

Abstract
Key Clinical Massage
We present a 27-month-old male infant with pseudohypoaldosteronism, with two novel α-subunits, epithelial sodium channel (ENaC) mutations. Despite the presence of the ENaC in the lungs, kidneys, and exocrine glands, he continues to only have renal and exocrine involvement, stressing differential effects of the mutation in each organ.
Keywords: ENaC, epithelial sodium channel, hyperkalemia, hyponatremia, pseudohypoaldosteronism, salt-wasting, SCNN1A
Introduction
Pseudohypoaldosteronism type 1 (PHA 1) presents in the first few weeks of life with life-threatening salt-wasting syndrome, hyperkalemia, and metabolic acidosis. PHA 1 includes both an autosomal dominant (AD) and a recessive form (AR). PHA 1 is rare, with a reported incidence ranging from 1:47,000 to 1:80,000 newborns [1,2]. PHA 1(AD) is caused by mutations in the mineralocorticoid receptor encoded by the NR3C2 gene (chr4:148999915-149363672; NM_000901), with the salt-wasting syndrome involving only the renal tubules. Typically, the extent of salt wasting improves over time, with resolution of the disease [3–5].
In contrast, PHA 1(AR) is caused by loss of function of the amiloride-sensitive epithelial sodium channel (ENaC). This is a highly conserved channel which is expressed in the kidneys, lungs, colon, salivary and sweat glands. In the kidneys, ENaC regulates sodium reabsorption and electrolyte balance, while in the lungs it regulates lung fluid. The ENaC is a trimeric channel, consisting of 3 genetically unique subunits assembled as alpha (α), beta (β), and gamma (γ) (encoded by the genes SCNN1A, SCNN1B and SCNN1G, respectively). Each subunit has two transmembrane segments, an extracellular loop, and an intracellular N- and C- terminus region. The α-subunit has been described as the main sodium conductor, while the β and γ-subunits are involved in channel flow regulation [6–9].
The classic presentation of PHA 1(AR) is thought to be lifelong and severe. In addition to severe salt wasting, affected patients may have symptoms very similar to cystic fibrosis (chronic pulmonary symptoms, elevated sweat chloride secretion, and failure to thrive) [10,3]. The availability of genetic testing has shown that regardless of which subunit is mutated, there appears to be variability of phenotypic presentation. Here, we report a new case of PHA 1(AR) with 2 novel mutations and discuss its clinical and genetic characteristics and compare it to previously described cases.
Clinical Report
A 27-month-old boy presented at 7 days of age with severe hyperkalemia, hyponatremia, and dehydration. He was born via C-section due to premature rupture of membranes at 36 weeks, with birth weight of 2.021 kg (3.5% ile), length of 40.6 cm (0.2% ile, and head circumference of 33 cm (55% ile). The parents were nonconsanguineous, of Caucasian descent, and mother was gravida 4 para 1 (the other 3 pregnancies ended in elective abortions). At birth, the baby had no respiratory difficulty, had normal electrolyte panel (Na 141 mmol/L, K 5.1 mmol/L, HCO3 22 mmol/L), and remained under routine care due to prematurity. At 1 week of age, he was noted to have mottling, jaundice, and difficulty feeding which were associated with marked electrolyte disturbances (Na 123 mmol/L, K 9.0 mmol/L, HCO3 17 mmol/L). Urine electrolytes analysis revealed Na of 69 mmol/L, K 2.6 mmol/L, Cl 42 mmol/L. The patient's sepsis work-up and metabolic newborn screening including cystic fibrosis were negative. Electrocardiography, renal function, renal ultrasound, and VCUG examinations were normal. He was treated with an IV hyperkalemia cocktail consisting of sodium bicarbonate, calcium gluconate, insulin, and glucose. He also received sodium polystyrene, increased sodium delivery, hydrocortisone, and fludrocortisone. Further investigations revealed a cortisol level of 12.7 mcg/dL (2–11 mcg/dL), 17-hydroxyprogesterone 20 ng/dL (11–170 ng/dL), progesterone 3.13 ng/dL (7–52 ng/dL), plasma renin activity 93.96 ng ml/h (2–35 ng ml/h), and aldosterone 587.6 ng/dL (<217 ng/dL) (Table 1). A preliminary diagnosis of pseudohypoaldosteronism was then made. By day 12 of life, the patient stabilized clinically while receiving sodium supplementation at 8.5 mmol kg/day. On day 25 of life, he then had recurrence of poor feeding and severe electrolyte imbalance (Na 126 mmol/L, K 10 mmol/L) in addition to failure to thrive (2.085 kg). Patient was put on the acute hyperkalemia cocktail, aggressive sodium and fluid resuscitation. Due to a markedly increased urine output volume (16 cc kg/h with urine Na 175 mmol/L, urine K < 2), urine replacement with normal saline was set at 1:1. During this time, the sodium intake was approximately 61 mmol kg/day, resulting in normalization of his sodium levels. The patient continued to tolerate oral feeds, and both hemodynamic and respiratory functions remained stable. Once a new steady-state had been achieved, the patient's daily sodium requirement remained at 42.5 mmol kg/day (total of 118 mmol Na given orally, with 56 mmol from oral sodium chloride, 32 mmol from oral sodium citrate, and 32 mmol from oral sodium polystyrene.). For emergency access and ease of medication administration, a gastrostomy tube was placed. At 40 days of age, the patient developed tachypnea and lowered oxygen saturation requiring oxygen supplementation via nasal cannula. Chest X-ray examination showed mild pulmonary congestion. He responded quickly to a small dose of hydrochlorothiazide. While the congestion may have been the result of high salt and fluid intake, a systemic form of PHA was entertained. A sweat chloride test was obtained and repeated. Both measurements were markedly elevated at 104 and 118 mmol/L (normal <29 mmol/L).
Table 1.
Laboratory findings in the patient
| Normal values | Day 2 | Day 7 | Day 40 | |
|---|---|---|---|---|
| Na (mmol/L) | 135–145 | 141 | 123 | 145 |
| K (mmol/L) | 3.5–4.5 | 5.1 | 9 | 4.4 |
| HCO3 (mmol/L) | 20–28 | 22 | 17 | 23 |
| Urine Na (mmol/L) | 69 | 265 | ||
| Urine K (mmol/L) | 2.6 | <2 | ||
| Cortisol (mcg/dL) | 2–11 | 12.7 | ||
| 17-OH progesterone (ng/dL) | 11–170 | 20 | ||
| Plasma renin activation (ng/mL/h) | 2–35 | 93.96 | ||
| Aldosterone (ng/dL) | <217 | 587.6 | ||
| Sweat chloride (<29 mmol/L) | <29 | 104; 118 on repeat |
Day 7 of life represents the age of presentation. On day 40, the infant started to have respiratory difficulty, despite normalization of electrolytes. The multisystem form of PHA was genetically confirmed shortly after.
He was discharged at 3 months of age on an oral regimen of sodium chloride, sodium citrate, and sodium polystyrene (total 17 mmol kg/day). He is currently 27 months old and has not required hospitalization despite occasional self-limited upper respiratory tract infections. Minimal dose adjustments have been made on his medication regimen. His dietary management of sodium and potassium has been complicated by the family's preference of a vegan diet. He is gaining weight well, although continues to be small for his age, and is attaining appropriate milestones.
Due to abnormally elevated sweat chloride despite normal IRT (immunoreactive trypsinogen) levels on state newborn screening for cystic fibrosis, CFTR mutation analysis using the CFTR-InPlex™ 40-mutation panel (Hologic-Third Wave, Madison, WI, USA) was done. No mutation was found in this limited panel offered as a second tier test by the state of Kansas newborn screening laboratory. In addition, the patient did not have cutaneous lesions (such as milaria rubra), nasal discharge or gastrointestinal problems. Mutation analysis of the α-ENaC gene encoded by SCNN1A [chr12:6456009-6484905, NM_001038] revealed two potentially deleterious novel mutations: c.416G>A (p.Arg139Lys) and c.1360 + 1G>T. Since the mutations lie within the exon/intron splice consensus sequence, the mutations were predicted to destroy the normal splicing of exon 2 and 8 donor sites, respectively (Fig.1). This was not confirmed via RT-PCR since we did not have access to the patient's RNA. Since the mutation being described is an AR one, neither parent was tested as they were expected to be asymptomatic carriers.
Figure 1.
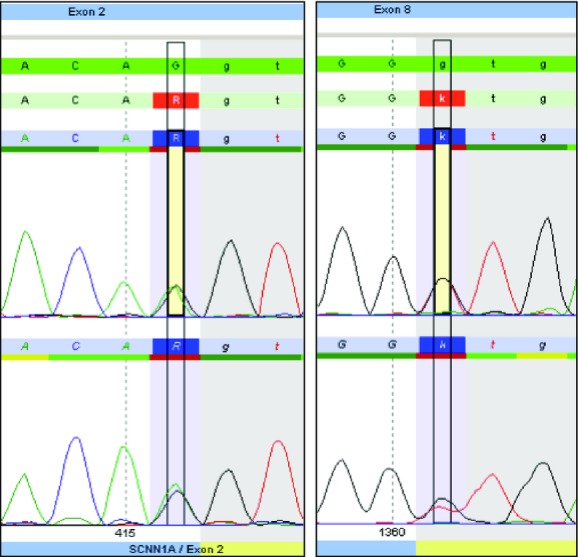
Genomic DNA analysis of SCNN1A in the proband. Analysis shows a missense mutation in exon 2 (c.416G>A [p.Arg139Lys], left panel) and a mutation in exon 8 (c.1360 + 1G>T, right panel), both of which are predicted to destroy splicing resulting in exon skipping. Mutated nucleotide is highlighted and its position is indicated at the bottom of the tracing which is shown in duplicate.
Discussion
Over the past decade, the number of patient reports on PHA 1(AR) had increased somewhat significantly, allowing better understanding of the disorder. In 2005, Edelheit et al. reported 3 new patients, with 22 independent mutations in the coding region of the αENaC subunit known worldwide. Abnormal phenotypes have been reported in all 3 subunits ranging from mild to severe disease, but α-subunit involvement was most common. Most of the mutations reported were typically single-base deletion/insertion, or splice site mutation, all of which were associated with severe phenotypes. Those patients with missense mutations seemed to have a milder course [11,12]. Numerous other novel mutations have been since been reported in the literature, with varying disease severity [13,14]. In our patient, two novel splice site mutations in SCNN1A were identified, both of which are predicted to cause exon skipping and a shorter unstable transcript. As expected, and while the patient had an abnormal sweat chloride test, CFTR mutation analysis was negative, therefore excluding cystic fibrosis as a possible explanation. In addition, his newborn screen for cystic fibrosis using IRT was normal. This illustrates the fact that patients with PHA 1(AR) are not expected to be identified via abnormal newborn screening for cystic fibrosis. CFTR gene analysis was not performed on the whole gene, but this was not felt to be necessary, as the gold standard for the diagnosis of CF remains to be a compatible clinical diagnosis plus abnormal sweat chloride test. The clinical presentation (electrolyte abnormalities), negative newborn screening (IRT) test as well negative CFTR mutation panel strongly argue against CF as a diagnosis.
The disease severity has been defined in the literature as requiring frequent hospitalizations, persistent critical salt wasting and potentially life-threatening hyperkalemia, significant respiratory dysfunction and growth failure [11]. The respiratory dysfunction is due to the inability to absorb airway fluid properly, with resulting increased airway liquid volume, and impaired mucociliary function. This results in a high incidence of lower respiratory tract involvement, with repeated episodes of coughing, cyanosis, dyspnea, wheezing, and fever, similar to cystic fibrosis [15,16]. Based on our experience and review of the literature, we propose to define severity based on the presence of sustained pulmonary disease and electrolyte imbalance, as the outcome and long-term management can be drastically different. Our patient had a severe initial presentation; however, it was predominantly an electrolyte issue. While still continuously needing sodium supplementation (chloride, bicarbonate, and polysterene), he did not need medication adjustments for almost 2 years. The patient has had a few mild upper respiratory tract infections from which he recovered fully within a few days and without hospitalization, typical of any healthy infant.
Absence of significant pulmonary involvement in a patient with severe salt wasting illustrates the complexity of ENaC. Traditionally, the α-subunit is considered to be the major component of the channel. For example, α-subunit knockout mice die within 40 h of birth secondary to respiratory distress [17]. However, transgenic manipulation of the α-subunit in these knockout mice resulted in low levels of ENaC that were sufficient to avoid pulmonary demise yet, severe salt wasting persisted [10]. Clearly, while ENaC is a major determinant of fluid and electrolyte balance in both lungs and kidneys, the presence of other mechanisms unique to each organ may alter the effect of a mutation [18,17].
To go one step further in evaluating the broad phenotypic spectrum of PHA 1(AR), the experience with spontaneous clinical amelioration has been described. Hanukoglu described a patient with a missense mutation, who had clinical improvement after infancy, with smaller sodium chloride supplementation requirements after 9 years of age [12]. Dirlewanger et al. reported a preterm infant with a homozygous missense mutation of the α-ENaC and salt-wasting syndrome which resolved completely by 6 months of age [19]. Interestingly, that patient's sibling, born at term with the same mutation was completely asymptomatic. A possible important compensatory mechanism responsible for the clinical improvement was demonstrated in a patient with mild AR-PHA, where investigators found an increased expression of the thiazide-sensitive transporter (NCC), leading to increased absorption of sodium in upstream nephrons [20].
Overall, our experience, in addition to evidence assessed in the literature, indicates a dichotomy of disease severity and effect between renal and pulmonary manifestations. While PHA1 (AR) is typically associated with multiorgan involvement, this may not be the case in every patient, and the lack of pulmonary involvement certainly improves long-term prognosis. Reporting of additional patients with PHA1 (AR, AD) and long-term follow up, in vitro studies of various ENac subunit mutations and the identification of other genetic modifiers will likely help us better understand this potentially life-threatening disease and improve its management. Multiorgan involvement should always be considered in infants presenting with Type 1 PHA, and a positive sweat test supports this AR form.
Acknowledgments
The authors wish to thank Steven J. Steinberg for providing the SCNNA1 mutation electroperogram, Colleen Peterson for helping with the CFTR mutation analysis, and the KU Neonatology team for providing outstanding care for the patient. Also, we wish to thank the family of our patient.
Conflict of Interest
None declared.
References
- 1.Amin N, Alvi NS, Barth JH, Field HP, Finlay E, Tyerman K, et al. Pseudohypoaldosteronism type 1: clinical features and management in infancy. Endocrinol. Diabetes Metab. Case Rep. 2013:130010. doi: 10.1530/EDM-13-0010. August 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Genetics Home Reference Website. 2011. Pseudohypoaldosteronism type 1. Available at http://ghr.nlm.nih.gov/condition (accessed 29 June 2014)
- 3.Geller DS, Zhang J, Zennaro MC, VAllo-Boado A, Rodriguez-Soriano J, Furu L, et al. Autosomal dominant pseudohypoaldosteronism type I: mechanisms, evidence for neonatal lethality, and phenotypic expression in adults. J. Am. Soc. Nephrol. 2006;17:1429–1436. doi: 10.1681/ASN.2005111188. [DOI] [PubMed] [Google Scholar]
- 4.Hanukoglu A. Type I pseudohypoaldosteronism includes two clinically and genetically distinct entities with either renal or multiple target organ defects. J. Clin. Endocrinol. Metab. 1993;73:936–944. doi: 10.1210/jcem-73-5-936. [DOI] [PubMed] [Google Scholar]
- 5.Riepe FG. Clinical and molecular features of type I pseudohypoaldosteronism. Horm. Res. 2009;72:1–9. doi: 10.1159/000224334. [DOI] [PubMed] [Google Scholar]
- 6.Arai K, Zachman K, Shibasaki T, Chrousos G. Polymorphisms of amiloride sensitive sodium channel subunits in five sporadic cases of pseudohypoaldosteronism: do they have pathologic potential? J. Clin. Endocrinol. Metab. 1999;84:2434–2437. doi: 10.1210/jcem.84.7.5857. [DOI] [PubMed] [Google Scholar]
- 7.Chang S, Grunder S, Hanukoglu A, Rosler A, Matthew PM, Hanukoglu I, et al. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalemic acidosis, pseudohypoaldosteronism type I. Nat. Genet. 1996;12:248–253. doi: 10.1038/ng0396-248. [DOI] [PubMed] [Google Scholar]
- 8.Edelheit O, Hanukoglu I, Shriki Y, Tfilin M, Dascal N, Gillis D, et al. Truncated beta epithelial sodium channel (ENaC) subunits responsible for multi-system pseudohypoaldosteronism support partial activity of ENaC. J. Steroid Biochem. Mol. Biol. 2010;119:84–88. doi: 10.1016/j.jsbmb.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 9.Jasti J, Furukawa H, Gonzales EB, Gouaux E. Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature. 2007;449:316–323. doi: 10.1038/nature06163. [DOI] [PubMed] [Google Scholar]
- 10.Bonny O, Hummler E. Dysfunction of epithelial sodium transport: from human to mouse. Kidney Int. 2001;57:1313–1318. doi: 10.1046/j.1523-1755.2000.00968.x. [DOI] [PubMed] [Google Scholar]
- 11.Edelheit O, Hanukoglu I, Gizewska M, Kandemir N, Tenenbaum-Rakover Y, Yurdakok M, et al. Novel mutations in epithelial sodium channel (ENaC) subunit genes and phenotypic expression of multisystem pseudohypoaldosteronism. J. Clin. Endocrinol. 2005;62:547–553. doi: 10.1111/j.1365-2265.2005.02255.x. [DOI] [PubMed] [Google Scholar]
- 12.Hanukoglu A, Edelheit O, Shriki Y, Gizewska M, Dascal N, Hanukoglu I. Renin-aldosterone response, urinary Na/K ratio and growth in pseudohypoaldosteronism patient with mutations in epithelial sodium channel (ENaC) subunit genes. J. Steroid Biochem. Mol. Biol. 2008;111:268–274. doi: 10.1016/j.jsbmb.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 13.Belot A, Ranchin B, Fichtner C, Pujo L, Rossier BC, Liutkus A, et al. Pseudohypoaldosteronism, report on a 10-patient series. Nephrol. Dial. Transplant. 2008;23:1636–1641. doi: 10.1093/ndt/gfm862. [DOI] [PubMed] [Google Scholar]
- 14.Schweiger B, Moriarty MW, Cadnapaphornchai MA. Case report: severe neonatal hyperkalemia due to pseudohypoaldosteronism type I. Curr. Opin. Pediatr. 2009;21:269–271. doi: 10.1097/MOP.0b013e328325a55f. [DOI] [PubMed] [Google Scholar]
- 15.Hanukoglu A, Bistritzer T, Rakover Y, Mandelberg A. Pseudohypoaldosteronism with increased sweat and saliva electrolyte values and frequent lower respiratory tract infections mimicking cystic fibrosis. J. Pediatr. 1994;125:752–755. doi: 10.1016/s0022-3476(94)70071-0. [DOI] [PubMed] [Google Scholar]
- 16.Kerem E, Bistritzer T, Hanukoglu A, Hofmann T, Zhou Z, Bennett W, et al. Pulmonary epithelial sodium-channel dysfunction and excess airway liquid in pseudohypoaldosteronism. N. Engl. J. Med. 1999;341:156–162. doi: 10.1056/NEJM199907153410304. [DOI] [PubMed] [Google Scholar]
- 17.Hummler E, Barker P, Gatzy J, Beerman F, Verdumo C, Schmidt A, et al. Early death due to defective neonatal lung liquid clearance in α-ENaC-deficient mice. Nat. Genet. 1996;12:325–328. doi: 10.1038/ng0396-325. [DOI] [PubMed] [Google Scholar]
- 18.Enuka Y, Hanukoglu I, Edelheit O, Vaknine H, Hanukoglu A. Epithelial sodium channels (ENaC) are uniformly distributed on motile cilia in the oviduct and the respiratory airways. Histochem. Cell Biol. 2012;137:339–353. doi: 10.1007/s00418-011-0904-1. [DOI] [PubMed] [Google Scholar]
- 19.Dirlewanger M, Huser D, Zennaro MC, Girardin E, Schild L, Schwitzgebel VM. A homozygous missense mutation in SCNN1A is responsible for a transient neonatal form of pseudohypoaldosteronism type I. Am. J. Physiol. Endocrinol. Metab. 2011;301:E467–E473. doi: 10.1152/ajpendo.00066.2011. [DOI] [PubMed] [Google Scholar]
- 20.Adachi M, Asakura Y, Muroya K, Tajima T, Fujieda K, Kuribayashi E, et al. Increased Na reabsorption via the Na-Cl cotransporter in autosomal recessive pseudohypoaldosteronism. Clin. Exp. Nephrol. 2010;14:228–232. doi: 10.1007/s10157-010-0277-0. [DOI] [PubMed] [Google Scholar]