

Abstract
This study aimed to determine the role of selective neutral sphingomyelinase (N-SMase) inhibition on arachidonic acid (AA) mediated inflammation following liver ischemia-reperfusion (IR) injury. Selective N-SMase inhibitor was administered via intraperitoneal injections. Liver IR injury was created by clamping blood vessels supplying the median and left lateral hepatic lobes for 60 min, followed by 60 min reperfusion. Levels of AA in liver tissue were determined by multiple reaction monitoring (MRM) using ultra fast-liquid chromatography (UFLC) coupled with tandem mass spectrometry (MS/MS). Phospholipase A2 (PLA2), cyclooxygenase (COX) and prostaglandin E2 (PGE2) were measured in liver tissue. Arachidonic acid levels, activity of PLA2, COX and PGE2 levels were significantly increased in postischemic liver tissue compared to nonischemic controls. N-SMase inhibition significantly decreased COX activity and PGE2 levels in postischemic liver. Future studies evaluating agents blocking N-SMase activity can facilitate the development of treatment strategies to alleviate inflammation in liver I/R injury.
Keywords: Arachidonic acid, liver, ischemia-reperfusion injury, neutral sphingomyelinase
Introduction
Interruption of hepatic flow is often necessary when liver surgery is performed. This interruption of blood flow is termed as “warm ischemia” and upon revascularization, when molecular oxygen is reintroduced, the organ undergoes a process called “reperfusion injury” that causes deterioration of organ function [1]. Although the mechanisms by which organ damage occurs in ischemia reperfusion (IR) injury are incompletely understood, ischemia results in the termination of oxidative phosphorylation and ATP production by aerobic respiration. Restoration of blood flow during reperfusion triggers the activation of Kupffer cells causing oxygen free radical formation, production of tumor necrosis factor-alpha (TNF-alpha) and interleukin-1 (IL-1) [2]. Elevated levels of the pro-inflammatory cytokines TNF-alpha and IL-1 promote PMN recruitment and activation which also generates reactive oxygen species (ROS) and leads to the release of proteases [3,4].
Adherence of circulating blood cells to vascular endothelium is modulated by polyunsaturated fatty acids (PUFAs). An increase in adherence and degranulation of neutrophils is observed when they are incubated with arachidonic acid (AA, C20: 4n-6) and dihomo-gamma-linolenic acid (DGLA, C20: 3n-6) [5]. Likewise, the ability of PUFAs to modulate endothelial activation is shown by a study in which docosahexaenoic acid (DHA, C22: 6n-3), when added to cultured endothelial cells before stimulation with cytokines reduces adhesion of monocytes and endothelial expression of VCAM-1, E-selectin and ICAM-1 [6].
Experimental studies have been performed on rats for the prevention of hepatic IR injury by administering omega-3 PUFA-rich diet [7,8]. It has been shown that omega-3 PUFA treatment effectively reduced hepatic steatosis and consequently attenuated hepatic IR injury in rats [7]. Diet enriched with omega-3 PUFAs has also been shown to have a preconditioning effect reducing liver IR injury in rats [8]. A recent study performed on sixty-six liver transplant patients has shown that post-transplant parenteral nutritional support combined with omega-3 fatty acids can significantly improve liver injury and shorten post-transplant hospital stay [9].
Accumulation of ceramide and alteration of sphingolipid metabolism during liver ischemia reperfusion (IR) injury have been previously demonstrated [10,11]. Ceramide can be generated through de novo synthesis, hydrolysis of sphingomyelin by sphingomyelinase (SMase) and breakdown of glycosphingolipids [12]. Among the different types of SMase that are present, acid and neutral SMase (N-SMase) enzymes have been investigated extensively in response to cellular stress [13]. In vivo administration of an acid SMase inhibitor, imipramine, or acid SMase knockdown by siRNA decreased ceramide generation during IR, and attenuated serum ALT levels, hepatocellular necrosis, cytochrome c release, and caspase-3 activation [11].
Although the effect of acid SMase inhibition on liver IR injury has been extensively studied; the effect of neutral SMase inhibition on liver IR injury and on changes in endogenous PUFA levels following liver IR injury has not been investigated. The aim of this study was to investigate the effect of neutral SMase inhibition on liver PUFAs, phospholipase A2, cyclooxygenase and prostaglandin E2 levels following warm IR injury.
Materials and methods
Animals
All experimental protocols conducted on rats were performed in accordance with the standards established by the Institutional Animal Care and Use Committee at Akdeniz University Medical School. Male Wistar rats weighing 350-450 g were housed in stainless steel cages and were allowed free access to standard rat chow (Korkutelim Yem, Antalya, Turkey) containing 6.05% crude fat which included linoliec acid, linolenic acid, saturated fatty acids and monounsaturated fatty acids. The Animals were maintained at 12 h light-dark cycles and a constant temperature of 23±1°C at all times. Neutral sphingomyelinase (N-SMase) inhibition was initiated 72 hours before liver IR injury was carried out. A selective N-SMase inhibitor, GW 4869 (1.25 mg/kg), was administered daily via intraperitoneal (ip) injections. GW4869 hydrate (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in saline with 2.5% DMSO. It was previously reported that this dose of GW4869 does not produce any adverse effects, does not alter the sphingomyelin or ceramide content of heart, skeletal muscle or liver and enters into brain in sufficient quantity to inhibit neutral sphingomyelinase activity [14]. Dimethyl sulfoxide treated rats were-injected with the same volume of vehicle used to dissolve GW4869 (2.5% DMSO).
Rat model of hepatic ischemia-reperfusion injury
Animals were fasted 12 h before surgery, but allowed to drink tap water ad libitum. Rats were anesthetized with urethane anesthesia (1.2 g/kg subcutaneously). A model of lobar (70%) hepatic warm ischemia was performed according to a method previously described [15,16]. After shaving and disinfecting the abdomen with betadine, a complete midline incision was made. The portal vein was exposed and vessels supplying the median and left lateral hepatic lobes were clamped for 60 min. Reperfusion followed for 60 min via removal of the microvascular clip. The caudal and right lobes retained an intact portal and arterial blood flow, in addition to venous outflow. These lobes served as control and also prevented intestinal congestion. The abdomen was kept closed throughout the experimental period and body temperature was maintained by placing rats under warming lamps. Blood samples were obtained before and after the experiment, from the tail vein and the right ventricle, respectively. At the end of the experimental period, liver was perfused with 0.9% NaCl injected from the left ventricle en route for inferior vena cava. Tissue samples obtained from the left and median lobes of the liver accounted for I/R while dissected right lateral and caudate lobes served as nonischemic group. Obtained liver tissues were either flash frozen in liquid nitrogen and stored at -70°C; or fixed for histological evaluation.
The rat model of hepatic IR injury performed in our study allowed us to obtain both control and IR-injured tissue from the same liver. Tissue samples obtained from the left and median lobes of the liver accounted for IR, whereas dissected right lateral and caudate lobes served as the nonischemic group. From this standpoint, control and IR livers (n=8) were obtained from rats that underwent IR injury. GW and GW+IR livers were obtained from rats that were treated with GW4869 and underwent IR injury (n=10). DMSO and DMSO+IR livers were obtained from rats that were treated with 2.5% DMSO and underwent IR injury (n=4). Sham livers (n=4) were obtained from rats in which only laparotomy was performed.
Histopathological evaluation of liver sections
Paraffin sections stained with hematoxylin and eosin were evaluated by a pathologist blinded to the experimental condition. 20 high-power fields (HPF, 200×) were evaluated in all sections for congestion, intracellular edema and necrosis as previously described [17]. Congestion and intracellular edema was scored as follows: 0=none, 1=present in zone III, 2=present in zones II-III, 3=present in zones I-II-III. Necrosis was scored as follows: 0=none, 1=single or focal necrosis, 2=submassive necrosis, 3=massive necrosis+infarction. Total histopathological score was obtained by summation of all scores given to each parameter.
Measurement of serum alanine aminotransferase
Serum alanine aminotransferase activity was measured via an alanine transaminase assay kit (Cayman Chemical, Cat No: 700260, Ann Arbor, MI, USA). The rate of NADH oxidation was monitored by a coupled reaction system using lactate dehydrogenase (LDH). One unit of enzyme activity was defined as the amount of enzyme that caused the oxidation of 1 μmol of NADH to NAD+ per minute at 37°C.
Electrospray ionization mass spectrometry
Standards for AA (C20: 4n-6), DGLA (C20: 3n-6), EPA (C20: 5n-3) and DHA (C22: 6n-3) were purchased from Sigma-Aldrich (St. Louis MO, USA). Deuterium labeled AA-d8 internal standard (5, 6, 8, 9, 11, 12, 14, 15-AA-d8) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Solutions of AA, DGLA, EPA, DHA and AA-d8 standards were prepared in analytical grade methanol (Merck, Darmstadt, Germany). An optimized multiple reaction monitoring (MRM) method was developed using ultra-fast liquid chromatography (UFLC) coupled with tandem mass spectrometry (MS/MS). A UFLC system (LC-20 AD UFLC XR, Shimadzu Corporation, Japan) was coupled to a LCMS-8040 triple quadrupole mass spectrometer (Shimadzu Corporation, Japan). Chromatographic separations were carried out using Inertsil HPLC column (ODS-4, 2.1x100 mm, 3 μm; GL Sciences Inc. Tokyo, Japan) maintained at 40°C. DHA, EPA, AA and DGLA were separated using a gradient elution with a flow rate of 0.45 ml/min. Mobile phase solvent A was 10 mM ammonium acetate (Sigma-Aldrich, St. Louis, MO, USA) in water and solvent B was acetonitrile (Sigma-Aldrich, St. Louis, MO, USA). Gradient program was solvent B, 70% (0 min), 90% (3 min), 100% (3.01-4 min) and 70 % (4.01-8 min). MRM transitions and responses were automatically optimized for individual compounds in negative electrospray ionization (ESI). In the negative ESI-MS mode the precursor and product m/z values for AA, DHA, EPA, DGLA and AA-d8 are given in the results section. Responses to AA, DHA, EPA and DGLA were optimized to a linear calibration range from 100 ng/ml to 30 μg/ml and a sample analysis time of 8 minutes.
Sample preparation for LC-MS/MS
Samples were prepared for LC-MS/MS analysis via a modified protocol as previously described [18,19]. All tissues were weighed and homogenized in ice-cold 50 mmol/L sodium phosphate buffer (pH 7.4). Homogenates were centrifuged (10,000 g for 15 min at 4°C) and supernatants were stored at -80°C. Briefly, in a glass test tube, 200 μl tissue supernatant was added to 200 μl AA-d8 internal standard solution. 1 ml of acetonitril/37% hydrochloric acid (Cayman, Ann Arbor, MI, USA) was added to the mixture in a 4:1 v/v. Tubes were capped with reusable teflon liner screw caps and samples were hydrolyzed by incubating at 90°C for 2 hours in a heating block (VLM, Bielefeld, Germany). After cooling down to room temperature, fatty acids were extracted with 2 ml of hexane. Samples were vortex-mixed for 20 seconds, left at room temperature for 5 minutes and centrifuged at 3000 rpm for 1 minute. The upper phase containing free fatty acids were transferred to glass tubes and evaporated at room temperature under a constant stream of nitrogen with height adjustable gas distribution unit (VLM, Bielefeld, Germany). Fatty acids were dissolved in 200 μl methanol-water (180:20, v/v) filtered via 0.2 μm polytetrafluoroethylene (PTFE) syringe filters (Whatman, GE Healthcare Bio-Sciences, Pittsburgh, USA) and transferred to autosampler vials (Vertical Ch-s romatography, Nonthaburi, Thailand).
Measurement of liver total phospholipase A2
Activity of liver PLA2 was measured via a PLA2 assay kit (Abcam, Cat No: ab133090, Cambridge, MA, USA). Liver tissues were weighed and homogenized in ice-cold 50 mmol/L sodium phosphate buffer (pH 7.4) containing 1 mM EDTA. Homogenates were centrifuged (10, 000 g for 15 min at 4°C) and supernatants were stored at -80°C. Before performing the assay, low molecular weight contaminants were removed from the samples using an ultrafiltration unit via centrifugation through a 10-kDa molecular mass cut-off filter (Amicon, Millipore Corporation, Bedford, MA, USA) for 30 minutes at 25°C. Samples were reconstituted with 50 mmol/L sodium phosphate buffer (pH 7.4) containing 1 mM EDTA. Arachidonoyl thio-PC synthetic substrate was used to detect PLA2 activity. Hydrolysis of the arachidonoyl thioester bond releases a free thiol which was detected by 5,5’-dithiobis-(2-nitrobenzoic acid (DTNB). One unit of enzyme activity was defined as the amount of enzyme that hydrolyzes one μmol of arachidonoyl thio-PC per minute at 25°C.
Measurement of liver cyclooxygenase activity
Liver tissues were weighed and homogenized in 0.1 M ice-cold Tris-HCl buffer at pH 7.8 containing 1 mM EDTA. Tissue homogenates were centrifuged at 10,000 g for 15 minutes at 4°C and supernatants were kept at -80°C until assayed. Cyclooxygenase (COX) activity was measured using a COX activity assay kit (Cayman Chemical, Cat No: 760151 Ann Arbor, MI, USA) according to manufacturer’s instructions. The COX activity assay kit measures enzyme activity colorimetrically by monitoring the appearance of oxidized N,N,N’,N’-tetramethyl-p-phenylenediamine (TMPD) at 590 nm. One unit of enzyme activity was defined as the amount of enzyme that caused the oxidation of 1 nmol of TMPD per minute at 25°C.
Measurement of prostaglandin E2
Prostaglandin E2 (PGE2) was measured in tissue samples by a commercial enzyme immunoassay test kit (Cayman Chemical, Cat No: 514010 Ann Arbor, MI, USA) according to manufacturer’s instructions. Liver tissues were weighed and homogenized in 0.1 M ice-cold phosphate buffer at pH 7.4 containing 1 mM EDTA and 10 μM indomethacin. Tissue homogenates were centrifuged at 10,000 g for 15 minutes at 4°C and supernatants were kept at -80°C until assayed. Briefly, PGE2 present in the sample competes with acetylcholinesterase-labeled PGE2 antibody for binding sites on a goat polyclonal anti-mouse antibody. Following a wash to remove unbound materials, a substrate solution is added to the wells to determine the bound enzyme activity. The color development is stopped, and the absorbance is read at 412 nm. The intensity of the color is inversely proportional to the concentration of PGE2 in the sample. A standard curve of absorbance values of known PGE2 standards was plotted as a function of the logarithm of PGE2 standard concentrations (pg/ml) using the GraphPad Prism Software program for windows version 5.03. (GraphPad Software Inc). PGE2 concentrations in the samples were calculated from their corresponding absorbance values via the standard curve.
Protein measurements
Protein concentrations were measured at 595 nm by a modified Bradford assay using Coomassie Plus reagent with bovine serum albumin as a standard (Pierce Chemical Company, Rockford, IL).
Statistical analysis
Data were analyzed using Sigma Stat (version 2.03) statistical software for Windows, and a P value < 0.05 was considered statistically significant. Statistical analysis for each measurement is described within the figure and table legends.
Results
Analysis of liver ischemia-reperfusion injury
Hepatic photomicrographs of a representative rat from each group are shown in Figure 1. Histopathological scores of liver IR injury are given in Table 1. Congestion, intracellular edema, necrosis, and total histopathological score were significantly greater (P < 0.05) in IR and DMSO+IR when compared to control, GW, sham and DMSO groups. GW treatment in IR injury caused a decrease in intracellular edema, necrosis and total histopathological score however it did not reach statistical significance. Biochemical results of liver IR injury are given in Table 2. Serum ALT levels were significantly increased in all IR groups confirming the presence of hepatic injury.
Figure 1.

Hematoxylin and eosin staining of liver sections. Hepatic photomicrographs of representative rat are shown from each of the experimental groups. IR, ischemia-reperfusion; CV, central vein. Magnification: 20×. IR, ischemia-reperfusion; GW, animals treated with GW 4869; DMSO, group treated with dimethyl sulfoxide.
Table 1.
Histopathological scores of liver sections
| Group | Congestion | Intracellular edema | Necrosis | Total score |
|---|---|---|---|---|
| Control (n=4) | 1.00±0.00 | 0.25±0.50 | 0.00±0.00 | 1.25±0.50 |
| IR (n=4) | 2.25±0.50a | 3.00±0.00b | 2.75±0.50c | 8.00±0.00a |
| DMSO (n=4) | 0.50±0.58 | 0.00±0.00 | 0.00±0.00 | 0.50±0.58 |
| DMSO+IR (n=4) | 2.75±0.50a | 3.00±0.00b | 2.50±1.00c | 8.25±1.50a |
| GW (n=4) | 0.75±0.50 | 0.00±0.00 | 0.50±0.58 | 1.25±0.96 |
| GW+IR (n=4) | 2.50±0.58a | 2.00±0.00 | 2.00±0.82 | 6.50±1.29a |
| Sham (n=4) | 0.00±0.00 | 0.50±0.58 | 0.75±0.50 | 0.50±0.58 |
Values are mean ± SD. IR, ischemia-reperfusion; GW, animals treated with GW 4869; DMSO, group treated with dimethyl sulfoxide. Statistical analysis for congestion, and total score were performed by one way analysis of variance and all pairwise multiple comparisons were via Tukey test. Statistical analysis for intracellular edema and necrosis was done by Kruskal-Wallis one-way analysis of variance and all pairwise multiple comparisons were by Dunn’s method.
P < 0.05 vs. control, sham, GW and DMSO.
P < 0.05 vs. control, GW and DMSO.
P < 0.05 vs. control and DMSO.
Table 2.
Plasma activity of alanine aminotransferase
| Group | n | Plasma ALT (U/L) |
|---|---|---|
| Control | 8 | 23.51 ± 4.42 |
| IR | 8 | 231.84 ± 48.84* |
| DMSO | 4 | 29.17 ± 1.49 |
| DMSO+IR | 4 | 195.75 ± 22.63* |
| GW | 10 | 29.80 ± 5.09 |
| GW+IR | 10 | 147.67 ± 30.11* |
| Sham | 4 | 24.91 ± 4.56 |
All values are mean ± SEM. IR, ischemia-reperfusion; GW, animals treated with GW 4869; DMSO, group treated with dimethyl sulfoxide. Statistical analysis was performed by Kruskal-Wallis one-way analysis of variance on ranks and all pairwise multiple comparisons were via Dunn’s method.
P < 0.05 vs. control.
ESI-MS spectra
The precursor and product m/z values for analyzed polyunsaturated fatty acids were as follows: DGLA (C20: 3n6), precursor m/z: 304.80, product m/z: 59.00, 260.70; AA (C20: 4n6), precursor m/z: 303.10, product m/z: 59.00, 258.90; EPA (C20: 5n3), precursor m/z: 301.10, product m/z: 59.10, 256.70; DHA (C22: 6n3): precursor m/z: 327.10, product m/z: 59.10, 283.20; AA-d8, precursor m/z: 310 1.10, product m/z: 59.10, 97.90, 267.10. Figure 2A, shows representative negative ion mode spectra. As shown in the Figure 2A, retention time of time of EPA (C20: 5n3), DHA (C22: 6n3), AA (C20: 4n6), AA-d8 and DGLA (C20: 3n6) was 1.869, 2.131, 2.391, 2.329 and 2.911 minutes, respectively. Figure 2B shows tandem mass spectra ob0tained by collision-induced dissociation of precursor ions. The m/z values of product ions correspond to endogenous C20: 5n3, C20: 4n6, C20: 3n6 and C22: 6n3. The deuterium-labeled internal standard fatty acid peak is indicated at m/z values 267.1.
Figure 2.
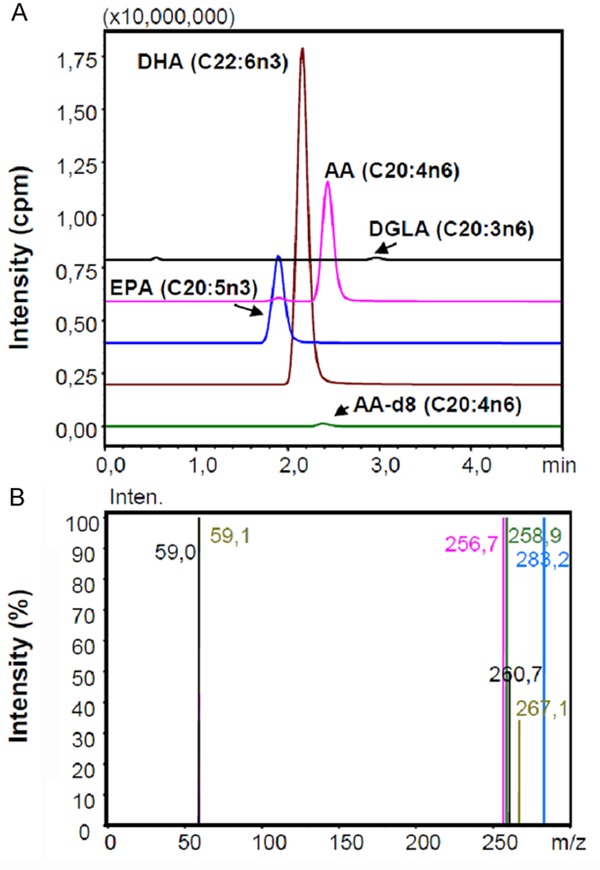
A: Representative negative ion mode spectra; B: Representative tandem mass spectra. IR, ischemia-reperfusion; GW, animals treated with GW 4869; DMSO, group treated with dimethyl sulfoxide. AA, Arachidonic acid; EPA, Eicosapentaenoic acid; DHA, Docosahexaenoic acid; DGLA, Dihomo-gamma-linolenic acid.
Levels of polyunsaturated fatty acids
Levels of endogenous PUFAs measured in liver tissue are given in Table 3. DGLA, AA, EPA and DHA were increased in all IR injury groups when compared to control, GW, DMSO and sham groups. No significant difference in AA/DHA and AA/EPA ratio was observed among the experimental groups.
Table 3.
Analysis of polyunsaturated fatty acids in liver tissue
| Parameter | Control (n=8) | Sham (n=4) | IR (n=8) | GW (n=10) | GW+IR (n=10) | DMSO (n=4) | DMSO+IR (n=4) |
|---|---|---|---|---|---|---|---|
| DGLA (C20: 3n6) | 3.37±1.45 | 4.30±1.39 | 8.62±2.03a | 3.50±2.98 | 6.67±2.52b | 4.17±1.30 | 7.49±1.30b |
| AA (C20: 4n6) | 25.00±9.99 | 31.42±10.29 | 67.54±15.32a | 24.83±19.12 | 52.89±16.94c | 35.17±11.69 | 68.62±5.92a |
| EPA (C20: 5n3) | 0.64±0.31 | 0.75±0.11 | 2.57±0.66d | 0.75±0.51 | 1.95±0.69c | 0.88±0.23 | 1.54±0.33 |
| DHA (C22: 6n3) | 11.27±4.57 | 10.19±2.46 | 22.26±7.40d | 11.59±3.69 | 18.38±5.49 | 13.97±4.60 | 24.30±2.94d |
| AA/DHA | 2.77±0.63 | 3.11±0.74 | 3.53±1.01 | 2.24±1.05 | 2.93±0.42 | 2.61±0.14 | 2.94±0.25 |
| AA/EPA | 34.99±10.71 | 25.58±2.26 | 31.19±7.76 | 32.50±7.06 | 29.36±9.24 | 39.42±4.69 | 35.57±7.46 |
Values are expressed as mg fatty acid/g tissue protein. Data are reported as mean ± SD. IR, ischemia-reperfusion; GW, animals treated with GW 4869; DMSO, group treated with dimethyl sulfoxide. DGLA, Dihomo-gamma-linolenic acid; AA, Arachidonic acid; EPA, Eicosapentaenoic acid; DHA, Docosahexaenoic acid. Statistical analysis for DGLA, AA and DHA was performed by one way analysis of variance and all pairwise multiple comparisons were via Tukey test. Statistical analysis for EPA was done by Kruskal-Wallis one-way analysis of variance and all pairwise multiple comparisons was by Dunn’s method.
P < 0.05 vs. control, sham, GW and DMSO.
P < 0.05 vs. control.
P < 0.05 vs. control and GW.
P < 0.05 vs. control, sham, GW.
Liver total phospholipase A2 activity
Total PLA2 activity measured in all IR tissue homogenates were significantly higher compared to control, sham, GW, and DMSO groups (Figure 3A). No significant difference was observed between control, sham, GW and DMSO groups.
Figure 3.
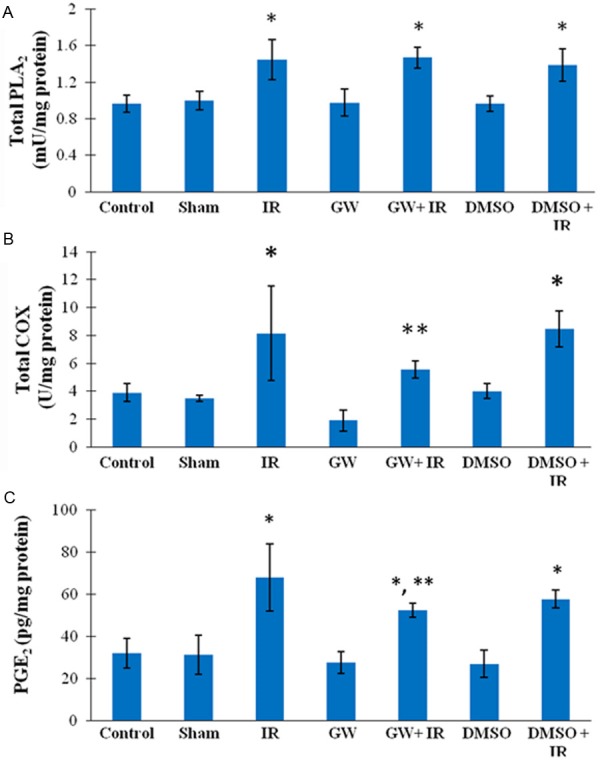
A: Liver total phospholipase A2 activity. IR, ischemia-reperfusion; GW, animals treated with GW 4869; DMSO, group treated with dimethyl sulfoxide. All values are mean ± SD. Statistical analysis was performed by one way analysis of variance and all pairwise multiple comparisons were via Tukey test. *, P < 0.01 vs. Control, Sham, GW and DMSO; B: Liver cyclooxygenase activity. All values are mean ± SD. Statistical analysis was done by Kruskal-Wallis one-way analysis of variance and all pairwise multiple comparisons were by Dunn’s method. *=P < 0.05 vs. Control, Sham, GW and DMSO; **, P < 0.05 vs. GW; C: Liver prostaglandin E2 levels. All values are mean ± SD. Statistical analysis was performed by one way analysis of variance and all pairwise multiple comparisons were via Tukey test. *, P ≤ 0.001 vs. Control, Sham, GW and DMSO; **, P=0.005 vs. IR.
Liver cyclooxygenase activity
Total COX activity measured in IR and DMSO+IR tissue homogenates were significantly higher compared to control, sham and DMSO groups. (Figure 3B). Treatment with GW decreased total COX levels in IR injury and thus no significant difference was observed between GW+IR group vs. control, sham and DMSO groups.
Liver prostaglandin E2 levels
Liver PGE2 contents are shown in Figure 3C. PGE2 measured in all IR samples were significantly higher compared to control, sham, GW, and DMSO groups. Treatment with GW significantly decreased PGE2 levels in IR injury and thus a significant difference was observed between GW+IR vs. IR group.
Discussion
This study investigated the effect of N-SMase inhibition on endogenous PUFA levels following liver IR injury in rats without omega-3 or omega-6 diet supplementation. To the best of our knowledge, this study is the first to investigate the effect of N-SMase inhibition on AA levels (C20: 4n-6) and downstream inflammatory pathways in liver IR injury.
Liver AA (C20: 4n-6), DGLA (C20: 3n-6), EPA (C20: 5n-3) and DHA (C22: 6n-3) were significantly increased following IR injury compared to control, sham, GW and DMSO groups. No significant difference was observed in AA/DHA and AA/EPA ratio among the experimental groups. Eicosanoids, derived mainly from AA (C20: 4n-6) are key mediators and regulators of inflammation. They include prostaglandins (PGs), thromboxanes (TXs) and leukotrienes (LTs) [20]. Elevated liver AA (C20: 4n-6) levels may thus be a source of pro-aggregatory substances in liver IR injury [21]. In this context, it is important to note that decreased levels of prostacyclin (PGI2) and increased levels of TXs and LTs is associated with abnormalities in the ratio of vasodilator to vasoconstrictor mediators in liver IR injury [22].
Previous studies have demonstrated a marked release of prostanoids from hepatic tissue after liver transplantation [23]. Increased eicosanoid synthesis is shown to be regulated at the level of key enzymes [24]. The present study has addressed changes of the local availability of these enzymes after warm liver IR injury. We have observed that total PLA2 activity measured in IR tissue homogenates were significantly higher compared to control, sham, GW and DMSO groups. Accumulating evidence has revealed that PLA2 plays an important role in IR injury [25,26]. Infact, PLA2 inhibitor, LY329722, was shown to attenuate hepatic IR injury caused by 2-hr total hepatic vascular exclusion in dogs [26]. Phospholipase A2 de-grades cell membrane phospholipids and plays an important role in the synthesis of pro-inflammatory lipid mediators such as AA (C20: 4n-6) and cytokines during IR injury after liver transplantation [26]. Phospholipase A2 comprises a large group of enzymes that include secretory (sPLA2), cytosolic PLA2 (cPLA2), and calcium independent PLA2 families [27]. These enzymes hydrolyze the phospholipid bond at the sn-2 position. Cytosolic PLA2 and calcium independent PLA2 are localized inside the cell and involved in the breakdown of intracellular membranes, whereas sPLA2 is secreted during inflammatory events [28]. PLA2 accelerates breakdown of membrane phospholipids in the liver and other organs under warm ischemia and releases free fatty acids including AA (C20: 4n-6) and lysophospholipids [29]. Increased total PLA2 activity observed in our experimental model may therefore explain increased levels of measured PUFAs in liver tissues. Released free fatty acids via the action of PLA2 are metabolized into PGs, TXs, LTs and platelet-activating factor [30]. These lipid derivatives have pro-inflammatory and vasoconstrictive actions and contribute to postischemic organ dysfunction. A role for cytosolic phospholipase A2 (cPLA2) has been proposed in coupling TNF-alpha action to the activation of N-SMase because cells that are deficient in cPLA2 do not show hydrolysis of sphingomyelin in response to TNF-alpha, and their responsiveness is largely restored by the expression of cPLA2 [31]. Activation of N-SMase can occur in response to TNFalpha stimulation [32] which is reported to increase in liver IR injury [2]. How cPLA2 is coupled to N-SMase requires further investigation. It is important to note that although cPLA2 is required for TNF-alpha mediated activation of N-SMase, we have found that inhibition of N-SMase does not block increased PLA2 activity in liver IR injury.
Activity of COX, the initial enzyme of prostaglandin synthesis was also measured in liver tissue following IR injury. Cyclooxygenase (COX) is the rate-limiting enzyme in the production of prostanoids from arachidonic acid. Research has showed that the COX/prostanoid pathway is activated in hepatic diseases and liver stress reaction, such as alcoholic liver disease [33] liver fibrogenesis [34], viral hepatitis C [35] and liver IR injury [36] causing liver damage manifested as inflammation, necrosis and fatty liver. In agreement with previous reports, we have also observed significantly increased activity of COX following liver IR injury compared to control, sham, DMSO and GW groups suggesting that the formation of prostanoids via the COX/prostanoid pathway also takes part in observed tissue damage. A significant decrease was observed in COX activity via inhibition of N-SMase. This finding supports previous studies that have shown ceramide regulates the transcription of COX-2 [37]. The ceramide signaling pathway is activated by SMase-mediated hydrolysis of cell membrane sphingomyelin to ceramide. Treatment of cells with neutral SMase or C2- or C6-ceramide enhances PGE2 synthesis and increases levels of COX-2 protein and mRNA [37]. In line with these findings we have seen that inhibition of N-SMase attenuates both PGE2 synthesis and total COX activity.
We have measured significantly increased liver PGE2 levels following IR in agreement with previous reports [23]. Arachidonic acid is a precursor of PGE2 synthesis and production of PGs occur by stereospecific lipid-oxidizing enzymes. In the liver, endogenous PGE2 is produced mainly by activated Kupffer cells during hepatic injury [38]. Previous studies have demonstrated that both endogenous and exogenous PGE2 are protective against liver injury caused by IR [39]. This effect may be due to increased inhibition of platelet aggregation, liver perfusion or direct cytoprotection by PGE2 [40]. PGE2 has also been suggested to restore liver damage through the regulation of cytokine cascades [38].
In conclusion, the present study revealed that liver IR injury significantly increased the concentration of AA (C20: 4n-6), DGLA (C20: 3n-6), EPA (C20: 5n-3) and DHA (C22: 6n-3) in liver tissue specimens and had no effect on hepatic ratio of AA/DHA and AA/EPA. The observed increase of endogenous PUFA levels in the liver following IR injury was accompanied by increased activity of key enzymes PLA2 and COX, which are involved in the production of prostanoids. N-SMase inhibition significantly decreased COX activity and PGE2 levels in postischemic liver. Future studies evaluating agents blocking N-SMase activity can facilitate the development of treatment strategies to alleviate inflammation in liver IR injury.
Acknowledgements
This study was supported by a grant (No: 2013.01.0103.004) from Akdeniz University Research Foundation, Turkey.
Disclosure of conflict of interest
None.
References
- 1.Hasselgren PO. Prevention and treatment of ischemia of the liver. Surg Gynecol Obstet. 1987;164:187–96. [PubMed] [Google Scholar]
- 2.Dogan S, Aslan M. Hepatic ischemia-reperfusion injury and therapeutic strategies to alleviate cellular damage. Hepatol Res. 2011;41:103–17. doi: 10.1111/j.1872-034X.2010.00765.x. [DOI] [PubMed] [Google Scholar]
- 3.Colletti LM, Kunkel SL, Walz A, Burdick MD, Kunkel RG, Wilke CA, Strieter RM. The role of cytokine networks in the local liver injury following hepatic ischemia/reperfusion in the rat. Hepatology. 1996;23:506–14. doi: 10.1002/hep.510230315. [DOI] [PubMed] [Google Scholar]
- 4.Jaeschke H, Farhood A, Smith CW. Neutrophils contribute to ischemia/reperfusion injury in rat liver in vivo. FASEB J. 1990;4:3355–9. [PubMed] [Google Scholar]
- 5.Bates EJ, Ferrante A, Smithers L, Poulos A, Robinson BS. Effect of fatty acid structure on neutrophil adhesion, degranulation and damage to endothelial cells. Atherosclerosis. 1995;116:247–59. doi: 10.1016/0021-9150(95)05553-9. [DOI] [PubMed] [Google Scholar]
- 6.De Caterina R, Liao JK, Libby P. Fatty acid modulation of endothelial activation. Am J Clin Nutr. 2000;71:213S–23S. doi: 10.1093/ajcn/71.1.213S. [DOI] [PubMed] [Google Scholar]
- 7.Marsman HA, Heger M, Kloek JJ, Nienhuis SL, ten Kate FJ, van Gulik TM. Omega-3 fatty acids reduce hepatic steatosis and consequently attenuate ischemia-reperfusion injury following partial hepatectomy in rats. Dig Liver Dis. 2011;43:984–90. doi: 10.1016/j.dld.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 8.Coelho AM, Machado MC, Takahashi HK, Sampietre SN, Stefano JT, Leite AZ, Curi R, D’Albuquerque LA. Ischemic preconditioning-like effect of polyunsaturated fatty acid-rich diet on hepatic ischemia/reperfusion injury. J Gastrointest Surg. 2011;15:1679–88. doi: 10.1007/s11605-011-1648-x. [DOI] [PubMed] [Google Scholar]
- 9.Zhu XH, Wu YF, Qiu YD, Jiang CP, Ding YT. Liver-protecting effects of omega-3 fish oil lipid emulsion in liver transplantation. World J Gastroenterol. 2012;18:6141–7. doi: 10.3748/wjg.v18.i42.6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhai ST, Liu GY, Xue F, Sun GP, Liang L, Chen W, Xu GD, Li JJ, Yang J, Liang TB. Changes of sphingolipids profiles after ischemia-reperfusion injury in the rat liver. Chin Med J (Engl) 2009;122:3025–31. [PubMed] [Google Scholar]
- 11.Llacuna L, Marí M, Garcia-Ruiz C, Fernandez-Checa JC, Morales A. Critical role of acidic sphingomyelinase in murine hepatic ischemia-reperfusion injury. Hepatology. 2006;44:561–72. doi: 10.1002/hep.21285. [DOI] [PubMed] [Google Scholar]
- 12.Wu BX, Clarke CJ, Hannun YA. Mammalian neutral sphingomyelinases: regulation and roles in cell signaling responses. Neuromolecular Med. 2010;12:320–30. doi: 10.1007/s12017-010-8120-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pavoine C, Pecker F. Sphingomyelinases: their regulation and roles in cardiovascular pathophysiology. Cardiovasc Res. 2009;82:175–83. doi: 10.1093/cvr/cvp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tabatadze N, Savonenko A, Song H, Bandaru VV, Chu M, Haughey NJ. Inhibition of neutral sphingomyelinase-2 perturbs brain sphingolipid balance and spatial memory in mice. J Neurosci Res. 2010;88:2940–51. doi: 10.1002/jnr.22438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dogan S, Ozlem Elpek G, Kirimlioglu Konuk E, Demir N, Aslan M. Measurement of intracellular biomolecular oxidation in liver ischemia-reperfusion injury via immuno-spin trapping. Free Radic Biol Med. 2012;53:406–14. doi: 10.1016/j.freeradbiomed.2012.05.028. [DOI] [PubMed] [Google Scholar]
- 16.Curek GD, Cort A, Yucel G, Demir N, Ozturk S, Elpek GO, Savas B, Aslan M. Effect of astaxanthin on hepatocellular injury following ischemia/reperfusion. Toxicology. 2010;267:147–53. doi: 10.1016/j.tox.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 17.Yilmaz S, Ates E, Tokyol C, Pehlivan T, Erkasap S, Koken T. The protective effect of erythropoietin on ischaemia/reperfusion injury of liver. HPB (Oxford) 2004;6:169–73. doi: 10.1080/13651820410026077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aslan M, Aslan I, Özcan F, Eryılmaz R, Ensari CO, Bilecik T. A pilot study investigating early postoperative changes of plasma polyunsaturated fatty acids after laparoscopic sleeve gastrectomy. Lipids Health Dis. 2014;13:62. doi: 10.1186/1476-511X-13-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aslan M, Özcan F, Aslan I, Yücel G. LC-MS/MS analysis of plasma polyunsaturated fatty acids in type 2 diabetic patients after insulin analog initiation therapy. Lipids Health Dis. 2013;12:169. doi: 10.1186/1476-511X-12-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wall R, Ross RP, Fitzgerald GF, Stanton C. Fatty acids from fish: the anti-inflammatory potential of long-chain omega-3 fatty acids. Nutr Rev. 2010;68:280–9. doi: 10.1111/j.1753-4887.2010.00287.x. [DOI] [PubMed] [Google Scholar]
- 21.Setty BN, Dampier CD, Stuart MJ. Arachidonic acid metabolites are involved in mediating red blood cell adherence to endothelium. J Lab Clin Med. 1995;125:608–17. [PubMed] [Google Scholar]
- 22.Besse T, Gustin T, Claeys N, Schroeyers P, Lambotte L. Effect of PGI2 and thromboxane antagonist on liver ischemic injury. Eur Surg Res. 1989;21:213–7. doi: 10.1159/000129026. [DOI] [PubMed] [Google Scholar]
- 23.Post S, Goerig M, Otto G, Manner M, Senninger N, Kommerell B, Herfarth C. Prostanoid release in experimental liver transplantation. Transplantation. 1990;49:490–4. doi: 10.1097/00007890-199003000-00002. [DOI] [PubMed] [Google Scholar]
- 24.Post S, Goerig M, Otto G, Manner M, Foltis C, Hofmann W, Herfarth C. Rapid increase in the activity of enzymes of eicosanoid synthesis in hepatic and extrahepatic tissues after experimental liver transplantation. Transplantation. 1991;51:1058–65. doi: 10.1097/00007890-199105000-00025. [DOI] [PubMed] [Google Scholar]
- 25.Koike K, Yamamoto Y, Hori Y, Ono T. Group IIA phospholipase A2 mediates lung injury in intestinal ischemia-reperfusion. Ann Surg. 2000;232:90–7. doi: 10.1097/00000658-200007000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogata K, Jin MB, Taniguchi M, Suzuki T, Shimamura T, Kitagawa N, Magata S, Fukai M, Ishikawa H, Ono T, Furukawa H, Fujita M, Todo S. Attenuation of ischemia and reperfusion injury of canine livers by inhibition of type II phospholipase A2 with LY329722. Transplantation. 2001;71:1040–6. doi: 10.1097/00007890-200104270-00004. [DOI] [PubMed] [Google Scholar]
- 27.Murakami M, Kudo I. Phospholipase A2. J Biochem. 2002;131:285–92. doi: 10.1093/oxfordjournals.jbchem.a003101. [DOI] [PubMed] [Google Scholar]
- 28.Menschikowski M, Hagelgans A, Siegert G. Secretory phospholipase A2 of group IIA: is it an offensive or a defensive player during atherosclerosis and other inflammatory diseases? Prostaglandins Other Lipid Mediat. 2006;79:1–33. doi: 10.1016/j.prostaglandins.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 29.Dan P, Nitzan DW, Dagan A, Ginsburg I, Yedgar S. H2O2 renders cells accessible to lysis by exogenous phospholipase A2: a novel mechanism for cell damage in inflammatory processes. FEBS Lett. 1996;383:75–8. doi: 10.1016/0014-5793(96)00227-x. [DOI] [PubMed] [Google Scholar]
- 30.Murakami M, Nakatani Y, Atsumi G, Inoue K, Kudo I. Regulatory functions of phospholipase A2. Crit Rev Immunol. 1997;17:225–83. doi: 10.1615/critrevimmunol.v17.i3-4.10. [DOI] [PubMed] [Google Scholar]
- 31.Jayadev S, Hayter HL, Andrieu N, Gamard CJ, Liu B, Balu R, Hayakawa M, Ito F, Hannun YA. Phospholipase A2 is necessary for tumor necrosis factor alpha-induced ceramide generation in L929 cells. J Biol Chem. 1997;272:17196–203. doi: 10.1074/jbc.272.27.17196. [DOI] [PubMed] [Google Scholar]
- 32.Liu B, Obeid LM, Hannun YA. Sphingomyelinases in cell regulation. Semin Cell Dev Biol. 1997;8:311–322. doi: 10.1006/scdb.1997.0153. [DOI] [PubMed] [Google Scholar]
- 33.Nanji AA, Miao L, Thomas P, Rahemtulla A, Khwaja S, Zhao S, Peters D, Tahan SR, Dannenberg AJ. Enhanced cyclooxygenase-2 gene expression in alcoholic liver disease in the rat. Gastroenterology. 1997;112:943–51. doi: 10.1053/gast.1997.v112.pm9041257. [DOI] [PubMed] [Google Scholar]
- 34.Planagumà A, Clària J, Miquel R, López-Parra M, Titos E, Masferrer JL, Arroyo V, Rodés J. The selective cyclooxygenase-2 inhibitor SC-236 reduces liver fibrosis by mechanisms involving non-parenchymal cell apoptosis and PPAR-gamma activation. FASEB J. 2005;19:1120–2. doi: 10.1096/fj.04-2753fje. [DOI] [PubMed] [Google Scholar]
- 35.Waris G, Siddiqui A. Hepatitis C virus stimulates the expression of cyclooxygenase-2 via oxidative stress: role of prostaglandin E2 in RNA replication. J Virol. 2005;79:9725–34. doi: 10.1128/JVI.79.15.9725-9734.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Kim SH, Lee SM. Expression of hepatic vascular stress genes following ischemia/reperfusion and subsequent endotoxemia. Arch Pharm Res. 2004;27:769–75. doi: 10.1007/BF02980147. [DOI] [PubMed] [Google Scholar]
- 37.Subbaramaiah K, Chung WJ, Dannenberg AJ. Ceramide regulates the transcription of cyclooxygenase-2. Evidence for involvement of extracellular signal-regulated kinase/c-Jun N-terminal kinase and p38 mitogen-activated protein kinase pathways. J Biol Chem. 1998;273:32943–9. doi: 10.1074/jbc.273.49.32943. [DOI] [PubMed] [Google Scholar]
- 38.Wanner GA, Müller P, Ertel W, Busch CJ, Menger MD, Messmer K. Differential effect of cyclooxygenase metabolites on proinflammatory cytokine release by Kupffer cells after liver ischemia and reperfusion. Am J Surg. 1998;175:146–51. doi: 10.1016/S0002-9610(97)00275-4. [DOI] [PubMed] [Google Scholar]
- 39.Arai M, Peng XX, Currin RT, Thurman RG, Lemasters JJ. Protection of sinusoidal endothelial cells against storage/reperfusion injury by prostaglandin E2 derived from Kupffer cells. Transplantation. 1999;68:440–5. doi: 10.1097/00007890-199908150-00017. [DOI] [PubMed] [Google Scholar]
- 40.Masaki N, Ohta Y, Shirataki H, Ogata I, Hayashi S, Yamada S, Hirata K, Nagoshi S, Mochida S, Tomiya T. Hepatocyte membrane stabilization by prostaglandins E1 and E2: favorable effects on rat liver injury. Gastroenterology. 1992;102:572–6. doi: 10.1016/0016-5085(92)90105-8. [DOI] [PubMed] [Google Scholar]