

Abstract
Exposure to clinical doses of the glucocorticoid dexamethasone increases brain activity and causes seizures in normoxic preterm fetal sheep without causing brain injury. In contrast, the same treatment after asphyxia increased brain injury. We hypothesised that increased injury was in part mediated by a mismatch between oxygen demand and oxygen supply. In preterm fetal sheep at 0.7 gestation we measured cerebral oxygenation using near-infrared spectroscopy, electroencephalographic (EEG) activity, and carotid blood flow (CaBF) from 24 h before until 72 h after asphyxia induced by 25 min of umbilical cord occlusion. Ewes received dexamethasone intramuscularly (12 mg 3 ml–1) or saline 15 min after the end of asphyxia. Fetuses were studied for 3 days after occlusion. During the first 6 h of recovery after asphyxia, dexamethasone treatment was associated with a significantly greater fall in CaBF (P < 0.05), increased carotid vascular resistance (P < 0.001) and a greater fall in cerebral oxygenation as measured by the difference between oxygenated and deoxygenated haemoglobin (delta haemoglobin; P < 0.05). EEG activity was similarly suppressed in both groups. From 6 to 10 h onward, dexamethasone treatment was associated with a return of CaBF to saline control levels, increased EEG power (P < 0.005), greater epileptiform transient activity (P < 0.001), increased oxidised cytochrome oxidase (P < 0.05) and an attenuated increase in [delta haemoglobin] (P < 0.05). In conclusion, dexamethasone treatment after asphyxia is associated with greater hypoperfusion in the critical latent phase, leading to impaired intracerebral oxygenation that may exacerbate neural injury after asphyxia.
Key Points
Mothers at risk of preterm delivery are routinely given synthetic glucocorticoids such as dexamethasone to help mature fetal lungs and improve survival after birth.
We have previously shown that dexamethasone given after an acute episode of asphyxia in preterm fetal sheep is associated with greater brain injury.
In this study we found that fetal exposure to dexamethasone after asphyxia in preterm fetal sheep was associated with reduced intracerebral oxygenation during the critical latent phase of recovery.
In the secondary phase, maternal dexamethasone was associated with increased epileptiform transient activity and evidence of greater mitochondrial oxidation.
These findings suggest that fetal exposure to the synthetic glucocorticoid dexamethasone is associated with a critical mismatch between the brain's demand for oxygenation and the supply of oxygen that may contribute to greater brain injury.
Introduction
Neurodevelopmental disability after premature birth is multifactorial (Mallard et al. 2014). Metabolic acidosis measured from cord blood and the need for resuscitation at birth are common among preterm babies and associated with an adverse neonatal course (Gezer et al. 2013) and increased risk of white matter injury (Reid et al. 2014). Among these cases, overt asphyxia occurs in approximately 20/1000 preterm infants, and is associated with a high risk of death, neural injury on modern imaging and disability among survivors (Low et al. 2003; Kerstjens et al. 2012; Sukhov et al. 2012; Corchia et al. 2013). The majority of preterm fetuses at risk of delivery are now exposed to maternal treatment with synthetic glucocorticoids such as dexamethasone or betamethasone (Lundqvist et al. 2009), which has significantly reduced perinatal morbidity and mortality (Roberts & Dalziel, 2006). Surprisingly, there is little direct information on how exposure to maternal glucocorticoid treatment affects fetal adaptation to asphyxia. Nearly all our knowledge is based on postnatal rodent studies, which largely suggest that steroids protect against hypoxic–ischaemic injury (as recently reviewed by Bennet et al. 2012b). Exposure to dexamethasone was, however, not protective in near-term fetal sheep against neural injury induced by acute reversible carotid artery occlusion (Elitt et al. 2003). Moreover, in contrast to postnatal rodent studies, we found that exposure to maternal dexamethasone after a severe acute asphyxial insult increased both white and grey matter injury in preterm fetal sheep (Koome et al. 2013).
The specific reasons for these conflicting results are unknown, but potentially in our recent study, dexamethasone may have impaired key protective fetal cerebral metabolic responses during recovery from asphyxia. There is now considerable evidence that preterm brain injury evolves over time after asphyxia (Bennet et al. 2010). For approximately the first 6 h after the end of the insult, there is a ‘latent’ phase during which mitochondrial oxidative metabolism recovers at least partly to normal (Bennet et al. 2006). During this time electroencephalographic (EEG) activity is suppressed and cerebral perfusion is reduced (Quaedackers et al. 2004), probably coupled to reduced cerebral metabolism (Jensen et al. 2006; Yan et al. 2009). Further, although EEG amplitude is suppressed during the latent phase, there can be a transient increase in spectral edge frequency mediated by epileptiform transients (low amplitude, high frequency events). Greater epileptiform transient activity in the latent phase after asphyxia in preterm fetal sheep is associated with greater neural injury (Dean et al. 2006; Bennet et al. 2010). Of particular interest, epileptiform transient activity is associated in time with reduced intracerebral oxygenation measured by near-infrared spectroscopy (NIRS) (Bennet et al. 2006).
The latent phase is followed by the secondary phase, which extends over several days and is characterised by secondary loss of cerebral oxidative metabolism (Bennet et al. 2006). This phase is associated with high amplitude, stereotypic evolving seizures and a partial restoration of cerebral perfusion (Bennet et al. 2006; Davidson et al. 2014). NIRS studies have shown that the fetal brain is well oxygenated during the secondary phase, consistent with the concept of luxury perfusion where oxygen delivery significantly exceeds cerebral metabolic demand (Bennet et al. 2006).
We have previously shown, in healthy, normoxic fetal sheep, that exposure to maternal dexamethasone was associated with transient EEG hyperexcitability, with marked induction of seizures and seizure-like activity (Davidson et al. 2014). After fetal asphyxia, maternal dexamethasone treatment was associated with increased epileptiform transients during the latent phase, although there was no increase in the burden of seizures in the secondary phase (Koome et al. 2013). Thus, we wished to test the hypothesis that dexamethasone may cause loss of protective endogenously mediated neural suppression during the latent phase of recovery from asphyxia in preterm fetal sheep at 0.7 of gestation leading to increased cerebral hypoxia. At this age, brain development is broadly equivalent to human brain development at 27–30 weeks’ gestation (McIntosh et al. 1979).
Methods
Fetal surgery
All procedures were approved by the Animal Ethics Committee of the University of Auckland. Fifty-eight singleton Romney/Suffolk fetal sheep were surgically instrumented at 98–100 days of gestation (term = 147 days) as previously described (Bennet et al. 1999b, 2006; Davidson et al. 2011; Koome et al. 2013). Ewes were anaesthetised by intravenous injection of propofol (5 mg kg−1; AstraZeneca Limited, Auckland, New Zealand) and general anaesthesia was maintained with 2–3% isoflurane in oxygen. A midline incision was made to expose the uterus, and the fetus was partially exteriorised for instrumentation. Polyvinyl catheters were placed in the left femoral artery (with the catheter positioned to sit in the aorta) and vein (with the catheter positioned to sit in the inferior vena cava) to measure blood pressures and right brachial artery for pre-ductal blood sampling. An additional catheter was placed into the amniotic sac for measurement of amniotic fluid pressure.
An ultrasound flow probe (size 3S; Transonic Systems, Ithaca, NY, USA) was placed around the left carotid artery to measure carotid artery blood flow (CaBF) as an index of cerebral blood flow (van Bel et al. 1994; Gratton et al. 1996; Gonzalez et al. 2005). Two pairs of electrodes (Cooner Wire, Chatsworth, CA, USA) were placed over the parietal cortex bilaterally, 10 mm lateral to bregma and 5 mm and 10 mm anterior to measure EEG activity. A reference electrode was sewn over the occiput. A pair of electrodes was placed across the fetal chest to measure the fetal electrocardiogram from which fetal heart rate (FHR) was derived.
In addition, in a subset of 21 fetuses, two small flexible fibre-optic probes used for NIRS recordings were placed biparietally on the skull 3.0–3.5 cm apart, 1.5 cm anterior to the bregma in fetuses assigned to the sham–saline, asphyxia–saline and asphyxia–dexamethasone groups, and secured using rapid setting dental cement (Rocket Red; Dental Adventures of America Inc., Anaheim, CA, USA) (Bennet et al. 1999b, 2006; Drury et al. 2012). An inflatable silicone occluder was placed around the umbilical cord to facilitate umbilical cord occlusions (In Vivo Metric, Healdsburg, CA, USA). All fetal leads were exteriorised through the maternal flank, and a maternal long saphenous vein was catheterised for postoperative care.
Gentamicin was administered into the amniotic sac (80 mg gentamicin; Pharmacia & Upjohn, Rydalmere, New South Wales, Australia) before the uterus was closed. Ewes were given 5 ml of streptocin (Stockguard Labs Ltd., Hamilton, New Zealand) intramuscularly 30 min before surgery for prophylaxis. The maternal midline skin incision was infiltrated with a local analgesic, 10 ml 0.5% bupivacaine plus adrenaline (AstraZeneca Ltd., Auckland, New Zealand).
Postoperative care
Following surgery, ewes were housed together in separate metabolic cages with ad libitum access to food and water. Rooms were temperature and humidity controlled (16 ± 1°C, humidity 50 ± 10%) with a 12 h light/dark cycle (light 06.00–18.00 h). Ewes were given daily intravenous antibiotics [600 mg Crystapen (Biochemie, Vienna, Austria) and 80 mg gentamicin (Pharmacia & Upjohn)] for 4 days after surgery. Fetal catheters were maintained patent with continuous infusion of heparinised saline (20 U ml−1 at 0.15 ml h−1). Experiments began 4–5 days after surgery.
Data recording
Fetal mean arterial blood pressure (MAP), central venous pressure, CaBF, FHR, EEG and NIRS parameters were recorded continuously from 24 h before until 72 h after umbilical cord occlusion (Davidson et al. 2011; Koome et al. 2013). All signals were digitised at a sampling rate of 4096 Hz and decimated to lower rates for data analysis. Data were stored and processed using the program Labview (National Instruments, Austin, TX, USA). Fetal blood pressures were recorded using Novatrans II, MX860 pressure transducers (Medex Inc., Hilliard, OH, USA) and corrected for maternal movement by subtraction of amniotic fluid pressure. The blood pressure signals were collected at 64 Hz and low-pass filtered at 30 Hz. CaBF was measured continuously using a two-channel Transonic T-206 Flowmeter (Transonic Systems Inc.) and data were 10 Hz low-pass filtered with a second order Butterworth filter. The raw electrocardiogram signal was analogue filtered with a first order high-pass filter set at 0.05 Hz and an eight order low-pass Bessel filter set at 100 Hz.
EEG signals were recorded via leads through a head-stage with an overall gain of 10,000. Signals were then processed with a sixth order low-pass Butterworth filter set to 500 Hz. Total EEG power (μV2) was calculated on the power spectrum between 1 Hz and 20 Hz. EEG spectral edge frequency was calculated as the frequency below which 90% of EEG power was present. EEG power was log transformed for presentation [dB, 20× log (power) (Szeto, 1990; Williams & Gluckman, 1990)]. EEG signals were saved at 1024 Hz for analysis of epileptiform transient activity. Concentration changes in fetal cerebral deoxyhaemoglobin ([Hb]), oxyhaemoglobin ([HbO2]) and cytochrome oxidase ([CytOx]) were measured using a NIRO 500 spectrophotometer (Hamamatsu Photonics KK, Hamamatsu City, Japan) as previously described (Bennet et al. 1999b, 2006; Drury et al. 2012). Data were collected as 10 s averages. The NIRS measures obtained were relative changes from zero not absolute changes (Bennet et al. 1999b, 2006; Drury et al. 2012). Signals were zeroed at the start of baseline recordings and are displayed relative to the average of the 24 h baseline period.
Experimental protocol
Experiments were conducted at 103–104 days of gestation. Fetuses were randomly assigned to sham asphyxia–saline (sham–saline, n = 13), sham asphyxia–dexamethasone (sham–dexamethasone, n = 12), asphyxia–saline (n = 18) or asphyxia–dexamethasone (n = 15) groups. Fetal asphyxia was induced by complete umbilical cord occlusion for 25 min (Bennet et al. 2006). Occlusions were started between 10.00 and 10.30 h. Sham asphyxia fetuses received no occlusion. To maximise group sizes to ensure statistical power, and in keeping with our animal ethics commitment to the 3Rs principle of minimizing animal experiments, we have included animals from previously published studies in the following groups: sham–saline, n = 13, five with NIRS; six previous animals including four with NIRS (Bennet et al. 2006) and seven new animals including one with NIRS, asphyxia–saline, n = 18, nine with NIRS; seven previous animals including three with NIRS (Bennet et al. 2006; Drury et al. 2013; Koome et al. 2013) and 11 new animals including six with NIRS, and asphyxia–dexamethasone, n = 15, seven with NIRS; eight previous animals (Koome et al. 2013) and seven new animals all with NIRS. All animals used in this study were judged as healthy and within the ranges considered normal for our laboratory after assessment of blood gases and physiological parameters before starting experiments.
Fifteen minutes after the end of occlusion or sham occlusion, ewes received a 3 ml intramuscular injection of either dexamethasone (12 mg dexamethasone sodium phosphate; David Bull Laboratories, Mulgrave, Victoria, Australia) or the equivalent volume of saline. Fetal arterial blood samples (0.3 ml) were taken 15 min before occlusion, at 5 and 17 min during occlusion, and at 2, 4 and 6 h post-occlusion, then daily thereafter between 08.30 and 09.30 h. Blood samples were analysed for pH and blood gases (blood gas analyser 845 and co-oximeter; Ciba-Corning Diagnostics, Cambridge, MA, USA) and glucose and lactate levels (YSI 2300, YSI Life Sciences, Yellow Springs, OH, USA). Three days after occlusion, ewes and fetuses were killed by an overdose of sodium pentobarbital intravenously to the ewe (9 g Pentobarb 300; Chemstock International, Christchurch, New Zealand).
Data analysis
Offline analysis of the physiological data was carried out using customised Labview programs (National Instruments). Changes in cerebral [HbO2], [Hb] and [CytOx] were calculated from a modified Lambert–Beer law using a previously established algorithm that describes optical absorption in a highly scattering medium (Wray et al. 1988; Bennet et al. 1999b, 2006; Drury et al. 2012). [DHb] was calculated as the difference between [HbO2] and [Hb], as a measure of net intracerebral oxygenation (Brun et al. 1997; Bennet et al. 2006). Carotid vascular resistance (CaVR) was calculated using the formula (MAP – mean venous pressure)/carotid blood flow (mmHg min−1 ml−1).
Continuous raw EEG traces in both asphyxia groups were analysed each hour after occlusion to determine the percentage of time spent making repetitive rhythmic slow wave transient epileptiform activity, as previously described (Koome et al. 2013). These waveforms were defined by a period of 200–350 ms from trough to peak, with waveforms forming consistent events lasting >10 s. These waveforms were often seen in conjunction with sharp and fast wave epileptiform transients, characterised as individual or multiple waveforms with a duration of >70 ms and <300 ms (Davidson et al. 2012).
Statistical analysis
Statistical analysis was performed using SPSS v22 (SPSS Inc., Chicago, IL, USA). The time course of changes in cardiovascular, haemodynamic, EEG and NIRS parameters after asphyxia were analysed using hourly averages in the following three phases: the latent phase (2–6 h after occlusion), early secondary phase (7–24 h after occlusion) and late secondary phase (25–72 h after occlusion). The first hour after occlusion was omitted from statistical analysis because dexamethasone was given 15 min after occlusion. The baseline period was taken as the mean of the 24 h before occlusion. Treatment effects on MAP, FHR, EEG power, EEG spectral edge frequency, CaBF and CaVR were evaluated by three-way ANOVA with asphyxia and dexamethasone as independent factors and changes over time treated as repeated measures. Treatment effects on NIRS parameters were evaluated by two-way ANOVA with group as the independent factor and changes over time treated as repeated measures. Treatment effects on fetal biochemistry were evaluated in the following three phases: during occlusion (5 and 17 min during occlusion), latent phase (2–6 h after occlusion) and secondary phase (24–72 h after occlusion) and assessed using three-way ANOVA as above.
If there was a significant interaction effect of combined treatment or of treatment with time after three-way ANOVA, or a significant effect of group after two-way ANOVA then the effect of group was further investigated using the Fisher's protected least significant difference (LSD) post-hoc test. Additional time intervals were tested as appropriate, including the nadir of [DHb], which was compared to baseline values by one-way ANOVA and the P value corrected for number of comparisons. The changes in post-occlusion interictal EEG activity were compared between the two asphyxia groups by two-way ANOVA with group as the independent factor and changes over time treated as repeated measures. Statistical significance was accepted when P < 0.05. Data are presented as means ± SEM.
Results
Fetal biochemistry
Umbilical cord occlusion was associated with profound hypoxia, mixed respiratory and metabolic acidosis and a fall in plasma glucose levels (Table 1, P < 0.001), which resolved after release of occlusion. Over the entire recovery period after occlusion, asphyxia was associated with a small decrease in 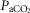 (P < 0.005) while dexamethasone treatment was associated with lower pH (P < 0.05). Both asphyxia and dexamethasone were independently associated with increased
(P < 0.005) while dexamethasone treatment was associated with lower pH (P < 0.05). Both asphyxia and dexamethasone were independently associated with increased 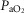 (P < 0.05), increased glucose levels (P < 0.05) and increased lactate levels (P < 0.001) on all three days of recovery after occlusion.
(P < 0.05), increased glucose levels (P < 0.05) and increased lactate levels (P < 0.001) on all three days of recovery after occlusion.
Table 1.
Fetal blood gases, pH and metabolites after fetal asphyxia and maternal dexamethasone treatment
| 5 min | 17 min | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Group | Baseline | occlusion | occlusion | +2 h | +4 h | +6 h | +24 h | +48 h | +72 h | |
| pH | SS | 7.36 ± 0.01 | 7.37 ± 0.01 | 7.38 ± 0.01 | 7.39 ± 0.01 | 7.39 ± 0.00 | 7.38 ± 0.01 | 7.37 ± 0.00 | 7.37 ± 0.01 | 7.37 ± 0.01 |
| SD | 7.37 ± 0.01 | 7.37 ± 0.01 | 7.38 ± 0.01 | 7.37 ± 0.01‡ | 7.35 ± 0.01‡ | 7.36 ± 0.01‡ | 7.37 ± 0.01‡ | 7.33 ± 0.02‡ | 7.36 ± 0.02‡ | |
| AS | 7.37 ± 0.01 | 7.04 ± 0.01† | 6.83 ± 0.01† | 7.35 ± 0.02 | 7.41 ± 0.01 | 7.39 ± 0.01 | 7.37 ± 0.01 | 7.37 ± 0.01 | 7.38 ± 0.01 | |
| AD | 7.37 ± 0.01 | 7.04 ± 0.01† | 6.87 ± 0.03† | 7.32 ± 0.02‡ | 7.36 ± 0.01‡ | 7.37 ± 0.01‡ | 7.38 ± 0.01‡ | 7.33 ± 0.02‡ | 7.32 ± 0.03‡ | |
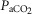 (mmHg) (mmHg) |
SS | 49.3 ± 1.2 | 47.4 ± 0.8 | 47.5 ± 1.1 | 47.4 ± 1.0 | 48.3 ± 1.1 | 48.6 ± 1.1 | 47.3 ± 1.5 | 48.8 ± 1.3 | 46.2 ± 1.4 |
| SD | 51.1 ± 1.3 | 47.1 ± 0.8 | 47.2 ± 1.1 | 52.7 ± 1.6 | 50.4 ± 1.7 | 47.6 ± 1.4 | 50.5 ± 1.9 | 50.1 ± 1.7 | 47.2 ± 1.5 | |
| AS | 49.3 ± 1.0 | 101.5 ± 3.2† | 147.7 ± 3.7† | 43.8 ± 1.0† | 44.5 ± 1.0† | 47.0 ± 0.8† | 46.3 ± 1.1† | 46.5 ± 1.3† | 45.6 ± 1.0† | |
| AD | 48.1 ± 1.1 | 97.0 ± 2.4† | 133.0 ± 4.3† | 44.5 ± 1.1† | 44.3 ± 0.8† | 44.9 ± 1.1† | 42.4 ± 0.9† | 44.2 ± 0.9† | 46.3 ± 1.1† | |
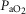 (mmHg) (mmHg) |
SS | 23.8 ± 0.6 | 23.9 ± 0.6 | 23.7 ± 0.7 | 23.0 ± 0.6 | 23.6 ± 0.6 | 23.4 ± 0.5 | 23.4 ± 0.7 | 23.5 ± 0.8 | 23.9 ± 0.6 |
| SD | 23.8 ± 0.6 | 23.6 ± 0.6 | 23.5 ± 0.7 | 22.4 ± 1.1‡ | 24.0 ± 1.4‡ | 23.8 ± 0.9‡ | 25.1 ± 0.9‡ | 24.4 ± 1.7‡ | 24.3 ± 1.2‡ | |
| AS | 23.2 ± 0.7 | 5.6 ± 0.4† | 7.0 ± 0.4† | 24.6 ± 0.8† | 23.6 ± 1.6† | 24.6 ± 1.5† | 26.0 ± 0.9† | 26.9 ± 1.1† | 26.7 ± 0.9† | |
| AD | 24.5 ± 0.4 | 7.8 ± 0.7† | 9.3 ± 0.9† | 28.2 ± 0.9†,‡ | 27.5 ± 0.9†,‡ | 27.4 ± 0.8†,‡ | 30.9 ± 0.6†,‡ | 32.6 ± 0.9†,‡ | 32.0 ± 1.5†,‡ | |
| Hct (%) | SS* | 25.6 ± 0.9 | 25.6 ± 0.4 | 25.8 ± 0.5 | 25.4 ± 0.7 | 26.2 ± 0.6 | 26.4 ± 0.4 | 25.6 ± 0.7 | 27.0 ± 0.9 | 28.4 ± 1.1 |
| SD | 27.1 ± 1.0 | 25.9 ± 0.7 | 26.2 ± 0.8 | 30.7 ± 2.2 | 29.2 ± 1.5 | 30.0 ± 1.4 | 25.5 ± 1.9 | 26.4 ± 1.5 | 26.4 ± 2.0 | |
| AS* | 25.8 ± 1.0 | 28.1 ± 1.0 | 27.4 ± 0.9 | 27.2 ± 1.0 | 28.1 ± 1.2 | 27.3 ± 1.4 | 28.2 ± 1.3 | 27.3 ± 1.1 | 28.1 ± 1.2 | |
| AD* | 23.0 ± 0.9 | 27.0 ± 0.9 | 25.4 ± 0.9 | 26.4 ± 1.0 | 26.3 ± 0.8 | 27.3 ± 1.0 | 25.4 ± 1.3 | 25.3 ± 1.8 | 26.3 ± 2.8 | |
| O2ct (mmol l−1) | SS* | 3.7 ± 0.1 | 3.8 ± 0.2 | 3.8 ± 0.2 | 3.7 ± 0.2 | 3.9 ± 0.1 | 3.9 ± 0.1 | 3.4 ± 0.2 | 3.6 ± 0.2 | 3.9 ± 0.2 |
| SD | 3.9 ± 0.2 | 3.6 ± 0.2 | 3.7 ± 0.1 | 4.0 ± 0.4 | 4.0 ± 0.3 | 4.2 ± 0.3 | 4.1 ± 0.2 | 3.7 ± 0.3 | 3.7 ± 0.3 | |
| AS* | 3.5 ± 0.2 | 0.5 ± 0.0† | 0.4 ± 0.0† | 4.0 ± 0.2 | 4.0 ± 0.3 | 4.1 ± 0.3 | 4.2 ± 0.2† | 4.1 ± 0.3† | 4.5 ± 0.2† | |
| AD* | 3.5 ± 0.1 | 0.5 ± 0.0† | 0.4 ± 0.0† | 4.1 ± 0.1 | 4.2 ± 0.2 | 4.2 ± 0.1 | 4.3 ± 0.1† | 4.3 ± 0.2† | 4.3 ± 0.4† | |
| Lactate (mmol l−1) | SS | 0.6 ± 0.0 | 0.7 ± 0.0 | 0.7 ± 0.0 | 0.7 ± 0.0 | 0.7 ± 0.0 | 0.7 ± 0.0 | 0.7 ± 0.0 | 0.7 ± 0.0 | 0.8 ± 0.1 |
| SD | 0.7 ± 0.1 | 0.7 ± 0.0 | 0.7 ± 0.0 | 0.9 ± 0.1‡ | 1.4 ± 0.1 ‡ | 1.6 ± 0.2‡ | 0.8 ± 0.1‡ | 1.1 ± 0.2‡ | 0.9 ± 0.1‡ | |
| AS | 0.8 ± 0.0 | 4.0 ± 0.1† | 7.0 ± 0.2† | 3.3 ± 0.3† | 1.9 ± 0.3† | 1.9 ± 0.2† | 1.1 ± 0.1† | 1.0 ± 0.1† | 0.8 ± 0.1† | |
| AD | 0.8 ± 0.0 | 4.0 ± 0.1† | 6.5 ± 0.3† | 4.2 ± 0.4†,‡ | 4.2 ± 0.4†,‡ | 4.7 ± 0.4†,‡ | 1.6 ± 0.1†,‡ | 0.9 ± 0.1†,‡ | 0.8 ± 0.1†,‡ | |
| Glucose (mmol l−1) | SS | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.1 |
| SD | 0.9 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.2 ± 0.1‡ | 1.7 ± 0.1‡ | 2.0 ± 0.2‡ | 1.3 ± 0.1‡ | 1.0 ± 0.1‡ | 1.0 ± 0.1‡ | |
| AS | 1.0 ± 0.1 | 0.5 ± 0.2† | 0.6 ± 0.1† | 1.3 ± 0.1† | 1.2 ± 0.1† | 1.4 ± 0.1† | 1.2 ± 0.1† | 1.1 ± 0.1† | 1.1 ± 0.1† | |
| AD | 1.0 ± 0.1 | 0.3 ± 0.0† | 0.7 ± 0.1† | 1.5 ± 0.1†,‡ | 2.3 ± 0.1†,‡ | 3.2 ± 0.1†,‡ | 2.1 ± 0.1†,‡ | 1.3 ± 0.1†,‡ | 1.1 ± 0.1†,‡ |
Data are means ± SEM. Abbreviations: AD, asphyxia–dexamethasone; AS, asphyxia–saline; Hct, haematocrit, O2ct, arterial blood oxygen content; 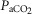 , arterial pressure of carbon dioxide;
, arterial pressure of carbon dioxide; 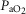 , arterial pressure of oxygen; SD, sham–dexamethasone; SS, sham–saline. *Data only shown for fetuses with near-infrared spectroscopy measurements. †Effect of asphyxia, P < 0.05. ‡Effect of dexamethasone treatment, P < 0.05.
, arterial pressure of oxygen; SD, sham–dexamethasone; SS, sham–saline. *Data only shown for fetuses with near-infrared spectroscopy measurements. †Effect of asphyxia, P < 0.05. ‡Effect of dexamethasone treatment, P < 0.05.
These three later parameters all showed a significant interaction between asphyxia and dexamethasone treatment (Table 1, P < 0.05), such that post-hoc analysis suggested that the asphyxia–dexamethasone group had a higher 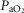 (P < 0.005, LSD test) and higher glucose levels (P < 0.001, LSD test) compared to all other groups over the recovery period. The sham–dexamethasone group had higher glucose levels than the two saline-treated groups on the first day of recovery (P < 0.01, LSD test), but lower levels than the asphyxia–dexamethasone group (P < 0.001, LSD test). The asphyxia–dexamethasone group also had higher lactate levels than all other groups during recovery (P < 0.05, LSD test).
(P < 0.005, LSD test) and higher glucose levels (P < 0.001, LSD test) compared to all other groups over the recovery period. The sham–dexamethasone group had higher glucose levels than the two saline-treated groups on the first day of recovery (P < 0.01, LSD test), but lower levels than the asphyxia–dexamethasone group (P < 0.001, LSD test). The asphyxia–dexamethasone group also had higher lactate levels than all other groups during recovery (P < 0.05, LSD test).
Post-occlusion mean arterial pressure and fetal heart rate
Asphyxia was independently associated with a moderate increase in MAP from 2 to 24 h after occlusion (Fig. 1, P < 0.05) and a significant tachycardia from 2 to 6 h (P < 0.005). Dexamethasone treatment was associated with an independent (i.e. additive) increase in MAP from 2 to 6 h (P < 0.001) as well as a small increase in FHR from 6 to 24 h (P < 0.05). Thereafter there were no significant effects of either treatment on MAP or FHR.
Figure 1. Time sequence of changes in MAP (mmHg) and FHR (bpm) from 24 h before until 72 h after umbilical cord occlusion in sham–saline (n = 13), sham–dexamethasone (n = 12), asphyxia–saline (n = 18) and asphyxia–dexamethasone (n = 15) groups.
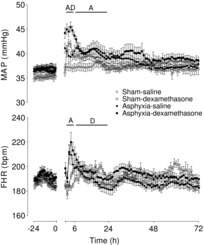
Data are 1 h means ± SEM. A, effect of asphyxia, P < 0.05; D, effect of dexamethasone treatment, P < 0.05; FHR, fetal heart rate; MAP, mean arterial pressure.
Post-occlusion carotid blood flow and carotid vascular resistance
Asphyxia was independently associated with significantly reduced CaBF (P < 0.001) and increased CaVR (P < 0.005) for the entire recovery period after occlusion (Fig. 2). From 2 to 6 h dexamethasone treatment was independently associated with reduced CaBF (P < 0.001) and increased CaVR (P < 0.001). Further, from 2 to 6 h, there was also a significant interaction between asphyxia and dexamethasone treatment on these parameters (P < 0.01), such that post-hoc analysis suggested that the asphyxia–dexamethasone group had significantly lower CaBF (P < 0.05, LSD test) and greater CaVR (P < 0.001, LSD test) compared to all other groups from 2 to 6 h. From 7 to 24 h dexamethasone treatment was associated with an apparent trend toward increased CaVR (P = 0.06). Finally, there was a complex interaction between asphyxia, dexamethasone treatment and time on CaBF from 7 to 24 h (P < 0.005), such that CaBF in the combined asphyxia–dexamethasone group rose over this time to asphyxia–saline levels.
Figure 2. Time sequence of changes in EEG power (dB, change from baseline), spectral edge frequency (Hz), CaBF (ml min−1) and CaVR (mmHg ml−1min−1) from 24 h before until 72 h after umbilical cord occlusion in sham–saline (n = 13), sham–dexamethasone (n = 12), asphyxia–saline (n = 18) and asphyxia–dexamethasone (n = 15) groups.
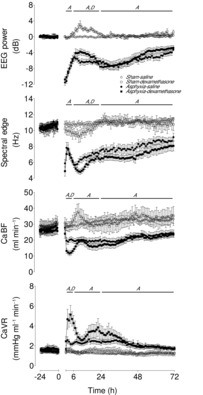
Data are 1 h means ± SEM. A, effect of asphyxia, P < 0.05; CaBF, carotid blood flow; CaVR, carotid vascular resistance; D, effect of dexamethasone treatment, P < 0.05.
Post-occlusion electroencephalographic power and spectral edge frequency
Asphyxia was associated with significant independent suppression of both EEG power (P < 0.001) and spectral edge frequency (P < 0.001) for the 72 h recovery period (Fig. 2). During the secondary phase from 7 to 24 h there was an independent effect of dexamethasone treatment to increase EEG power (P < 0.005) and decrease spectral edge frequency (P < 0.05).
Post-occlusion epileptiform and interictal activity
The increase in EEG power and accompanying decrease in spectral edge frequency in the asphyxia–dexamethasone group corresponded with the appearance of rhythmic slow wave and high amplitude sharp wave transient activity on the continuous EEG records (Fig. 3). These waveforms occurred in a markedly greater proportion of the EEG recordings in the asphyxia–dexamethasone group from 7 to 24 h after occlusion (Fig. 4, P < 0.001, two-way ANOVA). There were no differences between groups after 24 h of recovery (data not shown).
Figure 3. Examples of raw EEG activity from sham–saline controls and after asphyxia and dexamethasone treatment.
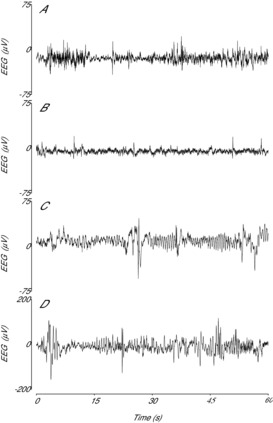
A, example of the normal discontinuous, mixed amplitude and frequency EEG activity from the sham–saline group. B, example of post-asphyxial EEG activity 4 h after asphyxia in the asphyxia–saline group, showing suppressed background activity interspersed with the presence of sharp and fast wave transients. C, effect of dexamethasone on post-asphyxial EEG activity 4 h after asphyxia in the asphyxia–dexamethasone group, showing markedly higher background amplitude and the presence of prolonged abnormal slow wave rolling activity. D, effect of dexamethasone on normal EEG activity in the sham–dexamethasone group, showing the presence of similar low frequency, high amplitude activity after combined asphyxia and dexamethasone treatment. Note the different scale used in (D).
Figure 4. Time sequence of changes in percentage time per hour of prolonged slow wave interictal activity and levels of oxidised CytOx (μmol 100 g−1) for the first 24 h after umbilical cord occlusion in asphyxia–saline and asphyxia–dexamethasone groups.
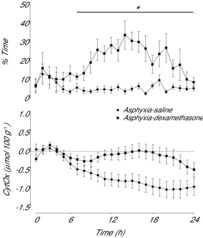
Data are 1 h means ± SEM. *Asphyxia–saline vs. asphyxia–dexamethasone, P < 0.05. CytOx, cytochrome oxidase.
Post-occlusion near-infrared spectroscopy parameters
Umbilical cord occlusion was associated with an increase in [Hb] and a profound fall in [HbO2], which resolved rapidly after release of occlusion [similar to our previous report (Bennet et al. 2007), data not shown]. During recovery there was a significant effect of group on [Hb] over the entire period until the end of the experiment (Fig. 5, P < 0.001) and on [HbO2] from 2 to 24 h after occlusion (P < 0.05). Post-hoc analysis showed that [Hb] was similarly decreased in both asphyxia groups compared to the sham–saline group for the entire recovery period (P < 0.005, LSD test). [HbO2] was significantly decreased in both asphyxia groups compared to the sham–saline group from 2 to 6 h (P < 0.001, LSD test) and this reduction was significantly greater in the asphyxia–dexamethasone group compared to the asphyxia–saline group (P < 0.001, LSD test). Afterwards, [HbO2] in the asphyxia–saline group returned to sham–saline levels from 7 to 24 h, whereas it remained lower in the asphyxia–dexamethasone group compared with both the sham–saline and the asphyxia–saline groups (P < 0.05, LSD test). From 24 h onward, thereafter there were no significant differences between groups for [HbO2].
Figure 5. Time sequence of changes in Hb (μmol 100 g−1) and HbO2 (μmol 100 g−1) from 24 h before until 72 h after umbilical cord occlusion in sham–saline (n = 5), asphyxia–saline (n = 9) and asphyxia–dexamethasone (n = 7) groups.
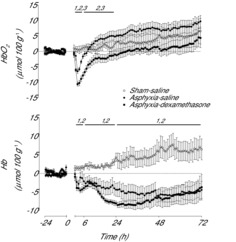
Data are 1 h means ± SEM. 1, asphyxia–saline vs. sham–saline, P < 0.05; 2, asphyxia–dexamethasone vs. sham–saline, P < 0.05; 3, asphyxia–saline vs. asphyxia–dexamethasone, P < 0.05; Hb, deoxyhaemoglobin; HbO2, oxyhaemoglobin.
There was a significant effect of group on [DHb] over the entire recovery period after occlusion (Fig. 6, P < 0.001). During the latent phase, [DHb] fell below baseline levels in both asphyxia groups (P < 0.005, one-way ANOVA) to a nadir at 3 h before recovering back to baseline levels. Post-hoc analysis showed [DHb] over the entire latent phase was significantly lower in the asphyxia–dexamethasone group compared to both the sham–saline and asphyxia–saline groups (P < 0.05, LSD test). From 7 to 24 h [DHb] gradually increased above sham–saline levels in both asphyxia groups (P < 0.05, LSD test), but this increase was significantly greater in the asphyxia–saline group compared to the asphyxia–dexamethasone group (P < 0.05, LSD test). After 24 h, [DHb] remained significantly elevated in both the asphyxia–saline (P < 0.001, LSD test) and asphyxia–dexamethasone (P < 0.005, LSD test) groups compared to the sham–saline group, with no significant difference between the two asphyxia groups.
Figure 6. Time sequence of changes in DHb (μmol 100 g−1) and levels of oxidised CytOx (μmol 100 g−1) from 24 h before until 72 h after umbilical cord occlusion in sham–saline (n = 5), asphyxia–saline (n = 9) and asphyxia–dexamethasone (n = 7) groups.
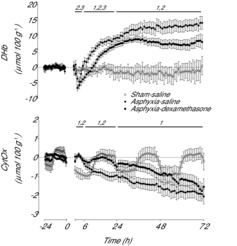
Data are 1 h means ± SEM. 1, Asphyxia–saline vs. sham–saline, P < 0.05; 2, asphyxia–dexamethasone vs. sham–saline, P < 0.05; 3, asphyxia–saline vs. asphyxia–dexamethasone, P < 0.05; CytOx, cytochrome oxidase; DHb, delta haemoglobin.
There was a significant effect of group on [CytOx] over the entire recovery period (Fig. 5, P < 0.05). Post-hoc analysis showed that [CytOx] was significantly higher in both asphyxia groups compared to the sham–saline group from 2 to 6 h (P < 0.05, LSD test). From 7 to 24 h [CytOx] was lower in the asphyxia–saline group compared to both the sham–saline and asphyxia–dexamethasone groups (P < 0.05, LSD test), with no significant difference between the sham–saline and asphyxia–dexamethasone groups. After 24 h, [CytOx] was lower in the asphyxia–saline group than in the sham–saline group (P < 0.05, LSD test). The asphyxia–dexamethasone group showed intermediate values that were not significantly different to either group. Over the final 12 h of the experiment both asphyxia groups had lower [CytOx] levels than the sham–saline group (P < 0.05, LSD test), with no significant difference between the two asphyxia groups.
Discussion
The present study demonstrates that exposure to maternal dexamethasone treatment starting 15 min after severe asphyxia in preterm fetal sheep was associated with a significantly greater fall in carotid blood flow and intracerebral oxygenation during the latent phase of recovery from asphyxia. From approximately 6 h onwards, asphyxia was associated with a progressive increase in intracerebral oxygenation above sham–saline levels, consistent with relative luxury perfusion (Bennet et al. 2006). Intriguingly, this late increase was both slower and less pronounced after dexamethasone treatment, and was associated with increased abnormal background (i.e. interictal) EEG activity and greater CytOx levels. Together these findings suggest that exposure to dexamethasone was associated with impaired intracerebral oxygenation during the latent phase followed by increased post-asphyxial cerebral activity and mitochondrial oxidation during the secondary phase that may have contributed to increased neural injury (Koome et al. 2013).
Regulation of cerebral blood flow after severe asphyxia is complex and only partly understood. Despite rapid recovery of cephalic blood flow and intracerebral oxygenation with reperfusion after asphyxia, this is followed by delayed hypoperfusion in many studies during the latent phase (Bennet et al. 1999b, 2006, 2012a). This hypoperfusion is not associated with hypotension, but rather is actively mediated by increased vascular resistance (Quaedackers et al. 2004). Studies in adult and fetal animals have shown that this cerebral hypoperfusion is closely coupled to reduced cerebral metabolism and suppression of EEG activity (Gold & Lauritzen, 2002; Bennet et al. 2006; Jensen et al. 2006; Yan et al. 2009). In the present study, there was a striking interaction between dexamethasone treatment and asphyxia such that fetuses exposed to both showed a greater reduction in CaBF during the latent phase than any other group. This greater fall in CaBF was not compensated for by a further reduction in EEG activity, resulting in a greater fall in intracerebral oxygenation. Given the stimulatory effect of dexamethasone on the fetal EEG (Davidson et al. 2011) further EEG suppression may not have been possible. Alternatively, EEG activity may have been maximally suppressed at this stage of recovery, preventing further adaptive responses.
These data suggest that there was uncoupling between cerebral metabolism and oxygen supply in the asphyxia–dexamethasone group. Although we did not measure tissue oxygenation directly in the present study, the greater fall in DHb in the asphyxia–dexamethasone group is highly consistent with impaired oxygen delivery (Soul et al. 2000) and thus may have contributed to the greater injury we observed in our previous study (Koome et al. 2013). An acute fall in umbilical blood flow has been observed after dexamethasone treatment in term fetal sheep, which was not associated with a fall in fetal 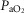 (Jellyman et al. 2004). This is in contrast to an earlier study that reported a transient reduction in
(Jellyman et al. 2004). This is in contrast to an earlier study that reported a transient reduction in 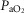 (Bennet et al. 1999a). Given that there was no reduction in fetal oxygen tension during recovery in the present study, changes in placental or umbilical perfusion cannot explain the fall in intracerebral oxygenation.
(Bennet et al. 1999a). Given that there was no reduction in fetal oxygen tension during recovery in the present study, changes in placental or umbilical perfusion cannot explain the fall in intracerebral oxygenation.
The mechanisms mediating the greater fall in cerebral perfusion after dexamethasone exposure in the present study are unclear, but may reflect altered control or activity of vascular constrictors and dilators. The sympathetic nervous system plays a key role in regulating blood flow after adverse events such as ischaemia (ter Laan et al. 2013). We have demonstrated that hypoperfusion in the latent phase is, at least in part, mediated by sympathetic activity (Quaedackers et al. 2004). Exposure to glucocorticoids can potentiate sympathetic vasoconstriction (Ullian, 1999), and thus could have indirectly augmented cerebral vasoconstriction. Consistent with this hypothesis, in the current study FHR was increased in the combined asphyxia–dexamethasone group during the latent phase.
Similarly, dexamethasone can enhance the sensitivity to the vasoconstrictor endothelin. In rats, glucocorticoids increase the rate of preproendothelin-1 gene transcription in vascular smooth muscle cells and the aorta (Provencher et al. 1998). In the fetus, Docherty et al. (2001) have shown in 0.75 of gestation fetal sheep that dexamethasone infusion to the fetus is associated with increased peripheral vascular resistance, which may be mediated by increased responsiveness of endothelin-1 receptors. Supporting this observation, Molnar and colleagues showed in fetal sheep that repeated maternal injections of dexamethasone at 0.7, 0.75 and 0.8 of gestation were associated with normal vascular vasodilatory responses but enhanced sensitivity to endothelin (Molnar et al. 2002), which persisted after birth (Molnar et al. 2003). However, in exploratory experiments we were unable to reverse latent phase hypoperfusion with either endothelial inhibition or nitric oxide donors, suggesting that delayed cerebral hypoperfusion after asphyxia is strongly neurally mediated (Bennet et al. 2012a). Endothelial function experiments with and without dexamethasone are required to dissect the effects of glucocorticoid treatment on post-asphyxial vascular control.
Given that dexamethasone was administered to the ewe, the effect of glucocorticoids on placental perfusion must also be considered. We did not measure the placental transfer of dexamethasone in the current study, but we and others have shown in near-term fetal sheep that dexamethasone levels peak in the ewe 1 h after injection, and in the fetus after 2 h, with levels still detectable 24 h later (Bennet et al. 1999a; Jellyman et al. 2009). Further, Kutzler et al. (2003) have shown that dexamethasone increases the sensitivity of fetal placental blood vessels to endothelin, which could have potentially altered placental function. However, as discussed above, in the current study any changes in placental or umbilical perfusion did not lead to a fall in fetal 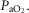
In term fetuses, the secondary phase of injury is associated with cerebral oedema, loss of mitochondrial function and reduced cerebral oxidative metabolism, but increased cerebral blood flow, blood volume and oxygenation (Wassink et al. 2014). Thus, in this phase the brain has relative luxury perfusion with an excess of oxygen delivery over cerebral utilization. In contrast, in the preterm fetus while there is a secondary loss of oxidative metabolism, there is no cortical oedema and no phase of hyperaemia (Bennet et al. 2006). Despite this there is still a significant rise in cerebral oxygenation as measured by NIRS, consistent with luxury perfusion as seen in term fetuses. Dexamethasone treatment altered this response and significantly attenuated the rise in cerebral oxygenation during the secondary phase. Again, this does not reflect reduced peripheral blood oxygenation, as the dexamethasone-treated fetuses had a greater pre-ductal 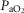 at this time. This observation along with the observation that there were no differences in CaBF between asphyxia groups strongly suggests that there was an increase in cerebral metabolism in the dexamethasone group.
at this time. This observation along with the observation that there were no differences in CaBF between asphyxia groups strongly suggests that there was an increase in cerebral metabolism in the dexamethasone group.
Consistent with this hypothesis, there was a greater increase in EEG activity in the asphyxia–dexamethasone group during the secondary phase, characterised by high amplitude, low frequency activity, which was similar to EEG hyperactivity observed in normoxic preterm fetal sheep after maternal dexamethasone treatment (Davidson et al. 2011). We speculate that a possible mediator of the increased EEG activity was enhanced glutamate activity (Abraham et al. 1996), potentially augmented by impaired reuptake of glutamate due to decreased expression of the GLT-1 glutamate transporter (Chang et al. 2013). Moreover, glucocorticoid exposure is associated with increased intracellular calcium levels during depolarization (Sze & Yu, 1995; Bhargava et al. 2002), and thus may augment neuronal responses to excitation. We have previously reported that post-asphyxial seizure activity was decreased by dexamethasone treatment after asphyxia (Koome et al. 2013). Thus, the increase in metabolism appears to reflect the increased background interictal activity.
Further supporting the hypothesis of increased cerebral activity, we observed greater fetal CytOx levels during the secondary phase after dexamethasone compared to saline treatment. Greater oxidization of CytOx could potentially reflect restricted supply of reducing equivalents down the oxidative phosphorylation pathway (Newman et al. 2000), or alternatively greater cellular consumption of high energy phosphates (Springett et al. 2003). The asphyxia–saline group showed a progressive loss of CytOx, consistent with evolving mitochondrial injury (van Bel et al. 1993; Marks et al. 1996; Bennet et al. 2006). In the present study, given that CaBF was not different between the two asphyxia groups in the secondary phase, increased CytOx in the asphyxia–dexamethasone group suggests that there was increased utilization of oxygen and reducing equivalents, leading to greater mitochondrial activity compared to the asphyxia–saline group. Thus, collectively these data suggest that increased neural activity during the secondary phase may not be adequately supported by increased oxygen delivery, and so may promote further neural cell death.
Our data also suggest, intriguingly, that the normal decline in CytOx during the secondary phase is not only a passive function of mitochondrial death, but may also reflect active suppression of cerebral metabolism, many days after asphyxia. We may speculate that the increase in fetal neural activity associated with maternal dexamethasone imposed an additional stress on damaged mitochondria, increasing oxidative activity and promoting the propagation of programmed cell death pathways (Drury et al. 2014). In rat pups, postnatal dexamethasone was associated with reduced brain growth, but these effects were ameliorated by co-administration of antioxidants (Camm et al. 2011). Whether such interventions can offset the deleterious effects of antenatal dexamethasone after asphyxia in the preterm brain remains to be explored.
The mechanisms of increased white and grey matter injury in this paradigm are probably multifactorial. For example, glucose levels were higher in the asphyxia–dexamethasone group throughout the 3 day recovery period. The fetus is an obligatory glucose user (Hay et al. 1983), and so increased glucose and an associated rise in insulin levels may drive an increase in cerebral metabolic rate and oxygen demand, as is speculated to occur after betamethasone exposure in term fetal sheep (McCallum et al. 2008). This in turn may have contributed to increased neural activity. Jellyman et al. (2009) also found that dexamethasone enhanced cerebral glucose delivery, which would be consistent with an increase in cerebral metabolism at this time. Increased activity and cerebral metabolism at this time would stress already injured cells, thereby limiting the chances of their survival.
Clinically hyperglycaemia is associated with increased risk of death and morbidity among preterm infants; however, it is not known whether controlling glucose improves outcomes (Mehta, 2003; Heald et al. 2012). Studies in newborn piglets have shown that hyperglycaemia during hypoxia–ischaemia (HI) exacerbated neural injury (LeBlanc et al. 1993) and may have impaired neural metabolism up to 2 h after HI (Park et al. 2001), but did not worsen injury when induced after HI (LeBlanc et al. 1994). Further studies are required to evaluate the effects of glucose alone, and to dissect out the effect of glucose on the brain independently from the changes in cerebral activity and perfusion.
Finally, this study evaluated the effects of dexamethasone and as yet we do not know if the same effects would be seen with betamethasone. There is some limited clinical evidence that maternal betamethasone treatment may be associated with better outcomes from preterm birth (for a review, see Bennet et al. 2012b). However, unlike dexamethasone, betamethasone is associated with cerebral hypoperfusion and increased cerebral metabolic activity in normoxic animals (Schwab et al. 2000; McCallum et al. 2008) and growth-restricted fetal sheep (Miller et al. 2007). Recently, Yawno et al. (2014) have shown that betamethasone has adverse effects on neural development of the preterm sheep fetus and that this is in part related to impaired neurosteroid production. Neurosteroids play an important role in regulating neural activity of the normal brain and are upregulated during the latent phase as part of the processes mediating endogenous neuroprotection (Nguyen et al. 2004; Yawno et al. 2007). Thus, it is probable that betamethasone may be associated with similar if not greater effects on the preterm brain after asphyxia.
Perspectives and significance
The majority of preterm infants are now exposed to maternal glucocorticoid treatment, and so it is vital to understand how antenatal glucocorticoids modulate evolving neural injury if given after perinatal asphyxia. In this study we found that dexamethasone exposed the damaged brain to cerebral deoxygenation in the critical latent phase, and subsequently to increased abnormal interictal EEG activity during the secondary phase, with evidence of greater mitochondrial oxidative activity, at a time when it is normally highly suppressed. We speculate that these events were associated with significant cellular metabolic stresses for the injured brain, and so increased secondary neural cell death.
Glossary
- CaBF
carotid blood flow
- CaVR
carotid vascular resistance
- CytOx
cytochrome oxidase
- DHb
delta haemoglobin
- FHR
fetal heart rate
- Hb
deoxyhaemoglobin
- HbO2
oxyhaemoglobin
- Hct
haematocrit
- HI
hypoxia–ischaemia
- MAP
mean arterial pressure
- NIRS
near-infrared spectroscopy
- O2ct
arterial blood oxygen content
Additional information
Competing interests
The authors declare that there are no competing interests.
Author contributions
These experiments were conducted in the Fetal Physiology and Neuroscience Group laboratory, at the University of Auckland. L.B., J.S.Q. J.O.D. and A.J.G. conceived the hypotheses, experimental design and analysis protocols. L.B. C.A.L. M.E.K., J.O.D., P.P.D., J.S.Q. and R.G. were responsible for the data collection. L.B., A.J.G., C.A.L. and M.E.K. were responsible for the analysis. All authors were involved in data interpretation, and were involved in writing and editing the paper. All authors qualify for authorship on the paper, and are listed on the paper. All authors approved the final version of the manuscript.
Funding
This study was funded by grants from the Health Research Council of New Zealand (grant numbers 12/613 and 14/216), the Auckland Medical Research Foundation (grant number 1108004) and New Zealand Lottery Grants Board (grant numbers 209214 and 340855). Christopher Lear was supported by an Auckland Medical Research Foundation Doctoral Scholarship (grant number 1213003).
References
- Abraham I, Juhasz G, Kekesi KA, Kovacs KJ. Effect of intrahippocampal dexamethasone on the levels of amino acid transmitters and neuronal excitability. Brain Res. 1996;733:56–63. doi: 10.1016/0006-8993(96)00538-0. [DOI] [PubMed] [Google Scholar]
- Bennet L, Booth LC, Drury PP, Quaedackers JS, Gunn AJ. Preterm neonatal cardiovascular instability: does understanding the fetus help evaluate the newborn. Clin Exp Pharmacol Physiol. 2012a;39:965–972. doi: 10.1111/j.1440-1681.2012.05744.x. [DOI] [PubMed] [Google Scholar]
- Bennet L, Booth L, Gunn AJ. Potential biomarkers for hypoxic-ischemic encephalopathy. Semin Fetal Neonatal Med. 2010;15:253–260. doi: 10.1016/j.siny.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennet L, Davidson JO, Koome M, Gunn AJ. Glucocorticoids and preterm hypoxic-ischemic brain injury: the good and the bad. J Pregnancy. 2012b;2012:751694. doi: 10.1155/2012/751694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennet L, Kozuma S, McGarrigle HHG, Hanson MA. Temporal changes in fetal cardiovascular, behavioural, metabolic and endocrine responses to maternally administered dexamethasone in the late gestation fetal sheep. Br J Obstet Gynaecol. 1999a;106:331–339. doi: 10.1111/j.1471-0528.1999.tb08270.x. [DOI] [PubMed] [Google Scholar]
- Bennet L, Roelfsema V, Dean J, Wassink G, Power GG, Jensen EC, Gunn AJ. Regulation of cytochrome oxidase redox state during umbilical cord occlusion in preterm fetal sheep. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1569–R1576. doi: 10.1152/ajpregu.00743.2006. [DOI] [PubMed] [Google Scholar]
- Bennet L, Roelfsema V, Pathipati P, Quaedackers J, Gunn AJ. Relationship between evolving epileptiform activity and delayed loss of mitochondrial activity after asphyxia measured by near-infrared spectroscopy in preterm fetal sheep. J Physiol. 2006;572:141–154. doi: 10.1113/jphysiol.2006.105197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennet L, Rossenrode S, Gunning MI, Gluckman PD, Gunn AJ. The cardiovascular and cerebrovascular responses of the immature fetal sheep to acute umbilical cord occlusion. J Physiol. 1999b;517:247–257. doi: 10.1111/j.1469-7793.1999.0247z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhargava A, Mathias RS, McCormick JA, Dallman MF, Pearce D. Glucocorticoids prolong Ca2+ transients in hippocampal-derived H19-7 neurons by repressing the plasma membrane Ca2+-ATPase-1. Mol Endocrinol. 2002;16:1629–1637. doi: 10.1210/mend.16.7.0861. [DOI] [PubMed] [Google Scholar]
- Brun NC, Moen A, Borch K, Saugstad OD, Greisen G. Near-infrared monitoring of cerebral tissue oxygen saturation and blood volume in newborn piglets. Am J Physiol Heart Circ Physiol. 1997;273:H682–H686. doi: 10.1152/ajpheart.1997.273.2.H682. [DOI] [PubMed] [Google Scholar]
- Camm EJ, Tijsseling D, Richter HG, Adler A, Hansell JA, Derks JB, Cross CM, Giussani DA. Oxidative stress in the developing brain: effects of postnatal glucocorticoid therapy and antioxidants in the rat. PLoS One. 2011;6:e21142. doi: 10.1371/journal.pone.0021142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KH, Yeh CM, Yeh CY, Huang CC, Hsu KS. Neonatal dexamethasone treatment exacerbates hypoxic-ischemic brain injury. Mol Brain. 2013;6:18. doi: 10.1186/1756-6606-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corchia C, Ferrante P, Da Fre M, Di Lallo D, Gagliardi L, Carnielli V, Miniaci S, Piga S, Macagno F, Cuttini M. Cause-specific mortality of very preterm infants and antenatal events. J Pediatr. 2013;162((6)):1125–1132. doi: 10.1016/j.jpeds.2012.11.093. [DOI] [PubMed] [Google Scholar]
- Davidson JO, Drury PP, Green CR, Nicholson LF, Bennet L, Gunn AJ. Connexin hemichannel blockade is neuroprotective after asphyxia in preterm fetal sheep. PLoS One. 2014;9:e96558. doi: 10.1371/journal.pone.0096558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson JO, Green CR, Nicholson LF, O'Carroll SJ, Fraser M, Bennet L, Gunn AJ. Connexin hemichannel blockade improves outcomes in a model of fetal ischemia. Ann Neurol. 2012;71:121–132. doi: 10.1002/ana.22654. [DOI] [PubMed] [Google Scholar]
- Davidson JO, Quaedackers JS, George SA, Gunn AJ, Bennet L. Maternal dexamethasone and EEG hyperactivity in preterm fetal sheep. J Physiol. 2011;589:3823–3835. doi: 10.1113/jphysiol.2011.212043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean JM, Gunn AJ, Wassink G, George S, Bennet L. Endogenous alpha2-adrenergic receptor-mediated neuroprotection after severe hypoxia in preterm fetal sheep. Neuroscience. 2006;142:615–628. doi: 10.1016/j.neuroscience.2006.06.066. [DOI] [PubMed] [Google Scholar]
- Docherty CC, Kalmar-Nagy J, Engelen M, Koenen SV, Nijland M, Kuc RE, Davenport AP, Nathanielsz PW. Effect of in vivo fetal infusion of dexamethasone at 0.75 GA on fetal ovine resistance artery responses to ET-1. Am J Physiol Regul Integr Comp Physiol. 2001;281:R261–R268. doi: 10.1152/ajpregu.2001.281.1.R261. [DOI] [PubMed] [Google Scholar]
- Drury PP, Bennet L, Booth LC, Davidson JO, Wassink G, Gunn AJ. Maturation of the mitochondrial redox response to profound asphyxia in fetal sheep. PLoS One. 2012;7:e39273. doi: 10.1371/journal.pone.0039273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drury PP, Davidson JO, van den Heuij LG, Tan S, Silverman RB, Ji H, Blood AB, Fraser M, Bennet L, Gunn AJ. Partial neuroprotection by nNOS inhibition during profound asphyxia in preterm fetal sheep. Exp Neurol. 2013;250C:282–292. doi: 10.1016/j.expneurol.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drury PP, Gunn ER, Bennet L, Gunn AJ. Mechanisms of hypothermic neuroprotection. Clin Perinatol. 2014;41:161–175. doi: 10.1016/j.clp.2013.10.005. [DOI] [PubMed] [Google Scholar]
- Elitt CM, Sadowska GB, Stopa EG, Pinar H, Petersson KH, Stonestreet BS. Effects of antenatal steroids on ischemic brain injury in near-term ovine fetuses. Early Hum Dev. 2003;73:1–15. doi: 10.1016/s0378-3782(03)00030-6. [DOI] [PubMed] [Google Scholar]
- Gezer A, Parafit-Yalciner E, Guralp O, Yedigoz V, Altinok T, Madazli R. Neonatal morbidity mortality outcomes in pre-term premature rupture of membranes. J Obstet Gynaecol. 2013;33:38–42. doi: 10.3109/01443615.2012.729620. [DOI] [PubMed] [Google Scholar]
- Gold L, Lauritzen M. Neuronal deactivation explains decreased cerebellar blood flow in response to focal cerebral ischemia or suppressed neocortical function. Proc Natl Acad Sci U S A. 2002;99:7699–7704. doi: 10.1073/pnas.112012499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez H, Hunter CJ, Bennet L, Power GG, Gunn AJ. Cerebral oxygenation during post-asphyxial seizures in near-term fetal sheep. J Cereb Blood Flow Metab. 2005;25:911–918. doi: 10.1038/sj.jcbfm.9600087. [DOI] [PubMed] [Google Scholar]
- Gratton R, Carmichael L, Homan J, Richardson B. Carotid arterial blood flow in the ovine fetus as a continuous measure of cerebral blood flow. J Soc Gynecol Investig. 1996;3:60–65. doi: 10.1016/1071-5576(95)00047-X. [DOI] [PubMed] [Google Scholar]
- Hay WW, Jr, Myers SA, Sparks JW, Wilkening RB, Meschia G, Battaglia FC. Glucose and lactate oxidation rates in the fetal lamb. Proc Soc Exp Biol Med. 1983;173:553–563. doi: 10.3181/00379727-173-41686. [DOI] [PubMed] [Google Scholar]
- Heald A, Abdel-Latif ME, Kent AL. Insulin infusion for hyperglycaemia in very preterm infants appears safe with no effect on morbidity, mortality and long-term neurodevelopmental outcome. J Matern Fetal Neonatal Med. 2012;25:2415–2418. doi: 10.3109/14767058.2012.699115. [DOI] [PubMed] [Google Scholar]
- Jellyman JK, Gardner DS, Fowden AL, Giussani DA. Effects of dexamethasone on the uterine and umbilical vascular beds during basal and hypoxemic conditions in sheep. Am J Obstet Gynecol. 2004;190:825–835. doi: 10.1016/j.ajog.2003.09.046. [DOI] [PubMed] [Google Scholar]
- Jellyman JK, Gardner DS, McGarrigle HH, Fowden AL, Giussani DA. Antenatal glucocorticoid therapy increases glucose delivery to cerebral circulations during acute hypoxemia in fetal sheep during late gestation. Am J Obstet Gynecol. 2009;201:e81–88. doi: 10.1016/j.ajog.2009.01.012. 82. [DOI] [PubMed] [Google Scholar]
- Jensen EC, Bennet L, Hunter CJ, Power GC, Gunn AJ. Post-hypoxic hypoperfusion is associated with suppression of cerebral metabolism and increased tissue oxygenation in near-term fetal sheep. J Physiol. 2006;572:131–139. doi: 10.1113/jphysiol.2005.100768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerstjens JM, Bocca-Tjeertes IF, de Winter AF, Reijneveld SA, Bos AF. Neonatal morbidities and developmental delay in moderately preterm-born children. Pediatrics. 2012;130:e265–272. doi: 10.1542/peds.2012-0079. [DOI] [PubMed] [Google Scholar]
- Koome ME, Davidson JO, Drury PP, Mathai S, Booth LC, Gunn AJ, Bennet L. Antenatal dexamethasone after asphyxia increases neural injury in preterm fetal sheep. PLoS One. 2013;8:e77480. doi: 10.1371/journal.pone.0077480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutzler MA, Molnar J, Schlafer DH, Kuc RE, Davenport AP, Nathanielsz PW. Maternal dexamethasone increases endothelin-1 sensitivity and endothelin a receptor expression in ovine foetal placental arteries. Placenta. 2003;24:392–402. doi: 10.1053/plac.2002.0920. [DOI] [PubMed] [Google Scholar]
- LeBlanc MH, Huang M, Vig V, Patel D, Smith EE. Glucose affects the severity of hypoxic-ischemic brain injury in newborn pigs. Stroke. 1993;24:1055–1062. doi: 10.1161/01.str.24.7.1055. [DOI] [PubMed] [Google Scholar]
- LeBlanc MH, Huang M, Patel D, Smith EE, Devidas M. Glucose given after hypoxic ischemia does not affect brain injury in piglets. Stroke. 1994;25:1443–1447. doi: 10.1161/01.str.25.7.1443. [DOI] [PubMed] [Google Scholar]
- Low JA, Killen H, Derrick EJ. Antepartum fetal asphyxia in the preterm pregnancy. Am J Obstet Gynecol. 2003;188:461–465. doi: 10.1067/mob.2003.37. [DOI] [PubMed] [Google Scholar]
- Lundqvist P, Kallen K, Hallstrom I, Westas LH. Trends in outcomes for very preterm infants in the southern region of Sweden over a 10-year period. Acta Paediatr. 2009;98:648–653. doi: 10.1111/j.1651-2227.2008.01155.x. [DOI] [PubMed] [Google Scholar]
- Mallard C, Davidson JO, Tan S, Green CR, Bennet L, Robertson NJ, Gunn AJ. Astrocytes and microglia in acute cerebral injury underlying cerebral palsy associated with preterm birth. Pediatr Res. 2014;75:234–240. doi: 10.1038/pr.2013.188. [DOI] [PubMed] [Google Scholar]
- Marks KA, Mallard EC, Roberts I, Williams CE, Sirimanne ES, Johnston B, Gluckman PD, Edwards AD. Delayed vasodilation and altered oxygenation after cerebral ischemia in fetal sheep. Pediatr Res. 1996;39:48–54. doi: 10.1203/00006450-199601000-00007. [DOI] [PubMed] [Google Scholar]
- McCallum J, Smith N, Schwab M, Coksaygan T, Reinhardt B, Nathanielsz P, Richardson BS. Effects of antenatal glucocorticoids on cerebral substrate metabolism in the preterm ovine fetus. Am J Obstet Gynecol. 2008;198:e101–109. doi: 10.1016/j.ajog.2007.05.007. 105. [DOI] [PubMed] [Google Scholar]
- McIntosh GH, Baghurst KI, Potter BJ, Hetzel BS. Foetal brain development in the sheep. Neuropathol Appl Neurobiol. 1979;5:103–114. doi: 10.1111/j.1365-2990.1979.tb00664.x. [DOI] [PubMed] [Google Scholar]
- Mehta S. The glucose paradox of cerebral ischaemia. J Postgrad Med. 2003;49:299–301. [PubMed] [Google Scholar]
- Miller SL, Chai M, Loose J, Castillo-Melendez M, Walker DW, Jenkin G, Wallace EM. The effects of maternal betamethasone administration on the intrauterine growth-restricted fetus. Endocrinology. 2007;148:1288–1295. doi: 10.1210/en.2006-1058. [DOI] [PubMed] [Google Scholar]
- Molnar J, Howe DC, Nijland MJ, Nathanielsz PW. Prenatal dexamethasone leads to both endothelial dysfunction and vasodilatory compensation in sheep. J Physiol. 2003;547:61–66. doi: 10.1113/jphysiol.2002.032565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnar J, Nijland MJ, Howe DC, Nathanielsz PW. Evidence for microvascular dysfunction after prenatal dexamethasone at 0.7, 0.75, and 0.8 gestation in sheep. Am J Physiol Regul Integr Comp Physiol. 2002;283:R561–R567. doi: 10.1152/ajpregu.00031.2002. [DOI] [PubMed] [Google Scholar]
- Newman JP, Peebles DM, Harding SR, Springett R, Hanson MA. Hemodynamic and metabolic responses to moderate asphyxia in brain and skeletal muscle of late-gestation fetal sheep. J Appl Physiol. 2000;88:82–90. doi: 10.1152/jappl.2000.88.1.82. [DOI] [PubMed] [Google Scholar]
- Nguyen P, Yan EB, Castillo-Melendez M, Walker DW, Hirst JJ. Increased allopregnanolone levels in the fetal sheep brain following umbilical cord occlusion. J Physiol. 2004;560:593–602. doi: 10.1113/jphysiol.2004.069336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park WS, Chang YS, Lee M. Effects of hyperglycemia or hypoglycemia on brain cell membrane function and energy metabolism during the immediate reoxygenation-reperfusion period after acute transient global hypoxia-ischemia in the newborn piglet. Brain Res. 2001;901:102–108. doi: 10.1016/s0006-8993(01)02295-8. [DOI] [PubMed] [Google Scholar]
- Provencher PH, Villeneuve A, Morin C. Glucocorticoids increase preproendothelin-1 expression in rat aorta. Endocr Res. 1998;24:737–741. doi: 10.3109/07435809809032679. [DOI] [PubMed] [Google Scholar]
- Quaedackers JS, Roelfsema V, Heineman E, Gunn AJ, Bennet L. The role of the sympathetic nervous system in post-asphyxial intestinal hypoperfusion in the preterm sheep fetus. J Physiol. 2004;557:1033–1044. doi: 10.1113/jphysiol.2004.062554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid SM, Dagia CD, Ditchfield MR, Carlin JB, Meehan EM, Reddihough DS. An Australian population study of factors associated with MRI patterns in cerebral palsy. Dev Med Child Neurol. 2014;56:178–184. doi: 10.1111/dmcn.12331. [DOI] [PubMed] [Google Scholar]
- Roberts D, Dalziel S. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst Rev. 2006;3:CD004454. doi: 10.1002/14651858.CD004454.pub2. [DOI] [PubMed] [Google Scholar]
- Schwab M, Roedel M, Anwar MA, Muller T, Schubert H, Buchwalder LF, Walter B, Nathanielsz PW. Effects of betamethasone administration to the fetal sheep in late gestation on fetal cerebral blood flow. J Physiol. 2000;528:619–632. doi: 10.1111/j.1469-7793.2000.00619.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soul JS, Taylor GA, Wypij D, Duplessis AJ, Volpe JJ. Noninvasive detection of changes in cerebral blood flow by near-infrared spectroscopy in a piglet model of hydrocephalus. Pediatr Res. 2000;48:445–449. doi: 10.1203/00006450-200010000-00005. [DOI] [PubMed] [Google Scholar]
- Springett RJ, Wylezinska M, Cady EB, Hollis V, Cope M, Delpy DT. The oxygen dependency of cerebral oxidative metabolism in the newborn piglet studied with 31P NMRS and NIRS. Adv Exp Med Biol. 2003;530:555–563. doi: 10.1007/978-1-4615-0075-9_53. [DOI] [PubMed] [Google Scholar]
- Sukhov A, Wu Y, Xing G, Smith LH, Gilbert WM. Risk factors associated with cerebral palsy in preterm infants. J Matern Fetal Neonatal Med. 2012;25:53–57. doi: 10.3109/14767058.2011.564689. [DOI] [PubMed] [Google Scholar]
- Sze PY, Yu BH. Glucocorticoid actions on synaptic plasma membranes: modulation of dihydropyridine-sensitive calcium channels. J Steroid Biochem Mol Biol. 1995;55:185–192. doi: 10.1016/0960-0760(95)00178-3. [DOI] [PubMed] [Google Scholar]
- Szeto HH. Spectral edge frequency as a simple quantitative measure of the maturation of electrocortical activity. Pediatr Res. 1990;27:289–292. doi: 10.1203/00006450-199003000-00018. [DOI] [PubMed] [Google Scholar]
- ter Laan M, van Dijk JM, Elting JW, Staal MJ, Absalom AR. Sympathetic regulation of cerebral blood flow in humans: a review. Br J Anaesth. 2013;111:361–367. doi: 10.1093/bja/aet122. [DOI] [PubMed] [Google Scholar]
- Ullian ME. The role of corticosteroids in the regulation of vascular tone. Cardiovasc Res. 1999;41:55–64. doi: 10.1016/s0008-6363(98)00230-2. [DOI] [PubMed] [Google Scholar]
- van Bel F, Dorrepaal CA, Benders MJ, Zeeuwe PE, van de Bor M, Berger HM. Changes in cerebral hemodynamics and oxygenation in the first 24 hours after birth asphyxia. Pediatrics. 1993;92:365–372. [PubMed] [Google Scholar]
- van Bel F, Roman C, Klautz RJ, Teitel DF, Rudolph AM. Relationship between brain blood flow and carotid arterial flow in the sheep fetus. Pediatr Res. 1994;35:329–333. doi: 10.1203/00006450-199403000-00011. [DOI] [PubMed] [Google Scholar]
- Wassink G, Gunn ER, Drury PP, Bennet L, Gunn AJ. The mechanisms and treatment of asphyxial encephalopathy. Front Neurosci. 2014;8:40. doi: 10.3389/fnins.2014.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CE, Gluckman PD. Real-time spectral intensity analysis of the EEG on a common microcomputer. J Neurosci Methods. 1990;32:9–13. doi: 10.1016/0165-0270(90)90066-o. [DOI] [PubMed] [Google Scholar]
- Wray S, Cope M, Delpy DT, Wyatt JS, Reynolds EO. Characterization of the near infrared absorption spectra of cytochrome aa3 and haemoglobin for the non-invasive monitoring of cerebral oxygenation. Biochim Biophys Acta. 1988;933:184–192. doi: 10.1016/0005-2728(88)90069-2. [DOI] [PubMed] [Google Scholar]
- Yan EB, Baburamani AA, Walker AM, Walker DW. Changes in cerebral blood flow, cerebral metabolites, and breathing movements in the sheep fetus following asphyxia produced by occlusion of the umbilical cord. Am J Physiol Regul Integr Comp Physiol. 2009;297:R60–R69. doi: 10.1152/ajpregu.00047.2009. [DOI] [PubMed] [Google Scholar]
- Yawno T, Mortale M, Sutherland AE, Jenkin G, Wallace EM, Walker DW, Miller SL. The effects of betamethasone on allopregnanolone concentrations and brain development in preterm fetal sheep. Neuropharmacology. 2014;85:342–348. doi: 10.1016/j.neuropharm.2014.05.031. [DOI] [PubMed] [Google Scholar]
- Yawno T, Yan EB, Walker DW, Hirst JJ. Inhibition of neurosteroid synthesis increases asphyxia-induced brain injury in the late gestation fetal sheep. Neuroscience. 2007;146:1726–1733. doi: 10.1016/j.neuroscience.2007.03.023. [DOI] [PubMed] [Google Scholar]