

Abstract
Objective
Diabetic hypertriglyceridemia is thought to be primarily driven by increased hepatic de novo lipogenesis. However, experiments in animal models indicated that insulin deficiency should decrease hepatic de novo lipogenesis and reduce plasma triglyceride levels.
Approach and Results
To address the discrepancy between human data and genetically altered mouse models, we investigated whether insulin deficient diabetic mice had triglyceride changes that resemble those in diabetic humans. Streptozotocin (STZ)–induced insulin deficiency increased plasma triglyceride levels in mice. Contrary to the mouse models with impaired hepatic insulin receptor signalling, insulin deficiency did not reduce hepatic triglyceride secretion and de novo lipogenesis-related gene expression. Diabetic mice had a marked decrease in postprandial TG clearance, which was associated with decreased lipoprotein lipase (LpL) and PPARα mRNA levels in peripheral tissues and decreased LpL activity in skeletal muscle, heart and brown adipose tissue. Diabetic heterozygous LpL knockout mice had markedly elevated fasting plasma triglyceride levels and prolonged postprandial TG clearance.
Conclusion
Insulin deficiency causes hypertriglyceridemia by decreasing peripheral lipolysis and not by an increase in hepatic TG production and secretion.
Keywords: lipoprotein lipase, hypertriglyceridemia, diabetes
Introduction
Dyslipidemia is a major risk factor for cardiovascular disease (CVD) in both type 1 diabetes mellitus (T1DM) and type 2 diabetes mellitus (T2DM)1, 2. Diabetic dyslipidemia is associated with high plasma triglycerides (TG), low HDL cholesterol and increased small dense LDL-cholesterol particles3. According to a National Health and Nutrition Examination Survey, over 30% of people with T2DM have TG levels >2.258 mmol/l (200 mg/dl)4. Patients with T1DM also have increased TG levels, especially with poor glucose management1. Possible causes of hypertriglyceridemia in patients with diabetes mellitus are increased hepatic VLDL production and/or defective removal of TG-rich lipoproteins (chylomicrons and VLDL).
A number of rodent models have been developed to explain the relationship between insulin actions and TG. It has been postulated that hyperinsulinemia associated with T2DM drives hepatic de novo TG synthesis via induction of sterol response element binding protein (SREBP)-1c5. Consistent with this hypothesis, lack of insulin action in the liver due to ablation of hepatic insulin receptors and Akt deficiency in mice prevented hepatic TG production, reduced liver TG secretion and led to low circulating TG levels6–8. According to this hypothesis, humans with poorly managed T1DM should show reduced hepatic TG production and plasma TG levels. Likewise, insulin therapy in T2DM should also drive greater liver TG production. However, the opposite has been found: In fact, plasma TG concentrations are increased in patients with T1DM9, 10. Moreover, treatment of T2DM patients with insulin results in systemic hyperinsulinemia, but reduced TG levels and decreased hepatic lipid accumulation11. Studies in diabetic rodents also conflict with conclusions derived from mice with genetic modifications in the insulin-signalling pathway. Viral destruction of pancreatic islet cells in mice leads to hypertriglyceridemia12 and re-feeding of insulin deficient mice increased lipogenic gene expression, suggesting that regulation of de novo synthesis is independent of insulin13. These data suggest that diabetic hypertriglyceridemia is not primarily caused by defective insulin signalling leading to increased hepatic fatty acid synthesis. The objective of this study was specifically to determine whether the effects of impaired insulin signalling on hepatic triglycerides production found with genetic modifications were also evident in mice with insulin deficiency.
In this report, we show that insulin deficiency in mice leads to increased plasma TG levels and defective removal of postprandial TG. This type of diabetic hypertriglyceridemia was not associated either with reduced mRNA levels of de novo TG synthesis-related genes or with decreased hepatic TG production. LpL mRNA was significantly reduced in skeletal muscle, white adipose tissue (WAT) and heart. Furthermore, LpL activity was decreased in skeletal muscle, brown adipose tissue (BAT) and heart. In addition, diabetes further increased plasma TG in animals with a genetic LpL defect. Our data support human studies and suggest that significant hypertriglyceridemia in insulin deficient diabetes is primarily due to changes in lipolysis and substrate return to the liver.signalling
Material and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
STZ-induced diabetes causes hypertriglyceridemia in mice
Two weeks after induction of insulin deficiency by intraperitoneal STZ administration, diabetic mice displayed marked hyperglycemia (6.66 ± 0.5 mmol/l vs. 25.55 ± 0.72 mmol/l) (Table 1A). Concomitantly, these mice had significantly elevated plasma TG levels (1.42 ± 0.09 versus 0.82 ± 0.03 mmol/l in non-diabetic mice). Hypertriglyceridemia persisted after 6 weeks of STZ diabetes (1.99 ± 0.18 versus 0.91 ± 0.06 mmol/l). In contrast, total plasma cholesterol levels and HDL cholesterol did not change at either time point. As expected, STZ-diabetic mice lost weight compared to non-diabetic control animals. Changes in TG were largely caused by increased VLDL TG (1.33 ± 0.09 mmol/l vs. 0.71 ± 0.02 mmol/l) (Table 1B). Plasma FFA were increased at both 3 and 6 weeks. Note that the baseline plasma FFA levels were higher in older mice. Plasma FFA showed a positive correlation with plasma TG levels in STZ-diabetic mice, whereas plasma FFA and TG did not significantly correlate with body weight (Supplement IA–C).
Table 1A.
Metabolic parameters in control and STZ-administered mice
| Parameter | 2nd week | 6th week | ||
|---|---|---|---|---|
|
| ||||
| non-diabetic control | STZ | non-diabetic control | STZ | |
| Glucose (mmol/l) | 6.66 ± 0.5 | 25.25 ± 0.72* | 9.44 ± 0.33 | 27.58 ± 1.28 * |
| Triglycerides (mmol/l) | 0.82 ± 0.03 | 1.42 ± 0.09* | 0.91 ± 0.06 | 1.99 ± 0.18† |
| Cholesterol (mmol/l) | 2.56 ± 0.13 | 2.79 ± 0.1 | 2.61 ± 0.1 | 2.69 ± 0.16 |
| FFA (mmol/l) | 0.88 ± 0.08 | 1.31 ± 0.17‡ | 1.58 ± 0.13 | 3.1 ± 0.34‡ |
| Body weight (g) | 27.5 ± 0.5 | 23.5 ± 0.5* | 27.2 ± 0.5 | 22.2 ± 0.8* |
| N | 24 | 22 | 14 | 12 |
p < 0.0001,
p < 0.001,
p<0.01 vs. non-diabetic control, Student’s t-test
Table 1B.
Cholesterol and triglycerides subfractions in control and STZ-administered mice
| Parameter | triglycerides | cholesterol | ||
|---|---|---|---|---|
|
| ||||
| non-diabetic control | STZ | non-diabetic control | STZ | |
| VLDL (mmol/l) | 0.71 ± 0.02 | 1.33 ± 0.09* | 0.36 ± 0.03 | 0.65 ± 0.05‡ |
| LDL (mmol/l) | 0.26 ± 0.02 | 0.27 ± 0.03 | 0.6 ± 0.08 | 0.62 ± 0.08 |
| HDL (mmol/l) | 0.20 ± 0.02 | 0.20 ± 0.03 | 1.50 ± 0.08 | 1.32 ± 0.1 |
p < 0.0001,
p<0.01 vs. non-diabetic control, Student’s t-test
Insulin deficiency does not change hepatic de novo lipogenesis and hepatic TG secretion
To assess whether insulin deficiency and circulating glucose levels affect hepatic TG production, we quantified TG secretion in mice treated with STZ and a lipase inhibitor, P407. STZ-induced diabetes did not alter TG secretion compared to non-diabetic wild type controls (Figure 1A). In line with this, hepatic gene expression of de novo lipogenesis genes (Fasn, Acc1, Dgat2, Scd1, Elovl6) showed no significant difference between the diabetic mice and healthy controls after 3 and 8 weeks (Figure 1B & Supplement ID). TG content in the liver was decreased (Figure 1C & Supplement IE), which was likely due to increased β-oxidation as indicated by increased gene expression of the rate limiting enzyme CPT1α (Figure 1B & Supplement ID). Thus, hypertriglyceridemia in insulin-deficient mice is not caused by increased de novo lipogenesis and hepatic TG secretion.
Figure 1. Insulin deficiency does not change hepatic de novo lipogenesis and hepatic TG secretion.

(A) TG secretion was measured in STZ-diabetic and non-diabetic C57BL/6 mice after i.p. injection of Poloxamer 407 (P407) (n = 5/group). (B) Hepatic gene expression of Fasn, Dgat2, Scd1, Acc1, Elovl6, Cpt1α and Acc2 was assessed using quantitative real-time PCR. Gene expression is expressed relative to fed non-diabetic control mice (n = 5/group). (C) Hepatic TG, FFA and cholesterol content (n = 5/group). *: p ≤ 0.05, **: p ≤ 0.01. Results are presented as mean ± SEM.
Hyperglycemia does not drive hypertriglyceridemia
Excess glucose in diabetes can be a substrate for hepatic de novo lipogenesis, thereby contributing to diabetic hypertriglyceridemia14. To study if hyperglycemia drives hypertriglyceridemia we treated control and STZ-diabetic mice with the glucose-lowering agent dapagliflozin for 3 weeks. Dapagliflozin selectively inhibits the sodium glucose co-transporter 2 in the kidney, thereby increasing urinary glucose excretion and decreasing plasma glucose levels without affecting insulin levels in STZ-diabetic mice as shown before15 and in our study (Figure 2D). Insulin levels in dapagliflozin treated control mice are reduced likely due to the reduction in circulating glucose levels. Despite a significant reduction in plasma glucose levels in STZ-diabetic mice neither plasma TG levels (Figure 2A & 2B) nor TG secretion (Figure 2C) decreased. Similar to the pharmacological approach, an antisense oligonucleotide against SGLT2 did not decrease plasma TG levels in both control and STZ-diabetic mice over the course of four weeks (Supplement II). These studies suggest that diabetic hypertriglyceridemia is not driven by excess glucose.
Figure 2. Hyperglycemia does not drive hypertriglyceridemia.

(A) STZ-diabetic and control C57BL/6 mice were treated with the SGLT2 inhibitor dapagliflozin or vehicle and plasma glucose was measured at indicated time points (n = 10/group). ***: p ≤ 0.001 vs. STZ + vehicle. (B) 4 hour fasting plasma TG levels over time (n = 10/group). ***: p ≤ 0.001 vs. non-diabetic controls. (C) Hepatic TG secretion was measured by i.p. injection of Poloxamer 407 (P407) (n = 5/group). (D) Plasma insulin levels after 4 hour fasting (n = 10/group). *: p ≤ 0.05, ***: p ≤ 0.001 vs. control + vehicle. Results are presented as mean ± SEM.
Insulin deficiency leads to increased postprandial lipemia
As hepatic de novo lipogenesis and TG secretion did not appear to contribute substantially to hypertriglyceridemia, we explored whether defective peripheral lipolysis is the culprit for increased plasma TG levels. After an olive oil gavage, STZ-diabetic mice displayed markedly higher plasma TG levels followed by delayed clearance of plasma TG compared to that of controls (Figure 3A). LpL mRNA levels were significantly reduced in skeletal muscle, WAT and heart after 3 weeks of diabetes (Figure 3B). At 8 weeks skeletal muscle LpL was reduced and tended to decrease in the WAT and heart of these mice (Supplement IIIA). However, mRNA levels of LpL do not always reflect LpL activity as LpL is regulated at transcriptional, translational and posttranslational levels15, 16. Heparin-releasable LpL activity in the heart, BAT and skeletal muscle was significantly reduced in STZ-diabetic mice (Figure 3C); there was no change in heparin-releasable activity of hepatic lipase and hepatic lipase mRNA in the liver (Supplement IIIB & IV). Hepatic mRNA expression of the LpL regulating proteins angiopoietin-like 3, 4 and 8 (Angptl 3, 4, and 8) and apolipoproteins apoA-V, ApoB and ApoC-III were not significantly altered at both 3 and 8 weeks. We also did not detect significant changes of Angptl4, lipase maturation factor 1 (LMF1) and glycosylphosphatidylinositol-anchored high-density binding protein 1 (GPIHBP1) in the skeletal muscle (Supplement Figure IV). However, we did find an increase in apoC-III in the plasma of STZ-diabetic mice (Figure 3D). LpL activity in postheparin plasma did not correlate with the changes in mRNA levels or heparin-released muscle LpL activity (Supplement IIIC). Taken together, insulin-deficient diabetes caused a marked reduction in postprandial TG removal associated with reduced expression and activity of skeletal muscle LpL.
Figure 3. Increased postprandial lipemia with insulin deficiency.

(A) TG turnover in mice after gavage of 10 ml/kg BW olive oil. Plasma TG levels were measured after 2,4 & 6 hours. (B) Lpl mRNA analysis by quantitative PCR in skeletal muscle, WAT and heart at 3 weeks. (C) Heparin-releasable (HR) LpL activity from freshly isolated heart, BAT and skeletal muscle (SM) using a radiolabeled triglyceride substrate. (D) Immunoblot of plasma for ApoC-III (E) Gene expression of PPARα, PPARδ, PPARγ and CD36 in the skeletal muscle after three weeks of STZ-diabetes using quantitative real-time PCR (n = 5/group). *: p ≤ 0.05, **: p ≤ 0.01, ***: p ≤ 0.001 Results are presented as mean ± SEM.
LpL deficiency is associated with decreased expression of PPARα and PPARδ
To explore how insulin deficiency affects LpL expression and activity we studied LpL regulating factors. LpL expression in several tissues has been linked to peroxisome proliferator-activated receptor (PPAR) expression and activation17–19. In our study, reduced LpL mRNA levels were associated with markedly lower PPARα mRNA in the skeletal muscle of diabetic mice after 3 weeks (Figure 3E). PPARδ mRNA was also decreased, while PPARγ was unchanged. We detected similar expression patterns of PPARs in the skeletal muscle after 8 weeks of STZ-diabetes (Supplement V).
LpL expression in the skeletal muscle modulates diabetic dyslipidemia
Fasted wild type mice have a rapid turnover of plasma TG. To study a more human-like model with less robust TG lipolysis, we induced diabetes in heterozygous LpL knockout mice (Lpl+/−). Our laboratory has previously reported that Lpl+/− mice on the ApoB overexpressing transgenic background have marked hypertriglyceridemia when treated with STZ20. STZ-treatment of Lpl+/− mice led to markedly increased plasma TG (2.45 ± 0.26 mmol/l) compared to non-diabetic Lpl+/− mice (1.389 ± 0.316 mmol/l) (Figure 4A & B). Similar to wild type mice, TG secretion was not altered in STZ-diabetic Lpl+/− mice after P407 treatment and hepatic TG content was reduced (Figure 4C & D). Again, after olive oil gavage STZ-diabetic Lpl+/− mice displayed a significant impairment of TG clearance (Figure 4E). The highest postprandial TG levels in STZ-treated mice were identical to those of diabetic wild type mice, but the rate of reduction between 4–6 hours was reduced compared to diabetic wild type mice (4.2 mmol/l/hour versus 7.7 mmol/l/hour).
Figure 4. STZ-diabetic heterozygous Lpl+/− mice display increased plasma TG and postprandial hypertriglyceridemia.
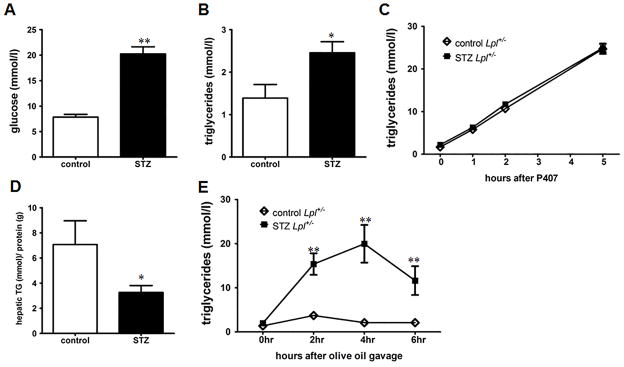
(A) 4 hour fasting glucose and (B) TG levels in STZ-diabetic and non-diabetic Lpl+/− mice at 4 weeks (n = 6–10/group). (C) Hepatic TG secretion in Lpl+/− mice was measured by i.p. injection of Poloxamer 407 (P407) (n = 6–10/group). (D) Hepatic TG content in STZ-diabetic and non-diabetic Lpl+/− mice (n = 5–7/group). (E) TG turnover was quantified in mice after gavage of 10 ml/kg BW olive oil. Plasma TG levels were measured after 2,4 & 6 hours (n = 5–7/group). *: p ≤ 0.05, **: p ≤ 0.01 and vs. non-diabetic Lpl+/− mice. Results are presented as mean ± SEM.
These data suggest that impaired TG clearance is the major cause for hypertriglyceridemia in STZ-diabetic mice. To test whether LpL overexpression would alter these diabetes-induced TG changes, we studied postprandial lipemia in mice expressing human LpL primarily in skeletal muscle; the transgene is denoted MCK-LpL. We note that the MCK-LpL transgene also leads to a small amount of LpL expression in the heart21. STZ-diabetic MCK-LpL/Lpl+/− mice had significantly lower fasting TG levels compared to STZ-diabetic Lpl+/− mice (Figure 5A). In addition, TG clearance after olive oil gavage was significantly improved in STZ-diabetic MCK-LpL/Lpl+/− mice compared to STZ-diabetic Lpl+/− mice (Figure 5B). Therefore, LpL overexpression corrects diabetic dyslipidemia in insulin deficient mice.
Figure 5. LpL overexpression in the skeletal muscle corrects diabetic dyslipidemia.
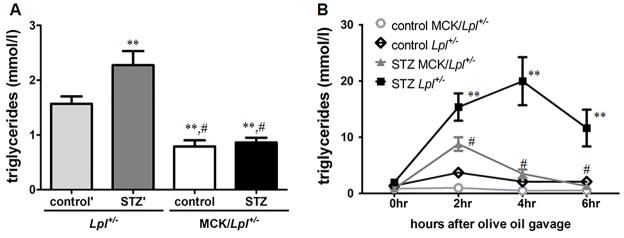
(A) 4 hour fasting plasma TG levels in STZ-diabetic and non-diabetic Lpl+/− and MCK/Lpl+/− mice (n ≥ 10/group). : p ≤ 0.01 vs. non-diabetic Lpl+/− control mice, #: p ≤ 0.001 vs. STZ-diabetic Lpl+/− mice. (B) TG turnover was quantified in mice after gavage of 10 ml/kg BW olive oil. Plasma TG levels were measured after 2,4 & 6 hours (n = 5–7/group). **: p ≤ 0.01 vs. non-diabetic Lpl+/− control mice, #: p ≤ 0.01 vs. STZ-diabetic Lpl+/− mice. Results are presented as mean ± SEM.
Discussion
In this report, we show that hypertriglyceridemia in insulin deficient diabetic mice is primarily due to changes in peripheral lipolysis and not due to changes in hepatic insulin signalling and hepatic TG secretion. Similar to humans with either T1DM or T2DM, STZ-induced diabetes in C57BL/6 mice led to a significant increase in plasma TG levels. This observation was similar to that described by others22, 23. The primary lipoprotein abnormality in human diabetes is an increase in VLDL, which was reproduced in our STZ-treated mice. There are two possible causes of diabetes-associated increase in TGs: elevated secretion of TGs from the liver and/or decreased clearance of TGs24, 25. 1) Hepatic overproduction of TGs has been attributed to compromised ApoB degradation due to the loss of insulin action, which eventually leads to increased VLDL assembly and secretion26, 27. Loss of insulin action also increases FFA flux to the liver and increases hepatic lipogenesis10. In addition, it has been suggested that hyperinsulinemia can drive de novo lipogenesis in the presence of increased plasma glucose levels and hepatic insulin resistance5. It should be noted that most human kinetic studies on TG production are carried out by injection of glycerol which cannot separate de novo lipogenesis from substrate driven TG production28–31. Therefore these studies cannot determine the contribution of hepatic de novo lipogenesis and substrate driven TG production to hypertriglyceridemia. 2) Diabetic humans exhibit a marked defect in the clearance of postprandial lipemia32, 33. These findings are in accordance with reports of decreased LpL activity in postheparin plasma34 and in skeletal muscle biopsies of diabetic patients35, 36. Accordingly, Taskinen et al. have shown that >20% of the hypertriglyceridemia in obese men is associated with increases in VLDL1 secretion but almost 50% is related to impaired fractional catabolic rate28. It is not clear, however, why only some patients develop hypertriglyceridemia with diabetes. For instance, in the ACCORD trial the average triglycerides level in all diabetic patients was 1.829 mmol/l (162 mg/dl), a level below the clinical threshold of hypertriglyceridemia (2.258 mmol/l (200 mg/dl))37. Therefore, it is likely that the minority of patients who develop severe hypertriglyceridemia have a defect in lipolysis.
Our objective was specifically to determine whether the effects of insulin signalling on hepatic triglyceride production found with genetic modifications were evident in mice with insulin deficiency. In our mouse model of STZ-induced diabetes, insulin deficiency did not lead to a significant reduction in hepatic TG secretion nor did the lack of insulin actions reduce expression of genes involved in de novo lipogenesis. As reported by others13, 23 hepatic TG content was significantly reduced and plasma FFA levels were increased in STZ-diabetic mice. The reduced hepatic TG content could be partially due to increased fatty acid oxidation as mRNA levels of the rate limiting enzyme of β-oxidation CPT1-α were increased. Jourdan et al. have also noted an increase in CPT1-α and reported increased β-oxidation of palmitate in the livers of STZ-treated mice 23. It is possible that without this increase in oxidation, more FFA would be converted to TG leading to greater hepatic TG secretion. Taken together, our data do not support the hypothesis that insulin deficiency reduces TG production and suggests that the hypertriglyceridemia in STZ-diabetic mice is primarily due to a catabolic defect.
No model of diabetes is perfect but the STZ model used in our studies is the one recommended by the NIH Animal Models of Diabetic Complications Consortium (AMDCC)38. The low dose STZ leads to partial insulin deficiency that causes hyperglycemia but allows sufficient insulin to prevent ketoacidosis and early death. This model does not lead to obesity and peripheral insulin resistance as would be seen in T2DM. In fact, STZ-treated mice have reduced adipose stores and the effects of insulin deficiency in obese animals are likely to be different. Specifically, greater adipose would be expected to lead to greater FFA release and high plasma FFA levels that are likely to drive greater liver production of TG. Nonetheless, we believe that STZ-treatment is more likely to reflect human disease than models that lead to non-physiologic modification of gene expression.
Independent of insulin action on hypertriglyceridemia, glucose also generates a signal that may modulate TG production and TG clearance39, 40. Glucose controls the expression of key genes involved in energy metabolism through the carbohydrate response element binding protein (CHREBP). CHREBP binds to the promoter region of glycolysis- (e.g. pyruvate kinase) and lipogenesis-associated genes40 and may drive diabetic hypertriglyceridemia by increasing transcription of de novo lipogenesis genes41. Glucose may also exert direct effects on TG clearance. Hyperglycemic conditions downregulate LpL activity in humans42, 43 and in cultured human adipocytes44. This suggests that hyperglycemia per se contributes to the decreased LpL activity and subsequent hypertriglyceridemia in poorly controlled diabetes. Until recently it has been difficult to separate the effects of hyperglycemia versus defective insulin actions on diabetic hypertriglyceridemia because glucose-reducing interventions were achieved by either insulin administration or by improved insulin sensitivity. By inhibiting SGLT2 in the kidney, which leads to increase glucose output in the urine, we were able to reduce plasma glucose levels without affecting insulin levels in STZ-diabetic mice, although we cannot rule out changes in insulin signalling45. Both the pharmacological inhibition by dapagliflozin and antisense oligonucleotide mediated inhibition of SGLT2 significantly reduced plasma glucose levels in STZ-diabetic mice, but did not change plasma TG levels. More importantly, hepatic TG secretion was similar in STZ-diabetic mice with normalized glucose levels making it unlikely that increased glucose levels contribute substantially to hypertriglyceridemia in STZ-diabetic mice.
Postprandial lipemia is markedly increased in humans with diabetes46–48. Accordingly, in our study STZ-diabetic mice displayed increased plasma TG levels and delayed clearance after gavage with olive oil. LpL is the rate-limiting enzyme for the hydrolysis of TGs in circulating chylomicrons and VLDL. LpL-mediated hydrolysis products, fatty acids and monoacylglycerol, are taken up by the tissues and stored as neutral lipids in WAT, or oxidized in skeletal and cardiac muscle15, 16. LpL is regulated at the transcriptional, post-transcriptional, and post-translational level in a tissue-specific manner49. Food-intake decreases LpL activity in skeletal muscle and increases it in WAT. Fasting increases LpL activity in the skeletal muscle and heart and decreases LpL in the WAT. We detected a significant decrease of LpL gene expression in the skeletal muscle, heart and WAT after 3 and 8 weeks of STZ-diabetes. Accordingly, heparin-releasable LpL activity in the skeletal muscle, heart and BAT of STZ-diabetic mice was significantly reduced; all these tissues are major sites for LpL synthesis16, 50. We were unable to study heparin-releasable LpL activity in WAT since STZ-diabetic mice were lipodystrophic, but the reduction in total adipose LpL, as we have shown before, likely contributes to the defective lipolysis51. Therefore these results show that decreased LpL activity in BAT, skeletal muscle and heart correlates with hypertriglyceridemia in STZ-diabetic mice. Increased apoC-III that we detected in plasma might also affect lipolysis or the changes in lipoproteins associated with LpL deficiency and diabetes might have caused a secondary increase in apoC-III. Perhaps this ApoC-III increase is also responsible for the defect in LDL uptake previously reported in diabetic LDL receptor knockout mice52.
LpL activity has been studied previously in diabetic humans and rodents. In patients with T2DM LpL mRNA, protein expression and activity were significantly decreased in skeletal muscle35, 36. Conflicting results have been reported in studies of LpL activity in the heart and the skeletal muscles in STZ-treated rodents: Both decreased53 and increased54 activity levels have been observed. The reported differences might relate to the length as well as severity of diabetes and methodological differences, e.g. measurement of LpL activity in homogenized tissue versus heparin-releasable LpL activity and data obtained from mice versus rats. In fact, although we found reduced heparin-releasable LpL activity in the muscles of diabetic mice, LpL activity measurements of tissue homogenates were not significantly different (data not shown).
In contrast to the decreased LpL activity in the skeletal muscle, postheparin lipase activity in the plasma did not differ between our STZ-diabetic and control mice (2.4 ± 0.16 μm FFA/ml/h versus 2.41 ± 0.10 μm FFA/ml/h respectively). Similarly, Pollare et al. did not detect differences in postheparin LpL activity in the plasma of T2DM patients but reported decreased LpL activity in the skeletal muscle of these patients36. It is questionable whether total postheparin plasma LpL activity accurately reflects changes in total lipolytic activity.
In order to explain the reduced levels of LpL we tested gene expression levels of two well-known transcriptional regulators of LpL, PPARα and PPARδ. Schoonjans et al. showed that the expression of LpL is regulated by PPARα (and PPARγ), which interact with a response element in the LpL promoter17. PPARδ agonist treatment increases LpL mRNA and protein levels in skeletal muscle cells18; PPARδ also increases expression of the LpL inhibitor Angplt455, which was not significantly decreased in our studies. A number of clinical studies suggested that the TG-lowering action of PPARα agonists is associated with an increase in LpL activity56–58. Others have shown that PPARα and PPARγ agonists enhance LpL expression and activity in human macrophages19. Considering these studies, we hypothesized that decreased LpL expression in the skeletal muscle of STZ-diabetic mice was due to a decreased transcription of PPARα. Insulin enhances the transcriptional activity of PPARα in vitro59. We found reduced expression of PPARα and PPARδ with insulin deficiency in skeletal muscle of STZ-diabetic mice. We hypothesize that the lack of insulin leads to a decrease primarily of PPARα followed by a decrease of LpL expression in the skeletal muscle. Surprisingly, we noted reduced CD36 expression in skeletal muscle of diabetic mice. While one might expect that myocytes would shift to greater fatty acid oxidation with insulin deficiency, we suspect that muscle metabolism in our animals was heavily dependent on glucose, but with glucose uptake primarily via non-insulin- and non-GLUT4-dependent pathways.
To study if changes in LpL expression would actually modify diabetes-induced hypertriglyceridemia we studied mice with a heterozygous deletion of the LpL gene (as homozygous LpL knockout mice are not viable). Lpl+/− mice exhibited marked hypertriglyceridemia after induction of STZ-diabetes. More importantly, TG secretion was again not significantly different in STZ-diabetic Lpl+/− mice but TG clearance was delayed after gavage with olive oil. Similar to our diabetic Lpl+/− mice, human diabetic carriers of dysfunctional LpL alleles are at risk for severe lipemia60. We hypothesized that increasing LpL expression would ameliorate diabetic hypertriglyceridemia in our animal model. Constitutive skeletal muscle-specific expression of human LpL eliminated hypertriglyceridemia in the STZ-diabetic Lpl+/− mice and TG clearance after olive oil gavage was reduced to almost normal non-diabetic levels. We presume that the constitutive expression of LpL increased lipolysis and VLDL remnant removal. Similarly, Shimada et al.61 reported that diabetes-mediated increases in TG were eliminated by overexpression of LpL in STZ-diabetic C57BL/6 mice. Thus, a major regulator of circulating TG in diabetes is LpL action regardless of changes in liver TG production.
In conclusion, hypertriglyceridemia in insulin deficient mice is primarily caused by a defect in peripheral lipolysis and not by an increase in hepatic TG secretion as suggested by mouse models of impaired hepatic insulin signalling. signalling. Most humans with diabetes do not have marked hypertriglyceridemia. However humans, like mice, that have a defect in LpL regulation are most likely to develop severe hypertriglyceridemia with diabetes60, 62,63. On the other hand, humans with robust LpL activity are likely to be protected from hypertriglyceridemia regardless of their degree of insulin deficiency or resistance. Therefore, methods to augment peripheral TG lipolysis are likely to be most efficacious in the treatment of diabetes-induced hypertriglyceridemia.
Supplementary Material
Significance.
In this report, we show that insulin deficiency in mice leads to increased plasma TG levels and defective removal of postprandial TG. Our data are most compatible with that in humans and suggests that significant hypertriglyceridemia in insulin deficient diabetes is primarily due to changes in lipolysis and not changes in hepatic insulin signalling.
Acknowledgments
The authors thank Yunying Hu for technical assistance.
Sources of funding: F.W. is supported by a postdoctoral fellowship of the German Research Foundation (DFG). P.R.N. is supported by a NIH Pathway to Independence Award (1K99HL122505-01). C.M.T. is supported by a predoctoral fellowship from the American Heart Association, Heritage Affiliate. This work is supported in part by NIH grants K99 HL11285 (K.D.), DK095684 and HL45095 and by the DiaComp Pilot and Feasibility Award #25034-37 (to I.J.G)
Nonstandard Abbreviations
- ASO
antisense oligonucleotide
- BAT
brown adipose tissue
- CHREBP
carbohydrate response element binding protein
- CVD
cardiovascular disease
- FFA
free fatty acid
- LpL
lipoprotein lipase
- PPARα/γ/δ
Peroxisome proliferator-activated receptor α/γ/δ
- SGLT2
sodium glucose co-transporter 2
- SREBP-1c
sterol response element binding protein-1c
- STZ
streptozotocin
- T1DM
type 1 diabetes mellitus
- T2DM
type 2 diabetes mellitus
- TG
Triglycerides
- WAT
white adipose tissue
Footnotes
Disclosures: None
References
- 1.Miller M, Stone NJ, Ballantyne C, Bittner V, Criqui MH, Ginsberg HN, Goldberg AC, Howard WJ, Jacobson MS, Kris-Etherton PM, Lennie TA, Levi M, Mazzone T, Pennathur S American Heart Association Clinical Lipidology T, Prevention Committee of the Council on Nutrition PA, Metabolism, Council on Arteriosclerosis T, Vascular B, Council on Cardiovascular N, Council on the Kidney in Cardiovascular D. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123:2292–2333. doi: 10.1161/CIR.0b013e3182160726. [DOI] [PubMed] [Google Scholar]
- 2.Goldberg IJ, Eckel RH, McPherson R. Triglycerides and heart disease: still a hypothesis? Arterioscler Thromb Vasc Biol. 2011;31:1716–1725. doi: 10.1161/ATVBAHA.111.226100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mooradian AD. Dyslipidemia in type 2 diabetes mellitus. Nat Clin Pract Endocrinol Metab. 2009;5:150–159. doi: 10.1038/ncpendmet1066. [DOI] [PubMed] [Google Scholar]
- 4.Resnick HE, Foster GL, Bardsley J, Ratner RE. Achievement of American Diabetes Association clinical practice recommendations among U.S. adults with diabetes, 1999–2002: the National Health and Nutrition Examination Survey. Diabetes Care. 2006;29:531–537. doi: 10.2337/diacare.29.03.06.dc05-1254. [DOI] [PubMed] [Google Scholar]
- 5.Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. 2008;7:95–96. doi: 10.1016/j.cmet.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 6.Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, Magnuson MA, Kahn CR. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Molecular cell. 2000;6:87–97. [PubMed] [Google Scholar]
- 7.Biddinger SB, Hernandez-Ono A, Rask-Madsen C, Haas JT, Aleman JO, Suzuki R, Scapa EF, Agarwal C, Carey MC, Stephanopoulos G, Cohen DE, King GL, Ginsberg HN, Kahn CR. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab. 2008;7:125–134. doi: 10.1016/j.cmet.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leavens KF, Easton RM, Shulman GI, Previs SF, Birnbaum MJ. Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metab. 2009;10:405–418. doi: 10.1016/j.cmet.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Purnell JQ, Hokanson JE, Marcovina SM, Steffes MW, Cleary PA, Brunzell JD. Effect of excessive weight gain with intensive therapy of type 1 diabetes on lipid levels and blood pressure: results from the DCCT. Diabetes Control and Complications Trial. JAMA. 1998;280:140–146. doi: 10.1001/jama.280.2.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Subramanian S, Chait A. Hypertriglyceridemia secondary to obesity and diabetes. Biochim Biophys Acta. 1821:819–825. doi: 10.1016/j.bbalip.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 11.Juurinen L, Tiikkainen M, Hakkinen AM, Hakkarainen A, Yki-Jarvinen H. Effects of insulin therapy on liver fat content and hepatic insulin sensitivity in patients with type 2 diabetes. Am J Physiol Endocrinol Metab. 2007;292:E829–835. doi: 10.1152/ajpendo.00133.2006. [DOI] [PubMed] [Google Scholar]
- 12.Renard CB, Kramer F, Johansson F, Lamharzi N, Tannock LR, von Herrath MG, Chait A, Bornfeldt KE. Diabetes and diabetes-associated lipid abnormalities have distinct effects on initiation and progression of atherosclerotic lesions. J Clin Invest. 2004;114:659–668. doi: 10.1172/JCI17867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsuzaka T, Shimano H, Yahagi N, Amemiya-Kudo M, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Tomita S, Sekiya M, Hasty A, Nakagawa Y, Sone H, Toyoshima H, Ishibashi S, Osuga J, Yamada N. Insulin-independent induction of sterol regulatory element-binding protein-1c expression in the livers of streptozotocin-treated mice. Diabetes. 2004;53:560–569. doi: 10.2337/diabetes.53.3.560. [DOI] [PubMed] [Google Scholar]
- 14.Foufelle F, Ferre P. New perspectives in the regulation of hepatic glycolytic and lipogenic genes by insulin and glucose: a role for the transcription factor sterol regulatory element binding protein-1c. Biochem J. 2002;366:377–391. doi: 10.1042/BJ20020430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Merkel M, Eckel RH, Goldberg IJ. Lipoprotein lipase: genetics, lipid uptake, and regulation. J Lipid Res. 2002;43:1997–2006. doi: 10.1194/jlr.r200015-jlr200. [DOI] [PubMed] [Google Scholar]
- 16.Wang H, Eckel RH. Lipoprotein lipase: from gene to obesity. Am J Physiol Endocrinol Metab. 2009;297:E271–288. doi: 10.1152/ajpendo.90920.2008. [DOI] [PubMed] [Google Scholar]
- 17.Schoonjans K, Peinado-Onsurbe J, Lefebvre AM, Heyman RA, Briggs M, Deeb S, Staels B, Auwerx J. PPARalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J. 1996;15:5336–5348. [PMC free article] [PubMed] [Google Scholar]
- 18.Dressel U, Allen TL, Pippal JB, Rohde PR, Lau P, Muscat GE. The peroxisome proliferator-activated receptor beta/delta agonist, GW501516, regulates the expression of genes involved in lipid catabolism and energy uncoupling in skeletal muscle cells. Mol Endocrinol. 2003;17:2477–2493. doi: 10.1210/me.2003-0151. [DOI] [PubMed] [Google Scholar]
- 19.Li L, Beauchamp MC, Renier G. Peroxisome proliferator-activated receptor alpha and gamma agonists upregulate human macrophage lipoprotein lipase expression. Atherosclerosis. 2002;165:101–110. doi: 10.1016/s0021-9150(02)00203-4. [DOI] [PubMed] [Google Scholar]
- 20.Kako Y, Masse M, Huang LS, Tall AR, Goldberg IJ. Lipoprotein lipase deficiency and CETP in streptozotocin-treated apoB-expressing mice. J Lipid Res. 2002;43:872–877. [PubMed] [Google Scholar]
- 21.Levak-Frank S, Radner H, Walsh A, Stollberger R, Knipping G, Hoefler G, Sattler W, Weinstock PH, Breslow JL, Zechner R. Muscle-specific overexpression of lipoprotein lipase causes a severe myopathy characterized by proliferation of mitochondria and peroxisomes in transgenic mice. J Clin Invest. 1995;96:976–986. doi: 10.1172/JCI118145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bishop JR, Foley E, Lawrence R, Esko JD. Insulin-dependent diabetes mellitus in mice does not alter liver heparan sulfate. J Biol Chem. 2010;285:14658–14662. doi: 10.1074/jbc.M110.112391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jourdan T, Djaouti L, Demizieux L, Gresti J, Verges B, Degrace P. Liver carbohydrate and lipid metabolism of insulin-deficient mice is altered by trans-10, cis-12 conjugated linoleic acid. J Nutr. 2009;139:1901–1907. doi: 10.3945/jn.109.111062. [DOI] [PubMed] [Google Scholar]
- 24.Reaven EP, Reaven GM. Mechanisms for development of diabetic hypertriglyceridemia in streptozotocin-treated rats. Effect of diet and duration of insulin deficiency. J Clin Invest. 1974;54:1167–1178. doi: 10.1172/JCI107860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Looney P, Irwin D, Briscoe P, Vahouny GV. Lipoprotein composition as a component in the lipoprotein clearance defect in experimental diabetes. J Biol Chem. 1985;260:428–432. [PubMed] [Google Scholar]
- 26.Malmstrom R, Packard CJ, Caslake M, Bedford D, Stewart P, Yki-Jarvinen H, Shepherd J, Taskinen MR. Defective regulation of triglyceride metabolism by insulin in the liver in NIDDM. Diabetologia. 1997;40:454–462. doi: 10.1007/s001250050700. [DOI] [PubMed] [Google Scholar]
- 27.Adiels M, Olofsson SO, Taskinen MR, Boren J. Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28:1225–1236. doi: 10.1161/ATVBAHA.107.160192. [DOI] [PubMed] [Google Scholar]
- 28.Taskinen MR, Adiels M, Westerbacka J, Soderlund S, Kahri J, Lundbom N, Lundbom J, Hakkarainen A, Olofsson SO, Orho-Melander M, Boren J. Dual metabolic defects are required to produce hypertriglyceridemia in obese subjects. Arterioscler Thromb Vasc Biol. 2011;31:2144–2150. doi: 10.1161/ATVBAHA.111.224808. [DOI] [PubMed] [Google Scholar]
- 29.Shojaee-Moradie F, Ma Y, Lou S, Hovorka R, Umpleby AM. Prandial hypertriglyceridemia in metabolic syndrome is due to an overproduction of both chylomicron and VLDL triacylglycerol. Diabetes. 2013;62:4063–4069. doi: 10.2337/db13-0935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adiels M, Taskinen MR, Packard C, Caslake MJ, Soro-Paavonen A, Westerbacka J, Vehkavaara S, Hakkinen A, Olofsson SO, Yki-Jarvinen H, Boren J. Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia. 2006;49:755–765. doi: 10.1007/s00125-005-0125-z. [DOI] [PubMed] [Google Scholar]
- 31.Siler SQ, Neese RA, Parks EJ, Hellerstein MK. VLDL-triglyceride production after alcohol ingestion, studied using [2–13C1] glycerol. J Lipid Res. 1998;39:2319–2328. [PubMed] [Google Scholar]
- 32.Lewis GF, O’Meara NM, Soltys PA, Blackman JD, Iverius PH, Pugh WL, Getz GS, Polonsky KS. Fasting hypertriglyceridemia in noninsulin-dependent diabetes mellitus is an important predictor of postprandial lipid and lipoprotein abnormalities. J Clin Endocrinol Metab. 1991;72:934–944. doi: 10.1210/jcem-72-4-934. [DOI] [PubMed] [Google Scholar]
- 33.De Man FH, Cabezas MC, Van Barlingen HH, Erkelens DW, de Bruin TW. Triglyceride-rich lipoproteins in non-insulin-dependent diabetes mellitus: post-prandial metabolism and relation to premature atherosclerosis. Eur J Clin Invest. 1996;26:89–108. doi: 10.1046/j.1365-2362.1996.114256.x. [DOI] [PubMed] [Google Scholar]
- 34.Brunzell JD, Porte D, Jr, Bierman EL. Reversible abnormalities in postheparin lipolytic activity during the late phase of release in diabetes mellitus (postheparin lipolytic activity in diabetes) Metabolism. 1975;24:1123–1137. doi: 10.1016/0026-0495(75)90149-3. [DOI] [PubMed] [Google Scholar]
- 35.Morino K, Petersen KF, Sono S, Choi CS, Samuel VT, Lin A, Gallo A, Zhao H, Kashiwagi A, Goldberg IJ, Wang H, Eckel RH, Maegawa H, Shulman GI. Regulation of mitochondrial biogenesis by lipoprotein lipase in muscle of insulin-resistant offspring of parents with type 2 diabetes. Diabetes. 2012;61:877–887. doi: 10.2337/db11-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pollare T, Vessby B, Lithell H. Lipoprotein lipase activity in skeletal muscle is related to insulin sensitivity. Arterioscler Thromb. 1991;11:1192–1203. doi: 10.1161/01.atv.11.5.1192. [DOI] [PubMed] [Google Scholar]
- 37.Group AS, Ginsberg HN, Elam MB, Lovato LC, Crouse JR, 3rd, Leiter LA, Linz P, Friedewald WT, Buse JB, Gerstein HC, Probstfield J, Grimm RH, Ismail-Beigi F, Bigger JT, Goff DC, Jr, Cushman WC, Simons-Morton DG, Byington RP. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362:1563–1574. doi: 10.1056/NEJMoa1001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brosius F. Animal Models of Diabetic Complications Consortium. [accessed Sepember 9, 2014];Low-Dose Streptozotocin Induction Protocol (Mouse) 2003 www.diacomp.org/shared/showFile.aspx?doctypeid=3&docid=19.
- 39.Towle HC. Glucose as a regulator of eukaryotic gene transcription. Trends Endocrinol Metab. 2005;16:489–494. doi: 10.1016/j.tem.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 40.Poupeau A, Postic C. Cross-regulation of hepatic glucose metabolism via ChREBP and nuclear receptors. Biochim Biophys Acta. 2011;1812:995–1006. doi: 10.1016/j.bbadis.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 41.Ferre P, Foufelle F. Hepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes, obesity & metabolism. 2010;12 (Suppl 2):83–92. doi: 10.1111/j.1463-1326.2010.01275.x. [DOI] [PubMed] [Google Scholar]
- 42.Jindrichova E, Kratochvilova S, Kovar J. Glucose administration downregulates lipoprotein lipase activity in vivo: a study using repeated intravenous fat tolerance test. Physiological research / Academia Scientiarum Bohemoslovaca. 2007;56:175–181. doi: 10.33549/physiolres.930953. [DOI] [PubMed] [Google Scholar]
- 43.Kovar J, Fejfarova V, Pelikanova T, Poledne R. Hyperglycemia downregulates total lipoprotein lipase activity in humans. Physiological research / Academia Scientiarum Bohemoslovaca. 2004;53:61–68. [PubMed] [Google Scholar]
- 44.Kern PA, Mandic A, Eckel RH. Regulation of lipoprotein lipase by glucose in primary cultures of isolated human adipocytes. Relevance to hypertriglyceridemia of diabetes. Diabetes. 1987;36:1238–1245. doi: 10.2337/diab.36.11.1238. [DOI] [PubMed] [Google Scholar]
- 45.Merovci A, Solis-Herrera C, Daniele G, Eldor R, Fiorentino TV, Tripathy D, Xiong J, Perez Z, Norton L, Abdul-Ghani MA, DeFronzo RA. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest. 2014;124:509–514. doi: 10.1172/JCI70704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen YD, Coulston AM, Zhou MY, Hollenbeck CB, Reaven GM. Why do low-fat high-carbohydrate diets accentuate postprandial lipemia in patients with NIDDM? Diabetes Care. 1995;18:10–16. doi: 10.2337/diacare.18.1.10. [DOI] [PubMed] [Google Scholar]
- 47.Ginsberg HN, Illingworth DR. Postprandial dyslipidemia: an atherogenic disorder common in patients with diabetes mellitus. Am J Cardiol. 2001;88:9H–15H. doi: 10.1016/s0002-9149(01)01831-8. [DOI] [PubMed] [Google Scholar]
- 48.Evans M, Anderson RA, Graham J, Ellis GR, Morris K, Davies S, Jackson SK, Lewis MJ, Frenneaux MP, Rees A. Ciprofibrate therapy improves endothelial function and reduces postprandial lipemia and oxidative stress in type 2 diabetes mellitus. Circulation. 2000;101:1773–1779. doi: 10.1161/01.cir.101.15.1773. [DOI] [PubMed] [Google Scholar]
- 49.Goldberg IJ, Merkel M. Lipoprotein lipase: physiology, biochemistry, and molecular biology. Frontiers in bioscience. 2001;6:D388–405. doi: 10.2741/goldberg. [DOI] [PubMed] [Google Scholar]
- 50.Kersten S. Physiological regulation of lipoprotein lipase. Biochim Biophys Acta. 2014;1841:919–933. doi: 10.1016/j.bbalip.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 51.Garcia-Arcos I, Hiyama Y, Drosatos K, Bharadwaj KG, Hu Y, Son NH, O’Byrne SM, Chang CL, Deckelbaum RJ, Takahashi M, Westerterp M, Obunike JC, Jiang H, Yagyu H, Blaner WS, Goldberg IJ. Adipose-specific lipoprotein lipase deficiency more profoundly affects brown than white fat biology. J Biol Chem. 2013;288:14046–14058. doi: 10.1074/jbc.M113.469270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ebara T, Conde K, Kako Y, Liu Y, Xu Y, Ramakrishnan R, Goldberg IJ, Shachter NS. Delayed catabolism of apoB-48 lipoproteins due to decreased heparan sulfate proteoglycan production in diabetic mice. J Clin Invest. 2000;105:1807–1818. doi: 10.1172/JCI8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aktin E, Meng HC. Release of clearing factor lipase (lipoprotein lipase) in vivo and from isolated perfused hearts of alloxan diabetic rats. Diabetes. 1972;21:149–156. doi: 10.2337/diab.21.3.149. [DOI] [PubMed] [Google Scholar]
- 54.Rauramaa R, Kuusela P, Hietanen E. Adipose, muscle and lung tissue lipoprotein lipase activities in young streptozotocin treated rats. Horm Metab Res. 1980;12:591–595. doi: 10.1055/s-2007-999207. [DOI] [PubMed] [Google Scholar]
- 55.Staiger H, Haas C, Machann J, Werner R, Weisser M, Schick F, Machicao F, Stefan N, Fritsche A, Haring HU. Muscle-derived angiopoietin-like protein 4 is induced by fatty acids via peroxisome proliferator-activated receptor (PPAR)-delta and is of metabolic relevance in humans. Diabetes. 2009;58:579–589. doi: 10.2337/db07-1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heller F, Harvengt C. Effects of clofibrate, bezafibrate, fenofibrate and probucol on plasma lipolytic enzymes in normolipaemic subjects. Eur J Clin Pharmacol. 1983;25:57–63. doi: 10.1007/BF00544015. [DOI] [PubMed] [Google Scholar]
- 57.Ikeda H, Taketomi S, Sugiyama Y, Shimura Y, Sohda T, Meguro K, Fujita T. Effects of pioglitazone on glucose and lipid metabolism in normal and insulin resistant animals. Arzneimittel-Forschung. 1990;40:156–162. [PubMed] [Google Scholar]
- 58.Weisweiler P. Low-dose colestipol plus fenofibrate: effects on plasma lipoproteins, lecithin:cholesterol acyltransferase, and postheparin lipases in familial hypercholesterolemia. Metabolism. 1989;38:271–274. doi: 10.1016/0026-0495(89)90086-3. [DOI] [PubMed] [Google Scholar]
- 59.Shalev A, Siegrist-Kaiser CA, Yen PM, Wahli W, Burger AG, Chin WW, Meier CA. The peroxisome proliferator-activated receptor alpha is a phosphoprotein: regulation by insulin. Endocrinology. 1996;137:4499–4502. doi: 10.1210/endo.137.10.8828512. [DOI] [PubMed] [Google Scholar]
- 60.Wilson DE, Hata A, Kwong LK, Lingam A, Shuhua J, Ridinger DN, Yeager C, Kaltenborn KC, Iverius PH, Lalouel JM. Mutations in exon 3 of the lipoprotein lipase gene segregating in a family with hypertriglyceridemia, pancreatitis, and non-insulin-dependent diabetes. J Clin Invest. 1993;92:203–211. doi: 10.1172/JCI116551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shimada M, Ishibashi S, Gotoda T, Kawamura M, Yamamoto K, Inaba T, Harada K, Ohsuga J, Perrey S, Yazaki Y, et al. Overexpression of human lipoprotein lipase protects diabetic transgenic mice from diabetic hypertriglyceridemia and hypercholesterolemia. Arterioscler Thromb Vasc Biol. 1995;15:1688–1694. doi: 10.1161/01.atv.15.10.1688. [DOI] [PubMed] [Google Scholar]
- 62.Nikkila EA, Huttunen JK, Ehnholm C. Postheparin plasma lipoprotein lipase and hepatic lipase in diabetes mellitus. Relationship to plasma triglyceride metabolism. Diabetes. 1977;26:11–21. doi: 10.2337/diab.26.1.11. [DOI] [PubMed] [Google Scholar]
- 63.Marcais C, Bernard S, Merlin M, Ulhmann M, Mestre B, Rochet-Mingret L, Revol A, Berthezene F, Moulin P. Severe hypertriglyceridaemia in Type II diabetes: involvement of apoC-III Sst-I polymorphism, LPL mutations and apo E3 deficiency. Diabetologia. 2000;43:1346–1352. doi: 10.1007/s001250051537. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.