

Abstract
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the human gut. Most sporadic GISTs have somatic gain-of-function mutations of the c-kit gene. The mutations are frequently found at exon 11, sometimes at exon 9 and rarely at exon 13 or 17. Recently, exon 8 c-kit gene mutations were reported in very minor proportion of sporadic GISTs. We also found 3 GISTs with exon 8 c-kit gene mutations in approximately 1,000 sporadic GISTs examined. In the present report, we showed the clinicopathological data of those GISTs. One case had a deletion of codon 419 of aspartate, and 2 cases had a substitution of 3 amino acids of codon 417 to codon 419 to tyrosine. The former was the same mutation recently reported in 2 GIST cases, but the latter has not been reported in any GISTs. All three cases occurred at extragastric sites and two of three showed distant metastasis. Since the remaining case was regarded as high risk for recurrence, imatinib adjuvant treatment has been done without evidence of metastasis. Our results confirmed the idea that exon 8 mutations are minor but actually existing abnormalities in sporadic GISTs, and suggested that such GISTs have a feature of extragastric development and a metastasis-prone nature. Since the exon 8 mutations appeared to be really sensitive to imatinib as shown in the present case study, accurate genotyping including exon 8 of the c-kit gene is necessary in GISTs to predict response to imatinib in both the unresectable/metastatic and adjuvant settings.
Keywords: c-kit gene, exon 8, gain-of-function mutation, GIST, imatinib, sunitinib
Introduction
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the human gut. Most GISTs express a receptor tyrosine kinase (TK), KIT [1,2], encoded by proto-oncogene c-kit [3-6]. Interstitial cells of Cajal (ICCs), which are present in the gastrointestinal (GI) wall and regulate the GI motility through their spontaneous impulse generation [7], are also positive for KIT. Since ICCs are the only proper cells in GI tract that express KIT, GISTs are now considered to originate from ICCs or precursor of ICCs.
KIT consists of an extracellular (EC) domain with five immunoglobulin-like repeats, a transmembrane domain, a juxtamembrane (JM) domain, and TK I and II domains split by the kinase insert [3-6]. A ligand for KIT is stem cell factor (SCF) [8-10]. The binding of SCF and KIT induces autophosphorylation of KIT, and its downstream pathways such as Ras-MAPK, PI3K-Akt and Jak-Stat systems are activated to induce proliferation and differentiation of ICCs, mast cells, melanocytes, germ cells and erythroblasts. On the other hand, gain-of-function mutations of the c-kit gene cause constitutive autophosphorylation of KIT without SCF stimulation, and subsequently activate the downstream signaling molecules. Thus, the gain-of-function mutations could be a cause of GISTs, mast cell neoplasms, seminomas, melanomas and acute leukemias [1,11-15]. In sporadic GISTs, most of them have somatic gain-of-function mutations of the c-kit gene [1]. The mutations are frequently found at exon 11 encoding JM domain (70-80%), at exon 9 encoding EC domain (approximately 10%) and rarely at exon 13 encoding TK I domain and exon 17 encoding TK II domain (less than 2% each) [16,17]. In approximately a half of c-kit gene mutation-negative GISTs, the mutations of platelet-derived growth factor receptor alpha (PDGFRA) gene which shows a quite similar structure to KIT are observed at exon 18 encoding TK II domain (approximately 10%), rarely at exon 12 encoding JM domain (less than 2%), and more rarely at exon 14 encoding TK I domain (less than 1%) [18,19]. On the other hand, several types of germline gain-of-function mutations of the c-kit gene have been detected in approximately 20 families with multiple GISTs [20-23]. Although the mutations in the familial GISTs are also most frequently detected at exon 11, one family with the mutation at exon 8 encoding EC domain, 3 families with the mutation at exon 13 and 4 families with the mutation at exon 17 have been reported [20-23]. Development of multiple GISTs with ICC hyperplasia is commonly observed in patients with the familial GISTs, but some families have mast cell neoplasms and/or hyperpigmentation of the digital, perioral and perineal regions.
Imatinib is a selective TK inhibitor for KIT and PDGFRA [24], which is clinically used for treatment of metastatic or unresectable GISTs [25]. It generally shows a remarkable effect on most GIST cases, but its effectiveness is well-known to be dependent on the types of the c-kit and PDGFRA gene mutations [26,27]. Most GISTs with exon 11 c-kit gene mutations show best response to imatinib treatment whereas the particular type of the PDGFRA gene mutation at exon 18, a substitution of aspartate to valine at codon 842 (Asp842Val), is resistant to imatinib [19]. Imatinib is now being used for GISTs after complete resection when they are regarded as high risk tumors for recurrence [28]. On the other hand, sunitinib and regorafenib are multi-targeted TK inhibitors. After the failure of imatinib treatment, sunitinib is administered as a second-line drug [29] and regorafenib as a third-line one in GISTs [30].
In contrast with sporadic GISTs, most of the c-kit gene mutations are present at exon 17 in sporadic mast cell neoplasms. However, some sporadic mast cell neoplasms have mutations at exon 9, at exon 11 and even at exon 8 [31]. Recently, moreover, exon 8 c-kit gene mutations were also reported in very minor proportion of sporadic GIST cases [32]. Types of exon 8 c-kit gene mutations reported in mast cell neoplasms are various [31], but deletion of aspartate at codon 419 (Del-Asp419) is the only type of exon 8 c-kit gene mutation reported in GISTs [32]. In the present study, we examined whether GISTs have exon 8 c-kit gene mutations in approximately 1,000 sporadic cases, and found 3 such cases. One case had Del-Asp419 and two had substitution of 3 amino acids of codon 417 to codon 419 to tyrosine, hereafter designated as ThrTyrAsp (417-419) Tyr. Here, we showed clinicopathological features of these GISTs.
Materials and methods
Analyses of c-kit and PDGFRA gene mutations
We have analyzed c-kit and PDGFRA gene mutations in approximately 1,000 authentic GIST cases. When fresh-frozen samples were available, total RNA was extracted using RNeasy Mini Kit (QIAGEN, Valencia, CA). Almost all coding regions of the c-kit and PDGFRA genes were amplified by polymerase chain reaction (PCR) after reverse transcription of the extracted RNA as described previously [1,19]. When fresh-frozen samples were not available, genomic DNA was extracted from paraffin sections using QIAamp DNA Mini Kit (QIAGEN). Exons 8, 9, 11, 13 and 17 of the c-kit gene and exons 12, 14 and 18 of the PDGFRA gene were amplified by PCR. Primers used for PCR were as described previously [17,19], and a forward primer of 5’-CCATTTCTGTTTTCCTGTAG-3’ and a reverse primer of 5’-CTCTGCATTATAAGCAGTGC-3’ were used for genomic DNA analysis at exon 8 of the c-kit gene. Direct sequencing of the amplified products was carried out with ABI BigDye terminator ver.3.1 (Applied Biosystems, Foster City, CA) and ABI Prism 3100-Avant Genetic Analyzer (Applied Biosystems). The informed consent for the present study was obtained, and the study was approved by the institutional review boards.
Histology and immunohistochemistry of GISTs
Resected GIST tissues were fixed with 10% formalin and embedded in paraffin. Sections (3 micrometer thick) were cut and used for hematoxylin and eosin staining and immunohistochemistry. Immunohistochemistry was performed using ENVISION+ KIT HRP (DAB) system (DAKO, Glostrup, Denmark). Rabbit polyclonal antibody against human KIT (A4502; DAKO) and mouse monoclonal antibody against human CD34 (Novocastra Laboratories, Newcastle upon Tyne, UK) were used as the primary antibodies.
Results
Detection of exon 8 c-kit gene mutations
We examined whether exon 8 c-kit gene mutations were detected in approximately 1,000 sporadic GISTs. Among them, one GIST had Del-Asp419 and 2 GISTs had ThrTyrAsp (417-419) Tyr (Figure 1A and 1B).
Figure 1.
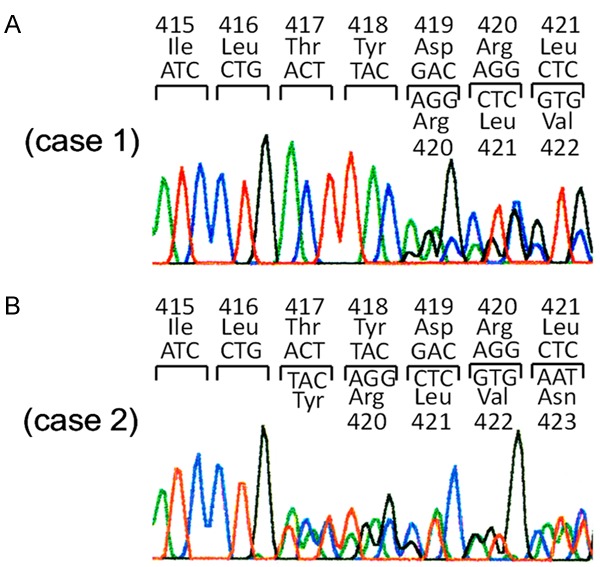
Detection of exon 8 c-kit gene mutations. A. Heterozygous mutation of Del-Asp419 is shown in case 1. B. Heterozygous mutation of ThrTyrAsp (417-419) Tyr is shown in case 2. Case 3 has the same heterozygous mutation of ThrTyrAsp (417-419) Tyr (data not shown).
Clinicopathologicl features of GISTs with exon 8 c-kit gene mutations
Brief clinicopathological features of 3 GIST cases with exon 8 c-kit gene mutations were shown in Table 1. Characteristically, all GISTs with the mutation occurred outside the stomach (2 at the duodenum and 1 at the small intestine). All three tumors consisted of spindle shaped cells (Figure 2A, 2D and 2G). Immunohistochemistry revealed that the tumor cells of all cases were diffusely and strongly positive for KIT (Figure 2B, 2E and 2H). CD34 was positive in cases 1 and 3 but negative in case 2 (Figure 2C, 2F and 2I). All three GISTs had high mitotic figures (Table 1), suggesting that they were highly aggressive. In fact, two of three GISTs showed metastasis. The patient of case 3 has been receiving imatinib adjuvant therapy until now, and no recurrence is evident.
Table 1.
Clinicopathological characteristics of GISTs with exon 8 c-kit gene mutations
| Case No. | Age | Sex | Size (cm) | Site | KIT | CD34 | Mitosis/50HPFs | Metastasis | Survival period (months)a |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 53 | M | 8.0 | SI | + | + | 51 | + (B) | 159 (dead) |
| 2 | 56 | M | 3.5 | D | + | _ | 13 | + (L, P) | 102 (dead) |
| 3 | 41 | M | 8.5 | D | + | + | 32 | _b | 29 (alive) |
HPF, high power field; M, male; SI, small intestine; D, duodenum; B, bone; L, liver; P, peritoneum.
Survival period after the first GIST resection;
This case is during adjuvant imatinib therapy.
Figure 2.
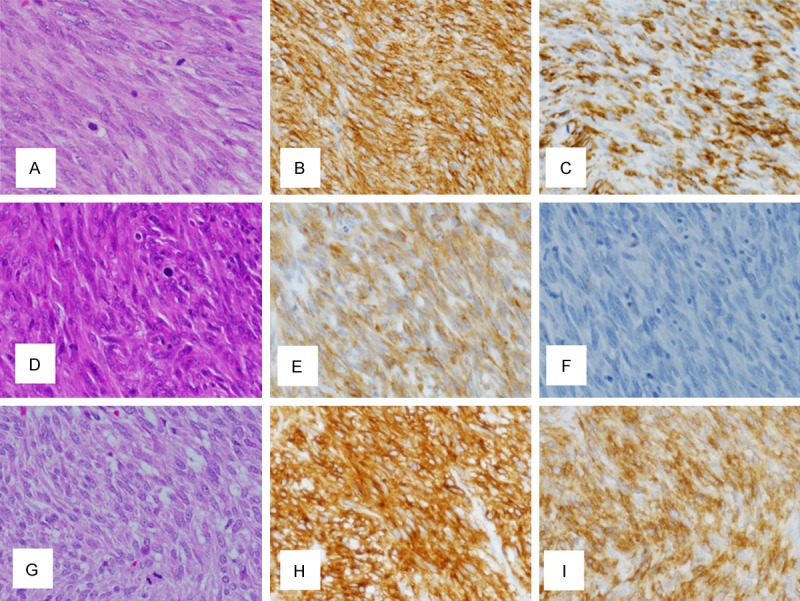
Histopathology and immunohistochemistry of 3 GISTs with exon 8 c-kit gene mutations. A. Hematoxylin and eosin staining reveals that the tumor cells in case 1 are spindle-shaped. B. Tumor cells in case 1 are immunohistochemically KIT-positive. C. Tumor cells in case 1 are also immunohistochemically CD34-positive. D. Tumor cells in case 2 are spindle-shaped by hematoxylin and eosin staining. E. Immunohistochemistry reveals that tumor cells in case 2 are KIT-positive. F. Immunohistochemistry reveals that tumor cells in case 2 are CD34-negative. G. Hematoxylin and eosin staining shows that the tumor cells in case 3 are spindle-shaped. H. Tumor cells in case 3 are immunohistochemically KIT-positive. I. Tumor cells in case 3 are immunohistochemically CD34-positive.
Clinical history of patients with exon 8 c-kit gene mutations
Case 1
A 53-year-old Japanese man received partial resection of the small bowel for small intestinal GIST (8.0 cm in diameter) which had Del-Asp419 at exon 8 of the c-kit gene. A focus of GIST metastasis developed at the 5th thoracic vertebra approximately 7 years after the complete resection of the primary tumor. He underwent partial resection of the metastatic lesion, but the remaining tumor at the metastatic site enlarged 9 months after the partial metastatectomy. Imatinib therapy (400 mg/day) was started, and size of the metastatic lesion decreased. However, the metastatic tumor regrew 22 months after the initial imatinib administration, and imatinib was changed to sunitinib. The imatinib-resistant lesion had been controlled during 16 months under sunitinib administration. The metastatic tumor grew again, and it was resected as far as possible. In spite of multidisciplinary therapy including reintroduction of imatinib or sunitinib, irradiation, and metastatectomy, the drug-resistant remaining lesion could not be controlled. He died 13 years and 3 months after the first operation for small intestinal GIST. Sequencing of c-kit cDNA derived from the resected samples revealed that the sunitinib-resistant lesion had second Asp822Lys at exon 17 in addition to Del-Asp419. Imatinib- and sunitinib-resistant characteristics are considered to be resulted from this second mutation.
Case 2
A 56-year-old Japanese man underwent partial resection of the duodenum for duodenal GIST (3.5 cm in diameter) which showed ThrTyrAsp (417-419) Tyr at exon 8 of the c-kit gene. Two years after the complete resection of duodenal GIST, multiple liver metastases were found by abdominal CT. Imatinib administration (400 mg/day) was started, and the metastatic foci had been controlled for 18 months. Because of regrowth of one of the metastatic foci in the liver, partial hepatectomy was performed. In spite of continuation of imatinib administration after the metastatectomy, peritoneal masses developed. Although the peritoneal lesions were resected, multiple peritoneal masses developed again. He received sunitinib therapy with 4 month control period. After the failure of sunitinib, regorafenib was administered on clinical trial, but it showed no apparent effect. He died 8 years and 4 months after the first operation for duodenal GIST. Sequencing of c-kit cDNA derived from the imatinib-resistant hepatic tumor and peritoneal tumor revealed that both lesions had Asp910Tyr at exon 18 in addition to ThrTyrAsp (417-419) Tyr at exon 8. This second mutation is considered to be a cause of imatinib-resistant character.
Case 3
A 41-year-old Japanese man received pancreaticoduodenectomy for duodenal GIST (8.5 cm in diameter) which had ThrTyrAsp (417-419) Tyr at exon 8 of the c-kit gene. Since it was judged as a high risk tumor for recurrence, adjuvant imatinib treatment (400 mg/day) has been done for 29 months after the operation. At present, he is under imatinib treatment and there is no evidence of recurrence.
Discussion
We reported here 3 cases of sporadic GISTs with exon 8 c-kit gene mutations. Frequency of the mutations is low (approximately 0.3%), but at least 3 GISTs really had two types of the mutations such as Del-Asp419 and ThrTyrAsp (417-419) Tyr.
Exon 8 c-kit gene mutations was first reported in acute myeloid leukemia in 1999 [33]. In acute myeloid leukemia [33,34], various types of exon 8 c-kit gene mutations are known including ThrTyr (417&418) His, ThrTyrAsp (417-419) Asn, ThrTyrAsp (417-419) Ile, ThrTyrAsp (417-419) Phe, ThrTyrAsp (417-419) Tyr, ThrTyrAsp (417-419) Val, ThrTyrAsp (417-419) ArgAla, ThrTyrAsp (417-419) ArgGly, TyrAsp (418&419) Gly, TyrAsp (418&419) Ser, Asp419Phe, Del-Asp419 and AspArg (419&420) PhePheAspGly. In pediatric mastocytosis, on the other hand, exon 8 c-kit gene mutations were reported in 2010 [31]. The types of the exon 8 c-kit gene mutations reported in pediatric mastocytosis are Del-Asp419, Ins-PhePhe between codon418 and codon419, ThrTyrAsp (417-419) Tyr and Cys443Tyr [31]. Many types of exon 8 c-kit gene mutations are common in acute myeloid leukemia and pediatric mastocytosis. Although acute myeloid leukemia does not appear to have any highly frequent mutation types, Del-Asp419 appears to be the most frequent mutation type in pediatric mastocytosis.
Exon 8 c-kit gene mutations in GISTs were first reported in familial GIST cases with germline c-kit gene mutation in 2005 [23], and the type of the mutation was Del-Asp419. The patients in the family members had not only multiple GISTs but also mastocytosis. Recently, 2 sporadic GIST cases with Del-Asp419 were found [32]. Therefore, Del-Asp419 was the only mutation type reported in GISTs with exon 8 c-kit gene mutation. In the present study, we reported one GIST case with Del-Asp419 and 2 GISTs cases with ThrTyrAsp (417-419) Tyr. Described above, ThrTyrAsp (417-419) Tyr has been reported in acute myeloid leukemia and sporadic pediatric mast cell neoplasms [31,33,34]. Thus, these are the first reported GIST cases with ThrTyrAsp (417-419) Tyr among sporadic and familial GIST cases.
In the previous report, 2 sporadic GISTs with exon 8 c-kit gene mutations occurred at the small bowel [32]. In the present study, 3 GISTs with exon 8 c-kit gene mutations were present at the duodenum in 2 cases and at the small intestine in 1 case. Since all of the reported GIST cases with exon 8 c-kit gene mutations developed at the small intestine or duodenum, those GISTs appear to arise from extragastric sites. However, the number of GIST cases with exon 8 c-kit gene mutations is too small to draw conclusions, and further effort to collect those GIST cases is needed.
In the previous report of sporadic GISTs with exon 8 c-kit gene mutations, one GIST developed multiple metastatic foci in the peritoneum [32]. The other case did not show metastasis. However, the patient had been receiving adjuvant imatinib therapy because the tumor was regarded as intermediate to high risk for recurrence [32]. In the present study, 2 cases of GISTs with exon 8 c-kit gene mutations showed distant metastasis; one was in bone and the other was in liver and peritoneum. The other case in this study does not show metastasis, but the patient also has been receiving adjuvant imatinib therapy because the tumor was classified as high risk for recurrence. These results suggested that GISTs with exon 8 c-kit gene mutations might be prone to be metastatic. In fact, all 3 cases in the present study had high mitotic figures, suggesting GISTs with exon 8 c-kit gene mutations might be aggressive. More cases with exon 8 c-kit gene mutations have to be collected to clarify the possibility.
In the previous report, exon 8 c-kit gene mutations such as Del-Asp419, ThrTyrAsp (417-419) Tyr, Cys443Tyr, and ThrTyrAsp (417-419) Ile were demonstrated to be ligand-independently autophosphorylated [25,31,34]. In the previous report, moreover, exon 8 c-kit gene mutations of Del-Asp419 and ThrTyrAsp (417-419) Ile were demonstrated to be sensitive to imatinib in vitro [25,34]. As described above, imatinib was reported to be administered to one sporadic GIST case with Del-Asp419 because of being regarded as intermediate to high risk for recurrence [32]. The patient did not show recurrence for 24 months under adjuvant imatinib treatment, but the case cannot clearly demonstrate whether the mutation is sensitive to imatinib in vivo. On the other hand, our 2 cases with distant metastasis showed apparent clinical effect of imatinib. Recently, imatinib tends to be selectively used only for GISTs with imatinib-sensitive mutations. In many GIST cases, the mutations are usually examined only at exons 9, 11, 13 and 17 of the c-kit gene and exon 12, 14, 18 of the PDGFRA gene. Therefore, GISTs with exon 8 c-kit gene mutation could be erroneously regarded as so-called wild-type GISTs which are usually resistant to imatinib, and those cases might not be considered to be candidates for imatinib treatment. We have to examine whether the exon 8 c-kit gene mutations are present in so-called wild-type GISTs to prevent loss of opportunity for imatinib treatment in those patients.
Asp816Val c-kit gene mutation at exon 17 is often observed in sporadic mast cell neoplasms [11,12]. However, any GISTs have not been reported to possess this mutation. Namely, there is tumor-type specificity in Asp816Val. However, this is an exceptional correlation between tumor-types and particular mutations. Many types of the c-kit gene mutations are common in mast cell neoplasms and GISTs. Indeed, both of mast cell neoplasms and GISTs have the same exon 8 c-kit gene mutations, at least Del-Asp419 and ThrTyrAsp (417-419) Tyr. The cause of tumor-type specificity in Asp816Val remains to be clarified.
Finally, we showed that GISTs with exon 8 c-kit gene mutations might have features of extragastric development and metastasis-prone nature. Since the exon 8 c-kit gene mutations appeared to be sensitive to imatinib, accurate genotyping including not only exons 9, 11, 13 and 17 but also exon 8 of the c-kit gene is necessary to predict response to imatinib in both unresectable/metastatic and adjuvant settings.
Acknowledgements
This work was partly supported by Grant-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science (Grant No. 23390094).
Disclosure of conflict of interest
Seiichi Hirota received research fund from Novartis Pharma K.K. Japan. The other authors have no conflict of interest.
References
- 1.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Tunio GM, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y. Gain-of-function mutations of c-kitin human gastrointestinal stromal tumors. Science. 1998;279:577–580. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 2.Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol. 1998;152:1259–1269. [PMC free article] [PubMed] [Google Scholar]
- 3.Yarden Y, Kuang WJ, Yang-Feng T, Coussens L, Munemitsu S, Dull TJ, Chen E, Schlessinger J, Francke U, Ullrich A. Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987;6:3341–3351. doi: 10.1002/j.1460-2075.1987.tb02655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Besmer P, Murphy JE, George PC, Qiu F, Bergold PJ, Lederman L, Snyder HW Jr, Broudeur D, Zuckerman EE, Hardy WD. A new acute transforming feline retrovirus and relationship of its oncogene v-kit with the protein kinase gene family. Nature. 1986;320:415–421. doi: 10.1038/320415a0. [DOI] [PubMed] [Google Scholar]
- 5.Qiu FH, Ray P, Brown K, Barker PE, Jhanwar S, Ruddle FH, Besmer P. Primary structure of c-kit: relationship with the CSF-1/PDGF receptor kinase family--oncogenic activation of v-kit involves deletion of extracellular domain and C terminus. EMBO J. 1988;7:1003–1011. doi: 10.1002/j.1460-2075.1988.tb02907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geissler EN, Ryan MA, Housman DE. The dominant-white spotting (W) locus of the mouse encodes the c-kitproto-oncogene. Cell. 1988;55:185–192. doi: 10.1016/0092-8674(88)90020-7. [DOI] [PubMed] [Google Scholar]
- 7.Thomsen L, Robinson TL, Lee JC, Farraway LA, Hughes MJ, Andrews DW, Huizinga JD. Interstitial cells of Cajal generate a rhythmic pacemaker current. Nat Med. 1998;4:848–851. doi: 10.1038/nm0798-848. [DOI] [PubMed] [Google Scholar]
- 8.Williams DE, Eisenman J, Baird A, Rauch C, Ness KV, March CJ, Park LS, Martin U, Mochizuki DY, Boswell HS, Burgess GS, Cosman D, Lyman SD. Identification of a ligand for the c-kitproto-oncogene. Cell. 1990;63:167–174. doi: 10.1016/0092-8674(90)90297-r. [DOI] [PubMed] [Google Scholar]
- 9.Flanagan JG, Leder P. The kit ligand: a cell surface molecule altered in steel mutant fibroblasts. Cell. 1990;63:185–194. doi: 10.1016/0092-8674(90)90299-t. [DOI] [PubMed] [Google Scholar]
- 10.Zsebo KM, Williams DA, Geissler EN, Broudy YC, Martin FH, Atkins HL, Hsu RY, Birkitt NC, Okino KH, Murdock DC, Jacobson FW, Langley KE, Smith KA, Takeishi T, Cattanach BM, Galli SJ, Suggs SV. Stem cell factor is encoded at the Sl locus of the mouse and is the ligand for the c-kittyrosine kinase receptor. Cell. 1990;63:213–224. doi: 10.1016/0092-8674(90)90302-u. [DOI] [PubMed] [Google Scholar]
- 11.Nagata H, Worobec AS, Oh CK, Chowdhury BA, Tannenbaum S, Suzuki Y, Metcalfe DD. Identification of a point mutation in the catalytic domain of the protooncogene c-kitin peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci U S A. 1995;92:10560–10564. doi: 10.1073/pnas.92.23.10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Longley BJ, Tyrrell L, Lu SZ, Ma YS, Langley K, Ding TG, Duffy T, Jacobs P, Tang LH, Modlin I. Somatic c-kitactivating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312–314. doi: 10.1038/ng0396-312. [DOI] [PubMed] [Google Scholar]
- 13.Tian Q, Frierson HF Jr, Krystal GW, Moskaluk CA. Activating c-kitgene mutations in human germ cell tumors. Am J Pathol. 1999;154:1643–1647. doi: 10.1016/S0002-9440(10)65419-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Went PT, Dirnhofer S, Bundi M, Mirlacher M, Schraml P, Mangialaio S, Dimitrijevic S, Kononen J, Lugli A, Simon R, Sauter G. Prevalence of KIT expression in human tumors. J. Clin. Oncol. 2004;22:4514–4520. doi: 10.1200/JCO.2004.10.125. [DOI] [PubMed] [Google Scholar]
- 15.Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 2006;24:4340–4346. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- 16.Rubin BP, Singer S, Tsao C, Duensing A, Lux ML, Ruiz R, Hibbard MK, Chen CJ, Xiao S, Tuveson DA, Demetri GD, Fletcher CD, Fletcher JA. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001;61:8118–8121. [PubMed] [Google Scholar]
- 17.Kinoshita K, Isozaki K, Hirota S, Nishida T, Chen H, Nakahara M, Nagasawa Y, Ohashi A, Shinomura Y, Kitamura Y, Matsuzawa Y. c-kitgene mutation at exon 17 or 13 is very rare in sporadic gastrointestinal stromal tumors. J Gastroenterol Hepatol. 2003;18:147–151. doi: 10.1046/j.1440-1746.2003.02911.x. [DOI] [PubMed] [Google Scholar]
- 18.Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A, Town A, Demetri GD, Fletcher CD, Fletcher JA. PDGFRA Activating Mutations in Gastrointestinal Stromal Tumors. Science. 2003;299:708–710. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]
- 19.Hirota S, Ohashi A, Nishida T, Isozaki K, Kinoshita K, Shinomura Y, Kitamura Y. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology. 2003;125:660–667. doi: 10.1016/s0016-5085(03)01046-1. [DOI] [PubMed] [Google Scholar]
- 20.Nishida T, Hirota S, Taniguchi M, Hashimoto K, Isozaki K, Nakamura H, Kanakura Y, Tanaka T, Takabayashi A, Matsuda H, Kitamura Y. Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat Genet. 1998;19:323–324. doi: 10.1038/1209. [DOI] [PubMed] [Google Scholar]
- 21.Isozaki K, Terris B, Belghiti J, Schiffmann S, Hirota S, Vanderwinden JM. Germline-activating mutation in the kinase domain of KIT gene in familial gastrointestinal stromal tumors. Am J Pathol. 2000;157:1581–1585. doi: 10.1016/S0002-9440(10)64795-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirota S, Nishida T, Isozaki K, Taniguchi M, Nishikawa K, Ohashi A, Takabayashi A, Obayashi T, Okuno T, Kinoshita K, Chen H, Shinomura Y, Kitamura Y. Familial gastrointestinal stromal tumors associated with dysphagia and novel type germline mutation of KIT gene. Gastroenterology. 2002;122:1493–1499. doi: 10.1053/gast.2002.33024. [DOI] [PubMed] [Google Scholar]
- 23.Hartmann K, Wardelmann E, Ma Y, Merkelbach-Bruse S, Preussner LM, Woolery C, Baldus SE, Heinicke T, Thiele J, Buettner R, Longley BJ. Novel germline mutation of KIT associated with familial gastrointestinal stromal tumors and mastocytosis. Gastroenterology. 2005;129:1042–1046. doi: 10.1053/j.gastro.2005.06.060. [DOI] [PubMed] [Google Scholar]
- 24.Buchdunger E, Cioffi CL, Law N, Stover D, Ohno-Jones S, Druker BJ, Lydon NB. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kitand platelet-derived growth factor receptors. J Pharmacol Exp Ther. 2000;295:139–145. [PubMed] [Google Scholar]
- 25.Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CD, Joensuu H. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 26.Chen H, Isozaki K, Kinoshita K, Ohashi A, Shinomura Y, Matsuzawa Y, Kitamura Y, Hirota S. Imatinib inhibits various types of activating mutant kit found in gastrointestinal stromal tumors. Int J Cancer. 2003;105:130–135. doi: 10.1002/ijc.11025. [DOI] [PubMed] [Google Scholar]
- 27.Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, McGreevey LS, Chen CJ, Van den Abbeele, Druker BJ, Kiese B, Eisenberg B, Roberts PJ, Singer S, Fletcher CDM, Silberman S, Dimitrijevic S, Flecther JA. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J. Clin. Oncol. 2003;21:4342–4349. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 28.Joensuu H, Eriksson M, Hall KS, Hartmann JT, Pink D, Schütte J, Ramadori G, Hohenberger P, Duyster J, Al-Batran SE, Schlemmer M, Bauer S, Wardelmann E, Sarlomo-Rikala M, Nilsson B, Sihto H, Monge OR, Bono P, Kallio R, Vehtari A, Leinonen M, Alvegård T, Reichardt P. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: randamized trial. JAMA. 2012;307:1265–1272. doi: 10.1001/jama.2012.347. [DOI] [PubMed] [Google Scholar]
- 29.Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, Bello CL, Huang X, Baum CM, Casali PG. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randamised controlled trial. Lancet. 2006;368:1329–1338. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 30.Demetri GD, Reicherdt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, Hohenberger P, Leahy M, von Mehren M, Joensuu H, Badalamenti G, Blackstein M, Le Cesne A, Schöffski P, Maki RG, Bauer S, Nguyen BB, Xu J, Nishida T, Chung J, Kappeler C, Kuss I, Laurent D, Casali PG GRID study investigators. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international multicentre randamised placebo-controlled phase 3 trial. Lancet. 2013;381:295–302. doi: 10.1016/S0140-6736(12)61857-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bodemer C, Hermine O, Palmérini F, Yang Y, Grandpeix-Guyodo C, Leventhal PS, Hadj-Rabia S, Nasca L, Georgin-Lavialle S, Cohen-Akenine A, Launay JM, Barete S, Feger F, Arock M, Catteau B, Sans B, Stalder JF, Skowron F, Thomas L, Lorette G, Plantin P, Bordigoni P, Lortholary O, de Prost Y, Moussy A, Sobol H, Dubreuil P. Pediatric mastocytosis is clonal disease associated with D816V and other activating c-KITmutations. J Invest Dermatol. 2010;130:804–805. doi: 10.1038/jid.2009.281. [DOI] [PubMed] [Google Scholar]
- 32.Huss S, Kunstlinger H, Wardelmann E, Kleine MA, Binot E, Merkelbach-Bruse S, Rüdiger T, Mittler J, Hartmann W, Büttner R, Schildhaus HU. A subset of gastrointestinal stromal tumors previously regarded as wild-type tumors carries somatic activating mutations in KIT exon 8 (p. D419del) Mod Pathol. 2013;26:1004–1012. doi: 10.1038/modpathol.2013.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gari M, Goodeve A, Wilson G, Winship P, Langabeer S, Linch D, Vandenberghe E, Peake I, Reilly J. c-kitproto-oncogene exon 8 in-frame deletion plus insertion mutations in acute myeloid leukaemia. Br J Haematol. 1999;105:894–900. doi: 10.1046/j.1365-2141.1999.01449.x. [DOI] [PubMed] [Google Scholar]
- 34.Goemans BF, Zwaan ChM, Miller M, Zimmermann M, Harlow A, Meshinchi S, Loonen AH, Hählen K, Reinhardt D, Creutzig U, Kaspers GJ, Heinrich MC. Mutations in KIT and RAS are frequent events in pediatric core-binding factor acute myeloid leukemia. Leukemia. 2005;19:1536–1542. doi: 10.1038/sj.leu.2403870. [DOI] [PubMed] [Google Scholar]