

Abstract
Prorenin receptor (PRR) has been implicated in the onset and progression of various renal diseases, though its possible association with immunoglobulin A (IgA) nephropathy remains unclear. In the present study, we tried to clarify expression and pathophysiological significance of PRR in IgA nephropathy. We immunohistochemically assessed PRR levels in renal biopsy specimens from 48 patients with IgA nephropathy and evaluated its relevance to the clinical and pathological features of the disease. PRR was detected mainly in renal tubular cells, which was confirmed at the subcellular level using immunoelectron microscopy. The PRR-positive area (%PRR area) correlated with daily urinary protein, which is known to reflect disease severity (r=0.286, P=0.049). PRR levels were weaker in tubular cells bordering areas of severe interstitial fibrosis, where α-smooth muscle actin-positive myofibroblasts were present. We also used immunohistochemical detection of microtubule-associated protein-1 light chain 3 (LC3) and electron microscopy to assess autophagy, a cytoprotective mechanism downstream of PRR. We noted an apparent coincidence between autophagy activation in tubular cells and PRR expression in the same cells. Taken together, our findings suggest that renal expression of PRR in IgA nephropathy may be a compensatory response slowing disease progression by preventing tubular cell death and subsequent fibrosis through activation of cytoprotective autophagic machinery. Further studies using different type of kidney diseases could draw conclusion if the present finding is a generalized observation beyond IgA nephropathy.
Keywords: IgA nephropathy, prorenin receptor, renal biopsy
Introduction
The prorenin receptor (PRR) was first identified as a receptor for both prorenin and renin in 2002 [1]. Binding to PRR induces not only enzymatic activation of prorenin and renin, but also angiotensin II-independent intracellular signaling. Moreover, it now appears that PRR is critically involved in the tissue renin-angiotensin system (RAS), and that it plays a key role in the development of organ damage in hypertension and diabetes. For example, PRR blockade significantly inhibits the development and progression of nephropathy in animal models of diabetes and hypertension [2,3]. Conversely, glomerulosclerosis develops in transgenic rats overexpressing PRR [4]. These findings suggest PRR may contribute to the pathophysiology underling the development and progression of kidney disease in humans.
Immunoglobulin A (IgA) nephropathy is the most common form of primary glomerulonephritis. In Asia, it typically represents 30-35% of all primary glomerular diseases, though it has been reported as high as 45% [5]. In Europe, the incidence of IgA nephropathy ranges from about 30% to 40%. IgA nephropathy was also recently reported to be the most common primary glomerulopathy among young adult Caucasians in the USA [6]. IgA nephropathy is diagnosed through kidney biopsy and is defined as the occurrence of mesangial IgA deposits within glomeruli and the presence of recurrent episodes of macroscopic hematuria with upper respiratory tract infection or microscopic hematuria and/or proteinuria [7,8]. This condition was initially thought to be benign, but is now recognized to be a major health problem, as 20% to 40% of patients progress to end-stage renal disease within 20 years after disease onset [9].
Despite the fact that PRR has been implicated in the onset and progression of various renal diseases and the critical importance of IgA nephropathy, information is entirely lacking on the possible involvement of PRR in IgA nephropathy. In an effort to address that issue, we evaluated the relevance of PRR expression to the clinical and pathological features in IgA nephropathy patients. We also examined the relevance of PRR expression to autophagy, as PRR is known to be an integral component of mammalian vacuolar-type H+-ATPase (V-ATPase), which is essential for activation of the autophagic process [10,11]. Autophagy is a cellular mechanism that enables cells to rid themselves of superfluous or damaged organelles and misfolded cytoplasmic proteins, and is a central biological pathway that functions to promote cell survival [12,13]. Here we describe the distribution of PRR in the kidneys of patients with IgA nephropathy and propose a hypothesis that renal PRR expression may be a compensatory mechanism functioning to mitigate disease severity in IgA nephropathy.
Materials and methods
Patients
We retrospectively reviewed the medical records of 48 patients (34 males and 14 females; median age, 48 years) with biopsy-proven IgA nephropathy diagnosed between February 2010 and January 2013. Clinical data, including age, gender, history of disease, medication, and blood and urine chemistry were obtained. The criteria used in the decision to perform a biopsy were recurrent macroscopic hematuria, persistent proteinuria of > 0.5 g/day, and persistent microscopic hematuria. The diagnosis of IgA nephropathy was based on the finding of sole or predominant glomerular IgA immunofluorescence in the biopsy specimen and the absence of clinical evidence for Henoch-Schoenlein purpura. All of the patients gave informed consent, and the study was performed in accordance with the Declaration of Helsinki. The ethical committee of Gifu University Graduate School of Medicine has approved this study.
Measurement of plasma prorenin
Plasma prorenin levels were measured using an ELISA kit (LINCO Research Inc., St. Charles, MO, USA).
Renal pathology
Four-μm-thick paraffin sections stained with hematoxylin-eosin, periodic acid-Schiff (PAS), Masson’s trichrome, periodic acid-methenamine-silver (PAM) and Sirius red were examined using a light microscope. Mesangial hypercellularity, increased mesangial matrix, glomerulosclerosis, extracapillary lesions (crescent formation), interstitial fibrosis and inflammation, tubular atrophy, tubulitis and arterial hyalinosis were evaluated. A single observer blinded to the study protocols performed grading of these features.
Mesangial hypercellularity was classified into the following four groups according to Oxford classification: normal (< 4 cells per mesangial zone), mild (4-5 cells per mesangial zone), moderate (6-7 cells per mesangial zone) or marked (8 or more cells per mesangial zone) [14]. The number of patients with global and segmental sclerotic glomeruli was noted, as was the number of patients with crescentic glomeruli. Mesangial matrix, tubular atrophy, tubulitis, interstitial inflammation and fibrosis were semiquantitatively graded into four groups (normal, mild, moderate or marked) modeled on the grading system used by Myllymaki et al. [15]. With this model, tubular atrophy was scored as normal (no tubular atrophy), mild (tubular atrophy in < 25% of cortical tubules), moderate (tubular atrophy in 25-50% of cortical tubules) or marked (tubular atrophy in > 50% of cortical tubules). Tubulitis was classified as absent (no inflammatory cells in tubules), mild (1-4 inflammatory cells per tubular cross section), moderate (5-10 inflammatory cells per tubular cross section) or marked (> 10 inflammatory cells per tubular cross section) [16]. Interstitial fibrosis and inflammation were graded as normal (< 5% of the cortical area is involved in interstitial fibrosis/inflammation), mild (< 25% of the cortical area is involved in interstitial fibrosis/inflammation), moderate (25-50% of the cortical area is involved in interstitial fibrosis/inflammation) or marked (> 50% of the cortical area is involved in interstitial fibrosis/inflammation) [15]. The grades (normal or absent, mild, moderate or marked) were assigned scores of 0, 1, 2 or 3, respectively.
Immunohistochemistry and immunofluorescence
After deparaffinization, 4-μm-thick sections were incubated with a primary antibody against prorenin receptor (PRR; diluted 1:1000 with 0.1 mol/l phosphate buffered saline, a kind gift from Dr. Kazuhito Totsune, Tohoku Fukushi University, Sendai, Japan) [17,18], α-smooth muscle actin (α-SMA; 1:500, Sigma-Aldrich) or microtubule-associated protein-1 light chain 3 (LC3; 1:100, MBL). A Vectastain Elite ABC system (Vector Laboratories) was then used to immunostain the sections; diaminobenzidine served as the chromogen, and the nuclei were counterstained with hematoxylin. For immunofluorescence, sections incubated with a primary antibody were labeled using Alexa 488 (green; Molecular Probes) and counterstained using Hoechst 33342. They were then analyzed under a confocal microscope (C2, Nikon). For double immunofluorescence, the sections incubated with primary antibodies were labeled with Alexa 488 and Alexa 568 (red; Molecular Probes) and counterstained with Hoechst 33342.
Quantitative assessment of the PRR immunopositive area was carried out in 5 or more randomly selected 400 m2 areas of renal cortex in HPFs (200×) using a multipurpose color image processor (Nireco, Tokyo, Japan). The edges of specimens were not used for this analysis to avoid possible inclusion of artificial stains. The HPF photographs were then processed using Image J software (NIH) for binarization (red, immunopositive area; white, immunonegative area), after which the % area of PRR-immunopositivity was calculated for each specimen.
As the control specimens for PRR immunohistochemistry, we employed 2 renal biopsy specimens from patients with thin basement membrane disease which lacked of proteinuria and histological abnormalities as a non-IgA nephropathy control.
Electron microscopy
Biopsy specimens were fixed for 4 h at 4 C in 2.5% glutaraldehyde in 0.1 mol/l phosphate-buffer. They were then postfixed in 1% buffered osmium tetroxide, dehydrated through a graded ethanol series, and embedded in Epon. Ultrathin sections (90 nm) were cut with a diamond knife, collected on 200-mesh nickel grids, and double stained with uranyl acetate and lead citrate before examination using an electron microscope (H-700, Hitachi, Tokyo, Japan).
Immunoelectron microscopy
Immunoelectron microscopy for PRR was performed using the pre-embedding method. In brief, 20-μm-thick cryosections of renal biopsies from 3 patients were mounted on slide glass and fixed in 0.1 mol/l phosphate buffer containing 0.5% glutaraldehyde and 2% paraformaldehyde for 4 h at 4°C. They were then incubated first with a primary antibody against PRR (1:500 dilution) overnight at 4°C, and then with horseradish peroxidase-conjugated F(ab’) 2 fragments (1:100, Abnova) for 1 h at room temperature. The sections were incubated in 3, 3’-diaminobenzidine (DAB) solution containing with 0.05% hydrogen peroxide for 5 min. Thereafter, the sections were postfixed with 1% osmium tetroxide for 30 min to produce electron-dense osmium black precipitates, dehydrated through a graded ethanol series, and embedded in epoxy resin. Ultrathin sections were lightly counterstained with lead citrate and examined under an electron microscope.
Statistical analysis
Results were analyzed using SPSS version 22, and were expressed as medians (minimum-maximum) for non-normally distributed data and as mean ± SD for normally distributed data. Group comparisons were made using analysis of variance with post hoc Student’s t test. Spearman’s analysis was used for determining the correlation between parameters. Values of P < 0.05 was considered significant.
Results
Clinical and pathological characteristics
The clinical characteristics of the study population are presented in Table 1. The mean age at the patients at the time of diagnosis was 48 years (18-80 years), and renal biopsy was performed about 2 years (0.1-20 years) after detection of urine abnormalities. Nineteen percent of the patients had recurrent macroscopic hematuria. Proteinuria was > 0.5 g/day in 56% of the patients and was > 1 g/day in 27% of the patients. The mean estimated glomerular filtration rate was 74 ml/min/1.73 m2, (median, 75). Nineteen percent of patients also had coexisting infections, and 40% had high blood pressure. High serum IgA levels were seen in 13% of patients. Plasma renin activity and plasma aldosterone were within the normal range in 57% and 88% of patients, respectively. Plasma prorenin level was 618 pg/ml ranging 303 to 5439 pg/ml. Thirty-eight percent of the patients received an angiotensin II type 1 receptor blocker (ARB), angiotensin converting enzyme inhibitors (ACEI) or a direct renin inhibitor, while none received an aldosterone blocker.
Table 1.
Clinical characteristics of 48 patients with IgA nephropathy
| Mean ± SD or median with range | |
|---|---|
| Age | 48 (18-80) |
| Gender (M/F) | 34/14 |
| Time from onset of symptoms to biopsy (years) | 2 (0.1-20) |
| Proteinuria (g/day) | 0.58 (0.06-4.3) |
| > 0.5 | 21 (44%) |
| ≤ 0.5 | 27 (56%) |
| Hematuria (RBC/f) | 25 (5-100) |
| Creatinine (mg/dl) | 0.84 (0.45-3.0) |
| BUN (mg/dl) | 15 (6-46) |
| eGFR (ml/min/1.73 m2) | 74±29 |
| Serum IgA Level (mg/ml) | 343±111 |
| Plasma Renin Activity (ng/ml/h)* | 1.8 (0.1-14.6) |
| Plasma Aldosterone (pg/ml)* | 86±39 |
| Plasma Prorenin (pg/ml)* | 618 (303-5439) |
| Medication | |
| Renin Inhibitor | 1 (2%) |
| ARB/ACEI | 18 (38%) |
| Anti-Aldosterone | 0 (0%) |
BUN, blood urea nitrogen; eGFR, estimated glomerular filtration rate; ARB/ACEI, angiotensin II type 1 receptor blockers/angiotensin converting enzyme inhibitors.
n=26.
Common findings in the patient biopsies were mild glomerular hypercellularity and mild mesangial matrix expansion, observed in 67% and 38% of patients, respectively. In addition, mild interstitial fibrosis was seen in 44%, mild tubular atrophy in 63%, mild interstitial inflammation in 54%, segmental glomerulosclerosis in 17% and global sclerosis in 75% of patients. Arteriosclerosis was seen in 18 biopsies. None of the biopsies showed tubulitis (Table 2).
Table 2.
Histopathological findings in the renal biopsies from patients with IgA nephropathy
| Glomeruli | |||
| Mesangial cellularity | |||
| 0: Normal 2.0% | 1: Mild 67% | 2: Moderate 25% | 3: Marked 6.3% |
| Mesangial matrix expansion | |||
| 0: Normal 35% | 1: Mild 38% | 2: Moderate 23% | 3: Marked 4.2% |
| Glomerulosclerosis (n) | |||
| Global 36 | Segmental 8 | Crescent 12 | |
| Tubulointerstitial | |||
| Tubular atrophy | |||
| 0: Normal 25% | 1: Mild 63% | 2: Moderate 13% | 3: Marked 0% |
| Tubulitis | |||
| 0: Absent 100% | 1: Mild 0% | 2: Moderate 0% | 3: Marked 0% |
| Interstitial Inflammation | |||
| 0: Normal 35% | 1: Mild 54% | 2: Moderate 6.3% | 3: Marked 4.2% |
| Interstitial fibrosis | |||
| 0: Normal 44% | 1: Mild 44% | 2: Moderate 13% | 3: Marked 0% |
Expression of PRR in renal biopsies
Immunohistochemical analysis revealed the presence of PRR in renal biopsies from all IgA nephropathy patients, which completely disappeared when the primary antibody was substituted with a nonspecific rabbit IgG. The PRR-positive cells were tubular cells, as judged from serial histological preparations stained with PAS (Figure 1A). After binarization was performed, the %PRR-positive (red) area was calculated to range from 1.7% and 27% (mean, 11%) in the immunohistochemical preparations. Immunostaining of PRR was observed not only at the plasma membrane but also throughout the cytoplasm. Expression of PRR in thin basement membrane disease of both 2 patients looked far weaker compared with IgA nephropathy (Figure 1B). The %PRR-positive area was 3.5% and 3.9% (mean, 3.7%).
Figure 1.

Immunohistochemical detection of PRR in renal biopsies. A. Renal biopsies from IgA nephropathy patients. The left and middle panels respectively contain low- and high-magnification photomicrographs of an example preparation; the right panels contain the binarization images highlighting the PRR-immunopositive area. The uppermost panels are the serial sections for the lower panels, which show specimens immunostained for PRR and stained with PAS. The middle row contains a negative control specimen treated with nonspecific rabbit IgG instead of the primary antibody against PRR. The bottom row shows a preparation immunostained (brown) for PRR. Bars, 100 μm; B. Renal biopsy from a patient with thin basement membrane disease without proteinuria and histological abnormalities as a non-IgA nephropathy control. The left panel, PAS stain; the middle panel, PRR immunostain; and the right panel, the binarization image. Bars, 100 μm; C. Immunoelectron microphotograph showing PRR-positive staining weakly in the cytoplasm and strongly at the plasma membrane and perinuclear regions (arrows) of proximal tubular cells. BM, basement membrane. Bar, 1 μm.
Immunoelectron microscopy confirmed the localization of PRR in the cytoplasm of tubular cells; PRR-positive staining was observed weakly in the cytoplasm and strongly at the plasma membrane and perinuclear regions of tubular cells (Figure 1C).
Relationship between PRR expression and clinicopathological parameters
We next investigated the possible association between PRR expression and the clinical and pathological features of IgA nephropathy. Table 3 shows the coefficients of correlation between the %PRR in biopsy specimens and clinicopathological parameters in the IgA nephropathy patients from whom the specimens were collected. We found that %PRR had no relation to age, illness duration, hematuria, serum creatinine, blood urea nitrogen (BUN), estimated glomerular filtration rate (eGFR), serum IgA level, plasma renin activity or plasma aldosterone concentration (Table 3). There was also no difference in the %PRR between males (12±5.1%) and females (10±5.5%, P=0.12) or between patients who were prescribed an ARB or ACEI (12±5.4%) and those who were not (11±5.4%, P=0.87). However, the %PRR showed a significant positive correlation with the daily urinary protein (r=0.286, P=0.049) (Figure 2).
Table 3.
Coefficients of correlation between the %PRR in biopsy specimens and clinicopathological variables
| %PRR | ||
|---|---|---|
| R | P | |
| Clinical Parameters | ||
| Age | 0.003 | 0.982 |
| Duration of illness (months) | -0.145 | 0.325 |
| Proteinuria (g/day) | 0.286 | 0.049 |
| Hematuria (RBC/f) | 0.052 | 0.727 |
| Creatinine (mg/dl) | 0.093 | 0.528 |
| BUN (mg/dl) | 0.044 | 0.766 |
| eGFR (ml/min/1.73 m2) | 0.002 | 0.989 |
| Serum IgA level (mg/ml) | 0.028 | 0.851 |
| Plasma renin activity (ng/ml/h)* | -0.036 | 0.863 |
| Plasma aldosterone (pg/ml)* | -0.177 | 0.387 |
| Plasma prorenin (pg/ml)* | -0.067 | 0.744 |
| Pathological parameters | ||
| Mesangial cellularity | -0.149 | 0.311 |
| Mesangial matrix expansion | -0.065 | 0.659 |
| Tubular atrophy | -0.140 | 0.342 |
| Interstitial inflammation | 0.094 | 0.527 |
| Interstitial fibrosis | -0.092 | 0.535 |
n=26.
Figure 2.

Plots showing the correlation between %PRR and the indicated clinical parameters. There was a positive correlation between %PRR and daily urinary protein (r=0.286, P=0.049).
Among the pathological variables tested, we detected no correlation between %PRR and mesangial cellularity, mesangial matrix expansion, tubular atrophy, interstitial inflammation or interstitial fibrosis (Table 3). However, comparison of serial sections immunostained for PRR and with Sirius red revealed an obvious weakness of PRR expression in tubular cells bordering areas of severe fibrosis (Figure 3A). Consistent with that observation, confocal microscopic examination revealed that PRR levels tended to be low in tubules bordering the interstitium, where α-smooth muscle actin-positive myofibroblasts were abundant (Figure 3B).
Figure 3.
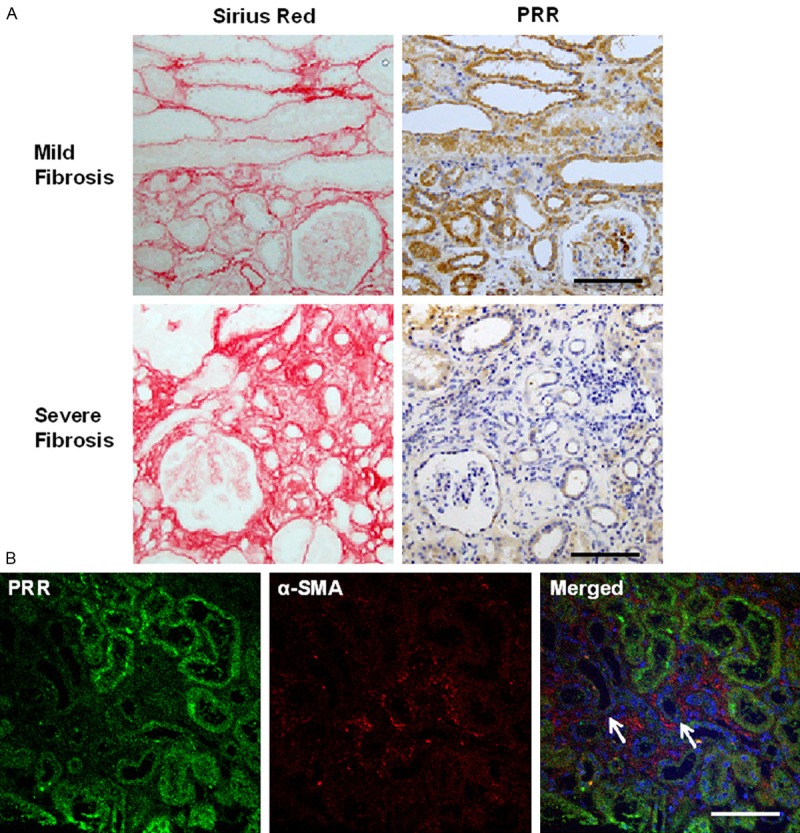
Local relationship between PRR expression and interstitial fibrosis. A. The left and right panels show, respectively, Sirius red-stained and PRR-immunostained serial sections. The upper and lower panels respectively show specimens with mild and severe fibrosis. Bars, 100 μm; B. Confocal micrographs showing double immunostaining for PRR (green) and α-smooth muscle actin (red); note the inverse relation between the signal intensities. The PRR signal tended to be weak in the tubules (arrows) bordering the interstitium where the α-smooth muscle actin signal from myofibroblasts was strong. Bar, 100 μm.
Relationship between PRR expression and autophagy
Immunohistochemical analysis of the renal biopsy specimens also revealed the presence of microtubule-associated protein 1 light chain 3 (LC3)-immunopositive dots localized almost exclusively in tubular cells, suggesting an increase in autophagic activity in those cells (Figure 4A). Using electron microscopy, we confirmed the presence of abundant autophagic vacuoles in the tubular cells (Figure 4B), and confocal microscopy showed the presence of both PRR-immunostaining and LC3 dots within tubular cells. It was also apparent that the LC3 dots tended to be more abundant in cells showing higher levels of PRR (Figure 4C).
Figure 4.
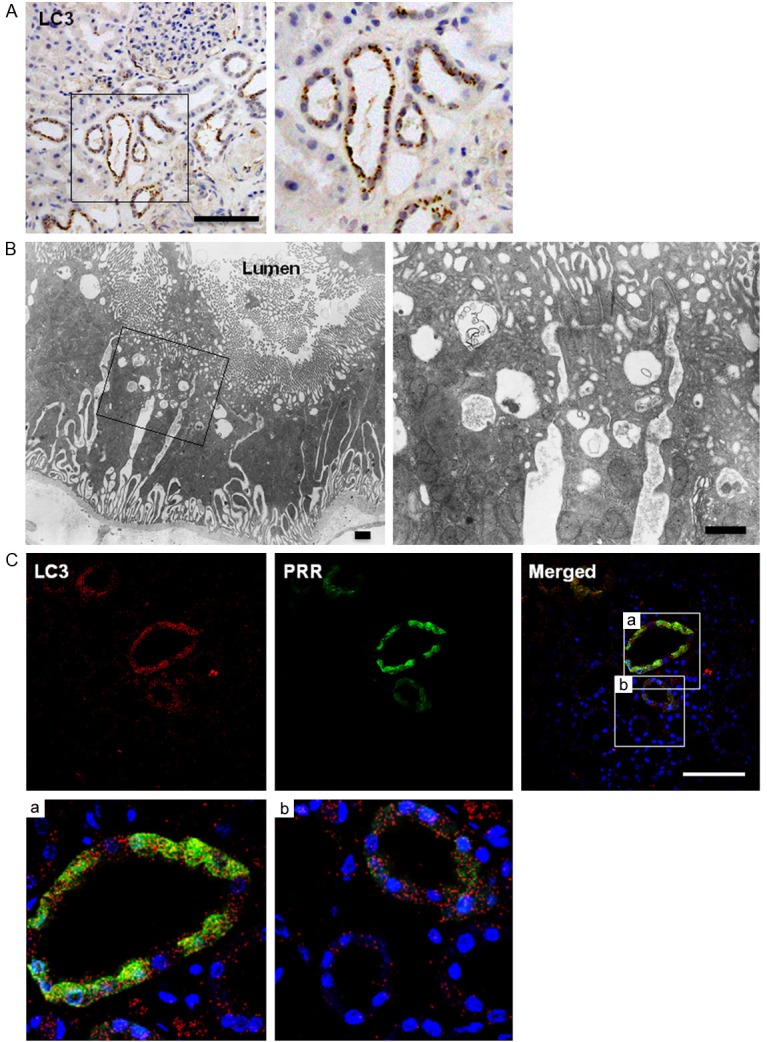
Relationship between PRR and autophagy. A. Immunostaining for LC3, which appears as brown dots. The boxed portion in the left panel is highly magnified in the right panel. Bar, 100 μm; B. Electron micrographs showing abundant autophagic vacuoles in the cytoplasm of proximal renal tubular cells. The boxed portion in the left panel is highly magnified in the right panel. Bars, 1 μm; C. Confocal micrographs showing double immunostaining of LC3 (red) and PRR (green). Note the parallel relation between the PRR and LC3 signals. Bar, 100 μm.
Discussion
It was previously reported that renal expression of PRR occurs in the macula densa [19], mesangial cells [1], glomerular epithelial cells [20], tubular cells [17,18], and collecting ducts (intercalated cells) [21]. The present study is the first examining the distribution of PRR in renal biopsy specimens from humans. We found that in IgA nephropathy patients, PRR was predominantly localized in the cytoplasm of renal tubular cells. Although PRR was once thought to be situated mainly at the plasma membrane, more recent studies have shown that most PRR is situated elsewhere within cells, particularly around the nucleus [22,23], and the trans-Golgi network [23]. Our results are consistent with those later reports. In addition, PRR expression looked more intense in the kidneys of IgA nephropathy compared with thin basement membrane disease (a non-IgA nephropathy control).
Interestingly, we detected a significant positive correlation between renal PRR levels and urinary protein, which reflects the severity of IgA nephropathy. We also noted a local inverse relationship between PRR and interstitial fibrosis; PRR levels appeared to be locally reduced in tubular cells bordering fibrotic areas, where collagen-producing myofibroblasts were present. Because PRR activates the RAS both proteolytically and nonproteolytically [1], it is reasonable to suggest that prorenin plays a significant role in the fibrosis and end-organ damage seen in diseases associated with activation of tissue RAS, including hypertension, diabetes, and preeclampsia [2,3,24]. In addition, PRR can also induce activation of the mitogen activated protein (MAP) kinases, extracellular signal-regulated kinase (ERK) 1/2 and p38, leading to upregulated expression of profibrotic and cyclooxygenase-2 genes, independently of the RAS. And these effects also appear to contribute to the pathophysiology of end-organ damage [2,3].
PRR was first cloned in 2002, and it soon became clear that it was the full-length form of a smaller protein previously reported to be associated with V-ATPase (vacuolar type H+-ATPase) [1,11]. The PRR gene was therefore named ATP6AP2 (ATPase 6 accessory protein 2)/PRR. V-ATPase is a multimeric proton pump involved in diverse and fundamental aspects of cellular physiology, including receptor-mediated endocytosis and recycling, processing of proteins and signaling molecules, membrane sorting and trafficking, and activation of lysosomal/autophagosomal enzymes [11,25]. In addition, PRR works as an adaptor protein between the Wnt receptor complex and V-ATPase [26]. PRR is thus a complex molecule with multiple functions. Autophagy in particular plays a pivotal role in the protection of cells and organs from stresses [12,13]. In the kidneys, for example, autophagy within podocytes is reportedly protective against glomerulosclerosis and age-related renal dysfunction, and also protects proximal tubular cells against acute cisplatin- and ischemia-reperfusion-induced injury [27-29]. In addition, more recent studies have reported a direct link between PRR and autophagy-mediated cytoprotection in podocytes [30-32].
Given the two facets of PRR function, activation of RAS and activation of autophagy, it is difficult to precisely determine the pathophysiological significance (beneficial or detrimental) of the relationship between PRR expression, the severity of IgA nephropathy reflected by daily urinary protein, and interstitial fibrosis. One possibility is that within tubular cells strongly expressing PRR, the resultant RAS activation leads to interstitial fibrosis, while PRR-mediated autophagy protects the cells from cell death, ultimately protecting the kidney from fibrosis. Consistent with that idea, we noted that PRR expression was parallel to autophagic activation in tubular cells, whereas it correlated inversely with fibrosis in neighboring areas. Therefore, although we are aware that no cause and effect relationship can be drawn in a human study, we hypothesize that renal PRR expression in IgA nephropathy may be a compensatory mechanism mitigating the severity of the disease through autophagy-mediated prevention of tubular cell death and subsequent fibrosis, which might be reparative for detrimental effects (if any) caused by RAS activation (Figure 5).
Figure 5.

Hypothesized pathophysiological role for PRR expression in the kidneys of IgA nephropathy patients. Renal PRR expression in IgA nephropathy may serve to slow disease progression through autophagy-mediated prevention of tubular cell death and the subsequent fibrosis. This may compensate for the detrimental effects of RAS activation, such as fibrosis.
Although we used renal biopsies from patients with thin basement membrane disease as a non-IgA nephropathy control, we could not employ ideal control specimens, i.e., biopsies from normal kidneys, because of the ethical reason. Moreover, biopsies were not included in the present study from patients with other proteinuric diseases and with interstitial fibrosis despite PRR expression was found correlated with proteinuria and fibrosis. Such incomplete controls make it impossible to determine whether the present findings are specific for IgA nephropathy or generalized in kidney diseases beyond IgA nephropathy. In addition, the number of patients studied was relatively small. Further studies of larger patient populations with different types kidney disease could provide a more precise understanding of the pathophysiological functions of PRR in kidney disease.
In sum, we described the distribution of PRR in renal biopsies obtained from IgA nephropathy patients, which correlated positively with daily urinary protein and autophagic activation and inversely with neighboring fibrosis. These results suggest renal expression of PRR in IgA nephropathy may be a compensatory mechanism slowing disease progression by preventing tubular cell death and subsequent fibrosis via cytoprotective autophagy.
Acknowledgements
We thank Drs. Kazuhito Totsune (Tohoku Fukushi University) and Takuo Hirose (Tohoku University) for providing anti-PRR antibody and Mss. Akiko Tsujimoto and Kazuko Goto (Gifu University) for technical assistance. Supported in part by research grants from Gifu University and Asahi University.
Disclosure of conflict of interest
None.
References
- 1.Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002;109:1417–27. doi: 10.1172/JCI14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ichihara A, Hayashi M, Kaneshiro Y, Suzuki F, Nakagawa T, Tada Y, Koura Y, Nishiyama A, Okada H, Uddin MN, Nabi AH, Ishida Y, Inagami T, Saruta T. Inhibition of diabetic nephropathy by a decoy peptide corresponding to the “handle” region for nonproteolytic activation of prorenin. J Clin Invest. 2004;114:1128–35. doi: 10.1172/JCI21398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ichihara A, Kaneshiro Y, Takemitsu T, Sakoda M, Nakagawa T, Nishiyama A, Kawachi H, Shimizu F, Inagami T. Contribution of nonproteolytically activated prorenin in glomeruli to hypertensive renal damage. J Am Soc Nephrol. 2006;17:2495–503. doi: 10.1681/ASN.2005121278. [DOI] [PubMed] [Google Scholar]
- 4.Kaneshiro Y, Ichihara A, Sakoda M, Takemitsu T, Nabi AH, Uddin MN, Nakagawa T, Nishiyama A, Suzuki F, Inagami T, Itoh H. Slowly progressive, angiotensin II-independent glomerulosclerosis in human (pro)renin receptor-transgenic rats. J Am Soc Nephrol. 2007;18:1789–95. doi: 10.1681/ASN.2006091062. [DOI] [PubMed] [Google Scholar]
- 5.Li PK, Ho KK, Szeto CC, Yu L, Lai FM. Prognostic indicators of IgA nephropathy in the Chinese-clinical and pathological perspectives. Nephrol Dial Transplant. 2002;17:64–9. doi: 10.1093/ndt/17.1.64. [DOI] [PubMed] [Google Scholar]
- 6.Nair R, Walker PD. Is IgA nephropathy the commonest primary glomerulopathy among young adults in the USA? Kidney Int. 2006;69:1455–8. doi: 10.1038/sj.ki.5000292. [DOI] [PubMed] [Google Scholar]
- 7.Berger J, Hinglais N. Intercapillary deposits of IgA-IgG. J Urol Nephrol (Paris) 1968;74:694–5. [PubMed] [Google Scholar]
- 8.Monteiro RC, Moura IC, Launay P, Tsuge T, Haddad E, Benhamou M, Cooper MD, Arcos-Fajardo M. Pathogenic significance of IgA receptor interactions in IgA nephropathy. Trends Mol Med. 2002;8:464–8. doi: 10.1016/s1471-4914(02)02405-x. [DOI] [PubMed] [Google Scholar]
- 9.Coppo R, D’Amico G. Factors predicting progression of IgA nephropathies. J Nephrol. 2005;18:503–12. [PubMed] [Google Scholar]
- 10.L’Huillier N, Sharp MGF, Dunbar DR, Mullins JJ. On the relationship between the renin receptor and the vacuolar proton ATPase membrane sector associated protein (M8-M9) In: Frolich ED, Richard NRE, editors. The Local Cardiac Renin Angiotensin-Aldosterone System. Heidelberg: Springer; 2006. pp. 17–34. [Google Scholar]
- 11.Burckl C, Bader M. Prorenin and its ancient receptor. Hypertension. 2006;48:549–51. doi: 10.1161/01.HYP.0000241132.48495.df. [DOI] [PubMed] [Google Scholar]
- 12.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Working Group of the International IgA Nephropathy Network and the Renal Pathology Society. Roberts IS, Cook HT, Troyanov S, Alpers CE, Amore A, Barratt J, Berthoux F, Bonsib S, Bruijn JA, Cattran DC, Coppo R, D’Agati V, D’Amico G, Emancipator S, Emma F, Feehally J, Ferrario F, Fervenza FC, Florquin S, Fogo A, Geddes CC, Groene HJ, Haas M, Herzenberg AM, Hill PA, Hogg RJ, Hsu SI, Jennette JC, Joh K, Julian BA, Kawamura T, Lai FM, Li LS, Li PK, Liu ZH, Mackinnon B, Mezzano S, Schena FP, Tomino Y, Walker PD, Wang H, Weening JJ, Yoshikawa N, Zhang H. The Oxford classification of IgA nephropathy: pathology definitions, correlations, and reproducibility. Kidney Int. 2009;76:546–56. doi: 10.1038/ki.2009.168. [DOI] [PubMed] [Google Scholar]
- 15.Myllymaki J, Honkanen T, Syrjanen JT, Helin H, Rantala I, Pasternack A, Mustonen J. Uric acid correlates with the severity of histopathological parameters in IgA nephropathy. Nephrol Dial Transplant. 2005;20:89–95. doi: 10.1093/ndt/gfh584. [DOI] [PubMed] [Google Scholar]
- 16.Topaloglu R, Orhan D, Bilginer Y, Karabulut E, Ozaltin F, Duzova A, Kale G, Besbas N. Clinicopathological and immunohistological features in childhood IgA nephropathy: a single-centre experience. Clin Kidney J. 2013;6:169–75. doi: 10.1093/ckj/sft004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirose T, Mori N, Totsune K, Morimoto R, Maejima T, Kawamura T, Metoki H, Asayama K, Kikuya M, Ohkubo T, Kohzuki M, Takahashi K, Imai Y. Gene expression of (pro)renin receptor is upregulated in hearts and kidneys of rats with congestive heart failure. Peptides. 2009;30:2316–22. doi: 10.1016/j.peptides.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi K, Yamamoto H, Hirose T, Hiraishi K, Shoji I, Shibasaki A, Kato I, Kaneko K, Sasano H, Satoh F, Totsune K. Expression of (pro)renin receptor in human kidneys with end-stage kidney disease due to diabetic nephropathy. Peptides. 2010;31:1405–8. doi: 10.1016/j.peptides.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 19.Kaneshiro Y, Ichihara A, Takemitsu T, Sakoda M, Suzuki F, Nakagawa T, Hayashi M, Inagami T. Increased expression of cyclooxygenase-2 in renal cortex of human prorenin receptor-gene transgenic rats. Kidney Int. 2006;70:641–6. doi: 10.1038/sj.ki.5001627. [DOI] [PubMed] [Google Scholar]
- 20.Ichihara A, Kaneshiro Y, Takemitsu T, Sakoda M, Itoh H. The (pro)renin receptor and the kidney. Semin Nephrol. 2007;27:524–8. doi: 10.1016/j.semnephrol.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 21.Advani A, Kelly DJ, Cox AJ, White KE, Advani SL, Thai K, Connelly KA, Yuen D, Trogadis J, Herzenberg AM, Kuliszewski MA, Leong-Poi H, Gilbert RE. The (pro)renin receptor: Site-specific and functional linkage to the vacuolar H+-ATPase in the kidney. Hypertension. 2009;54:261–9. doi: 10.1161/HYPERTENSIONAHA.109.128645. [DOI] [PubMed] [Google Scholar]
- 22.Schefe JH, Neumann C, Goebel M, Danser J, Kirsch S, Gust R, Kintscher U, Unger T, Funke-Kaiser H. Prorenin engages the (pro)renin receptor like renin and both ligand activities are unopposed by aliskiren. J Hypertens. 2008;26:1787–94. doi: 10.1097/HJH.0b013e3283060f2e. [DOI] [PubMed] [Google Scholar]
- 23.Cousin C, Bracquart D, Contrepas A, Corvol P, Muller L, Nguyen G. Soluble form of the (pro)renin receptor generated by intracellular cleavage by furin is secreted in plasma. Hypertension. 2009;53:1077–82. doi: 10.1161/HYPERTENSIONAHA.108.127258. [DOI] [PubMed] [Google Scholar]
- 24.Zhou A, Carrell RW, Murphy MP, Wei Z, Yan Y, Stanley PL, Stein PE, Broughton Pipkin F, Read RJ. A redox switch in angiotensinogen modulates angiotensin release. Nature. 2010;468:108–11. doi: 10.1038/nature09505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kinouchi K, Ichihara A, Sano M, Wei Z, Yan Y, Stanley PL, Stein PE, Broughton Pipkin F, Read RJ. The (pro)renin receptor/ATP6AP2 is essential for vacuolar H+-ATPase assembly in murine cardiomyocytes. Circ Res. 2010;107:30–4. doi: 10.1161/CIRCRESAHA.110.224667. [DOI] [PubMed] [Google Scholar]
- 26.Cruciat CM, Ohkawara B, Acebron SP, Karaulanov E, Reinhard C, Ingelfinger D, Boutros M, Niehrs C. Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science. 2010;327:459–63. doi: 10.1126/science.1179802. [DOI] [PubMed] [Google Scholar]
- 27.Hartleben B, Gödel M, Meyer-Schwesinger C, Liu S, Ulrich T, Köbler S, Wiech T, Grahammer F, Arnold SJ, Lindenmeyer MT, Cohen CD, Pavenstädt H, Kerjaschki D, Mizushima N, Shaw AS, Walz G, Huber TB. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. 2010;120:1084–96. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Periyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin XM, Dong Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int. 2008;74:631–40. doi: 10.1038/ki.2008.214. [DOI] [PubMed] [Google Scholar]
- 29.Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012;82:1271–83. doi: 10.1038/ki.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Riediger F, Quack I, Qadri F, Hartleben B, Park JK, Potthoff SA, Sohn D, Sihn G, Rousselle A, Fokuhl V, Maschke U, Purfürst B, Schneider W, Rump LC, Luft FC, Dechend R, Bader M, Huber TB, Nguyen G, Muller DN. Prorenin receptor is essential for podocyte autophagy and survival. J Am Soc Nephrol. 2011;22:2193–202. doi: 10.1681/ASN.2011020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oshima Y, Kinouchi K, Ichihara A, Sakoda M, Kurauchi-Mito A, Bokuda K, Narita T, Kurosawa H, Sun-Wada GH, Wada Y, Yamada T, Takemoto M, Saleem MA, Quaggin SE, Itoh H. Prorenin receptor is essential for normal podocyte structure and function. J Am Soc Nephrol. 2011;22:2203–12. doi: 10.1681/ASN.2011020202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meima ME, Danser AH. The prorenin receptor: what’s in a name. J Am Soc Nephrol. 2011;22:2141–3. doi: 10.1681/ASN.2011100981. [DOI] [PubMed] [Google Scholar]