffney B. J.; Bradshaw M. D.; Frausto S. D.; Wu F. Y.; Freed J. H.; Borbat P. Locating a Lipid at the Portal to the Lipoxygenase Active Site. Biophys. J. 2012, 103, 2134–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorins G.; Kuhnert L.; Johnson C. R.; Marnett L. J. (Carboxyalkyl) benzyl propargyl ethers as selective inhibitors of leukocyte-type 12-lipoxygenases. J. Med. Chem. 1996, 39, 4871–4878. [DOI] [PubMed] [Google Scholar]
- Brash A. R.; Ingram C. D.; Harris T. M. Analysis of a Specific Oxygenation Reaction of Soybean Lipoxygenase-1 with Fatty-Acids Esterified in Phospholipids. Biochemistry 1987, 26, 5465–5471. [DOI] [PubMed] [Google Scholar]
- Borbat P. P.; Freed J. H.. Pulse Dipolar Electron Spin Resonance: Distance Measurements. In Structural Information from Spin-Labels and Intrinsic Paramagnetic Centres in the Biosciences; Timmel C. R., Harmer J. R., Eds.; Springer: Heidelberg, Berlin, 2013; Vol. 152; pp 1–82. [Google Scholar]
- Stich T. A.; Myers W. K.; Britt R. D. Paramagnetic Intermediates Generated by Radical S-Adenosylmethionine (SAM) Enzymes. Acc. Chem. Res. 2014, 47, 2235–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji M.; Ruthstein S.; Saxena S. Paramagnetic Metal Ions in Pulsed ESR Distance Distribution Measurements. Acc. Chem. Res. 2014, 47, 688–695. [DOI] [PubMed] [Google Scholar]
- Kaminker I.; Tkach I.; Manukovsky N.; Huber T.; Yagi H.; Otting G.; Bennati M.; Goldfarb D. W-band orientation selective DEER measurements on a Gd3+/nitroxide mixed-labeled protein dimer with a dual mode cavity. J. Magn. Reson. 2013, 227, 66–71. [DOI] [PubMed] [Google Scholar]
- Hatmal M. M.; Li Y. Y.; Hegde B. G.; Hegde P. B.; Jao C. C.; Langen R.; Haworth I. S. Computer Modeling of Nitroxide Spin Labels on Proteins. Biopolymers 2012, 97, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagelueken G.; Ward R.; Naismith J. H.; Schiemann O. MtsslWizard: In Silico Spin-Labeling and Generation of Distance Distributions in PyMOL. Appl. Magn. Reson. 2012, 42, 377–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyhach Y.; Bordignon E.; Jeschke G. Rotamer libraries of spin labelled cysteines for protein studies. Phys. Chem. Chem. Phys. 2011, 13, 2356–2366. [DOI] [PubMed] [Google Scholar]
- Jeschke G.Interpretation of Dipolar EPR Data in Terms of Protein Structure. In Structural Information from Spin-Labels and Intrinsic Paramagnetic Centers in the Biosciences; Timmel C. R., Harmer J. R., Eds.; Springer: Heidelberg-Berlin, 2014; Vol. 152; pp 83–120. [Google Scholar]
- Bradshaw M. D.; Gaffney B. J. Fluctuations of an Exposed π-Helix Involved in Lipoxygenase Substrate Recognition. Biochemistry 2014, 53, 5102–5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheesbrough T. M.; Axelrod B. Determination of the Spin State of Iron in Native and Activated Soybean Lipoxygenase-1 by Paramagnetic-Susceptibility. Biochemistry 1983, 22, 3837–3840. [Google Scholar]
- De Groot J. J. M.; Veldink G. A.; Vliegenthart J. F. G.; Boldingh J.; Wever R.; Van Gelder B. F. Demonstration by EPR Spectroscopy of Functional Role of Iron in Soybean Lipoxygenase-1. Biochim. Biophys. Acta 1975, 377, 71–79. [DOI] [PubMed] [Google Scholar]
- Pavlosky M. A.; Zhang Y.; Westre T. E.; Gan Q. F.; Pavel E. G.; Campochiaro C.; Hedman B.; Hodgson K. O.; Solomon E. I. Near-Infrared Circular-Dichroism, Magnetic Circular-Dichroism, and X-Ray-Absorption Spectral Comparison of the Nonheme Ferrous Active-Sites of Plant and Mammalian 15-Lipoxygenases. J. Am. Chem. Soc. 1995, 117, 4316–4327. [Google Scholar]
- Yang A. S.; Gaffney B. J. Determination of Relative Spin Concentration in Some High-Spin Ferric Proteins Using E/D-Distribution in Electron-Paramagnetic Resonance Simulations. Biophys. J. 1987, 51, 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffney B. J.; Silverstone H. J.. Simulation of the EMR Spectra of High-Spin Iron in Proteins. In EMR of Paramagnetic Molecules; Berliner L. J., Ed.; Plenum: New York, 1993; pp 1–57. [Google Scholar]
- Gaffney B. J.; Silverstone H. J. Simulation methods for looping transitions. J. Magn. Reson. 1998, 134, 57–66. [DOI] [PubMed] [Google Scholar]
- Gaffney B. J.; Su C.; Oliw E. H. Assignment of EPR transitions in a manganese-containing lipoxygenase and prediction of local structure. Appl. Magn. Reson. 2001, 21, 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffney B. J.; Mavrophilipos D. V.; Doctor K. S. Access of Ligands to the Ferric Center in Lipoxygenase-1. Biophys. J. 1993, 64, 773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garreta A.; Val-Moraes S. P.; Garcia-Fernandez Q.; Busquets M.; Juan C.; Oliver A.; Ortiz A.; Gaffney B. J.; Fita I.; Manresa A.; Carpena X. Structure and interaction with phospholipids of a prokaryotic lipoxygenase from Pseudomonas aeruginosa. FASEB J. 2013, 27, 4811–4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobe M. J.; Neau D. B.; Mitchell C. E.; Bartlett S. G.; Newcomer M. E. The Structure of Human 15-Lipoxygenase-2 with a Substrate Mimic. J. Biol. Chem. 2014, 289, 8562–8569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffney B. J.EPR of Mononuclear Non-Heme Iron Proteins. In High Resolution EPR: Applications to Metalloenzymes and Metals in Medicine; Hanson G., Berliner L., Eds.; Springer: New York, 2009; pp 233–268. [Google Scholar]
- Hamberg M.; Su C.; Oliw E. Manganese lipoxygenase - Discovery of a bis-allylic hydroperoxide as product and intermediate in a lipoxygenase reaction. J. Biol. Chem. 1998, 273, 13080–13088. [DOI] [PubMed] [Google Scholar]
- Wennman A.; Oliw E. H.; Karkehabadi S. Crystallization and preliminary crystallographic analysis of manganese lipoxygenase. Acta Crystallogr., Sect. F 2014, 70, 522–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C.; Sahlin M.; Oliw E. H. Kinetics of manganese lipoxygenase with a catalytic mononuclear redox center. J. Biol. Chem. 2000, 275, 18830–18835. [DOI] [PubMed] [Google Scholar]
- Tabares L. C.; Gatjens J.; Un S. Understanding the influence of the protein environment on the Mn(II) centers in Superoxide Dismutases using High-Field Electron Paramagnetic Resonance. Biochim. Biophys. Acta 2010, 1804, 308–317. [DOI] [PubMed] [Google Scholar]
- Yu Z. Y.; Schneider C.; Boeglin W. E.; Marnett L. J.; Brash A. R. The lipoxygenase gene ALOXE3 implicated in skin differentiation encodes a hydroperoxide isomerase. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 9162–9167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristea M.; Oliw E. H. A G316A mutation of manganese lipoxygenase augments hydroperoxide isomerase activity - Mechanism of biosynthesis of epoxyalcohols. J. Biol. Chem. 2006, 281, 17612–17623. [DOI] [PubMed] [Google Scholar]
- Gaffney McFarland B.; Inoue Y.; Nakanishi K. The Reaction of Koshland’s Protein Reagent with Tryptophan. Tetrahedron 1969, 10, 857–860. [DOI] [PubMed] [Google Scholar]
- McConnell H. M.; Gaffney McFarland B. Physics and chemistry of spin labels. Q. Rev. Biophys. 1970, 3, 91–136. [DOI] [PubMed] [Google Scholar]