

Abstract
Oxidants are important signaling molecules known to increase endothelial permeability. Studies implicate reactive oxygen species (ROS) and the intrinsic apoptotic signaling cascades as mediators of vascular hyperpermeability. Here we report the protective effects of ulinastatin, a serine protease inhibitor with antiapoptotic properties, against oxidant-induced endothelial monolayer hyperpermeability. HUVECs were respectively pretreated with 10,000 and 50,000 u/l ulinastatin, followed by stimulation of 0.6 mM H2O2. Monolayer permeability was determined by transendothelial electrical resistance (TER); Mitochondrial release of cytochrome c was determined by enzyme-linked immunosorbent assay; Caspase-3 activity was measured by fluorometric assay; Adherens junction protein β-catenin was detected by immunofluorescense staining; Ratio of cell apoptosis was evaluated by Annexin-V/PI double stain assay; Mitochondrial membrane potential (Δψm) was determined with JC-1; Intracellular ATP content was assayed by a commercial kit; Bax and Bcl-2 expression were estimated by western blotting; Intracellular reactive oxygen species (ROS) level was measured by DCFH-DA. H2O2 exposure resulted in endothelial hyperpermeability and ROS formation (P < 0.05). The activation of mitochondrial intrinsic apoptotic signaling pathway was evidenced from BAX up-regulation, Bcl-2 down-regulation, mitochondrial depolarization, an increase in cytochrome c release, and activation of caspase-3 (P < 0.05). UTI (50,000 u/l) attenuated endothelial hyperpermeability, ROS formation, mitochondrial dysfunction, cytochrome c release, activation of caspase-3, and disruption of cell adherens junctions (P < 0.05). Together, these results demonstrate that UTI provides protection against vascular hyperpermeability by modulating the intrinsic apoptotic signaling.
Keywords: Endothelial barrier, adherens junctions, apoptosis, oxidative stress, mitochondria
Introduction
An increase in vascular permeability is a key hallmark of inflammation and has been implicated in the pathophysiology of many disease states including acute lung injury, ischemia-reperfusion injury, and sepsis [4,17,18,39]. Oxidants including superoxide and hydrogen peroxide (H2O2) generated by activated neutrophils and endothelial cells in response to inflammatory stimuli increase endothelial permeability [18,22,34].
Studies have demonstrated the involvement of mitochondrial “intrinsic” apoptotic signaling in endothelial cell dysfunction and vascular permeability [8,30,35]. Mitochondrial regulation of apoptosis is mediated through the release of cytochrome c, apoptosis-inducing factor (AIF), and second mitochondrial-derived activator of caspases (smac), ultimately caspases activation [5,26]. Caspase-3 has been shown to cleave β-catenin, thereby disrupting the vascular endothelial (VE)-cadherin-β-catenin complex, which serves as a key role of cell-cell adhesion in endothelial cells. Damage of (VE)-cadherin-β-catenin complex may lead to microvascular hyperpermeability [7,25,27].
Ulinastatin (UTI), a serine protease inhibitor, is mainly used in the treatment of sepsis, shock, pancreatitis, and ischemia-reperfusion injury [16,20,38]. Moreover, recent studies have revealed the protective effects of ulinastatin attenuate oxidation and apoptosis [12,13,31]. In present study, we hypothesized that UTI would improve oxidant-induced endothelial hyperpermeability by regulating the intrinsic apoptotic signaling cascade.
Materials and methods
Monolayer permeability
Primary human umbilical endothelial cell (HUVECs; ScienCell, USA) were maintained in DMEM/F12 containing 10% fetal bovine serum at 37°C in a humidified atmosphere with 5% CO2 -95% air. In all experiments, HUVECs were grown to 90% confluence and starved of serum for 12 hours before being stimulated with H2O2. The monolayers were respectively pretreated with 10,000 u/L and 50,000 u/L of UTI for 60 min, followed by stimulation of 0.6 mM H2O2. DMEM/F12 was used as vehicle control. Finally, endothelial permeability was measured 60 min at 15min-intervals.
Transendothelial electrical resistance (TER)
Transendothelial electrical resistance (TER) of HUVECs monolayer was determined using STX2 electrode and EVOM2 meter according to the instruction manual of manufacture (World Precision Instruments, Sarasota, FL, USA) [32]. HUVECs were seeded with number of 1 × 105/cm2 on fibronectin-coated, 6.5 mm Transwell filters (0.4 mm pore size) and were used until full confluence. Resistance values of multiple Transwell inserts of an experimental group were measured sequentially and the mean was expressed in the common unit (Ω cm2) after subtraction of the value of a blank cell-free filter.
Measurement of cytosolic cytochrome c
Cytosolic cytochrome c content was estimated with a cytochrome c ELISA kit. The cell homogenates were centrifuged (10,000 × g for 60 min at 4°C), and the supernatant (cytosolic fraction) was collected and subjected to protein estimation (bicinchoninic acid [BCA] method). Then, the samples were treated with a conjugate reagent, transferred to a cytochrome c antibody-coated microwell plate, and incubated at room temperature for 60 min. The wells were washed and treated with a substrate and incubated for 30 min, followed by addition of a stop solution. The optical density was read at 450 nm using an automatic microplate reader (SpectraMax M5; Molecular Devices, Sun-nyvale, CA, USA). A serial dilution of cytochrome c calibrator was subjected to the assay along with the samples, the values were plotted, and the concentration of cytochrome c was calibrated from the standard curve.
Measurement of caspase-3 activity
Caspase-3 activity was determined using a caspase-3 activity assay kit (Sigma, USA). The cells were lysed in caspase-3 sample lysis buffer provided in the kit. The homogenates were centrifuged at 10,000 g, and the supernatant was collected for protein estimation (BCA method) and for caspase-3 assay. The cell lysates were exposed to the DEVD substrate conjugate provided in the kit. The sample was measured in an automatic microplate reader (SpectraMax M5) at excitation 400 nm and emission 505 nm.
Measurement of cell apoptosis
Cell apoptosis were detected by an Annexin V-FITC apoptosis detection kit (BD Biosciences, USA). After induction with H2O2, cells were washed twice with PBS and suspended in 1× binding buffer at a concentration of approx 1 × 105 cells/ml. 5 μl of FITC-Annexin V and 10 μl of propidium iodide (PI, 50 μg/ml, Sigma) was added to cell suspension. After incubation at room temperature for 20 minutes at dark, the fluorescence of the cells was determined immediately with a flow cytometer (Becton Dickinson FACScan, San Jose, CA).
Measurement of mitochondrial membrane potential
The mitochondrial membrane potential (Δψm) was determined by flow cytometry, using the potential-sensitive fluorescent dye JC-1. The color of this dual-emission probe changed from red-orange to green as the mitochondrial membrane turned depolarized. The JC-1 (5 μmol/L) was loaded onto RLMVECs for 15 min at 37°C. The stained cells were washed with PBS, and analyzed by flow cytometry (Becton Dickinson FACScan). A minimum of 10,000 cells per sample was analyzed. JC-1 monomers emit at 527 nm and “J-aggregates” emit at 590 nm. The percentage of cells with abnormally low Δψm (green fluorescence) was determined.
Measurement of cellular ATP
Intracellular ATP was determined by a luciferase-based assay (CellTiter-Glo, Madison, WI), according to the manufacturer’s recommendation. After adding 100 μL of the CellTiter-Glo reagent to 100 μL of cells suspension containing 10,000 cells in each well of a standard opaque-walled 96-well plate, the plates were allowed to incubate at room temperature for 10 min and the luminescence was recorded in an automatic microplate reader (SpectraMax M5).
Measurement of ROS levels
Intracellular ROS levels were assessed usingDCFH-DA probe (Sigma, USA). Cells were treated with DCFH-DA (10 μM), following sham/burn serum, for 20 mins at 37°C. After incubation, the cells were washed and analyzed using an automatic microplate reader (Spectra Max, M5). The relative intensity of DCF fluorescence was determined at a wavelength of 535 nm as compared to sham group cells.
Measurement of Bcl-2 and Bax expression
The cells were homogenated and analyzed for Bcl-2 and Bax by western blotting. Protein concentrations were determined using the BCA method. An equal amount of protein was loaded onto 10% sodium dodecyl sulphate polyacrylamide gel for electrophoresis. After electrophoresis, proteins were electroblotted onto polyvinylidene fluoride membranes and blotted with primary antibodies against smac (Abcam, UK). Membranes were then incubated with the horseradish peroxidase-tagged secondary antibody (Tianjin Sungene Biotech Co., Ltd. Tianjin, China), and protein expression was detected using an enhanced chemiluminescence reagent.
Immunofluorescence staining for β-catenin
HUVECs were rinsed quickly with ice-cold PBS, and fixed with 4% paraformaldehyde. Cells were permeabilized for 30 min with 0.1% Triton X-100 and then incubated with β-catenin antibody (Abcam, UK) for 24 h at 4°C, followed by incubation with FITC-labeled secondary antibody (Tianjin Sungene Biotech Co.) for 2 h at room temperature. Cell nuclei were labeled with Hoechst33258 (Sigma, USA). Cells were examined using a confocal microscope (LSM780; Zeiss Microsystems, Jena, Germany). Images were collected using the software (Zeiss Microsystems, Germany).
Statistical analysis
All variables are presented as Means ± SD Differences between groups were determined using one-way ANOVA with the LSD multiple-comparison test and Student’s t-test when appropriate. Values were considered significant when P < 0.05.
Results
UTI attenuates oxidant-induced endothelial hyperpermeability
As shown in Figure 1, oxidant-induced monolayer hyperpermeability was evidenced by an obvious decrease in the TER of the cell monolayer treated with H2O2 compared to that of the cell monolayer in the control group; the hyperpermeability was significantly attenuated by pretreatment with 50,000 u/l of UTI but not 10,000 u/l of UTI. At 60 min, TER (normalized) of the HUVECs monolayer decreased from 1.03 ± 0.04 in the control group to 0.68 ± 0.07 in the vehicle group (Figure 1; P < 0.05); however, the TER were improved to 0.73 ± 0.05 (Figure 1; P > 0.05) and 0.90 ± 0.06 (Figure 1; P < 0.05) respectively by 10,000 u/l and 50,000 u/l of UTI.
Figure 1.
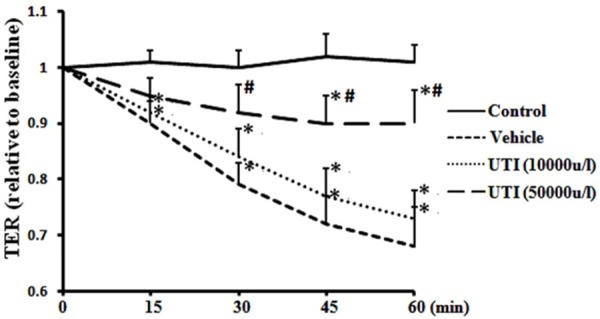
UTI attenuates oxidant-induced endothelial hyperpermeability. HUVECs were respectively pretreated with 10.000 and 50,000 u/l of UTI for 60 min, followed by H2O2 stimulation. The TER was recorded for 60 min at 15-min intervals. Data are presented as mean ± SD (n = 6 in each group). *P < 0.05, versus the control group; #P < 0.05, versus the vehicle group.
UTI prevents oxidant-induced cytochrome c release
Cytosolic cytochrome c levels were markedly increased in the vehicle group (49.3 ± 7.5 ng/mg protein) compared with the control group (18.1 ± 3.9 ng/mg protein) (Figure 2; P < 0.05). In contrast, cytochrome c levels in the UTI (50,000 u/l) treatment group were significantly lower (31.5 ± 6.1 ng/mg protein) than those in the vehicle group (Figure 2; P < 0.05). The findings indicate that UTI attenuated oxidant-induced release of cytochrome c from mitochondria to the cytosol.
Figure 2.
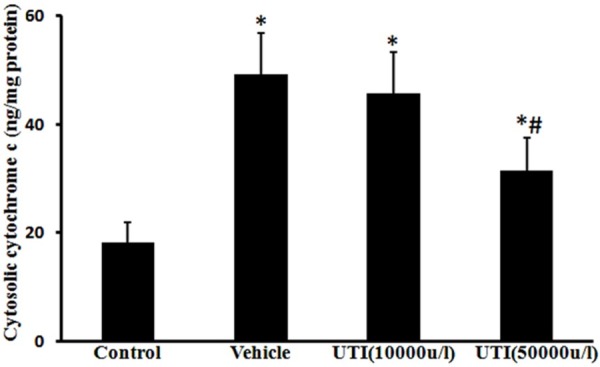
UTI inhibits oxidant-induced cytochrome c release. Cytosolic cytochrome c levels were estimated using a cytochrome c ELISA kit. Data are presented as mean ± SD (n = 6 in each group). *P < 0.05, versus the control group; #P < 0.05, versus the vehicle group.
UTI decreases oxidant-induced caspase-3 activation
Caspase-3 activity was obviously increased in the vehicle group compared to the control group (309% ± 42%, Figure 3; P < 0.05), which was prevented by UTI (50,000 u/l) treatment (192% ± 35%, Figure 3; P < 0.05).
Figure 3.
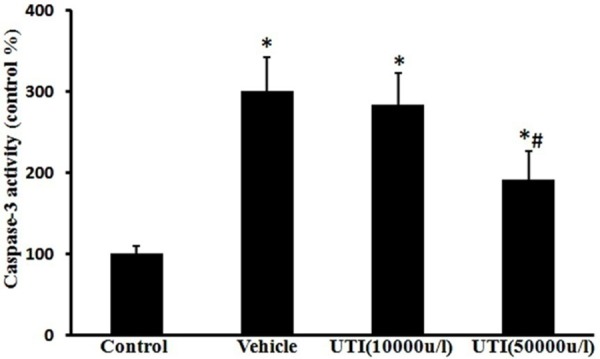
UTI decreases oxidant-induced caspase-3 activation. The caspase-3 activity was measured using the caspase-3 fluorometric assay kit. Data are presented as mean ± SD (n = 6 in each group). *P < 0.05, versus the control group; #P < 0.05, versus the vehicle group.
UTI inhibits oxidant-induced disruption of adherens junctions
Control cells showed strong and continuous β-catenin immunofluorescence at the cell-cell junctions, indicating an intact cell barrier. In contrast, treatment with H2O2 disrupted the cell junctions among cells as evidenced by the irregular and scattered β-catenin fluorescence observed. However, the alterations induced by H2O2 were improved by UTI (50,000 u/l) pretreatment (Figure 4).
Figure 4.
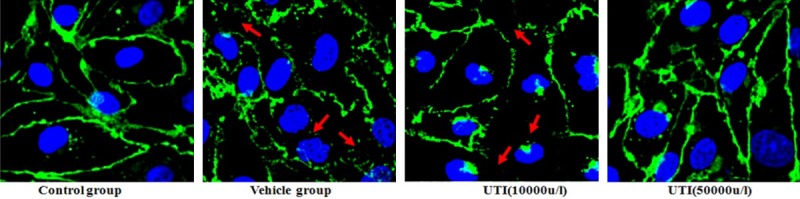
UTI inhibits oxidant-induced disruption of endothelial cell adherens junction. Adherens junction protein β-catenin was detected by immunofluorescense staining. Red arrow: disruption of the junctions evidenced by irregular and scattered β-catenin fluorescence.
UTI reduces oxidant-induced endothelial cell apoptosis
Rates of cell apoptosis were markedly increased in the vehicle group (19.4% ± 4.7%) compared with the control group (5.1% ± 1.1%) (Figure 5; P < 0.05). In contrast, the alterations were improved by the UTI (50,000 u/l) treatment group (10.4% ± 2.1%) (Figure 5; P < 0.05).
Figure 5.
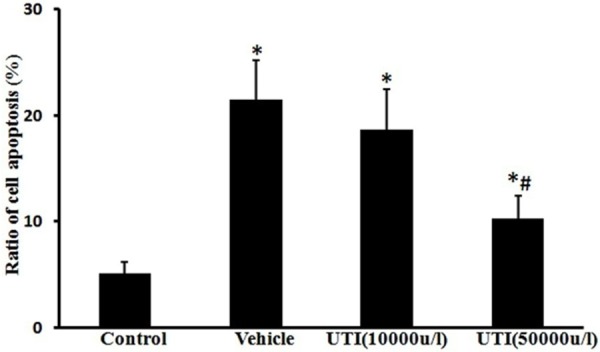
UTI inhibits oxidant-induced cell apoptosis. Ratio of cell apoptosis was evaluated by Annexin-V/PI double stain assay and analyzed by flow cytometry. Data are presented as mean ± SD (n = 6 in each group). *P < 0.05, versus the control group; #P < 0.05, versus the vehicle group.
UTI inhibits oxidant-induced loss of mitochondrial transmembrane potential
The percentage of low Δψm cells increased from 12.8 ± 3.8% in the control group to 57.3 ± 8.5% following H2O2 stimulation (Figure 6; P < 0.05). The percentage of cells with low mitochondrial potential decreased to 24.2 ± 6.6% by UTI (50,000 u/l) pretreatment (Figure 6; P < 0.05).
Figure 6.
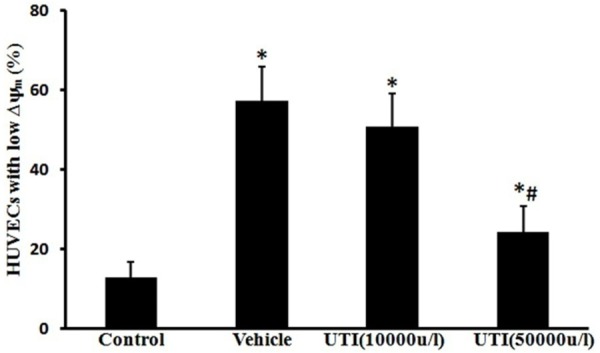
UTI inhibits oxidant-induced loss of mitochondrial membrane potential (Δψm). The Δψm was measured using the fluorescent probe JC-1 and analyzed by flow cytometry. Data are presented as mean ± SD (n = 6 in each group). *P < 0.05, versus the control group; #P < 0.05, versus the vehicle group.
UTI improves oxidant-induced mitochondrial dysfunction
In the vehicle group, the intracellular ATP level was found to be 56.0 ± 6.4% of that of the control group (Figure 7; P < 0.05 vs. control group), indicating HUVECs mitochondrial dysfunction following H2O2 stimulation. Following pretreatment with the UTI (50,000 u/l), the ATP level increased to 81.4 ± 10.3% of that of the control group (Figure 7; P < 0.05 vs. vehicle group).
Figure 7.
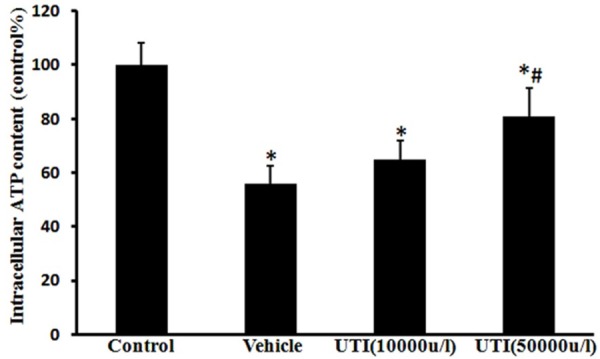
UTI improves oxidant-induced mitochondrial dysfunction. HUVECs’ ATP level was determined by a luciferase-based assay. Data are presented as mean ± SD (n = 6 in each group). *P < 0.05, versus the control group; #P < 0.05, versus the vehicle group.
UTI improves oxidant-induced Bax up-regulation and Bcl-2 down-regulation
In vehicle group cells, Bax and Bcl-2 expression were respectively 376% ± 51.8% and 45% ± 8.1% of normal values (Figure 8; P < 0.05). In UTI (50,000 u/l) pretreated cells, Bax and Bcl-2 expression were respectively185% ± 31.6% and 87% ± 13.3% of normal values (Figure 8; P < 0.05).
Figure 8.
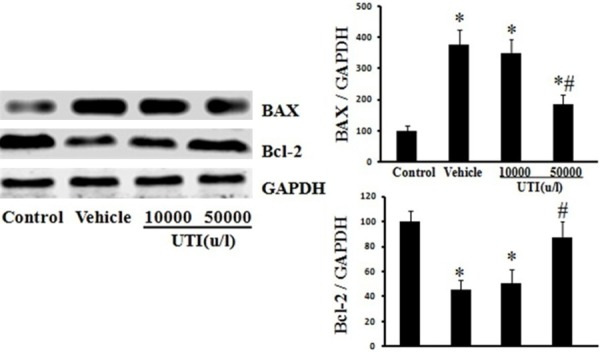
UTI improves oxidant-induced Bax up-regulation and Bcl-2 down-regulation. The Bax and Bcl-2 expression of HUVECs were detected by Western blot. Left, Representative Western blots for Bax and Bcl-2. Right, Protein quantification by densitometry. Data are presented as mean ± SD (n = 6 in each group). *P < 0.05, versus the control group; #P < 0.05, versus the vehicle group.
UTI reduces oxidant-induced ROS production
The ROS levels reflected by intensity of DCF fluorescence were decreased from 361% ± 49% of normal values in the vehicle group to 211% ± 32.0 % in UTI (50,000 u/l) treatment group (Figure 9; P < 0.05).
Figure 9.
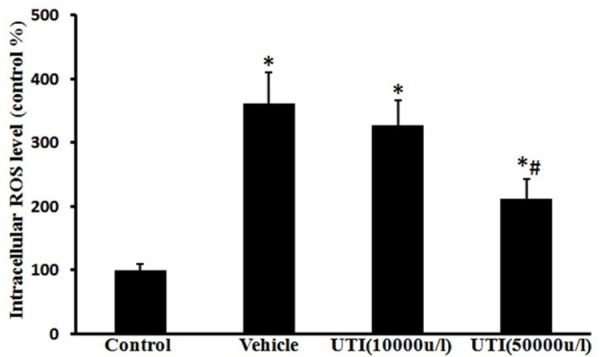
UTI reduces oxidant-induced oxidative stress. Intracellular ROS levels were assessed using DCFH-DA probe. Data are presented as mean ± SD (n = 6 in each group). *P < 0.05, versus the control group; #P < 0.05, versus the vehicle group.
Discussions
In present study, we have demonstrated that UTI attenuates oxidant-induced endothelial monolayer hyperpermeability. Further studies showed that UTI inhibited oxidant-induced ROS formation, preserved mitochondrial function, decreased the release of cytochrome c from mitochondria into the cytoplasm, inhibited the activation of caspase-3, and prevented disruption of adherens junctions.
The protective effects against inflammation, oxidative stress, and apoptosis have been well described [12,19,29,38]. Moreover, UTI has been shown prevented vascular endothelial injury in patients undergoing open heart surgery with cardiopulmonary bypass [21]. However, the effects of UTI on endothelial barrier functions and the underlying cellular and physiological mechanisms are not known. In this study, HUVECs were respectively pretreated with 10,000 and 50,000 u/l of UTI, followed by H2O2 stimulation. Results shown that UTI (50,000 u/l) significantly attenuates H2O2-induced endothelial hyperpermeability.
Studies demonstrated the activation of intrinsic apoptotic signaling, release of cytochrome c from mitochondria, and activation of caspase 3 in association with vascular hyperpermeability [8,28]. Thus, the main purpose of the present study was to test the effectiveness of UTI on oxidative stress and intrinsic apoptotic signaling. Our results suggest that UTI protected mitochondria, which is evident from the prevention of leakage of JC-1 dye from mitochondria into the cytoplasm, increase of the intracellular ATP level. The observation that UTI prevented oxidant-induced cytochrome c release further shows the protective effects of UTI on mitochondrial function. In normal cells, cytochrome c is located in the mitochondrial intermembrane/intercristae spaces. The mitochondrial outer membrane permeabilization that occurs in response to several proapoptotic stimuli can mobilize cytochrome c from cardiolipin leading to the release of cytochrome c to the cytosol [3,28,37]. Our results have demonstrated UTI treatment prevented membrane potential collapsed and cytochrome c release. After the release to the cytosol, cytochrome c mediates the allosteric activation of apoptotic protease activating factor 1, which is required for the proteolytic maturation of caspase 9 and caspase 3 [15,28,40]. So, the caspase 3 activation observed in our study may be explained as a result of an increased mitochondrial release of cytochrome c.
Our results show that UTI inhibited oxidant-induced ROS formation. Oxidative stress is one of the most important mediators of apoptotic signaling and subsequent cell death. Continuous mitochondrial oxidative stress caused by ROS is known to regulate mitochondrial release of cytochrome c [1,24]. The mitochondrial transition pore (mPT) is known to be activated by Ca2+ and ROS. Exogenous ROS also induced mPT opening and cytochrome c release in isolated mitochondria [10,33]. It is quite possible that in oxidant-induced endothelial permeability, mitochondrial oxidative stress played an important role in the release of cytochrome c to the cytoplasm. UTI, by its antioxidant activity, might have prevented this effect.
The intrinsic or mitochondrial-dependent mechanism of caspase activation depends on the Bcl-2 family of proteins, consists of anti-apoptotic (Bcl-2, Bcl-xL) and proapoptotic (BAK, BAX) factors, regulate mitochondrial outer membrane permeabilization [2,9,23]. The pro-apoptotic members of this family trigger the release of mitochondrial apoptogenic factors into the cytoplasm by regulating mPT. While the antiapoptotic members play a contrast role to prevent apoptosis [6,9]. In present study, BAX and Bcl-2 expression has been detdcted by western blotting. Results suggest that H2O2-induced BAX up-regulation and Bcl-2 down-regulation, which were improved by UTI treatment.
The activation of caspase 3 leads to the cleavage of a variety of cell adhesion proteins [25,36]. The major components of endothelial cell adherens junctions are the cadherin family of proteins, α-, β-, and γ-catenins. A stable cell-cell adherens junction requires the close interaction of the cytoplasmic domain of the cadherins with a group of intracellular proteins, the catenins [11]. β-Catenin, a member of the Armadillo repeat protein family, functions as a regulator of cadherin-mediated cell-cell adhesion in endothelial cells, and its absence may lead to fluid leakage [8,14]. Proteolytic cleavage of β-catenin occurs after the activation of procaspase 3, 6, or 8 [8]. Results of immunofluorescence staining showed that oxidant stimulation caused irregular and scattered β-catenin fluorescence, which was improved by UTI treatment. Thus, UTI mediated inhibition of caspase 3 activation, and subsequent prevention of microvascular endothelial cell-cell detachment is one of the possible mechanisms by which it protected barrier integrity and prevented oxidant-induced endothelial monolayer hyperpermeability.
In conclusion, our findings show that oxidant stimulation activates intrinsic apoptotic signaling and ultimately activates caspase-3, which leads to the disruption of cell adherens junctions and hyperpermeability. The protective effect of UTI on vascular barrier functions may be caused by its inhibitory effects on ROS formation and intrinsic apoptotic signaling pathway.
Disclosure of conflict of interest
None.
References
- 1.Atlante A, Calissano P, Bobba A, Azzariti A, Marra E, Passarella S. Cytochrome c is released from mitochondria in a reactive oxygen species (ROS)-dependent fashion and can operate as a ROS scavenger and as a respiratory substrate in cerebellar neurons undergoing excitotoxic death. J Biol Chem. 2000;275:37159–37166. doi: 10.1074/jbc.M002361200. [DOI] [PubMed] [Google Scholar]
- 2.Azad N, Iyer A, Vallyathan V, Wang L, Castranova V, Stehlik C, Rojanasakul Y. Role of oxidative/nitrosative stress-mediated Bcl-2 regulation in apoptosis and malignant transformation. Ann N Y Acad Sci. 2010;1203:1–6. doi: 10.1111/j.1749-6632.2010.05608.x. [DOI] [PubMed] [Google Scholar]
- 3.Barbu EM, Shirazi F, McGrath DM, Albert N, Sidman RL, Pasqualini R, Arap W, Kontoyiannis DP. An antimicrobial peptidomimetic induces Mucorales cell death through mitochondria-mediated apoptosis. PLoS One. 2013;8:e76981. doi: 10.1371/journal.pone.0076981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cai W, Torreggiani M, Zhu L, Chen X, He JC, Striker GE, Vlassara H. AGER1 regulates endothelial cell NADPH oxidase-dependent oxidant stress via PKC-delta: implications for vascular disease. Am J Physiol Cell Physiol. 2010;298:C624–C634. doi: 10.1152/ajpcell.00463.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Q, Liu XF, Zheng PS. Grape Seed Proanthocyanidins (GSPs) Inhibit the Growth of Cervical Cancer by Inducing Apoptosis Mediated by the Mitochondrial Pathway. PLoS One. 2014;9:e107045. doi: 10.1371/journal.pone.0107045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen YB, Aon MA, Hsu YT, Soane L, Teng X, McCaffery JM, Cheng WC, Qi B, Li H, Alavian KN, Dayhoff-Brannigan M, Zou S, Pineda FJ, O’Rourke B, Ko YH, Pedersen PL, Kaczmarek LK, Jonas EA, Hardwick JM. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J Cell Biol. 2011;195:263–276. doi: 10.1083/jcb.201108059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Childs EW, Tharakan B, Byrge N, Tinsley JH, Hunter FA, Smythe WR. Angiopoietin-1 inhibits intrinsic apoptotic signaling and vascular hyperpermeability following hemorrhagic shock. Am J Physiol Heart Circ Physiol. 2008;294:H2285–H2295. doi: 10.1152/ajpheart.01361.2007. [DOI] [PubMed] [Google Scholar]
- 8.Childs EW, Tharakan B, Hunter FA, Smythe WR. Apoptotic signaling induces hyperpermeability following hemorrhagic shock. Am J Physiol Heart Circ Physiol. 2007;292:H3179–H3189. doi: 10.1152/ajpheart.01337.2006. [DOI] [PubMed] [Google Scholar]
- 9.Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, Huang DC, Kluck RM, Adams JM, Colman PM. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell. 2013;152:519–531. doi: 10.1016/j.cell.2012.12.031. [DOI] [PubMed] [Google Scholar]
- 10.Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5:415–418. doi: 10.1023/a:1009616228304. [DOI] [PubMed] [Google Scholar]
- 11.Hinck L, Nathke IS, Papkoff J, Nelson WJ. Dynamics of cadherin/catenin complex formation: novel protein interactions and pathways of complex assembly. J Cell Biol. 1994;125:1327–1340. doi: 10.1083/jcb.125.6.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu CL, Xia JM, Cai J, Li X, Liao XX, Li H, Zhan H, Dai G, Jing XL. Ulinastatin attenuates oxidation, inflammation and neural apoptosis in the cerebral cortex of adult rats with ventricular fibrillation after cardiopulmonary resuscitation. Clinics (Sao Paulo) 2013;68:1231–1238. doi: 10.6061/clinics/2013(09)10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang N, Wang F, Wang Y, Hou J, Li J, Deng X. Ulinastatin improves survival of septic mice by suppressing inflammatory response and lymphocyte apoptosis. J Surg Res. 2013;182:296–302. doi: 10.1016/j.jss.2012.10.043. [DOI] [PubMed] [Google Scholar]
- 14.Huang QB. Barrier stabilizing mediators in regulation of microvascular endothelial permeability. Chin J Traumatol. 2012;15:105–112. doi: 10.3760/cma.j.issn.1008-1275.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lakhani SA, Masud A, Kuida K, Porter GA Jr, Booth CJ, Mehal WZ, Inayat I, Flavell RA. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–851. doi: 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leng YX, Yang SG, Song YH, Zhu X, Yao GQ. Ulinastatin for acute lung injury and acute respiratory distress syndrome: A systematic review and meta-analysis. World J Crit Care Med. 2014;3:34–41. doi: 10.5492/wjccm.v3.i1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li T, Cai S, Zeng Z, Zhang J, Gao Y, Wang X, Chen Z. Protective effect of polydatin against burn-induced lung injury in rats. Respir Care. 2014;59:1412–1421. doi: 10.4187/respcare.02831. [DOI] [PubMed] [Google Scholar]
- 18.Lum H, Roebuck KA. Oxidant stress and endothelial cell dysfunction. Am J Physiol Cell Physiol. 2001;280:C719–C741. doi: 10.1152/ajpcell.2001.280.4.C719. [DOI] [PubMed] [Google Scholar]
- 19.Luo HM, Du MH, Lin ZL, Hu Q, Zhang L, Ma L, Wang H, Wen Y, Lv Y, Lin HY, Pi YL, Hu S, Sheng ZY. Ulinastatin suppresses burn-induced lipid peroxidation and reduces fluid requirements in a Swine model. Oxid Med Cell Longev. 2013;2013:904370. doi: 10.1155/2013/904370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo HM, Hu S, Zhou GY, Bai HY, Lv Y, Wang HB, Lin HY, Sheng ZY. The effects of ulinastatin on systemic inflammation, visceral vasopermeability and tissue water content in rats with scald injury. Burns. 2012 doi: 10.1016/j.burns.2012.11.004. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 21.Miura M, Sugiura T, Aimi Y, Yasuda K, Ito S, Baba E, Katsuya H. [Effects of ulinastatin on PMNL and vascular endothelial injury in patients undergoing open heart surgery with CPB] . Masui. 1998;47:29–35. [PubMed] [Google Scholar]
- 22.Patterson EK, Fraser DD, Capretta A, Potter RF, Cepinskas G. Carbon monoxide-releasing molecule 3 inhibits myeloperoxidase (MPO) and protects against MPO-induced vascular endothelial cell activation/dysfunction. Free Radic Biol Med. 2014;70:167–173. doi: 10.1016/j.freeradbiomed.2014.02.020. [DOI] [PubMed] [Google Scholar]
- 23.Shore GC, Nguyen M. Bcl-2 proteins and apoptosis: choose your partner. Cell. 2008;135:1004–1006. doi: 10.1016/j.cell.2008.11.029. [DOI] [PubMed] [Google Scholar]
- 24.Sinha K, Das J, Pal PB, Sil PC. Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol. 2013;87:1157–1180. doi: 10.1007/s00204-013-1034-4. [DOI] [PubMed] [Google Scholar]
- 25.Steinhusen U, Badock V, Bauer A, Behrens J, Wittman-Liebold B, Dörken B, Bommert K. Apoptosis-induced cleavage of beta-catenin by caspase-3 results in proteolytic fragments with reduced transactivation potential. J Biol Chem. 2000;275:16345–16353. doi: 10.1074/jbc.M001458200. [DOI] [PubMed] [Google Scholar]
- 26.Takeda D, Hasegawa T, Ueha T, Imai Y, Sakakibara A, Minoda M, Kawamoto T, Minamikawa T, Shibuya Y, Akisue T, Sakai Y, Kurosaka M, Komori T. Transcutaneous carbon dioxide induces mitochondrial apoptosis and suppresses metastasis of oral squamous cell carcinoma in vivo. PLoS One. 2014;9:e100530. doi: 10.1371/journal.pone.0100530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tharakan B, Hellman J, Sawant DA, Tinsley JH, Parrish AR, Hunter FA, Smythe WR, Childs EW. β-Catenin dynamics in the regulation of microvascular endothelial cell hyperpermeability. Shock. 2012;37:306–311. doi: 10.1097/SHK.0b013e318240b564. [DOI] [PubMed] [Google Scholar]
- 28.Tharakan B, Hunter FA, Smythe WR, Childs EW. Alpha-lipoic acid attenuates hemorrhagic shock-induced apoptotic signaling and vascular hyperpermeability. Shock. 2008;30:571–577. doi: 10.1097/SHK.0b013e31816a7308. [DOI] [PubMed] [Google Scholar]
- 29.Tong Y, Tang Z, Yang T, Yang Y, Yang L, Shen W, Chen W. Ulinastatin preconditioning attenuates inflammatory reaction of hepatic ischemia reperfusion injury in rats via high mobility group box 1(HMGB1) inhibition. Int J Med Sci. 2014;11:337–343. doi: 10.7150/ijms.7861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walter DH, Haendeler J, Galle J, Zeiher AM, Dimmeler S. Cyclosporin A inhibits apoptosis of human endothelial cells by preventing release of cytochrome C from mitochondria. Circulation. 1998;98:1153–1157. doi: 10.1161/01.cir.98.12.1153. [DOI] [PubMed] [Google Scholar]
- 31.Wang H, Sun X, Gao F, Zhong B, Zhang YH, Sun Z. Effect of ulinastatin on growth inhibition, apoptosis of breast carcinoma cells is related to a decrease in signal conduction of JNk-2 and NF-kappaB. J Exp Clin Cancer Res. 2012;31:2. doi: 10.1186/1756-9966-31-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang L, Luo H, Chen X, Jiang Y, Huang Q. Functional characterization of S100A8 and S100A9 in altering monolayer permeability of human umbilical endothelial cells. PLoS One. 2014;9:e90472. doi: 10.1371/journal.pone.0090472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X, Song R, Chen Y, Zhao M, Zhao KS. Polydatin--a new mitochondria protector for acute severe hemorrhagic shock treatment. Expert Opin Investig Drugs. 2013;22:169–179. doi: 10.1517/13543784.2013.748033. [DOI] [PubMed] [Google Scholar]
- 34.Wang XM, Song SS, Xiao H, Gao P, Li XJ, Si LY. Fibroblast growth factor 21 protects against High Glucose Induced Cellular Damage and Dysfunction of Endothelial Nitric-Oxide Synthase in Endothelial Cells. Cell Physiol Biochem. 2014;34:658–671. doi: 10.1159/000363031. [DOI] [PubMed] [Google Scholar]
- 35.Whaley JG, Tharakan B, Smith B, Hunter FA, Childs EW. (-)-Deprenyl inhibits thermal injury-induced apoptotic signaling and hyperpermeability in microvascular endothelial cells. J Burn Care Res. 2009;30:1018–1027. doi: 10.1097/BCR.0b013e3181bfb825. [DOI] [PubMed] [Google Scholar]
- 36.Wu X, Deng G, Hao X, Li Y, Zeng J, Ma C, He Y, Liu X, Wang Y. A caspase-dependent pathway is involved in Wnt/beta-catenin signaling promoted apoptosis in Bacillus Calmette-Guerin infected RAW264.7 macrophages. Int J Mol Sci. 2014;15:5045–5062. doi: 10.3390/ijms15035045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiong S, Mu T, Wang G, Jiang X. Mitochondria-mediated apoptosis in mammals. Protein Cell. 2014 doi: 10.1007/s13238-014-0089-1. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu CE, Zhang MY, Zou CW, Guo L. Evaluation of the pharmacological function of ulinastatin in experimental animals. Molecules. 2012;17:9070–9080. doi: 10.3390/molecules17089070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, Zhang L, Li Y, Sun S, Tan H. Different contributions of clathrin- and caveolae-mediated endocytosis of vascular endothelial cadherin to lipopolysaccharide-induced vascular hyperpermeability. PLoS One. 2014;9:e106328. doi: 10.1371/journal.pone.0106328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]