

Abstract
Although acidic peptides compose a substantial portion of many proteomes, their less efficient ionization during positive polarity electrospray ionization (ESI) impedes their detection in bottom-up mass spectrometry workflows. We have implemented a derivatization strategy based on carbamylation which converts basic amine sites (Lys, N-termini) to less basic amides for enhanced analysis in the negative mode. Ultraviolet photodissociation (UVPD) is used to analyze the resulting peptide anions, as demonstrated for tryptic peptides from bovine serum albumin and Halobacterium salinarum in a high throughput liquid chromatography/tandem mass spectrometry (LC/MS/MS) mode. LC/UVPD-MS of a carbamylated H. salinarum digest resulted in 45% more identified peptides and 25% more proteins compared to the unmodified digest analyzed in the negative mode.
With the advent of modern mass spectrometric-based proteomics, hundreds or even thousands of proteins can be identified in a single experiment.1 Several hurdles still remain in the path to efficient sampling of a complete proteome in high-throughput applications. Chief among these hurdles is identification of underrepresented proteins (based on analysis of the corresponding peptides created upon proteolysis in the typical bottom-up approach). Underrepresented proteins include those that have fewer copies per cell (low abundance) as well as those for which the proteolytic peptides are undersampled due to a variety of factors. These factors include low protein solubility under the digestion conditions utilized (resulting in ineffective proteolysis and inefficient production of representative peptides), suboptimal peptide size (mass is too large or too small), and peptides being too hydrophilic or hydrophobic resulting in unsatisfactory chromatographic properties and/or poor ionization efficiencies. Moreover, peptides are routinely “missed” due to the stochastic nature of data dependent tandem mass spectrometry (MS). Several strategies have been developed to address undersampling due to low abundance. These approaches include reducing sample complexity by fractionation,2 enriching low abundance peptides3 (that could contain a targeted post translational modification, for example), preferential proteolysis and depletion of the most abundant proteins,4 and immunodepletion of abundant proteins.5 Fewer studies have reported means to improve the analysis of peptides that ionize poorly upon positive polarity electrospray ionization (ESI) after a conventional low pH liquid chromatography (LC) separation.6,7In silico digestion of whole proteomes typically result in a bimodal distribution of peptide isoelectric points (pI), even when performed with trypsin as the proteolytic agent (which leaves a basic site at both termini).8 This natural bimodal pI distribution of proteolytic peptides (as illustrated in Supporting Figure 1 in the Supporting Information) from several model proteomes justifies extra effort in targeting the substantial acidic portion of a given peptidome. Although rarely employed in high-throughput proteomics experiments, negative polarity mass spectrometry provides access to the acidic peptidome which is not well-suited for positive mode analysis. At neutral and slightly basic pH, deprotonation of glutamic and aspartic acid residues promotes the formation of peptide anions which can be readily detected and characterized in the negative mode. In order to achieve the most efficient deprotonation, high pH mobile phases are typically required for LC–MS experiments utilizing negative polarity ESI. Raising the pH of the mobile phase several units above the pKa of the amino acid side chains results in deprotonation; however, in practice high pH mobile phases are generally incompatible with standard silica based stationary phases and capillaries used in peptide separations.
Aside from the high pH required for efficient deprotonation of peptides, the ability to generate informative fragmentation patterns of peptide anions is also challenging. Negative mode analysis suffers from a dearth of options for efficient peptide fragmentation. While positive mode peptide analysis is proficiently accomplished using collision induced dissociation (CID),9 electron capture or electron transfer dissociation (ECD10 or ETD,11 respectively), infrared multiphoton dissociation12 (IRMPD), or some combination of the above methods, negative mode tandem mass spectrometry (MS/MS) analysis is more limited. Electron detachment dissociation13 (EDD), negative mode electron transfer dissociation14 (NETD), and 193 nm negative mode ultraviolet photodissociation15 (NUVPD) have been shown to provide diagnostic fragmentation of peptide anions, and the latter two methods have been implemented for the successful analysis of elaborate proteomic mixtures. Kjeldsen optimized EDD for the generation of diagnostic a and x fragment anions and demonstrated it for LC–MS/MS analysis of a simple single protein digest as well as for phosphopeptide identification from 12 model proteins.13 Using NETD and a mobile phase around pH 10, Coon and co-workers identified 1412 unique peptides from yeast proteins and showed 45% greater coverage of the acidic yeast GRX1 protein when compared to solely positive mode CID and ETD activation.6 Despite these positive gains on single proteins, NETD required lengthier (>100 ms) activation times which made it less compatible with high-throughput LC time scales. Madsen et al. reported the identification of over 2000 peptides and 659 proteins upon ultraviolet photodissociation (UVPD) analysis of HeLa cell lysates analyzed in the negative mode.7 UVPD at 193 nm is successfully implemented using a 2–10 ms activation period (to allow multiple laser pulses) and commonly produces multiple diagnostic ion series; most notably x-, a-, b-, and z-type fragments and occasionally c- and y-type ions. UVPD and NETD have been directly compared15 for LC–MS analyses of tryptic digests in the negative mode, with the finding that either method, when combined with complementary positive mode CID data, increased sequence coverages and peptide identifications compared to CID alone.16 UVPD has also proven to be particularly proficient for analysis of peptides with labile acidic post-translational modifications17 (PTMs), like phosphorylation18 and sulfation,19,20 as these PTMs are not lost during UVPD.
Here we introduce a highly efficient means to lower the pKa of the N-termini and lysine side-chains of peptides by converting the reactive amines to amides. This simple derivatization procedure is readily implemented on complex proteolytic mixtures and results in detection and identification of significantly more peptides in the negative mode by UVPD than obtained for noncarbamylated peptide mixtures, as demonstrated for whole cell lysates of Halobacterium salinarum.
Experimental Section
Materials
HPLC solvents and buffer components were obtained from Sigma-Aldrich (St. Louis, MO). Proteomics-grade trypsin was obtained from Promega (Madison, WI). All other reagents and solvents were obtained from ThermoFisher Scientific (Fairlawn, NJ). The model peptides DRVYIHPFHL and WAGGDASGE were obtained from American Peptide Company (Sunnyvale, CA). Bovine serum albumin was obtained from Sigma-Aldrich (St. Louis, MO). Halobacterium salinarum was obtained from American Type Culture Collection (ATCC, Manassas, VA). The bacteria were grown in the recommended medium (American Type Culture Collection medium 2185). Cells were suspended in 10 mM Tris-HCL, 10 mM KCl, 1.5 mM MgCl2 at pH 8 to swell and were lysed by dounce homogenization. The whole cell lysate was centrifuged to clarify the soluble lysate and to remove the insoluble pellet.
Sample Preparation
Both bovine serum albumin and proteins isolated from H. salinarum were digested at 37 °C overnight with trypsin. Prior to digestion, proteins were reduced in 5 mM dithiothreitol for 30 min at 55 °C and subsequently alkylated in 10 mM iodoacetamide at room temperature in the dark for 30 min. Alkylation was quenched with a second aliquot of dithiothreitol, thus bringing the final concentration of dithiothreitol to ∼10 mM. Trypsin was added to achieve a 1:20 enzyme-to-substrate ratio, and the solution was buffered at pH 8 in 150 mM ammonium bicarbonate. After digestion the sample was dried in a vacuum centrifuge for subsequent derivatization.
Derivatization
Carbamylation was performed as previously reported.21 Briefly, each sample was split into two aliquots; one for derivatization and one as a control. Each was resuspended in 200 mM Tris-HCl in the presence or absence of 8 M urea. Both samples were incubated at 80 °C for 4 h. Derivatized peptides were desalted using C18 spin columns (Pierce, Rockford, IL), then evaporated to dryness and resuspended in solvent to match the LC starting conditions (2% acetonitrile/98% water/0.05% acetic acid). The peptides DRVYIHPFHL and WAGGDASGE were derivatized as described above.
LC–MS/MS
The H. salinarum samples were analyzed on a Thermo Scientific Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) equipped with a 193 nm excimer laser (Coherent, Santa Clara, CA) and modified to allow UVPD activation in the HCD cell.25 Photodissociation was implemented in a manner described previously.7 Chromatographic separations were performed using water (A) and acetonitrile (B) mobile phases containing 0.05% acetic acid on an Eksigent Nanoultra 2D Plus liquid chromatography system (Redwood, CA). The trap (35 mm × 0.1 mm) and analytical column (with integrated emitter) (15 cm × 0.075 cm) were packed in-house using 3 μm Michrom Magic C18 packing (New Objective, Woburn, MA). Approximately 3 μg of digest was loaded onto the trap column at 2 μL/min for 20 min and separated with a gradient that changed from 0 to 35% B over the course of 240 min at a flow rate of 300 nL min–1. For nanospray, 2.1 kV was applied at a precolumn liquid voltage junction for negative polarity mode, and the tip–inlet distance was carefully adjusted to mitigate the occurrence of corona discharge. Survey and MS/MS scans were acquired by averaging one and three scans, respectively. Automated gain control targets were 1 000 000 for both survey MS and MSn scan modes. The maximum ion time was 200 ms for MS and MSn.
All data-dependent nano LC–MS methods on the Orbitrap involved an FT survey scan (m/z 400–2000) at a resolution of 120 000 followed by a series of MS/MS scans on the top 10 most abundant ions from the first survey. The minimum signal required for MS2 selection was 100 000, and the isolation width was fixed at 3 m/z. Dynamic exclusion was enabled for 30 s with a repeat count of one and a list size of 500 m/z values. For UVPD, three 2-mJ pulses were delivered during an activation period of 6 ms. Product ions from UVPD were detected in the Orbitrap at a resolution of 15 000.
The RAW data files collected on the mass spectrometer were converted to mzXML files by use of MassMatrix data conversion tools (v3.9, http://www.massmatrix.net/download). All data were searched using an in-house MassMatrix Web server (v2.4.2, http://www.massmatrix.net). The search parameters in MassMatrix employed were (i) enzyme, trypsin; (ii) missed cleavage, maximum 2; (iii) modifications, fixed iodoacetamide derivative of cysteine and variable oxidation of methionine (fixed carbamylation of n-term and lysine, when appropriate for modified samples); (iv) precursor ion mass tolerances, 15 ppm for Orbitrap data; (v) product ion mass tolerances, 0.02 Da for UVPD-MS data on Orbitrap; (vi) maximum number of modifications allowed for each peptide, 3; (vii) peptide length, 6–40 amino acid residues; (viii) score thresholds of 5.3 and 1.3 for the pp/pp2 and pptag scores, respectively. The Halobacterium_sp_nrc1 database was used for Halo data sets. Peptide and protein identifications were both filtered at a 1% false discovery rate. The peptide spectral matches were ranked by confidence and listed in descending order. As the percentage of matches to the decoy database approached one, all spectral matches below that point on the list were discarded.
pKa Calculation
The change in pKa between carbamylated and unmodified lysine residues and N-termini were calculated using Marvin (http://www.chemaxon.com/marvin/sketch/index.php), a widely available chemical visualization and property calculation tool (Marvin 14.7.7, 2014). The inverse log of Ka values were calculated using the default parameters.
In Silico Digestion
In silico digests were performed using freely available software (http://omics.pnl.gov/software/protein-digestion-simulator). FASTA files containing the proteomes for H. sapiens, S. cerevisiae, E. coli, and H. salinarum were downloaded from the Swiss-prot database (http://beta.uniprot.org/uniprot/? query=*&fil=reviewed%3Ayes) in their reviewed forms. Tryptic digests were performed allowing up to two missed cleavages.
Results and Discussion
In order to expand the depth and breadth of coverage in proteomics applications, in particular the ability to analyze intrinsically more acidic peptides which may be less effectively ionized in positive mode, negative mode offers an appealing option. Having previously demonstrated the capabilities of UVPD for analysis of peptides in both the positive and negative modes,7 we wished to further extend the proteome coverage by enhancing the range of peptides suitable for analysis in the negative mode. Owing to the often lower efficiency of electrospray ionization in the negative mode (which remains an area of active interest),22−24 the development of methods to make peptides more amenable to deprotonation is a key objective. In practice, this includes strategies to reduce the basicities of the most basic sites (such as lysines and the N-termini in peptides), thus suppressing protonation and/or increasing the acidities of acidic groups to enhance deprotonation. The high pKa of the primary amine functional groups is a particularly significant factor, which may strongly influence negative mode ESI efficiencies of peptides. The strategy reported here uses a simple and highly efficient carbamylation reaction which converts primary amines to amide groups, thus decreasing the pKa values of those functional groups (in particular the lysine side-chains and N-termini). The carbamylation reaction is shown schematically in Supporting Figure 2 in the Supporting Information, resulting in a mass shift of +43.0058 Da per carbamylation.
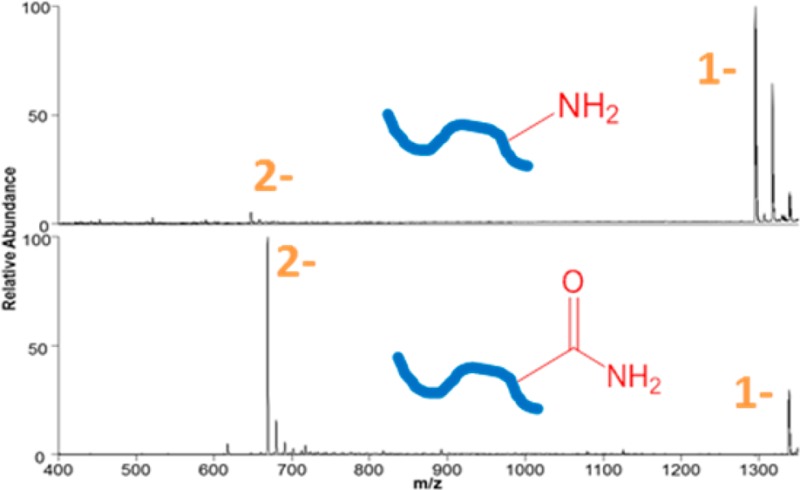
Feasibility experiments were undertaken using model peptides in order to optimize the carbamylation reaction (i.e., minimize the presence of partially reacted species) and cleanup procedure (i.e., minimize sample loss). The carbamylation and C18 spin column cleanup procedure was extremely simple and efficient for individual peptides and, in fact, translated remarkably well to complex mixtures of tryptic peptides, as described later. Reaction efficiencies were estimated based on examination of the abundances of carbamylated and unmodified peptides obtained from extracted ion chromatograms for individual peptides subjected to carbamylation. The ESI mass spectra obtained in the negative mode for one representative unmodified peptide (DRVYIHPFHL) and the same peptide after carbamylation are shown in Figure 1. The unmodified peptide is observed primarily as a singly deprotonated species; the carbamylated peptide is observed predominantly as a doubly deprotonated species and its abundance is nearly a factor of 10 greater than that of the unmodified peptide. Examples of the LC traces used to estimate reaction efficiency are shown in Supporting Figure 3 in the Supporting Information, in which the reaction efficiency of carbamylation was estimated to be 97% for peptide LVNELTEFAK (based on integration of the peak areas for the unmodified and carbamylated peptides). Carbamylation resulted in a modest shift in retention times of peptides because the ionizable amine groups are converted to more hydrophobic amide functionalities. It is estimated that the conversion of the primary amines (N-terminus and lysine side-chains) to amides changes the pKa of those groups from 9.5 and 10.5, respectively, to an estimated pKa of −1.7 (Marvin 14.7.7 2014 http://www.chemaxon.com). This is also consistent with the shift in charge state noted for the DRVYIHPFHL peptide (in Figure 1) as well as other peptides upon carbamylation.
Figure 1.
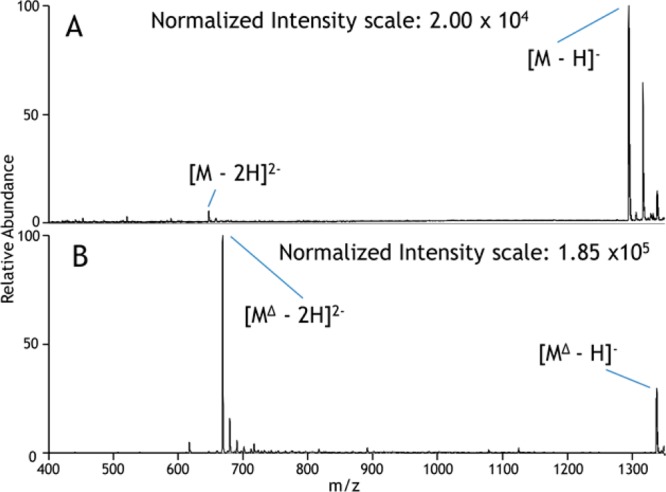
Negative ESI mass spectra of peptide DRVYIHPFHL: (A) the unmodified peptide and (B) the carbamylated peptide. Δ represents carbamylation.
In the negative ESI mode, the unmodified peptides typically are detected in low charge states, often as singly deprotonated species of modest abundance, whereas the corresponding carbamylated peptides are observed in higher charge states and with much greater abundances. It is well-known that CID of deprotonated peptides predominantly yields fragment ions resulting from uninformative neutral losses of water and CO2. In contrast, UVPD of deprotonated peptides results primarily in diagnostic a/x sequence ions in addition to lower abundances of b/y and c/z ions and charge-reduced precursors (i.e., via photoinduced electron detachment).14 Examples of the rich UVPD mass spectra of an unmodified peptide, GEEVTAEVADGPQSVIFDQAENR, and its carbamylated counterpart are shown in Figure 2. The relative abundances and types of fragment ions are similar for both the unmodified and carbamylated peptide, indicating that carbamylation does not suppress or significantly alter the UVPD process.
Figure 2.
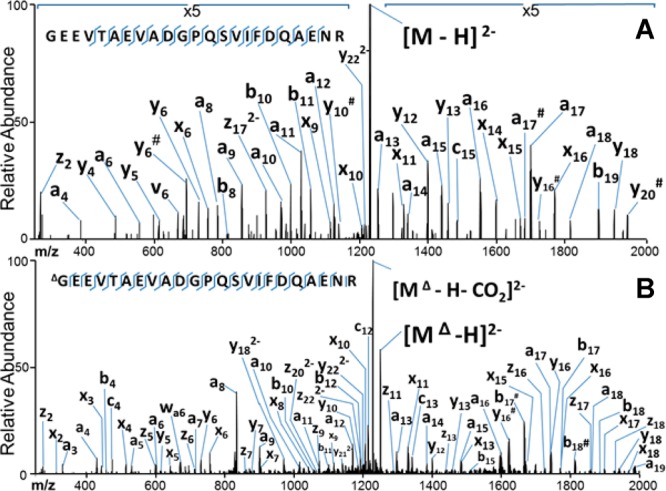
UVPD mass spectra of GEEVTAEVADGPQSVIFDQAENR from H. salinarum: (A) unmodified (2−) and (B) carbamylated (2−). # indicates the loss of water.
While these initial experiments were important for proving the feasibility of the method, to evaluate the scalability of the carbamylation reaction for more complex mixtures of tryptic peptides, BSA was digested and the resulting peptides were carbamylated. Extracted ion chromatograms (EIC) were generated to monitor the elution of both unmodified and the corresponding carbamylated peptides. The areas of the extracted ion peaks were used to measure the efficiency of carbamylation according to the following equation where A is chromatographic peak area:
In agreement with the reactions of individual model peptides, the reaction efficiencies of the measured BSA tryptic peptides averaged more than 97%. Other derivatization reagents (such as the popular 4-sulfophenyl isothiocyanate) have been used in the past successfully to enhance negative mode ionization, but their success for high-throughput proteomics applications have proven to be subpar due to the low reaction efficiencies for complex multicomponent mixtures.25 As also noted above, the dominant charge states of the resulting carbamylated tryptic peptides of BSA were typically shifted by one charge (e.g., from 1– to 2−), and the abundances increased by a factor of 7.6 on average relative to the unmodified peptides.
Trypsin was used as the protease of choice in this study because it is the enzyme most commonly used for mass spectrometric-based bottom-up proteomics applications. One advantage of using trypsin for conventional positive mode LC–MS studies is that it generally results in at least two very basic sites (N-terminus and C-terminal K or R residues) which enhances the formation of multiply charged peptide cations that are ideal for CID and database searches. Carbamylation reduces the pKa values of the lysine side-chain and N-terminus, thus reducing the basicity of those sites and making them less ionizable in the positive mode and overall making the peptides more amenable to negative mode ESI. By retaining the use of trypsin in the present study, the protein mixtures can be subjected to tryptic digestion, then split into two samples: one for traditional bottom-up/positive mode approach and the other processed in parallel using the carbamylation/negative mode UVPD strategy. This dual positive/negative MS/MS approach should extend the range of peptides (and therefore proteins) identified with confidence. Moreover, the nearly stoichiometric carbamylation reaction efficiencies observed for the model peptides and BSA digest allowed carbamylation of the Lys side-chains and N-termini to be treated as fixed modifications.
A summary of the observed carbamylated peptides and their corresponding peak areas is shown in Supporting Table 1 in the Supporting Information for carbamylated BSA tryptic peptides. In cases where peptides contain multiple primary amines (i.e., one or more lysine side-chains plus the N-terminus), the predominant products were the fully carbamylated species (see Supporting Figure 4 in the Supporting Information). Despite the reaction undertaken in somewhat basic conditions (pH 8), the pKa of the arginine side-chain is substantially greater (pKa 12.5) and thus the majority (>99%) of arginine side-chains remained protonated and unreactive.
To evaluate the carbamylation/UVPD strategy for a larger array of peptides, the method was applied to the analysis of the H. salinarum proteome. Many of the proteins in the H. salinarum proteome are naturally acidic, thus resulting in a large distribution of tryptic peptides possessing lower than average pI values (average 5.8) (Supporting Figure 5 in the Supporting Information). In fact, over 65% of the predicted tryptic peptides are expected to have pI values below 7. Carbamylation was used to further reduce the average peptide pI values by decreasing the pKa values of the N-termini and lysine residues. After tryptic digestion of the proteins extracted from the H. salinarum lysate, the peptides were incubated and carbamylated in 8 M urea and desalted. Upon comparison of the chromatograms obtained from the carbamylated and noncarbamylated tryptic peptides, the carbamylated peptides were retained on the column for between 5 and 15 min longer than their underivatized counterparts, and the degree of the retention time shift scaled with the number of carbamylated sites. This increase in hydrophobicity agrees with the findings mentioned earlier for the model peptides and BSA peptides and is consistent with replacement of the ionizable primary amines by the more hydrophobic amide moieties. Despite the increased retention times, most carbamylated peptides eluted when the mobile phase composition contained less than 35% (v/v) acetonitrile.
An example of an LC trace for a carbamylated tryptic digest of H. salinarum and a representative UVPD mass spectrum for one peptide (ΔDNVAAIIIGSR carbamylated at its N-terminus) is shown in Figure 3. The UVPD mass spectrum is dominated by a/x ions with lower abundances of z ions, and the sequence coverage for this peptide is very high (as is also the case for many of the other carbamylated tryptic peptides). Inspection of the charge state distributions of those peptides identified by UVPD for H. salinarum demonstrates a shift in average charge state for the carbamylated peptides, as summarized in Figure 4. Among the 789 carbamylated peptides identified by UVPD, a larger portion was detected as 3– and 2– charge states, whereas more were detected as 2– and 1– charge states among the 549 peptides identified for the noncarbamylated digest. More importantly, the average peptide abundances were higher for the carbamylated digest than the unmodified digest by a factor of 2.4, thus confirming the signal enhancement obtained by reducing the net pI values of the peptides upon carbamylation.
Figure 3.
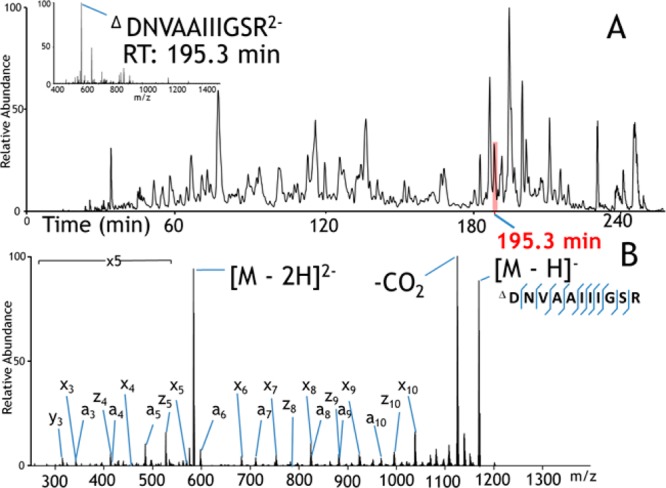
(A) Negative mode LC–MS trace (total ion chromatogram) of H. salinarum tryptic peptides. Inset: ESI mass spectrum acquired at 195.3 min. (B) UVPD mass spectrum of carbamylated peptide DNVAAIIIGSR (2−) eluting at 195.3 min.
Figure 4.
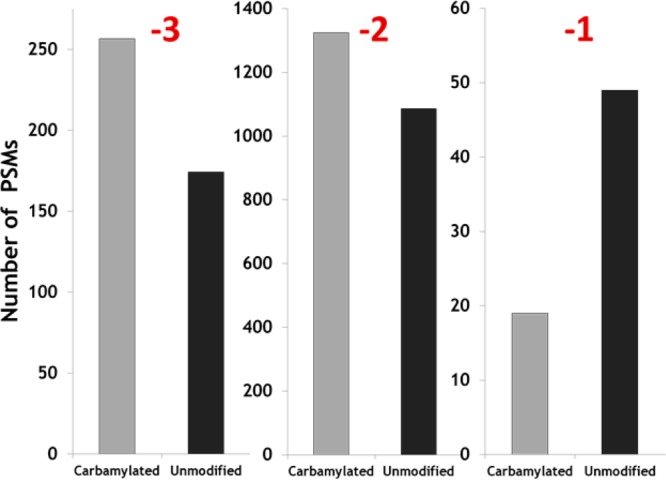
Distributions of charge states for carbamylated (light bars) and unmodified (dark bars) tryptic peptide spectral matches (PSMs) found for the digest of H. salinarum.
The UVPD fragmentation patterns of the more highly charged peptides give better peptide sequence coverage. For carbamylated peptides, 1.7 diagnostic ions per residue on average were generated for peptides in the 3– charge state, 1.3 diagnostic ions per residue for peptides in the 2– charge state, and 0.6 diagnostic ions per residue in the 1– charge state. The similarities in the average number and types of fragment ions for carbamylated versus noncarbamylated peptides offers assurance that the carbamylation reaction does not suppress or significantly alter the rich UVPD patterns generated for peptide anions. With respect to the types of fragment ions, the distributions are nearly identical for both the carbamylated and unmodified peptides: averaging 50% a/x ions, 30% b/y ions, and 20% c/z ions (see Supporting Figure 6 in the Supporting Information).
The average number and standard deviation of peptides and proteins identified from the H. salinarum proteome were calculated from triplicate negative mode LC–MS UVPD analyses of the carbamylated and the unmodified tryptic digests, as summarized in Figures 5 (histograms) and 6 (Venn diagrams). Combining three runs led to the identification of 1086 peptides for the carbamylated digest compared to 747 for the unmodified digest. Similarly, at the protein level, 430 proteins were identified based on the peptides found in the carbamylated digests compared to 348 proteins for the unmodified digests. Upon combining the results of three runs, 682 peptides were found uniquely for the carbamylated digest, 343 peptides were found exclusively for the unmodified digest, and surprisingly only 404 were found for both digests. At the protein level, 156 proteins were uniquely identified from the results of the carbamylated tryptic digest, whereas 74 were exclusively found for the unmodified digest, and 274 proteins were identified in both cases. The results show 45% more peptide identifications and 25% more protein identifications after carbamylation when compared to the unmodified digest. With respect to the charge states of the peptides that were identified, on average 267 carbamylated peptide spectral matches (PSMs) were found in the 3– charge state compared to 174 PSMs for unmodified peptides, 1325 carbamylated PSMs were found in the 2– charge state compared to 1086 PSMs for unmodified peptides, and 19 carbamylated PSMs were found in the 1– charge state compared to 49 PSMs for unmodified peptides.
Figure 5.
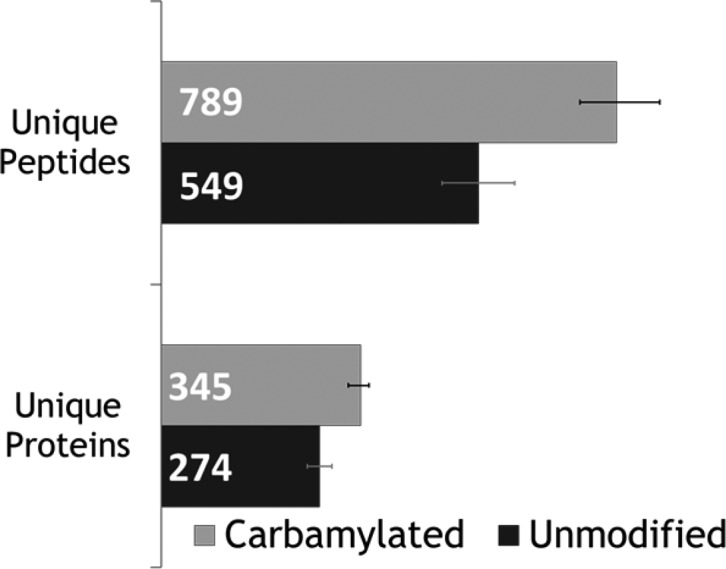
Average number of peptide and protein identifications from negative LC/UVPD-MS analyses of carbamylated and unmodified H. salinarum tryptic peptides (in triplicate).
Figure 6.
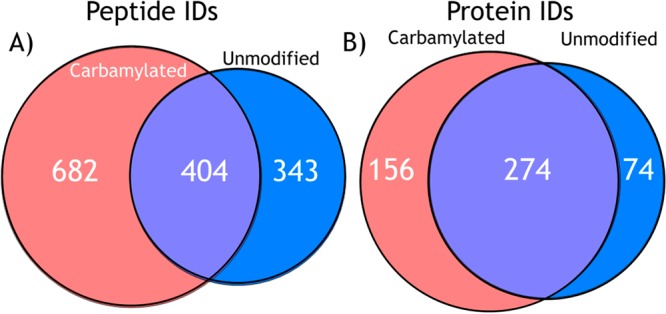
Combined number of (A) unique peptide and (B) protein identifications from LC/UVPD-MS analyses of carbamylated and unmodified tryptic peptides from H. salinarum in the negative mode in triplicate.
Interestingly a reasonably large number of peptides (343) were identified only from analysis of the unmodified digest. Given the stochastic nature of data dependent acquisition and bias toward the most abundant peptide precursor ions,26 many peptides are not selected for fragmentation in a routine mass spectrometry proteomics experiment.27 Closer inspection of those peptides identified only for the unmodified tryptic digests show larger peptides on average than ones commonly identified for the carbamylated digests (Supporting Figure 7 in the Supporting Information). On the basis of the retention times of these peptides (Supporting Figure 8 in the Supporting Information), they are also more hydrophobic (with longer elution times), and thus carbamylation of these large peptides would be expected to further increase their hydrophobicities and further delay elution. This may account in part for why this set of peptides was not identified for the corresponding carbamylated digest.
Conclusion
Carbamylation of lysine residues and N-termini was utilized to enhance the ionization of peptides by negative polarity ESI and improve the sensitivity of negative mode LC/UVPD-MS analyses. Results show a significant enhancement in negative mode ionization of carbamylated peptides compared to unmodified peptides, consistent with the significant decrease in pKa upon carbamylation of primary amines. Carbamylation of tryptic digests resulted in 45% more peptide identifications and 25% more protein identifications compared to that obtained for the unmodified digests, confirming the enhancement in sensitivity in the negative mode. The improvement in peptide identification metrics also arises from a shift to higher charge states, thus yielding more efficient UVPD. The carbamylation method could be combined with other proteases, like LysC, to ensure multiple modifications of each peptide.
Acknowledgments
Funding from the NIH (Grant R21GM099028) and the Robert A. Welch Foundation (Grant F1155) is gratefully acknowledged. J.R.C. acknowledges support from the NIH (Grant 1K12GM102745).
Supporting Information Available
pI distributions of in silico generated tryptic peptides from three proteomes, a reaction scheme showing the carbamylation of a peptide, extracted ion chromatograms and ESI mass spectra of carbamylated and unmodified peptides, the distributions of ion types arising from UVPD of carbamylated and unmodified tryptic peptides, mass distributions of peptides from carbamylated and unmodified tryptic digests, the peptides identified for an unmodified tryptic digest, and a summary of peptides for carbamylated and unmodified peptides from bovine serum albumin. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bensimon A.; Heck A. J. R.; Aebersold R. Annu. Rev. Biochem. 2012, 81, 379–405. [DOI] [PubMed] [Google Scholar]
- Gunaratne J.; Schmidt A.; Quandt A.; Neo S. P.; Sarac O. S.; Gracia T.; Loguercio S.; Ahrne E.; Xia R. L. H.; Tan K. H.; Lossner C.; Bahler J.; Beyer A.; Blackstock W.; Aebersold R. Mol. Cell. Proteomics 2013, 12, 1741–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gygi S.; Villén J. Nat. Protoc. 2008, 3, 1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonslow B. R.; Stein B. D.; Webb K. J.; Xu T.; Choi J.; Park S. L.; Yates J. R. Jr. Nat. Methods 2013, 10, 54–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn E.; Whiteaker J.; Mani D. R.; Jackson A.; Lei Z.; Pope M.; Smith D.; Rivera K.; Anderson N. L.; Skates S. J.; Pearson T. W.; Paulovich A. G.; Carr S. A. Mol. Cell. Proteomics 2012, 9, 184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister G. C.; Russell J. D.; Rumachik N. G.; Hebert A. S.; Syka J. E. P.; Geer L. Y.; Westphall M. S.; Pagliarini D. J.; Coon J. J. Anal. Chem. 2012, 84, 2875–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen J. A.; Xu H.; Robinson M. R.; Horton A. P.; Shaw J. B.; Giles D. K.; Kaoud T. S.; Dalby K. N.; Trent M. S.; Brodbelt J. S. Mol. Cell. Proteomics 2013, 12, 2604–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz R.; Ting C. S.; King J. Genome Res. 2001, 11, 703–709. [DOI] [PubMed] [Google Scholar]
- Wells J. M.; McLuckey S. A. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2005; Vol. 402, pp 148–185. [DOI] [PubMed] [Google Scholar]
- Cooper H. J.; Håkansson K.; Marshall A. G. Mass Spectrom. Rev. 2005, 24, 201–222. [DOI] [PubMed] [Google Scholar]
- Syka J. E. P.; Coon J. J.; Schroeder M. J.; Shabanowitz J.; Hunt D. F. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 9528–9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner M. A.; Ledvina A. R.; Smith S.; Madsen J.; Schwartz G. C.; Stafford G. C.; Coon J. J.; Brodbelt J. S. Anal. Chem. 2009, 81, 8109–8118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjeldsen F.; Hørning O. B.; Jensen S. S.; Giessing A. M. B.; Jensen O. N. J. Am. Soc. Mass Spectrom. 2008, 19, 1156–1162. [DOI] [PubMed] [Google Scholar]
- Coon J. J.; Shabanowitz J.; Hunt D. F.; Syka J. E. P. J. Am. Soc. Mass. Spectrom. 2005, 16, 880–882. [DOI] [PubMed] [Google Scholar]
- Madsen J. A.; Kaoud T. S.; Dalby K. N.; Brodbelt J. S. Proteomics 2011, 11, 1329–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw J. B.; Madsen J. A.; Xu H.; Brodbelt J. S. J. Am. Soc. Mass Spectrom. 2012, 23, 1707–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson J. C.; Fage C. D.; Cannon J. R.; Brodbelt J. S.; Keatinge-Clay A. T.; Trent M. S. ACS Chem. Biol. 2014, 9, 2382–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y.; Yogesha S. D.; Cannon J. R.; Yan W.; Brodbelt J. S.; Zhang Y. ACS Chem. Biol. 2013, 8, 2042–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S. W.; Lee S. W.; Bahar O.; Schwessinger B.; Robinson M. R.; Shaw J. B.; Madsen J. A.; Brodbelt J. S. Nat. Commun. 2012, 3, 1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.; Moore K.; Brodbelt J. S. J. Am. Soc. Mass Spectrom. 2014, 25, 1461–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angel P. M.; Orlando R. Rapid Commun. Mass Spectrom. 2007, 10, 1623–1634. [DOI] [PubMed] [Google Scholar]
- Allen S. J.; Schwartz A. M.; Bush M. F. Anal. Chem. 2013, 85, 12055–12061. [DOI] [PubMed] [Google Scholar]
- Douglass K. A.; Venter A. R. Anal. Chem. 2013, 85, 8212–8218. [DOI] [PubMed] [Google Scholar]
- Konermann L.; Ahadi E.; Rodriguez A. D.; Vahidi S. Anal. Chem. 2013, 85, 2–9. [DOI] [PubMed] [Google Scholar]
- Vasicek L. A.; Ledvina A. R.; Shaw J. B.; Griep-Raming J.; Westphall M. S.; Coon J. J.; Brodbelt J. S. J. Am. Soc. Mass Spectrom. 2011, 22, 1105–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Sadygov R. G.; Yates J. R. Anal. Chem. 2004, 76, 4193–4201. [DOI] [PubMed] [Google Scholar]
- Michalski A.; Cox J.; Mann M. J. Proteome Res. 2011, 10, 1785–1793. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.