

Abstract
GRIM-19 has been demonstrated as an important regulator for the normal tissue development. Recently, more evidences regarded GRIM-19 as the new tumor suppressor. However, the possible mechanisms underlying GRIM-19 suppressing cancer growth are unclear. In the present study, Paired hepatocellular carcinoma (HCC) and adjacent non-tumor liver tissues were obtained from 54 patients who underwent primary surgical HCC tissue resection. GRIM-19 protein expression in HCC tissues was performed by immunohistochemistry. Cells were transfected by lentiviruses plasmid expressing GRIM-19. RT-PCR and Western blot analyses were performed to confirm the expression of GRIM-19 mRNA or protein. Cell proliferation was assessed by MTT and FCM analyses. Mitochondrial membrane potential and apoptosis were respectively determined by using fluorescence microscopy and FCM analyses. AKT1, pAKT1, cyclinD1, CDK4, PCNA, Bax, Bcl-2, cleaved caspase-9, cleaved caspase-3, and cytochrome C were detected by Western blot and immunofluorescence. GRIM-19 protein expression was markedly lower in HCC than in paired adjacent non-tumor liver tissues. GRIM-19 overexpression in HCC cells significantly induced cell cycle arrest and enhanced apoptosis. We also found that AKT1 expression and phosphorylation were regulated by the expression of GRIM-19. Collectively, our study demonstrated that GRIM-19 overexpression suppressed HCC growth and downregulated AKT1 expression, suggesting that GRIM-19 might play a crucial role in hepatocarcinogenesis through negatively regulating the PI3K/AKT signaling pathway.
Keywords: GRIM-19, hepatocellular carcinoma, apoptosis, cell cycle, AKT1
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common cancer worldwide but the second most frequent cause of cancer death [1]. Curative treatment options chiefly consist of surgical resection or liver transplantation [2]. Despite many scientific advances, HCC prognosis remains poor due to the absence of early disease symptoms, rapid tumor progression, and invasion-related spreading during early tumor stages [3]. Therefore, identifying potential biological markers for liver cancer occurrence and development is critical for developing new therapeutic strategies.
Accumulating evidence shows notable similarities between normal tissue development and tumorigenesis. For example, the Wnt/β pathway plays a critical role in regulating growth in normal liver development and is also a major mutation target in hepatocellular carcinoma [4]. P53, a tumor suppressor gene, is important for liver development, and its inactivation has been involved in HCC formation [5]. Based on these findings, genes that are involved in normal tissue developmental processes, if dysregulated, might contribute to the tumorigenesis. Gene associated with retinoic and interferon-induced mortality (GRIM-19; also called NDUFA13) was originally isolated as a growth suppressor by screening for an interferon (IFN)/all-trans retinoic acid (RA) combination-induced cell death in human cancer cell lines [6]. It does not belong to any of the known cell death gene categories, such as the Bcl-xl, caspase, and death receptor families. GRIM-19 is a nuclear-encoded protein of mitochondrial respiratory chain complex I, which provides a link between the mitochondrion and its electron transport chain and apoptotic cell death [7,8]. GRIM-19 is closely correlated with mitochondrial metabolism, cell growth, embryonic development, and innate immunity [8-11]. Homologous GRIM-19 gene deletion resulted in embryonic lethality by day 9.5, with the retarded growth and abnormal mitochondrial structure, morphology, and distribution exhibited in the blastocysts [12]. Recent studies reported that GRIM-19 protein expression was lost or severely repressed in numerous human cancers, including carcinomas of the kidney, colorectal, prostate, cervix, glioblastomas, lung, Hurthle cell thyroid, and breast [13-19]. These observations were further corroborated by upregulating STAT3, a multifaceted growth mediator in the cells, and specifically revealing the carcinoma tissues [20]. This suggests that GRIM-19 is not only essential for normal embryonic development, but also a key protein for tumorigenesis. However, the mechanisms underlying GRIM-19 inducing tumorigenesis are still poorly investigated.
In the present study, we examined the protein expression of GRIM-19 in human hepatocellular carcinoma and paired adjacent non-tumor liver tissues. Additionally, we investigated GRIM-19’s roles in human liver cancer cell lines and its possible mechanisms in tumorigenesis.
Materials and methods
HCC patient specimens and cell lines
HCC tissues and adjacent non-tumor liver tissues were collected from 54 HCC patients who underwent liver resection between June 2012 and June 2013 at the First Affiliated Hospital of Jilin University (Changchun, China). No patients had adjuvant therapies like chemotherapy or radiotherapy before surgery. This research was authorized by the Ethics Committee of the First Affiliated Hospital of Jilin University, and all patients signed an informed consent form. The human liver cell line SMMC-7721 was obtained from Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China) and was cultured in RPMI-1640 medium (Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum and 100 u/ml penicillin.
Plasmids, lentiviral particles, and establishment of stable cells
The GRIM-19 expression plasmid pIRES-Puro2-GRIM-19-Myc and the empty plasmid pIRES-Puro2-Myc were kindly provided by Professor Jiadi Hu (Greenebaum Cancer Center, Department of Microbiology and Immunology, Molecular Biology Program, University of Maryland School Medicine, Baltimore, Maryland). Lentiviral particle preparations were performed as previously described [21]. Briefly, GRIM-19 inserts were cloned into the XhoI and XbaI sites of pLVX-Puro (Clontech, Inc). HEK-293T cells were transfected with 5 μg of transfer vector (pLVX-Puro-GRIM-19-Myc), 3.75 μg of packaging plasmid (pCMV-dR8.2 dvpr), and 1.5 μg of envelope plasmid (pVSV-G) (Addgene, Inc.) using Lipofectamine-Plus reagent (Invitrogen, Inc.) for 6 h in serum-free medium. Cells were then fed with regular growth medium. Viral particles from the growth medium were collected from 48-96 h after transfection. The medium was centrifuged to remove debris and filtered through a 0.45 μm filter. SMMC-7721 cells were transfected with the viral titers and then selected by puromycin. After 2 weeks, the number of infectious virus particles was estimated based on the number of puromycin-resistant colonies formed.
Immunohistochemistry
A total of 54 hepatocellular carcinoma tumors and adjacent non-tumor liver tissues were used to determine GRIM-19 expression. All samples were fixed with 4% formaldehyde, dehydrated, embedded, and cut into 3 μm serial sections. Endogenous peroxidase activity was quenched by incubation in 3% hydrogen peroxide in methanol for 15 min. The samples were washed twice with PBS and incubated with the appropriate primary antibody dilutions (1:300) at 4°C overnight. Mouse anti-human GRIM-19 antibody (Abcam, Hong Kong) was utilized. Samples were washed twice in PBS and incubated with the corresponding secondary antibody for 25 min at room temperature. Then, antigen-antibody complexes were washed with PBS and incubated for 1 min with high-sensitivity DAB (St. Louis, MO, USA) substrate. Finally, the sections were counterstained with hematoxylin and dehydrated, mounted, and examined by light microscopy. Briefly, the percentage of cells positively stained in each section was categorized as follows: negative (samples with > 5% positive cells), low (5-25% positive cells), moderate (25-50% positive cells), and strong (50-100% positive cells) were considered as -, +, ++, and +++, respectively, for these analyses.
Semi-quantitative RT-PCR
Total RNA was extracted from the cells using a Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. First-strand cDNA was synthesized by reverse transcription of 5 μg total RNA using a Takara RNA PCR Kit (TaKaRa, Japan). Primers used in semiquantitation RT-PCR were as follows: β-actin sense, 5’-ATATCGCTGCGCTGGTCGTC-3’ and antisense, 5’-AGGATGGCGTGAGGGAGAGC-3’; GRIM-19 sense, 5’-AGTATGGCGGCGTCAAAGGTG-3’ and antisense, 5’-TTTTTTCCA-GGTCTGCAGAGC-3’. The PCR products were separated by electrophoresis on 1% agarose gels containing ethidium bromide and visualized by a GIS Gelatum imaging system (Tanon, Shanghai, China). Absolute gene transcription was compared to that of β-actin.
Western blot assay
We performed western blot analysis to determine protein level changes. The lysates were run on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA, USA). The following antibodies were used: monoclonal cyclinD1, Akt1, pAkt1, GRIM-19, and proliferating cell nuclear antigen (PCNA), all of which were purchased from Abcam (California, USA). Bax, Bcl-2, cleaved caspase-9, and β-actin were purchased from Santa Cruz Biotechnology, and cleaved caspase-3, cytochrome C, and CDK4 were purchased from Cell Signaling Technology (Boston, MA, USA).
Cell proliferation assay
In 96-well plates 1.5×103 cells per well were seeded and incubated for the indicated period of time at 37°C. Then, cell growth was measured using the 4,5-dimethylthiazol-2-yl-2,5-diphenyltetrazolium bromide (MTT). The cells were harvested and incubated with DMSO for 30 min, and absorbance was measured using a plate reader in 570 nm of wavelength.
Flow cytometry analysis
Treated cells, including adherent and non-adherent cells, were trypsinized and centrifuged, washed, and stained using the Annexin-V-FITC Apoptosis Detection Kit (Beyotime, Shanghai, China) and Propidium Iodide (Beyotime, Shanghai, China). The samples were then analyzed for apoptosis and cell cycle by a FACScanflow cytometer (Becton Dickinson, Franklin Lakes, NJ).
Mitochondrial membrane potential (Δψm) assay
Mitochondrial stability was assessed using a mitochondrial membrane potential assay kit with JC-1 (5,5’,6’,6-tetrachloro-1,1’,3,3’-tetraethylbenzimidazolcarbocyanine iodide). Stable cells were cultured in 12-well plates. After 24 h treatment, the cells were incubated with 1 ml JC-1 fluorescent dye for 30 min in the dark at 37°C. Then, the cells were washed slowly twice with JC-1 dyeing buffer. The mitochondrial membrane potential was imaged using fluorescence microscope (Olympus, Tokyo, Japan) at 550 nm excitation and 570 nm emissions.
Immunofluorescence
Cells were cultured on sterile cover glass (Fisher Scientific, Inc.) in a 24-well tissue culture plate for 24 h before staining. They were fixed for 15 min using 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 in PBS, and blocked in 5% BSA. Primary antibodies were diluted in PBS containing 3% BSA (1:100) and incubated for 24 h at room temperature. After washing with PBS, cells were incubated with Alexa Fluor 555-labeled secondary antibodies (diluted 1:400 in blocking buffer) for 1 h and washed with PBS again. Thereafter, nuclei from the samples were counterstained with Hoechst 33258 (Invitrogen, Carlsbad, CA, USA) and inspected with a confocal microscope (Olympus, Tokyo, Japan).
Statistical analysis
All experiments were performed at least in triplicate. The data are expressed as the mean values±standard deviation (SD). Statistical analysis was performed using the SPSS19.0 statistical software. Comparisons between treatments were made using a paired Student’s t-test, or one-way ANOVA for multiple group comparisons. P < 0.05 was considered statistically significant.
Results
Immunohistochemical analysis of GRIM-19 expression in HCC tissues and adjacent non-tumor liver tissues
To investigate the profile of GRIM-19 in HCC tissue, we evaluated the protein expression in HCC tissues and adjacent non-tumor liver tissues using immunohistochemistry. GRIM-19 protein was found to be highly expressed in adjacent non-tumor liver tissues and decreased in HCC tissues (Figure 1). The case of negative GRIM-19 expression in adjacent non-tumor liver tissues was 0 and that of HCC tissues was 24; the case of low GRIM-19 expression in adjacent non-tumor liver tissues was 6 and that of HCC tissues was 30; the case of moderate GRIM-19 expression in adjacent non-tumor liver tissues was 29 and that of HCC tissues was 0; and the case of strong GRIM-19 expression in adjacent non-tumor liver tissues was 19 and that of HCC tissues was 0, as shown in Table 1.
Figure 1.
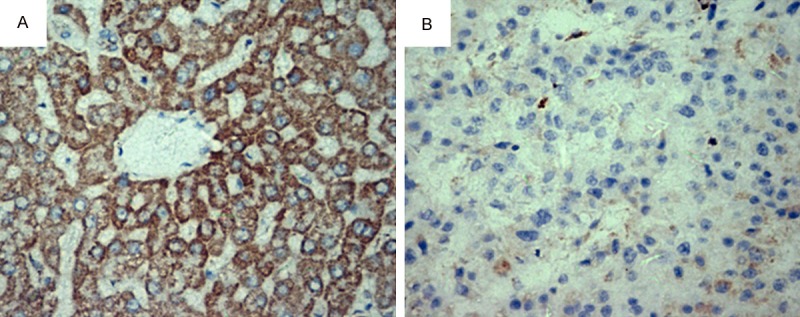
GRIM-19 expression in adjacent non-tumor liver tissues (A) and HCC tissues (B). Representative pictures of 54 hepatocellular carcinoma tumor samples and paired adjacent non-tumor liver tissues are shown. Original magnification: 200×.
Table 1.
A summary of GRIM-19 expression in all 54 HCC tissues and adjacent non-tumor liver tissues
| The expression level of GRIM-19 | ||||
|---|---|---|---|---|
|
|
||||
| - | + | ++ | +++ | |
| Hepatocellular carcinoma tissues (case) | 24 | 30 | 0 | 0 |
| Paried adjacent non-tumor tissues (case) | 0 | 6 | 29 | 19 |
GRIM-19 overexpression could inhibit tumor cell proliferation and induce G1/S cell cycle arrest and apoptosis in SMMC-7721 cells
Firstly, the stable cell line expressing wild-type GRIM-19 was established by transfecting lentiviral particles to hepatocellular cell line SMMC-7721 cells. It is demonstrated that GRIM-19 was effectively overexpressed in the lentiviral particles expressing wild-type GRIM-19 transfected cells (GRIM-19) compared to cells transfected with empty vector (EV). Furthermore, the mRNA and protein expression of GRIM-19 was significantly increased in the lentiviral particles expressing wild-type GRIM-19 transfected SMMC-7721 cells (Figure 2A, 2B).
Figure 2.
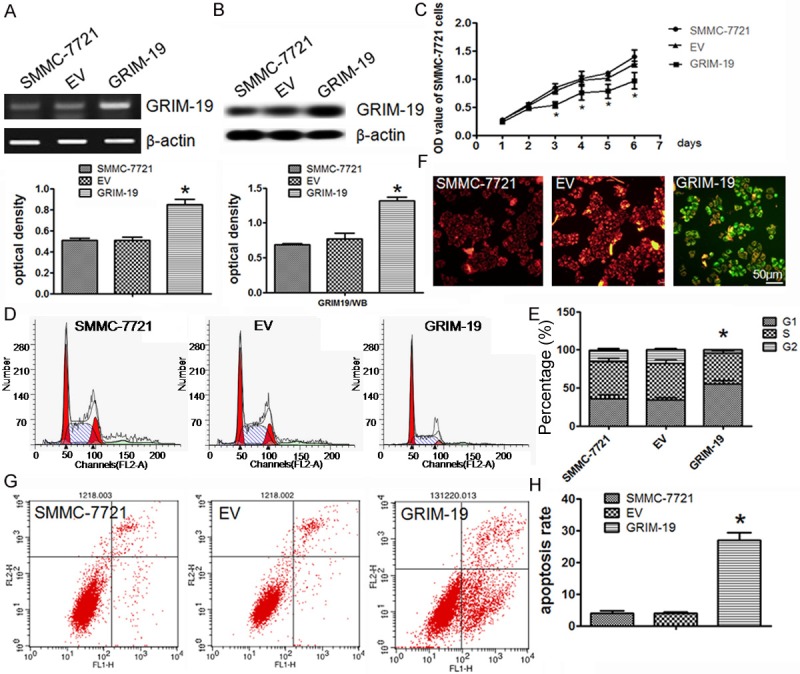
GRIM-19 overexpression controls cell proliferation, induces G1/S arrest, and enhances apoptosis in SMMC-7721 cells. SMMC-7721 cells were transfected with the lentiviral particles expressing wild-type GRIM-19 (GRIM-19) or an empty vector (EV); A. RT-PCR analysis of GRIM-19 mRNA overexpression; B. Western blot analysis of GRIM-19 protein overexpression; C. MTT assay showed GRIM-19 overexpression controls cell proliferation compared with the control; D. FCM raw data with PI staining for cell cycle arrest; E. Values represent the percentage of cells in each cell cycle phase; F. JC-1 staining evaluated exogenous GRIM-19’s effect on mitochondrial membrane potential. Red fluorescence is visible in cellular areas with high mitochondrial membrane potential, while green fluorescence of JC-1 monomer is present in cellular areas with low mitochondrial potential. Original magnification 400×; G. Representative FCM raw data with Annexin-V-FITC and PI staining for detecting cell death; H. Results from the different treatment conditions are presented as averages from the FCM assays. Data represent the mean±SD for three replicated experiments. *P < 0.05. Representative images of three separate experiments are shown in the figure.
Next, we analyzed whether elevated GRIM-19 expression would affect liver cancer cell proliferation, cell cycle or apoptosis. As shown in Figure 2C, overexpression of GRIM-19 markedly inhibited the SMMC-7721 cell growth compared to the same cells transfected with the empty vector. Meanwhile, we examined GRIM-19’s effects on the cell cycle distribution in SMMC-7721 using flow cytometry (FCM). In the SMMC-7721 cells transfected with GRIM-19, the percentage of cells in the G1 phase and the ratio of G1 cells to S phase cells were increased compared to the control (Figure 2D, 2E). Then, we investigated mitochondrial membrane potential (Δψm) with JC-1 staining, because loss of mitochondrial membrane potential was an important event during the mitochondrial-mediated apoptosis. Fluorescence from red to green was observed in response to GRIM-19 and EV treatment, respectively (Figure 2F). To quantify apoptosis, flow cytometry analysis of Annexin-V-FITC and propidium iodide staining were used. An increase in the apoptotic cell population in SMMC-7721 cells treated with ectopic GRIM-19 was observed (Figure 2G, 2H). Taken together, these results suggest that overexpression of GRIM-19 suppresses cell proliferation, induces G1/S cell cycle arrest, and increases apoptosis in SMMC-7721 cells.
Exogenous GRIM-19 induced cell cycle arrest by altering the cell cycle protein levels in SMMC-7721 cells
To elucidate the mechanism underlying the exogenous GRIM-19-mediated cell cycle arrest in HCC cells, various cell cycle regulatory proteins were examined by western blot analysis. The cells transfected with exogenous GRIM-19 showed a significant increase in G1 arrested cells, measured by the downregulation of cyclin D1, CDK4, and PCNA expression (Figure 3A). These results were supported by immunofluorescence analysis for cyclinD1, CDK4, and PCNA. The fluorescence intensity of these cell cycle proteins was significantly decreased in the SMMC-7721 cells transfected with GRIM-19 compared to control groups, respectively (Figure 3B), suggesting that GRIM-19 overexpression efficiently induces G1/S arrest th-rough lessening cyclinD1, CDK4, and PCNA protein expression.
Figure 3.
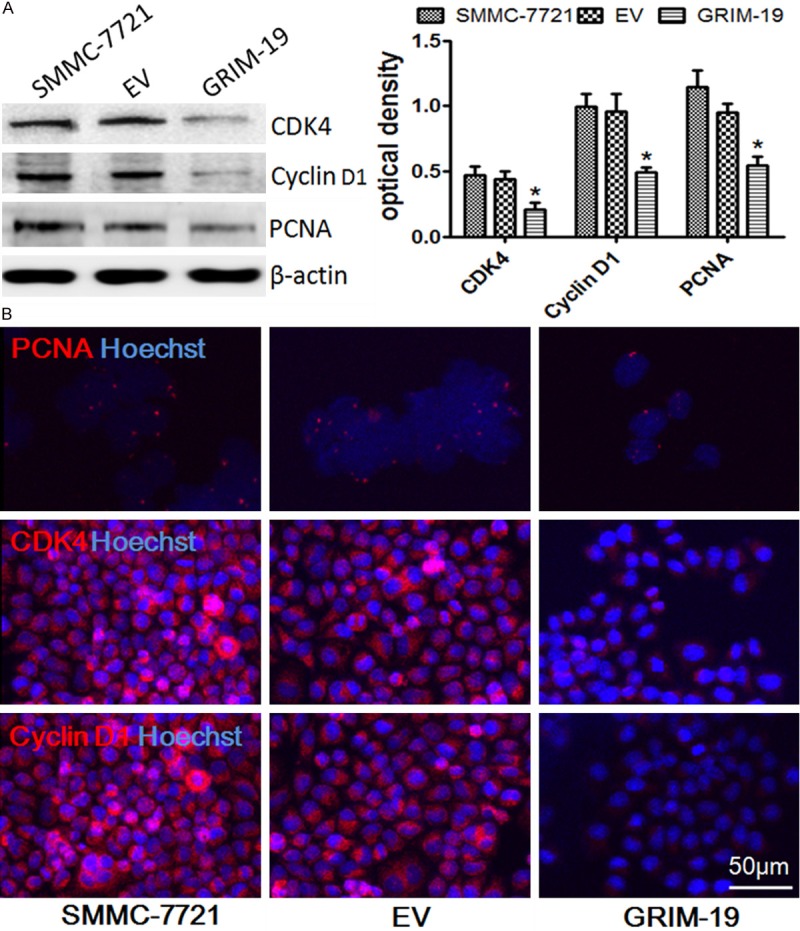
The effects of exogenous GRIM-19 on the expression of CyclinD1, CDK4, and PCNA. A. Western blot assay for protein expression of CyclinD1, CDK4, and PCNA after different treatments. *P < 0.05; B. SMMC-7721 cells transfected with lentiviral particles expressing wild-type GRIM-19 (GRIM-19) or an empty vector (EV) were immunostained for cyclinD1, CDK4, and PCNA with corresponding antibodies. Representative merged images with Hoechst 33258 labeled DNA are shown (400× magnifications).
Exogenous GRIM-19 enhanced HCC cell apoptosis by activating the mitochondrial apoptosis pathway
To determine the mechanism of enhanced HCC cell apoptosis by exogenous GRIM-19, we investigated the mitochondrial apoptosis pathway’s influence. We next analyzed the Bcl-2 family proteins, including the pro-apoptotic Bax and anti-apoptotic Bcl-2 proteins that regulated mitochondrial membrane potential and cytochrome C released from the mitochondrial inner membrane. We evaluated caspase-3 and caspase-9 expression by Western blot assay in the SMMC-7721 cells treated with lentiviral particles expressing wild-type GRIM-19. The cells transfected with the empty vector were used as a negative control. As shown in Figure 4A, exogenous GRIM-19 treatment significantly increased pro-apoptotic protein Bax expression and downregulated anti-apoptotic protein Bcl-2 expression in the SMMC-7721 cells. On the other hand, cytochrome c, cleaved caspase-3, and cleaved caspase-9 expression were upregulated. In accordance with these results, the cells transfected with exogenous GRIM-19 tended to have stronger staining for Bax, cytochrome C, cleaved caspase-3, and cleaved caspase-9 and weaker staining for Bcl-2 compared to the negative control (Figure 4B). These results reveal the enhanced HCC cell apoptosis by exogenous GRIM-19 is at least mediated by activating the mitochondrial apoptosis pathway.
Figure 4.
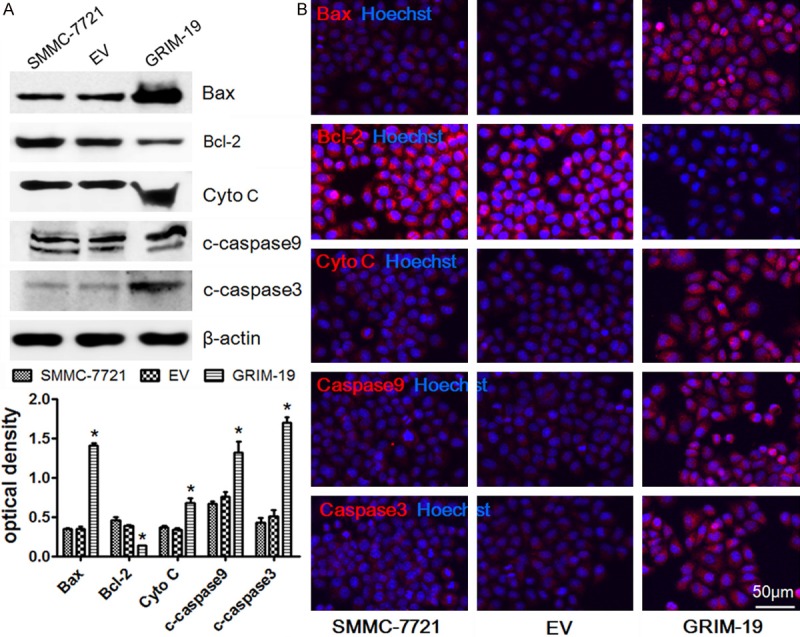
Exogenous GRIM-19 activates the mitochondrial apoptosis pathway to accelerate HCC cell apoptosis. A. Western blot assay for protein expression of Bax, Bcl-2, cytochrome C, cleaved caspase-3, and cleaved caspase-9 in cells transfected with exogenous GRIM-19 or the empty vector. *P < 0.05; B. The above-mentioned transfected HCC cells were immunostained for Bax, Bcl-2, cytochrome C, cleaved caspase-3, and cleaved caspase-9 with corresponding antibodies. Representative merged images with Hoechst 33258 labeled DNA are shown (400× magnifications).
GRIM-19 may negatively regulate PI3K/AKT signaling pathway to inhibit human liver cancer growth
The PI3K/AKT pathway plays a key role in hepatocarcinogenesis. To further research the possible mechanism involved in GRIM-19-mediated growth inhibition in SMMC-7721 cells, we surveyed GRIM-19’s effects on AKT1 protein expression and phosphorylation that is a key process in the above signaling pathway. As shown in Figure 5A, Western blot analysis revealed that exogenous GRIM-19 significantly lessened AKT1/pAKT1 expression. Immunofluorescence analysis showed AKT1 downregulation in the SMMC-7721 cells transfected with exogenous GRIM-19 compared to cells transfected with the empty vector (Figure 5B). These findings suggest that GRIM-19 restrictes hepatocellular carcinoma cell growth by potentially negatively regulating the PI3K/AKT signaling pathway.
Figure 5.
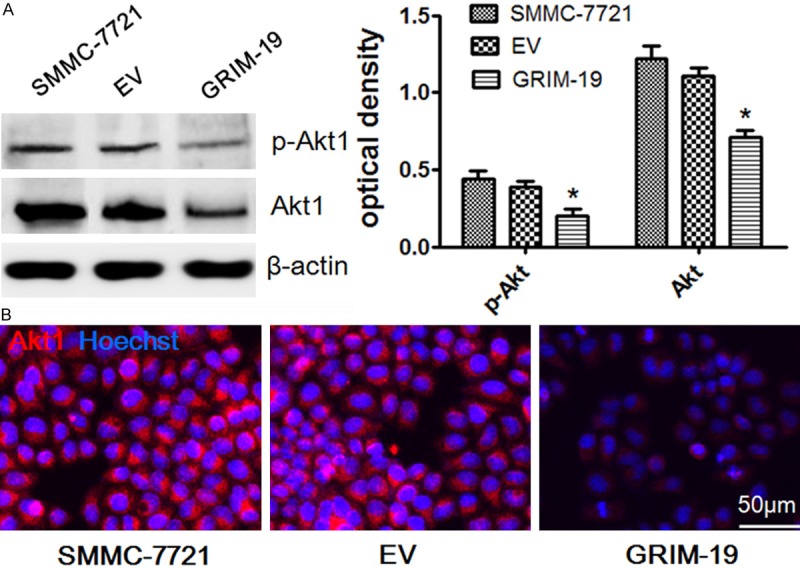
The possible mechanism that increased GRIM-19 expression suppresses tumor growth in SMMC-7721 cells. A. Western blotting of AKT1/pAKT1 in the control group (EV) and transfected with exogenous GRIM-19 group. AKT1 is a key molecule in the PI3K/AKT signaling pathway which plays an important role in liver cancer. *P < 0.05. B. SMMC-7721 cells transfected with GRIM-19 or EV were immunostained for AKT1 with corresponding antibody. Representative merged images with Hoechst 33258 labeled DNA are shown (400× magnifications).
Discussion
Recent Studies have shown that the main reasons for malignant tumors are closely related to genes that impact cellular differentiation, developmental processes, and senescence during normal growth [22]. More and more genes essential for normal tissues or organs growth and development have been identified being involved in hepatocellular carcinoma [23,24]. GRIM-19 gene is a novel gene essential for development. Our study showed GRIM-19 expression was decreased in most of the HCC samples in contrast to adjacent non-tumor liver tissues, which is in agreement with previous reports [25]. These results indicated that GRIM-19 might be involved in hepatocellular carcinoma pathogenesis. It has been reported that tumorigenesis was closely associated with GRIM-19 gene mutation or dysfunction [26]. Nevertheless, the exact mechanisms which cause GRIM-19 inactivation in HCC remain unknown.
More and more studies highlight that GRIM-19’s function as a tumor suppressor seems to occur at multiple levels. In previous study, Peng et al. proved that GRIM-19 could induce G1/S arrest of breast cancer cells by downregulation of CDK4 and cyclinD1 [27] . GRIM-19 has also been found to suppress cell growth in lung cancer cells and glioma cells via the stat3-associated signaling pathway, accompanied by upregulation of Bax and downregulation of Bcl-2 [18,19]. Depleting GRIM-19 accelerated hepatocellular carcinoma invasion [28]. However, we have gained insight into how GRIM-19 might control liver cancer growth. In this study, overexpressing GRIM-19 resulted in a significant decrease in SMMC-7721 cell growth. Cell cycle regulation is an important mechanism of normal cell growth. Normal cell progression from G1-S-G2-M-G0 during the division cycle is tightly controlled by the protein kinase activity of a class of serine-threonine kinases (CDK). CDK4, called the cyclin-dependent kinases, and cyclinD1, known as cyclins, are important factors during normal cell progression from G1-S-G2-M-G0 [29-31]. PCNA is a nuclear protein that is expressed in G-M phases of the cell cycle. Our results showed GRIM-19 overexpression efficiently induced G1/S arrest by lessening cyclinD1, CDK4, and PCNA protein expression in SMMC-7721 cells. These results were similar in MCF-7 cells and HeLa cells [27,32]. G1 arrest can prevent damaged DNA replication and is therefore helpful in limiting uncontrolled cancer cell proliferation.
Additionally, a previous study demonstrated that moderate GRIM-19 levels caused cycle arrest and high expression of GRIM-19 induced apoptosis in MCF-7 cells [6]. Moreover, apoptosis reportedly plays an important role during malignant normal cell growth transformation [33]. The relative balance of various pro-apoptotic (Bax, Bad) and anti-apoptotic (Bcl-2, Bcl-xL, Bcl-w, and Mcl-1) Bcl-2 family members is a critical cellular homeostasis determinant [34,35]. The anti-apoptotic Bcl-2 proteins interfere with engaging the mitochondrial apoptotic machinery by inhibiting Bax and Bak oligomerization [36]. Bax is translocated into mitochondria upon apoptosis induction. After translocation, Bax forms large oligomers, which insert into mitochondrial membranes and lead to cytochrome C release and cytotoxic activities. MMP’s collapse can initiate molecules being released from the space between the outer and inner mitochondrial membranes into the cytosol, which triggers the caspase cascade and other apoptotic processes [37]. In our study, we revealed that GRIM-19 overexpression promoted apoptosis by increasing the Bax/Bcl-2 ratio and cytochrome C, cleaved caspase-3, and cleaved caspase-9 protein to activate the intrinsic mitochondrial pathway, in addition to GRIM-19 induceing G1/S arrest. These discoveries imply that GRIM-19 dysregulation may disrupt the balance between proliferation and apoptosis and, as such, may represent an important pathogenic step in hepatocellular carcinoma development.
The PI3/AKT signaling pathway has been found to play important roles in regulating key cellular processes such as survival, proliferation, and differentiation [38]. AKT1 is a key molecule in the above signaling pathway. It is generally believed that AKT1 activation contributes to the development and progression of many types of cancer, including liver cancer. Phosphorylation on Ser473 is required for full activity. Previous studies have shown that AKT1 activation can promote cell survival, accelerate cell cycle progression, and inhibit cell apoptosis by re-gulateing its downstream target genes such as CDK4, cyclinD1, Bcl-2, and Bax [39,40]. Interestingly, a recent study demonstrated that pAKT1 was significantly expressed in HCC specimens [41]. Moreover, study has shown that GRIM-19 appears lower expression in human liver cancer tissues in comparison to the normal liver tissues [25]. These results suggest there may be some kind of relationship between GRIM-19 inactivation and AKT1 activation. GRIM-19 may control AKT1 expression and phosphorylation and then regulate cell cycle progression and cell apoptosis. To verify the hypothesis and further research the possible mechanism involved in GRIM-19-mediated growth inhibition, we surveyed its effects on AKT1/pAKT1 protein in SMMC-7721 cells. The results from this study demonstrate that exogenous GRIM-19 weakens AKT1 expression and phosphorylation, implying that GRIM-19 may negatively regulate PI3K/AKT signaling pathway to inhibit HCC growth.
In summary, we have shown that GRIM-19 downregulation occurs in hepatocellular cancer samples and that GRIM-19 suppresses liver cancer cell growth by inducing cell cycle arrest and promoting apoptosis. Furthermore, for the first time, we have provided evidence that GRIM-19 inhibited AKT1 expression and phosphorylation, which might alleviated the PI3K/AKT signaling pathway and then control liver cancer cell growth. There may be a couple of limitations in the present studies. For instance, since we have used only SMMC-72 cells in the present study, whether these results are also applicable to other liver cancer cell lines or other tumor cells remains unaddressed. How GRIM-19 affects AKT1 expression and phosphorylation remains unclear. Therefore, these interesting issues will be continually investigated in future studies. Considering GRIM-19’s role in hepatocarcinogenesis, GRIM-19 recovery may be a novel and potential strategy to treat hepatocellular cancer.
Acknowledgements
This work was funded by the National Natural Science Foundation of China (No. 81270529) and the Science & Technology Development Planning Project of Jilin Province (No. 20120953 and No. 20140414006GH).
Disclosure of conflict of interest
None.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global Cancer Statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Sotiropoulos GC, Molmenti EP, Losch C, Beckebaum S, Broelsch CE, Lang H. Meta-analysis of tumor recurrence after liver transplantation for hepatocellular carcinoma based on 1,198 cases. Eur J Med Res. 2007;12:527–534. [PubMed] [Google Scholar]
- 3.Song TJ, Ip EWK, Fong YM. Hepatocellular carcinoma: Current surgical management. Gastroenterology. 2004;127:S248–S260. doi: 10.1053/j.gastro.2004.09.039. [DOI] [PubMed] [Google Scholar]
- 4.Villanueva A, Newell P, Chiang DY, Friedman SL, Llovet JM. Genomics and signaling pathways in hepatocellular carcinoma. Semin Liver Dis. 2007;27:55–76. doi: 10.1055/s-2006-960171. [DOI] [PubMed] [Google Scholar]
- 5.Staib F, Hussain SP, Hofseth LJ, Wang XW, Harris CC. TP53 and liver carcinogenesis. Hum Mutat. 2003;21:201–216. doi: 10.1002/humu.10176. [DOI] [PubMed] [Google Scholar]
- 6.Angell JE, Lindner DJ, Shapiro PS, Hofmann ER, Kalvakolanu DV. Identification of GRIM-19, a novel cell death-regulatory gene induced by the interferon-beta and retinoic acid combination, using a genetic approach. J Biol Chem. 2000;275:33416–33426. doi: 10.1074/jbc.M003929200. [DOI] [PubMed] [Google Scholar]
- 7.Huang G, Lu H, Hao A, Ng DC, Ponniah S, Guo K, Lufei C, Zeng Q, Cao X. GRIM-19, a cell death regulatory protein, is essential for assembly and function of mitochondrial complex I. Mol Cell Biol. 2004;24:8447–8456. doi: 10.1128/MCB.24.19.8447-8456.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu H, Cao X. GRIM-19 is essential for maintenance of mitochondrial membrane potential. Mol Biol Cell. 2008;19:1893–1902. doi: 10.1091/mbc.E07-07-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Q, Wang L, Wang Z, Yang Y, Tian J, Liu G, Guan D, Cao X, Zhang Y, Hao A. GRIM-19 opposes reprogramming of glioblastoma cell metabolism via HIF1 destabilization. Carcinogenesis. 2013;34:1728–1736. doi: 10.1093/carcin/bgt125. [DOI] [PubMed] [Google Scholar]
- 10.Lufei C, Ma J, Huang G, Zhang T, Novotny-Diermayr V, Ong CT, Cao X. GRIM-19, a death-regulatory gene product, suppresses Stat3 activity via functional interaction. EMBO J. 2003;22:1325–1335. doi: 10.1093/emboj/cdg135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalvakolanu DV, Nallar SC, Kalakonda S. Cytokine-induced tumor suppressors: A GRIM story. Cytokine. 2010;52:128–142. doi: 10.1016/j.cyto.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang G, Chen Y, Lu H, Cao X. Coupling mitochondrial respiratory chain to cell death: an essential role of mitochondrial complex I in the interferon-beta and retinoic acid-induced cancer cell death. Cell Death Differ. 2007;14:327–337. doi: 10.1038/sj.cdd.4402004. [DOI] [PubMed] [Google Scholar]
- 13.Alchanati I, Nallar SC, Sun P, Gao L, Hu J, Stein A, Yakirevich E, Konforty D, Alroy I, Zhao X, Reddy SP, Resnick MB, Kalvakolanu DV. A proteomic analysis reveals the loss of expression of the cell death regulatory gene GRIM-19 in human renal cell carcinomas. Oncogene. 2006;25:7138–7147. doi: 10.1038/sj.onc.1209708. [DOI] [PubMed] [Google Scholar]
- 14.Gong LB, Luo XL, Liu SY, Tao DD, Gong JP, Hu JB. [Correlations of GRIM-19 and its target gene product STAT3 to malignancy of human colorectal carcinoma] . Ai Zheng. 2007;26:683–687. [PubMed] [Google Scholar]
- 15.Zhou Y, Li M, Wei Y, Feng DQ, Peng C, Weng HY, Ma Y, Bao L, Nallar S, Kalakonda S, Xiao WH, Kalvakolanu DV, Ling B. Down-Regulation of GRIM-19 Expression Is Associated With Hyperactivation of STAT3-Induced Gene Expression and Tumor Growth in Human Cervical Cancers. J Interferon Cytokine Res. 2009;29:695–703. doi: 10.1089/jir.2009.0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fusco A, Viglietto G, Santoro M. Point mutation in GRIM-19: a new genetic lesion in Hurthle cell thyroid carcinomas. Br J Cancer. 2005;92:1817–1818. doi: 10.1038/sj.bjc.6602556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang L, Gao LF, Li Y, Lin GM, Shao YT, Ji K, Yu H, Kalvakolanu DV, Kopecko DJ, Zhao XJ, Xu DQ, Hu J. Effects of plasmid-based stat3-specific short hairpin RNA and GRIM-19 on PC-3M tumor cell growth. Clin Cancer Res. 2008;14:559–568. doi: 10.1158/1078-0432.CCR-07-1176. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Hao H, Zhao S, Liu Q, Yuan Q, Ni S, Wang F, Liu S, Wang L, Hao A. Downregulation of GRIM-19 promotes growth and migration of human glioma cells. Cancer Sci. 2011;102:1991–1999. doi: 10.1111/j.1349-7006.2011.02059.x. [DOI] [PubMed] [Google Scholar]
- 19.Wang T, Yan XB, Zhao JJ, Ye J, Jiang ZF, Wu DR, Xiao WH, Liu RY. Gene associated with retinoid-interferon-induced mortality-19 suppresses growth of lung adenocarcinoma tumor in vitro and in vivo. Lung Cancer. 2011;72:287–293. doi: 10.1016/j.lungcan.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 20.Zhou T, Chao L, Rong GH, Wang CG, Ma R, Wang X. Down-regulation of GRIM-19 is associated with STAT3 overexpression in breast carcinomas. Hum Pathol. 2013;44:1773–1779. doi: 10.1016/j.humpath.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 21.Sun P, Nallar SC, Kalakonda S, Lindner DJ, Martin SS, Kalvakolanu DV. GRIM-19 inhibits v-Src-induced cell motility by interfering with cytoskeletal restructuring. Oncogene. 2009;28:1339–1347. doi: 10.1038/onc.2008.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zender L, Villanueva A, Tovar V, Sia D, Chiang DY, Llovet JM. Cancer gene discovery in hepatocellular carcinoma. J Hepatol. 2010;52:921–929. doi: 10.1016/j.jhep.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zender L, Xue W, Zuber J, Semighini CP, Krasnitz A, Ma B, Zender P, Kubicka S, Luk JM, Schirmacher P, McCombie WR, Wigler M, Hicks J, Hannon GJ, Powers S, Lowe SW. An Oncogenomics-Based In Vivo RNAi Screen Identifies Tumor Suppressors in Liver Cancer. Cell. 2008;135:852–864. doi: 10.1016/j.cell.2008.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J, Mizuno K, Hasegawa G, Kishimoto H, Iizuka M, Naito M, Enomoto K, Watanabe S, Mak TW, Nakano T. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest. 2004;113:1774–1783. doi: 10.1172/JCI20513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li F, Ren W, Zhao Y, Fu Z, Ji Y, Zhu Y, Qin C. Down-regulation of GRIM-19 is associated with hyperactivation of p-STAT3 in hepatocellular carcinoma. Med Oncol. 2012;29:3046–3054. doi: 10.1007/s12032-012-0234-8. [DOI] [PubMed] [Google Scholar]
- 26.Moreira S, Correia M, Soares P, Maximo V. GRIM-19 function in cancer development. Mitochondrion. 2011;11:693–699. doi: 10.1016/j.mito.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 27.Sun P, Nallar SC, Raha A, Kalakonda S, Velalar CN, Reddy SP, Kalvakolanu DV. GRIM-19 and p16 (INK4a) synergistically regulate cell cycle progression and E2F1-responsive gene expression. J Biol Chem. 2010;285:27545–27552. doi: 10.1074/jbc.M110.105767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hao H, Liu J, Liu G, Guan D, Yang Y, Zhang X, Cao X, Liu Q. Depletion of GRIM-19 accelerates hepatocellular carcinoma invasion via inducing EMT and loss of contact inhibition. J Cell Physiol. 2012;227:1212–1219. doi: 10.1002/jcp.24025. [DOI] [PubMed] [Google Scholar]
- 29.Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene. 2009;28:2925–2939. doi: 10.1038/onc.2009.170. [DOI] [PubMed] [Google Scholar]
- 30.Hochegger H, Takeda S, Hunt T. Cyclin-dependent kinases and cell-cycle transitions: does one fit all? Nat Rev Mol Cell Biol. 2008;9:910–U926. doi: 10.1038/nrm2510. [DOI] [PubMed] [Google Scholar]
- 31.Fung TK, Poon RY. A roller coaster ride with the mitotic cyclins. Semin Cell Dev Biol. 2005;16:335–342. doi: 10.1016/j.semcdb.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 32.Zhou Y, Wei Y, Zhu J, Wang Q, Bao L, Ma Y, Chen Y, Feng D, Zhang A, Sun J, Nallar SC, Shen K, Kalvakolanu DV, Xiao W, Ling B. GRIM-19 Disrupts E6/E6AP Complex to Rescue p53 and Induce Apoptosis in Cervical Cancers. PLoS One. 2011;6:e22065. doi: 10.1371/journal.pone.0022065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stefanec T. Endothelial apoptosis: Could it have a role in the pathogenesis and treatment of disease? Chest. 2000;117:841–854. doi: 10.1378/chest.117.3.841. [DOI] [PubMed] [Google Scholar]
- 34.Kim R. Unknotting the roles of Bcl-2 and Bcl-xL in cell death. Biochem Biophys Res Commun. 2005;333:336–343. doi: 10.1016/j.bbrc.2005.04.161. [DOI] [PubMed] [Google Scholar]
- 35.Mayorga M, Bahi N, Ballester M, Comella JX, Sanchis D. Bcl-2 is a key factor for cardiac fibroblast resistance to programmed cell death. J Biol Chem. 2004;279:34882–34889. doi: 10.1074/jbc.M404616200. [DOI] [PubMed] [Google Scholar]
- 36.Precht TA, Phelps RA, Linseman DA, Butts BD, Le SS, Laessig TA, Bouchard RJ, Heidenreich KA. The permeability transition pore triggers Bax translocation to mitochondria during neuronal apoptosis. Cell Death Differ. 2005;12:255–265. doi: 10.1038/sj.cdd.4401552. [DOI] [PubMed] [Google Scholar]
- 37.Valentijn AJ, Upton JP, Bates N, Gilmore AP. Bax targeting to mitochondria occurs via both tail anchor-dependent and -independent mechanisms. Cell Death and Differ. 2008;15:1243–1254. doi: 10.1038/cdd.2008.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase-AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 39.Zhou Q, Lui VWY, Yeo W. Targeting the PI3K/Akt/mTOR pathway in hepatocellular carcinoma. Future Oncol. 2011;7:1149–1167. doi: 10.2217/fon.11.95. [DOI] [PubMed] [Google Scholar]
- 40.Schulze-Bergkamen H, Brenner D, Krueger A, Suess D, Fas SC, Frey CR, Dax A, Zink D, Buchler P, Muller M, Krammer PH. Hepatocyte growth factor induces Mcl-1 in primary human hepatocytes and inhibits CD95-mediated apoptosis via Akt. Hepatology. 2004;39:645–654. doi: 10.1002/hep.20138. [DOI] [PubMed] [Google Scholar]
- 41.Zhou L, Huang Y, Li J, Wang Z. The mTOR pathway is associated with the poor prognosis of human hepatocellular carcinoma. Med Oncol. 2010;27:255–261. doi: 10.1007/s12032-009-9201-4. [DOI] [PubMed] [Google Scholar]