

Abstract
The determination of membrane protein (MP) structures has always trailed that of soluble proteins due to difficulties in their overexpression, reconstitution into membrane mimetics, and subsequent structure determination. The percentage of MP structures in the protein databank (PDB) has been at a constant 1-2% for the last decade. In contrast, over half of all drugs target MPs, only highlighting how little we understand about drug-specific effects in the human body. To reduce this gap, researchers have attempted to predict structural features of MPs even before the first structure was experimentally elucidated. In this review, we present current computational methods to predict MP structure, starting with secondary structure prediction, prediction of trans-membrane spans, and topology. Even though these methods generate reliable predictions, challenges such as predicting kinks or precise beginnings and ends of secondary structure elements are still waiting to be addressed. We describe recent developments in the prediction of 3D structures of both α-helical MPs as well as β-barrels using comparative modeling techniques, de novo methods, and molecular dynamics (MD) simulations. The increase of MP structures has (1) facilitated comparative modeling due to availability of more and better templates, and (2) improved the statistics for knowledge-based scoring functions. Moreover, de novo methods have benefitted from the use of correlated mutations as restraints. Finally, we outline current advances that will likely shape the field in the forthcoming decade.
Keywords: membrane proteins, protein structure, protein modeling, sequence-based methods, structure prediction, de novo folding, homology modeling, molecular dynamics simulations, alpha-helical membrane proteins, beta-barrel membrane proteins
Introduction
Experimental structure determination of MPs remains difficult
It is estimated that up to 30% of the human genome encodes membrane proteins1,2. Since MPs function as transporters, receptors, and enzymes, and since they are involved in critical functions such as cell adhesion, immune response, and signaling, it is not surprising that over 50% of drugs on the market target them3. However, knowledge of how these drugs operate at the molecular level remains sparse, chiefly due to a lack of atomic resolution structural information on the interaction of drugs with their target proteins. In fact, MPs are vastly under-represented in the protein databank (PDB): of the ∼100,000 protein structures currently in the PDB (May 2014) only ∼2,100 are MPs (FIG. 1). This scarcity is due to the challenges that are faced when trying to determine MP structures experimentally. Over-expression of sufficient amounts of protein often fails due to toxicity to the host cells or misfolding of the protein4. Expression in yeast, insect cells (baculovirus expression system), eukaryotic strains or cell-free expression are viable alternatives, but are far less commonly used than expression in E. coli. After expression, reconstitution into a suitable membrane mimetic that avoids structural perturbations of the protein and, at the same time, is ideal for structural studies, is required. Detergent micelles, bicelles, and recently also nanodiscs or amphipols are typically used for NMR studies5, whereas crystallographers typically use detergent micelles or lipids6 for protein reconstitution. Since a priori knowledge of the best membrane mimetic is unavailable for any particular protein, a laborious and time-consuming screening process is often required. Even then, protein engineering, such as deletion of long, flexible loops or insertion of other protein fragments - like Fab or T4-lysozyme for the crystallization of G-protein coupled receptors (GPCRs)7 - may be required to obtain crystals. Additionally, screening of suitable buffer conditions may be needed to enhance crystal quality and improve the resolution of the protein structure. For NMR studies the tumbling time of the protein-detergent complex sets an upper limit to the protein size that can be studied.
Figure 1.
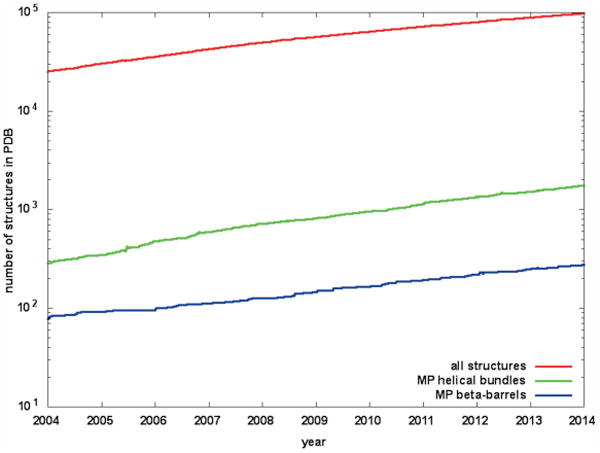
Growth of number of MP structures (green for α-helical bundles, blue for β-barrels) compared to soluble protein structures (red) during the past 10 years. Note the logarithmic scale.
As a result of these limitations, it can take years or even decades to determine structures of MPs with NMR or crystallographic methods. For example, NMR structure determination of DAGK, a homo-trimeric enzyme with 9 trans-membrane spans and a molecular weight of 39 kDa (adding up to 110 kDa with the detergent micelle), took 13 years to complete8. Comparably long time scales were required for the breakthroughs in GPCR structure determination, which started with the crystal structure of β-AR9, after many years of failed crystallization trials. In a recent example, the structure determination of a presenilin family aspartate protease required 160,000 crystallization trials to succeed10.
Advances in MP structure determination in the past two decades
The possibility to determine protein structures of this size and difficulty is owed to technical advancements over the past two decades. For crystallography, these include high-throughput crystallization and liquid handling robots. Recent structural highlights are the crystal structures of the 536 kDa proton pump complex 1 with 64 TM helices in 16 different subunits11, the sub-Angstrom resolution structure of aquaporin detailing interactions of the channel with individual water molecules12, the structure of the nine trans-membrane span presenilin homologue that provided insight into the function of aspartate protease implicated in Alzheimer's disease10, and the structure of the usher β-barrel FimD through which several other β-barrel subunits pass to elongate fibers during pilus biogenesis13.
Incredible progress has also been made in the field of NMR spectroscopy with the developments of higher field strengths, the TROSY technique14, perdeuteration15, selective labeling strategies16,17, multi-dimensional NMR18 up to 4 dimensions or higher, non-linear sampling18, cryo-probes19, Paramagnetic Relaxation Enhancements20 (PREs), and Residual Dipolar Couplings20 (RDCs). These methods allowed the determination of MP structures of up to 40 kDa. Most notable NMR structures are the previously mentioned DAGK8, the 7-trans-membrane span rhodopsin structures: sensory rhodopsin21, proteorhodopsin22, and bacteriorhodopsin23, the KcsA channel24, the mitochondrial uncoupling protein 225, and the voltage-dependent anion channel26. Notable are also the solid-state NMR structures of the YadA autotransporter β-barrel27 and the sensory rhodopsin trimer28.
The recent progress in image acquisition and processing for cryo-electron microscopy (Cryo-EM) continuously improved the resolution during the past decade down to 3-4 Å, even for single-particle reconstruction. Cryo-EM allowed the complete structure determination of the TRPV1 channel at 3.4 Å29 and of the Sec61 translocon engaged in membrane insertion of a nascent peptide at 6.9 Å30.
The need for faster MP structure determination also initiated the formation of 11 MP structure determination consortia as part of the Protein Structure Initiative31. For example, the GPCR network deposited 27 MP structures in the PDB since 2007, the Center for Structures of Membrane Proteins deposited 27 structures, 19 of which were non-homologous, and the New York Consortium for Membrane Protein Structure deposited 34 structures (14 non-homologous). The techniques developed or advanced through these initiatives have recently been used in a high-throughput manner to determine structures of 6 MPs (from 15 initial targets) within an 18 month time frame32. Even though this work did not rely on any ‘magic’ new techniques, the combination of larger-scale screening and the latest state-of-the-art techniques such as cell-free expression, combinatorial dual-labeling strategies, static light scattering, and PRE NMR enabled the structure determination of these proteins within such a short amount of time. The authors also screened another 150 MP candidates and identified 38 suitable targets for structure determination. Developments like these are truly exciting, especially in light of recent estimates from the same group of authors that stated that 100 well-picked MP targets for structure determination would increase the coverage of targets for homology modeling from currently 26% to 58%33.
Computational prediction aids in structure determination efforts
Since experimental structure determination of MPs is such a major endeavor, scientists have repeatedly tried to predict MP structures computationally. And in contrast to the challenges that are faced for experimental structure elucidation, prediction of MP structures has certain advantages over the prediction of soluble proteins. The major advantage for MPs is the smaller conformational search space that needs to be sampled since the membrane imposes strong folding constraints, with only two structural motifs allowed in the bilayer: α-helices and β- sheets. This advantage is somewhat offset by the larger size of the proteins. The challenge for MP structure prediction is the derivation of accurate scoring functions and molecular force field parameters to model the membrane, which are still under active development.
In the following paragraphs, we review methods currently available for MP structure determination (see Table I). We will start with sequence-based tools for prediction of secondary structure, trans-membrane spans, and topology, as well as surface accessibility and helix interaction motifs. Then, we discuss 3D structure prediction techniques such as homology modeling, fold-recognition, and de novo prediction methods. Since previous reviews have focused exclusively on either α-helical bundles34 or β-barrels35, we have decided to give a brief overview over the entire field. We will also report on recent successes, and discuss unsolved questions in this field.
Table I. Prediction methods and online servers for membrane proteins.
| Method name | REF | Laboratory | Comment |
|---|---|---|---|
| Secondary structure prediction | |||
| PsiPred | 37 | David Jones | three states: helix, strand, coil |
| BCL∷Jufo9D | 39 | Jens Meiler | combined withTMspan prediction |
| ProfPhd | 40 | BurkhardRost | part of Predict Protein suite |
| JPred | 41 | Geoffrey Barton | informs about homologous sequences |
| Topology prediction and TMspan prediction | |||
| BCL∷Jufo9D | 39 | Jens Meiler | SSPred and TMPred for helical bundles and beta-barrels |
| OCTOPUS | 54 | Arne Elofsson | TM helix prediction |
| TMMOD | 55 | GuangGao | TM helix prediction |
| TMHMM | 56 | Anders Krogh | TM helix prediction |
| Memsat-SVM | 57 | David Jones | TM helix prediction |
| MemBrain | 86 | James Chou | TM helix prediction, contact prediction |
| BOCTOPUS | 62 | ArneElofsson | TM beta-barrel prediction |
| BETAWARE | 63 | RitaCasadio | TM beta-barrel prediction |
| TMBeta-Net | 64 | Makiko Suwa | TM beta-barrel prediction |
| TMBHMM | 65 | Sikander Hayat | TM beta-barrel prediction and exposure |
| ProfTMB | 66 | BurkhardRost | TM beta-barrel prediction, part of PredictProtein suite |
| TMDET | 274 | Gabor Tusnady | membrane position from 3D structure |
| PPM | 275 | Henry Mosberg | membrane position from 3D structure |
| Other sequence-based predictors | |||
| AmphipaSeek | 69 | GilbertDeleage | amphipathic helix prediction |
| HeliQuest | 70 | Bruno Antonny | amphipathic helix prediction |
| TMkink | 71 | James Bowie | prediction of TM kinks |
| ASAP | 73 | Rohan Teasdale | solvent accessibility for MPs (α and β) and soluble proteins |
| MPRAP | 191 | Arne Elofsson | solvent accessibility for helical bundles |
| aTMX | 74 | Sikander Hayat | solvent accessibility for helical bundles |
| bTMX | 74 | Sikander Hayat | solvent accessibility for beta-barrels |
| PRIMSILPR | 75 | Volkhard Helms | prediction of pore-lining residues |
| LIPS | 77 | Jie Liang | lipid accessibility for helical bundles |
| PREDDIMER | 88 | Roman Efremov | dimerization of TM helices |
| PRALINETM | 91 | JaapHeringa | multiple sequence alignments for MPs |
| PHAT | 92 | Steven Henikoff | substitution matrices for MPs |
| JSUBST | 94 | Kenji Mizuguchi | substitution matrices for MPs |
| MP-T | 93 | Charlotte Deanne | sequence-structure alignment |
| Databases | |||
| PDBTM | 276 | Gabor Tusnady | MPs from PDB, transformed into membrane coordinates, bilayer thickness, updated weekly |
| OPM | 275 | Henry Mosberg | topology database with bilayer thickness |
| TOPDB | 59 | Gabor Tusnady | topology database |
| ExTopoDB | 60 | Stavros Hamodrakas | topology database |
| TOPDOM | 61 | Gabor Tusnady | topology, sequence motifs, domains |
| MeMotif | 84 | Michael Schroeder | sequence motifs in helical bundles |
| HOMEP | 277 | Barry Honig | homologous MP dataset |
| CGDB | 95 | Mark Sansom | coarse-grained molecular dynamics models |
| 3D structure prediction | |||
| RosettaMembrane | 96, 97 | Vladimir Yarov-Yarovoy Patrick Barth | homology modeling, de novo prediction of helicalbundles |
| MODELLER | 100 | Andrej Sali | homology modeling (no focus on MPs) |
| MEDELLER | 102 | Charlotte Deane | homology-based coordinate generation |
| MEMOIR | 103 | Charlotte Deane | homology modeling, fold-recognition |
| HHPred | 104 | Johannes Soeding | homology modeling (no focus on MPs) |
| Swissmodel | 105 | TorstenSchwede | homology modeling (no focus on MPs) |
| GoMoDo | 278 | Alejandro Giorgetti | GPCR modeling and docking |
| FREAD | 279 | Charlotte Deane | loop building from MP fragments |
| FUGUE | 94 | Kenji Mizuguchi | fold-recognition |
| iTASSER | 106 | Yang Zhang | fold-recognition (no focus on MPs) |
| BCL∷MP-Fold | 114 | Jens Meiler | de novo prediction of MP helical bundles |
| FILM3 | 116 | David Jones | de novo prediction of MP helical bundles with correlated mutations |
| EVfold_membrane | 115 | Deborah Marks | de novo prediction of MP helical bundles with correlated mutations |
| transFold | 147 | Peter Clote | MP beta-barrel predictor: SSPred, topology, contacts |
| partiFold | 148 | Bonnie Berger | MP beta-barrel predictor |
| TMBPro | 150 | Pierre Baldi | de novo prediction of MP beta-barrels |
| TOBMODEL | 151 | Arne Elofsson | de novo prediction of MP beta-barrels |
| TMBB-Explorer | 152 | Jie Liang | de novo prediction of MP beta-barrels |
| RosettaNMR | 155 | David Baker | de novo prediction with NMR restraints |
| CABS-fold | 157 | AndrzejKolinski | de novo prediction, can use NMR restraints |
| GeNMR | 161 | David Wishart | structure prediction with NMR restraints |
| CS-Rosetta | 163 | Oliver Lange | structure prediction using Chemical Shift NMR restraints |
| ProQM | 197 | BjoernWallner | quality assessment of 3D models |
| MD simulations | |||
| MARTINI | 209 | Siewert-Jan Marrink | coarse-grained MD force field for lipids and proteins |
| AMBER | 179 | Peter Kollman | all-atom force field |
| OPLS | 180 | William Jorgensen | all-atom force field |
| CHARMM | 178 | Martin Karplus | all-atom force field |
| GROMOS | 280 | Hermann Berendsen | all-atom force field |
| GROMACS | 230 | Hermann Berendsen | MD software package |
| NAMD | 231 | Klaus Schulten | MD software package |
| Desmond | 232 | DE Shaw | MD software package |
| CHARMM | 228 | Martin Karplus | MD software package |
Sequence-Based Prediction
Secondary structure prediction
The first step in studying protein structure both experimentally as well as computationally is the identification of membrane spanning regions (TMPred) and prediction of protein secondary structure (SSPred). Early attempts in secondary structure prediction attempted to correlate database-derived single residue statistics36 to the different types of secondary structure. Since these attempts emerged at the same time or even before the first protein structures were determined, their accuracies were only slightly better than random (for an excellent review about these early techniques see Rost and Sander36). Second generation predictors derived their statistics from longer segments (typically 11-21 residues) instead of single residues. The breakthrough, however, came with the utilization of evolutionary information from multiple sequence alignments (MSA) combined with artificial intelligence techniques. These third generation methods are still used to date. To our knowledge, the currently highest accuracy predictor is PsiPred37 from the Jones group, which uses Position-Specific Scoring Matrix (PSSM) profiles as input to an Artificial Neural Network (ANN). The output of this network is then fed into a second layer network for noise reduction. Another method, Jufo38 also uses an ANN and has recently been adapted to predict secondary structure and trans-membrane spans at the same time39. Prediction accuracies are comparable to PsiPred and other highest-quality predictors such as Octopus (see below). Other methods are Prof PhD as part of the Predict Protein suite40 developed by the Rost group, and JPred41, which informs the user about homologous sequences that are found in the PDB to enhance SSPred. Except BCL∷Jufo9D39, all other secondary structure prediction methods were trained on soluble proteins.
Hydrophobicity scales as a basis for trans-membrane span prediction
Similar to the use of amino acid propensities in SSPred, early attempts to predict trans-membrane (TM) spans relied on partitioning energies of amino acids from water into a non-polar solvent (such as octanol or cyclohexane). These partitioning energies could be determined experimentally, for instance, by measuring antigenic sites on the protein surface42, by measuring partitioning energies of host-guest peptides43, or by considering the energetics of helix-helix interactions44. Other hydrophobicity scales (reviewed by Koehler et al. 45) were derived by a consensus approach combining the advantages of several different scales46,47,48 or by using a knowledge-based approach from known protein structures45,49,50. The experimental scales, that previously used single residues or small peptides, were recently expanded by measuring partitioning energies of amino acids in fully folded proteins: (1) the biological hydrophobicity scale developed by White and von Heijne measures the partitioning of individual trans-membrane helices by the translocon51. This was accomplished by inserting helix segments into a helical MP with glycosylation sites on either side of the segment. The transfer free energies were computed from the ratio of singly glycosylated (membrane inserted helix segment) versus doubly glycosylated (translocated) helix segments. The authors also measured transfer free energies dependent on the position in the inserted helix segment. (2) Moon and Fleming derived a side-chain hydrophobicity scale for amino acids in the folded β-barrel OmpLA52. Guanidine hydrochloride unfolding curves were measured for all mutants of a residue at the center of the membrane. It was known from the structure of the protein that the particular side-chain faces the lipid bilayer. The penalty of burying an arginine residue at the center of the membrane was smaller than MD simulations initially suggested. Further, burial of two adjacent arginine residues led to cooperative effects with an even smaller penalty than for two separate arginines.
Different hydrophobicity scales were typically compared based on their ability to predict TM helices. A recent comparison53 has shown that von Heijne's biological scale and knowledge-based scales such as the Universal Hydrophobicity Scale (UHS) have highest prediction accuracies. Interestingly, the UHS performs well in predicting protein topology (inside/outside orientation) even though topology information was ignored when this scale was derived. Performance differences of the various scales in different publications (comparing to reference45) are likely due to distinct parameter sets such as window size, window function, datasets used, and choice of quality measures.
Prediction of trans-membrane spans and protein topology
For the early methods of TM span identification, the transfer free energies were summed over a number of residues (typically 10-25), and stretches with high hydrophobicity were predicted to be in the membrane. This simple method worked only for helical proteins, where all residues in the TM spanning region have the same sign on the hydrophobicity scale. For TM β-strands with alternating signs of hydrophobicities (i. e. β-barrels), this scheme was bound to fail45. Using this simple window averaging scheme, per-residue prediction accuracies on helical bundles were below 60% in the three state scenario that includes an interface region45.
Similar to the developments in SSPred, the inclusion of evolutionary information from multiple sequence alignments and the use of machine learning approaches increased prediction accuracies drastically. However, since the sequence features of TM helices and TM strands differ, prediction algorithms typically specialized on either one, but not both. An exception to the rule is the recently improved SSPred tool Jufo that has been extended to predict TM spans of both helical and strand nature in BCL∷Jufo9D39. To our knowledge, this is the first method that provides predictions irrespective of secondary structure type (helix or strand) and protein type (soluble or MP). Prediction accuracies are over 70% in the nine-state scenario (compare to 11.1% for a random prediction), 73% for three-state secondary structure, and almost 95% for three-state TM span. BCL∷Jufo9D is also able to predict some higher-resolution features such as kinks and re-entrant helices.
Predicting TM spans and topology of α-helical bundles
Several tools are available for predicting TM helices and topologies (i.e. defining the inside/outside orientation of residues in MPs). For OCTOPUS54, four separately trained ANNs for the membrane, interface, loops, and globular residues were combined globally using a Hidden Markov Model (HMM), and it achieves accuracies above 90% in the two-state scenario (residue is or is not part of a TM helix). Other TM helix prediction methods use HMMs (TMMOD55, TMHMM56) or SVMs (Memsat-SVM57) with varying accuracies. The principle however, is typically the same: the evolutionary information from the MSA serves as a basis to extract sequence features that are used to build models (for HMMs), to train networks (ANNs), or for classification (for SVMs). The structural information from a database of protein structures serves as a comparison to the predicted output for supervised learning. Topology prediction methods additionally use the “positive-inside” rule58 (that states that there is a positive charge bias on the cytosolic part of the protein) to classify the protein topology for training. Alternatively, databases that collect experimental information, such as protein fusion with reporter enzymes, glycosylation studies, accessibilities to proteases, and immunolocalization techniques, to pinpoint protein topology (such as TOPDB59 or ExTopoDB60) or that identify sequence motifs consistently located only on one side of the membrane (TOPDOM61), are useful resources.
Predicting TM spans and topology of β-barrels
For TM β-barrels, the group that developed OCTOPUS developed a method called BOCTOPUS62. It uses an SVM for local predictions that are then combined into an HMM for global prediction. The method is trained on 36 structures with 10-fold cross-validation and the authors report an accuracy of 87% in the three-state prediction (inside, outside, membrane). Other TM β-barrel predictors are the recently developed method BETAWARE63, TMBetaNet64 (an ANN trained on 13 outer membrane proteins), TMBHMM65 and ProfTMB66 (which is a profile-based HMM that is part of the Predict Protein40 server developed by the Rost group).
Predicting TM spans: What are we missing?
Even though these tools provide reliable predictions, several improvements related mostly to higher-resolution predictions would be useful and are likely to be implemented in the near future:
The prediction of re-entrant helices as seen in voltage-gated potassium channels, chloride channels, SecY and other proteins. Several tools such as OCTOPUS, Memsat-SVM, and BCL∷Jufo9D are able to predict some of them, but prediction accuracies well below 50% highlight the challenges that are faced. As more MP structure are determined, larger training datasets will likely facilitate the predictions and provide more statistics on these helices' different amino acid compositions.
A similar issue is the prediction of half-helices that insert into the membrane as in peripheral MPs, or the prediction of helices that differ from the “standard” TM helix length of 19 or 21 residues. Prediction tools such as OCTOPUS use fully spanning TM helices for their training, remove shorter helices in a post-processing step, or output only TM helices of a defined length. However, given the structural diversity of MPs with helix and strand lengths that span or insert into the membrane with different lengths, prediction algorithms need to be able to identify these features.
The prediction of amphipathic helices (helices that are lying flat on the membrane surface) from sequence information, even though one of most used methods in bioinformatics by the experimental community, still seems to be ‘stuck’ in the 20th century. The very few tools available still use a sliding window approach67 and hydrophobic moments to identify amphipathic helices. Some methods rely solely on helical wheel plots or wenxiang diagrams68. Servers like Amphipa Seek69 or HELIQUEST70 are either error-prone or lack user-friendliness. Moreover, none of the few methods state accuracies for predicting amphipathic helices.
The prediction of TM helix kinks: The Bowie lab trained an ANN, TMkink71, specifically for this task and they report a prediction sensitivity of 70% and specificity of 89%. Depending on the dataset and the kink definition, this prediction can drop to about 50%39. Interestingly, in proline-induced kinks the proline residue may not be conserved even if the kink is72. Again, larger structural databases will promote higher quality predictions in this area.
Most of the MP β-barrel predictors are exclusively trained on outer membrane proteins (OMPs) where the barrel is formed by a single chain. The performance of these tools on β-barrel porins, such as hemolysins and other pore-forming toxins, where the barrel is formed by multiple chains, is questionable.
Prediction of surface accessibility and membrane-facing regions
The few available methods that predict surface accessibility of MPs from sequence are mostly SVM classifiers. ASAP73, a method for both MP helical bundles as well as β-barrels has reported accuracies of 70-80%, depending on the type (helical or barrel) and probe radius. According to the reduced specificity, the method likely over-predicts accessibility. Similarly, the methods aTMX and bTMX74 are SVM classifiers that predict the burial status of residues in α-helical bundles as well as β-barrels with accuracies of almost 80%. Both methods for helical bundles, however, exclude pore-forming proteins or cap the pores such that they are inaccessible to the spherical probes used for estimation of solvent-accessible surface area. Therefore, it is unclear how these methods would perform on these types of proteins. Another recently developed SVM classifier, PRIMSIPLR75, has been trained on 90 helical MPs proteins specifically to predict pore-lining residues. The article shows an accuracy of 86% with possible under-prediction of residues. A different approach was used by Adamian & Liang who developed a lipophilicity scale76 that was later used for lipophilicity prediction (i. e. lipid accessibility) by their LIPS server77. The scoring function is based on the Shannon entropy and combines the average entropy and lipophilicity of different helical faces into a single score. The algorithm predicts lipid-exposed sides of TM helices with an accuracy of 88%. Other predictors are available for surface accessibility prediction, but are not trained on MPs (such as NetSurfP78 or SABLE79 as part of Predict Protein40).
Identifying helix interactions and other motifs from sequence
The first and most well known trans-membrane helix interaction motif, the GxxxG motif, was found at the beginning of the 1990's by studying the interaction surface in glycophorin A80. Since then, more interaction motifs between TM helices have been described. Senes et al. 81 examined over- and underrepresentation of residue pairs and triplets in sequences and found that the GxxxG motif frequently occurs with β-branched amino acids such as Val or Ile. Other motifs that were found typically involve small amino acids such as Gly, Ser, and Ala in the first and fourth position whereas Ile, Leu, and Val are in neighboring positions. Excellent reviews about trans-membrane helix interaction motifs with corresponding examples are Curran & Engelman82 and Langosch & Arkin83.
Marsico et al. created the MeMotif database84 that describes different sequence motifs found in helical MPs. Characteristics such as protein-protein interaction motifs in TM complexes (30% of the motifs), helix-helix interaction motifs (15%), and specific lipid/cholesterol binding motifs (12%) are represented there. The database server can be used to identify motifs in a sequence of interest.
Walters et al. investigated helix-packing motifs by clustering techniques. They found that 75% of helix interactions can be assigned to one of five clusters85 and that all of these clusters follow simple principles of helix-helix85 packing. For structure prediction, the MemBrain server86 combines correlated mutations with multiple machine learning classifiers to predict TM helix-helix contacts from sequence. The contact prediction accuracy is increased dramatically compared to other methods – it achieves 64%. These predicted helix contacts can also be used in conjunction with threading techniques, such as TASSER87, to predict 3D structures of the proteins. Another recently developed algorithm, PREDDIMER88, can predict structures of TM helix dimers (homo- as well as hetero-dimers) from the sequences of short helices. Alternatively, the tool can be used to analyze hydrophobic properties and contacting regions for a known structure.
A recent review from Li, Wimley, and Hristova89 challenges the notion of simple interaction motifs between TM helices. Since more and more MP structures become available, it has become clear that TM helix interactions are more complex and do not always follow the simple rules outlined above. The authors discuss examples that obey previously described interaction rules, and others that do not. Since it remains difficult to obtain high-resolution MP structures that are required for a deeper understanding of these interactions, we are just at the beginning to recognize these complex relationships between sequence and structure.
Going to Three Dimensions
Whereas the previous paragraphs have focused on sequence-based techniques that yield information about protein secondary structure and may be able to provide hints about tertiary contacts, the second half of the review discusses methods for protein tertiary structure prediction. These are ordered by increasing difficulty (and decreasing accuracy achievable) and are highlighted in FIG. 2.
Figure 2.
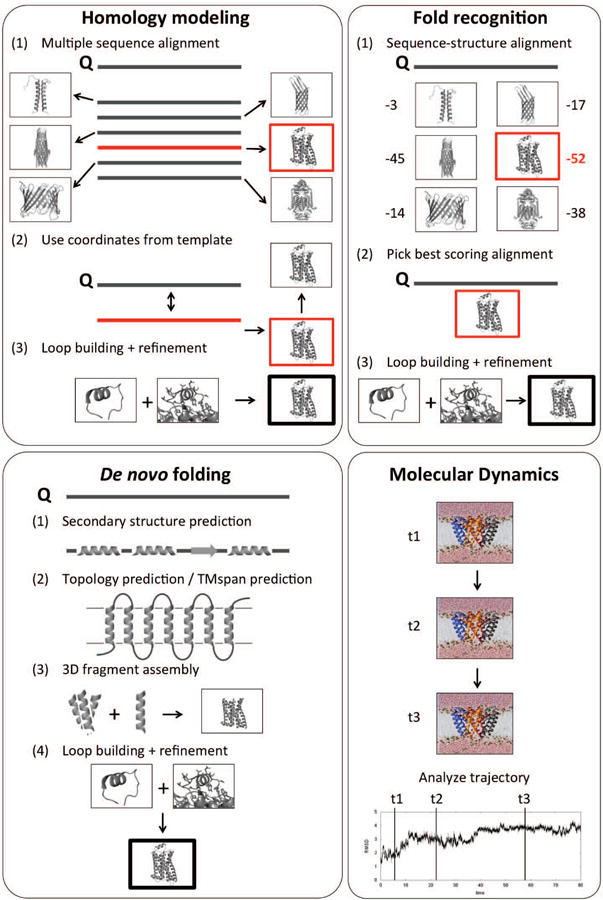
Methods for predicting 3D protein structures. (Top left) Homology modeling requires a template to be found by multiple sequence alignment to the query sequence (Q). (Top right) Fold-recognition is used for low sequence similarities that prevent template identification solely based on multiple sequence alignments. A sequence-structure alignment of the query sequence with a database of structures and subsequent scoring is necessary to identify a suitable template. (Bottom left) De novo (or ab initio) folding is used when no template is available and/or for novel protein folds. (Bottom right) MD simulations are currently unable to fold proteins larger than ∼80 residues, they are used to study dynamics and molecular processes of proteins along a time trajectory.
Homology modeling and fold recognition for membrane proteins
The past years have shown a steady increase in the use of homology modeling techniques, especially for MPs. Contributing factors are the increase of determined MP structures and therefore the availability of suitable templates, while at the same time experimental structure determination is still prohibitively slow or challenging. Critical drug targets for a wide variety of diseases, such as GPCRs and ion channels, are prime targets for homology modeling and heavily benefit from the increase in available template structures. For instance, according to PubMed, over 130 papers have been published on homology modeling of GPCRs, half of them published in the past 3 years.
Homology modeling requires a template structure with high sequence similarity to the target sequence: typically the higher the sequence similarity, the higher the accuracy of the resulting model. Sequence similarities of ∼70% can yield models with an RMSD of 1-2 Å whereas highest-quality models with sequence similarities of 25% to the template typically have RMSDs of 3-4 Å90. As a consequence, the quality of the sequence alignment also influences the accuracy of the resulting model. One of the alignment methods that have been developed specifically for MPs is PRALINETM91, which uses MP specific substitution matrices (PHAT matrices92) instead of BLOSUM or PAM and incorporates TM span prediction to improve sequence alignments. MP-T is a sequence-structure alignment tool93 that also uses MP specific substitution matrices (JSUBST matrices94) and environment parameters such as secondary structure, accessibility, and membrane depth from the Coarse Grained DataBase95 of MD models for each template.
After a high-quality alignment has been obtained, the alignment and the template can be submitted to a homology modeling tool. For some tools, the alignment step is already included in the calculation and the target sequence and template structure are sufficient for modeling. Several tools are available, RosettaMembrane96,97 being one of the earliest and most reliable (FIG. 3). It has been used to model the open and closed states of the Kv1.2 and KvAP potassium channels98. Recently, RosettaMembrane was used to model the HERG potassium channel99 and docking simulations were used to identify possible binding pockets of inhibitors or activators. Another popular tool in the modeling and experimental community is MODELLER100 that predicts the structure by satisfaction of spatial restraints derived from the alignment. Even though MODELLER does not have a membrane mode per se, it has been used for modeling MPs as well101. MEDELLER102, is a coordinate-generation tool designed for MPs that outperforms MODELLER for all types of targets from easy to difficult. It has been used in conjunction with other tools designed for MPs as the MEMOIR server103 that has been shown to outperform HHP red104 or Swiss Model105.
Figure 3.
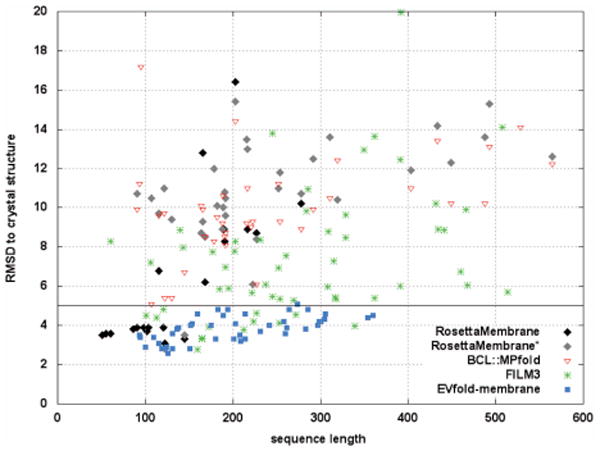
Comparison of different de novo structure prediction methods using RMSDs to crystal structures versus sequence lengths of the modeled proteins. The data is taken from the following references: RosettaMembrane [Yarov-Yarovoy, 2006, ProtStructFuncBioInfo; Barth, 2007, PNAS], RosettaMembrane* [Weiner, 2013, Structure], BCL∷MP-Fold [Weiner, 2013, Structure], FILM3 [Nugent, 2012, PNAS], EVfold_membrane [Hopf, 2012, Cell].
Homology modeling is a powerful tool for high sequence similarities between the target and the template, but it is not suitable for low (∼25%) to very low (5-10%) sequence similarities. For these regions, fold-recognition methods (or ‘threading’) are the tools of the trade. Threading methods rely on the principle of aligning the target sequence to all template structures in a database. The sequence-structure alignment is then scored with a knowledge-based scoring function that contains terms like secondary structure, environment, mutatability, and residue pairing, among others. One of the best performing methods is iTASSER from the Zhang group106 for which unfortunately no ‘membrane mode’ is available. It has, however been used in conjunction with MemBrain86 to model 13 GPCRs. Comparing the models to the crystal structures, inclusion of MemBrain contact prediction led to a significant drop in RMSDs: on average by 3 Å, in three cases even up to ∼10 Å. The fold recognition method FUGUE94 was combined with membrane-specific substitution matrices JSUBST and the coordinate-generation method MEDELLER102. Models in the sequence identity range between 15-35% had average RMSDs of 3.4 Å to the crystal structure. Another method, TMFR107 has recently been published, which uses topology features in sequence profiles combined with solvent-accessibility. The group developed individual methods for MP helical bundles and β-barrels and report increases in accuracies of up to 10% compared to HH align108.
De novo structure prediction of α-helical MP bundles
It is estimated that about 80% of the integral MPs are α-helical bundles109,110. Even though a variety of sequence-based techniques exists for this subclass of proteins, de novo structure prediction remains challenging, especially for large MPs. One of the first successful approaches was the RosettaMembrane96 software that uses protein fragments assembled by a Monte-Carlo search and scored by knowledge-based scoring functions. The lipid bilayer is represented implicitly and the low-resolution scoring function contains energy terms probing residue-residue interactions, residue environment, packing density, and steric clashes. RosettaMembrane penalizes non-spanning TM helices and non-helical torsion angles within the membrane. An all-atom refinement protocol was later added with a high-resolution score function97 based on an implicit solvent potential for MPs, IMM1111. Rosetta uses 3 and 9 residue fragments for assembly and was originally developed to fold small soluble proteins. For improved sampling of the extensive conformational search space of larger proteins, the fragment assembly approach of the BioChemicalLibrary112,113 used in BCL∷MP fold114 uses complete secondary structure elements and outperforms Rosetta for most MPs and some large soluble proteins. Recently, the concept of correlated mutations (i. e. covaration of amino acid residues) has been applied to de novo MP structure prediction115. Even though covariation for protein structure prediction had been tested repeatedly in the past, contact prediction accuracies were rather limited. The reason for these low accuracies are transitive correlations, meaning that covariations between residues (A and B), and (B and C) do not necessarily imply a correlation between (A and C). Hopf et al. recently described EVfold_membrane that uses a maximum entropy approach that is based on the inverse Ising problem, to eliminate this noise of false positives. Restraints derived from the covariation patterns were used in conjunction with the structure calculation software CNS, which lead to substantial improvements in prediction accuracies and consistently achieved RMSDs below ∼5 Å for proteins up to ∼360 residues115. Residue covariance has also been applied by Nugent & Jones in their FILM3 software116 that uses secondary structure prediction and topology prediction to guide fragment selection and assembles these fragments using correlated mutations.FIG. 3 shows the RMSDs vs. sequence lengths from best scoring models predicted by all four software algorithms. Whereas Rosetta predicts models as accurate as <5 Å RMSD for small MPs, BCL∷MPfold creates higher-accuracy models for larger MPs. The addition of correlated mutation restraints leads to an increased performance of FILM3 compared to BCL∷MPfold. However, all methods are outperformed by EVfold_membrane. These are impressive prediction accuracies, and given that three of the four methods are newly developed, further improvements in de novo folding algorithms are likely to happen.
Structure prediction of specific classes of membrane proteins: GPCRs and K+ channels
De novo MP structure prediction methods have also been developed for specific targets, such as GPCRs. The idea is to make specific assumptions about the target from a template structure or use experimental data as restraints for structure prediction (see below).
Even though GPCR structures are difficult to determine experimentally, they have the advantages of (1) sharing a similar fold with 7 TM spans; (2) challenges in expression and protein engineering have been mainly resolved, and (3) the GPCR Network117 as a structure determination consortium is solely dedicated to this task, bringing together the experts in the field. (4) Additionally, GPCRs are still within the size limit for NMR spectroscopy (even though it is still challenging to obtain usable spectra).
The PREDICT118 program makes general assumptions about helix arrangement and GPCR structure to create a coarse-grained model, later optimizes helix orientations, and uses MD simulations for a full-atom refinement. Ultimately, experimental data from ligand-binding studies is used for further refinement. With this method, the structure of rhodopsin was predicted to 2.9 Å to the crystal structure. The MembStruk algorithm119 used a similar approach by predicting TM spans and optimizing individual helices with MD simulations. The helices were later assembled into a bundle using the electron density map of bovine rhodopsin. After refining helix orientations, loop modeling, and full-atom refinement using MD simulations, the RMSDs to the crystal structures were 3.1 Å for bovine rhodopsin and 6.2 Å for bacteriorhodopsin. Fold GPCR120 starts from a helix bundle that has been assembled using distance restraints from a template structure with information about helix bundle topology, conserved residue contacts, and ligand binding geometry. The MD approach includes simulated annealing and a replica-exchange protocol for refinement. The RMSD to the crystal structure for the β2-adrenergic receptor is 2.1 Å. The TASSER121 method differs from the previous approaches by not using any restraints or experimental data to reduce the conformational search space, but rather identifying template fragments from the PDB that are assembled using a replica-exchange Monte-Carlo protocol. Iterative clustering, modeling and then refinement make their threading algorithm (aka fold-recognition) one of the best servers for protein structure prediction. In 2006, the TASSER group generated models for all 907 GPCRs in the human genome122 and achieved an RMSD to the bovine rhodopsin crystal structure of 4.6 Å.
Unfortunately, large tetrameric potassium channels, whose family is smaller than the GPCR family123 and whose members are expressed in various tissues, do not share the advantages that GPCRs have. Diseases associated with ion channels include cardiac arrhythmias (irregular heartbeat & sudden death), epilepsy, cystic fibrosis, and hearing loss, and blockers of potassium channels can be used as local anesthetics. Expression levels of potassium channels are very low124, which sometimes requires special in vitro expression systems125. Additionally, their folds vary between channels formed by oligomeric association of 2 TM spans (inward rectifiers Kir, KATP channels, G-protein regulated GIRK channels, K2P channels), 6 TM spans (Kv, calcium-regulated KCa), and 7 TM spans (Slo)126. There are several structures available to date that are commonly used as templates for homology modeling: KcsA127, KvAP128, Kv1.2129, Kv1.2 chimera, Kir124, KirBac130, and MthK131. The challenges for homology modeling are to find templates with a reasonably high sequence identity to the target – at least 30% are required to obtain a model of acceptable quality. Furthermore, loop regions are usually less conserved and often need to be modeled de novo. Since the loops typically contain the ligand-binding interfaces, accurate modeling is required to carry out high-confidence ligand-docking studies. If experimental constraints from ligand-binding assays are available, they can be used to restrict flexible loop conformations.
Other approaches have been used to study potassium channels: MD simulations132, Brownian dynamics133, and potentials of mean force134 among others were used to try to explain certain features, such as ion selectivity, channel gating, voltage sensing, and ligand binding. To focus on the pharmacological aspects of ion channels135 (or drug targets in general), quantitative structure activity relationships (QSAR) can be used136, a ligand-based approach that uses chemical or 3D descriptors of the ligand to predict biological activities against a target protein. QSAR can be combined with pharmacophore maps137, i. e. maps of pharmacologically relevant features or functional groups of ligand molecules, to perform virtual high-throughput screening (vHTS)138. Interestingly, vHTS can also be carried out without knowing the structure of the target protein by training machine learning approaches with the chemical or 3D descriptors and using experimentally tested protein activity data as output for training and testing139.
In recent years, the hERG potassium channel has received much attention both from the pharmaceutical industry as well as academia because many potassium channel blockers have shown to also non-specifically inhibit hERG as a side-effect, leading to Long-QT syndrome and possibly sudden death140. This inhibition has led the pharmaceutical industry to use hERG as an antitarget to probe drug safety. Since the structure of hERG is unknown, scientists rely on homology models of the channel99,141. As a model for drug binding, toxins such as scorpion toxin142,143 and tarantula toxin144 have been used because of their specificity towards the less conserved voltage-sensing part of the channel145. Currently, many drugs specifically target the highly conserved pore-forming part leading to side effects such as hERG blockage.
De novo structure prediction of MP β-barrels
Even though β-barrels are much less common among MPs than α-helical bundles (an estimate is 2-3% of the genome of gram-negative bacteria146), there has been some effort predicting their structures de novo. Early methods started with sequence-based prediction of topology or residue contacts, and only recently have algorithms emerged that attempt to predict the 3D fold of the barrel. The transFold147 algorithm is a sequence-based approach that predicts the ‘supersecondary’ structure of MP β-barrels, such as secondary structure, TM topology, residue contacts, side-chain orientation, and strand angles with respect the membrane. The underlying algorithm uses inter-strand statistical potentials in conjunction with grammars from formal language theory to build models that are subsequently evaluated using a dynamic programming algorithm. The program partiFold148 goes one step further in modeling ensembles of β-barrels by computing the Boltzmann partition function to estimate inter-strand residue contact probabilities and crystal structure B-values. The BetaBarrelPredictor149 uses a graph-theoretic approach to classify β-barrels and model their supersecondary structure.
The TMBpro150 software suite encompasses several prediction tools for secondary structure, side-chain orientation, residue contacts, and tertiary structure. The latter uses a simulated annealing protocol with a specified move set to assemble 3D folds from fragments of template structures. The algorithm predicted β-barrel structures under 6 Å RMSD to the native for 9 out of 14 proteins and RMSDs under 5 Å for 6 out of 14 proteins. Another predictor, TOBMODEL151, uses topology prediction as a starting point and assembles the β-sheets at different angles in the membrane. The resulting models are scored with ZPRED, which estimates the distance of each residue from the membrane center. On their test set, TOBMODEL correctly predicted the topology in more cases than TMBpro and also had a lower average RMSD to the crystal structure (7.2 vs 8.8 Å). Another recent method, 3D-SPOT152 (as part of the TMBB explorer) is probably the most accurate method for 3D structure prediction of β-barrel MPs with RMSDs below 4 Å. It models the physical interactions of strong H-bonds (inter- strand backbone interactions), weak H-bonds, and side-chain interactions between neighboring strands in the membrane. Whereas the topology is not predicted explicitly and the strands can vary in length, the balance between inter-strand interactions and an entropy-based loop term defines the length of the strands vs. loop lengths. To arrive at a low-scoring model, strands are allowed to slide up and down with respect to the previously inserted strand, which result in different strand registers and H-bonding patterns.
Incorporating experimental restraints into modeling – early approaches for soluble proteins
In the mid 1990's computational protein structure prediction produced models with a more or less ‘random’ structure and succeeded only in rare cases for very small proteins153. The computer scientists developing new algorithms were often disconnected from the experimentalists who determined structures with distance geometry methodologies and measured NMR restraints. The datasets for NMR structure calculations were (and still are) typically ‘rich’ datasets with 10 or more restraints per residue. It was realized that incorporating experimental restraints into structure prediction was a crucial step to obtain higher quality models. Additionally, scientists understood the challenge of obtaining rich experimental restraint datasets and aimed to develop methods that produce high-quality models with sparse datasets of one restraint per residue or less.
An early success was the MONSSTER154 method from Skolnick et al. which used secondary structure elements and fragment assembly with a few long-range distance restraints. Even though this was as early as 1997 and the method was not designed for MPs, the algorithm was able to determine the structure of the 146 residue protein myoglobin to 5.7 Å RMSD with only 20 distance restraints. In 2000 the Rosetta NMR155 algorithm combined de novo prediction with sparse restraints from chemical shifts and NOE data. Using one restraint per residue produced consistently better results than traditional distance geometry approaches; moreover, the produced models were closer to the crystal structures than the corresponding NMR structures. As early as in 2003, unassigned NMR data was used in conjunction with Rosetta to determine the folds of proteins up to 150 residues156. The group of Kolinski and coworkers developed the CABS-fold program157 in 2007 that uses secondary structure from chemical shift index data and TALOS dihedral angle restraints to fold proteins up to 140 residues. Their algorithm uses a simplified representation of protein residues combined with replica-exchange MD simulations.
These methods were continuously improved to (1) use less data, (2) use data that was easier to obtain, (3) fold larger proteins, and (4) increase the accuracy of the models: by 2010 Rosetta NMR could predict proteins up to 25kDa using backbone-only restraints from chemical shifts, RDCs, and NOEs158. Recently, backbone-only chemical shifts and distance restraints from homologous structures were used to model proteins of that size159. Another interesting idea is the RASREC protocol160 that resamples low-energy features from intermediate-stage models, thereby taking advantage of ‘good’ already-built substructures that can be recycled and combined in later stages. This method therefore provides crosstalk between the otherwise independent folding trajectories leading to improved sampling.
The Wishart group combined the use of various data (sequence data, NOEs, chemical shifts) and databases to predict protein structures with Rosetta and refine them with Xplor. They have shown that protein structures below 3 Å RMSD can be obtained even for non-homologous proteins. Their webserver GeNMR161 generates 10 models per query.
Using experimental data to model MPs
Since 3D modeling of MPs didn't emerge until the mid 2000s, studies that use restraints in structural modeling have emerged only recently. Shortly after RosettaMembrane was developed, Barth et al. 162 reported the use of few (down to a single) specific helix-helix packing restraints in MP de novo folding. Twelve MPs from 190 to 300 residues were folded, and the restraints promoted an enrichment of native structures and models below 4 Å RMSD. The RASREC protocol and Rosetta CS in combination with sparse restraints allowed model building of MPs up to 40kDa163. In half of the cases, the Rosetta model was closer to the crystal structure than the NMR structure. Large MPs can be modeled with BCL∷MPfold developed in the Meiler group114. When supplemented with NMR data such as NOEs, chemical shifts, and RDCs, 65 out of 67 proteins in a test set had the correct topology164. Incorporating restraints achieved an average decrease in RMSD100 (RMSD normalized to a protein size of 100 residues) of 2.5 Å, and the best-scoring protein model of 565 residues had an RMSD of ∼5 Å.
Since experimental structure determination of MPs is laborious and cumbersome, there have been attempts to predict the required number165 and location166 of spin labels for PRE NMR or EPR measurements. These distances can be used in modeling167, however, translating the spin-label distances into accurate backbone restraints is not straightforward due to the flexibility of the spin label168,169.
Other types of restraints that have been used are density maps from CryoEM170,171 and SAXS172,173. Whereas CryoEM is becoming a well-established tool and medium to high-resolution density maps become increasingly available, progress in the use of SAXS data is still slow. An emerging hybrid approach is the use of mass-spectromety or cross-linking data in modeling and docking174,175. Its success is most likely owed to the character of the restraints: distances of short range in 3D space but long range in sequence restrict the folding space drastically. Two beautiful examples of these hybrid approaches are (1) the structures of active and inactive ribosome-SecY channel complexes by combining CryoEM with homology modeling176, and (2) the structure of the ribosome using CryoEM, homology modeling, mass-spectrometry, and cross-linking data177.
Scoring functions for membrane proteins
Predicting the structures of proteins is intimately connected to the development of accurate scoring functions that are able to discriminate native and near native from non-native models. Scoring functions can either be physics-based, knowledge-based, learning-based or a combination. Examples of physics-based potentials[1] are the ones used by CHARMM178, Amber179, or OPLS180.
Physics-based potentials
Physics-based potentials typically include energy terms describing bonds, angles, dihedral angles, electrostatics, and van der Waals interactions. Interactions with the solvent can be modeled in two different ways: (1) explicitly, i. e. interactions of each protein atom with each solvent atom are modeled individually, or (2) implicitly, i. e. the solvent is represented as a continuous field with average solvent properties with which the protein atoms are interacting. To improve sampling and tackle the high cost of modeling water explicitly, implicit solvation models have been proposed and developed. The simplest way to model solvation is by considering the solvent-accessible-surface-area (SASA) of atoms or residues in a protein that are associated with a specific solvation parameter. This model requires accurate solvation parameters to be chosen and measured. Another model is the Poisson-Boltzmann (PB) formulation that represents the solvent as a dielectric continuum and describes the electrostatic interactions between the protein and the solvent implicitly. This model is computationally expensive and has been approximated by the generalized Born (GB) formulation. A different model, the effective energy function EEF1, has been developed181 that approaches solvent exclusion by Gaussian statistics.
To model the membrane bilayer as an implicit solvent, Lazaridis extended the EEF1 solvation potential to the lipid environment: the solvation term was parameterized based on experimental observation of energy transfer from water to cyclohexane, and a depth-dependence was introduced into reference energies and the dielectric potential. The resulting potential IMM1111, was used in combination with the Rosetta knowledge-based scores for soluble proteins to create the high-resolution RosettaMembrane97 scoring function. This score function complements the low-resolution scoring function of RosettaMembrane96 that consisted solely of knowledge-based potentials modeling residue pairs, solvation, and packing, and that were added to the standard Rosetta scoring function for soluble proteins.
Others have extended the generalized Born theory of solvation to include the membrane as a planar hydrophobic region. This approach, first developed by Spassov et al., improves sampling by 1-2 orders of magnitude compared to explicit lipid bilayer simulations182. In combination with replica exchange techniques, Spassov's work has allowed the first unguided protein folding simulations of small membrane-spanning polypeptide segments using classical MD or MC methods with standard molecular mechanics force fields (CHARMM, OPLS, GROMOS)183,184. The downsides of these models are the oversimplification of the chemically complex lipid bilayer interface and the inability to capture membrane deformations caused by partitioning of charged sidechains.
Another physics-based force field is that of MARTINI, originally developed to model lipid bilayers, it has now been extended to model proteins in the membrane185. This coarse-grained potential bundles four atoms of a lipid or amino acid into a super-atom, thereby achieving an increase in speed of 3-4 orders of magnitude. MARTINI has recently been combined with the GROMOS atomistic force field to model the lipid bilayer in coarse-grained mode while retaining the ability to model the protein atomistically186.
Knowledge-based potentials
The small number of determined MP structures have also been used to develop knowledge-based scoring functions, mostly for helical bundles. Adamian et al. examined interacting residues in TM helices187 and found that pairs between an aromatic residue and a basic residue (such as WR, WH, YK) are the most probable interactions. By investigating packing, they also found that large residues are most likely to occur in pockets whereas small residues are least likely. Additionally, the number of contacts correlates with residue size. A packing score to model helix-helix interactions was developed by Fleishman & Ben-Tal188 that rewards small residues in the interface and penalizes large ones. From a test set of 11 proteins, 73% of the predicted structures had an RMSD <= 2 Å. A potential with similar scoring ability was developed by Wendel et al. from a database of 71 MPs189. A purely distance dependent score for oligomeric helix-helix interfaces was also developed by Park et al. 190. Their lowest energy conformations of glycophorin A, ErbB2, and phospholamban matched experimental data.
Knowledge-based potentials that probe the interaction with the solvent can be used as a guide for modeling or for filtering out erroneous models. MPRAP191 is a sequence-based predictor of SASA that was developed by the Elofsson lab. It is a support vector machine that has been trained on databases of both soluble proteins as well as MPs and overcomes the limitations of other methods, that perform well in either solution or the membrane, but not both. The Liang group developed a lipid propensity scale76 that was used to train their LIPS server77, a method to predict lipid accessible surfaces from protein sequence alone. A much more fine-grained scoring function was derived from the Lazaridis group192 that describes binding of peptides to the membrane bilayer dependent on pH. Components of the binding free energy were computed from MD trajectories. The same group later derived potentials of mean force for ionizable side-chains at the water-bilayer interface region193.
Comparison of different scoring functions
Which scoring functions are good for which scenarios? Since the ruggedness of the energy landscape depends on the granularity with which the protein is modeled, the ‘correct’ scoring functions should be used in the appropriate scenarios. Forrest & Woolf194 studied loop conformations in MPs and compared several physics-based potentials in their ability to discriminate native-like conformations. They found that effective energy functions can distinguish native-like conformations under the assumption of a homogeneous environment and that continuum-solvent models and finite-difference PB models are the most powerful ones. MP loop conformations were also examined by Gao et al., who tested several high-resolution potentials on homology modeling decoys195. They found that the scoring functions of PLOP, Rosetta, and DFIRE have the most discrimination ability, followed by AMBER/GBSA, RAPDF, and MODELLER. It should be noted, however, that the homology models were built with PLOP and MODELLER, possibly biasing the test to the compatible score function. The Lazaridis group tested the discrimination ability of several physics-based scoring functions on decoys generated by RosettaMembrane in dependence on the membrane thickness196. They found that IMM1 in combination with CHARMM36 gives best results at a membrane thickness of 25.4 Å, similar to GB with simple switching, which includes a higher-order Coulomb field approximation and uses overlapping van der Waals spheres to represent the dielectric surface. Using GB with simple switching at a membrane thickness of 28.5 Å produced identical results.
The Wallner group developed ProQM197, a learning based scoring function consisting of a support vector machine. The SVM was trained on structural characteristics of the models, namely atom-atom contacts, residue-residue contacts, SASA, secondary structure, TM spans, membrane depth, and evolutionary information. The method produced higher correlation coefficients than any other methods and, combined with RosettaMembrane low-resolution scoring function, led to a 7-fold enrichment in near-native models. Surprisingly, this learning based method was outperformed by a recently developed simple scoring function based on known residue-residue interactions198. The IQ-score is the sum of two scoring terms that considers the number of interactions (hydrophobic interactions, hydrogen bonds, ionic bonds, disulfides) and their distribution compared to a native protein structure. Based on its performance on three datasets, it outperforms ProQM on average by ∼7%.
Molecular Dynamics (MD) simulations – advances and challenges
Another popular technique in the computational modeling field is MD simulations, which have traditionally been used in conjunction with NMR restraints to compute protein structures. MD simulations have also been used to uncover a wealth of atomic detail information on the molecular mechanisms underlying protein function, such as ion conduction, selectivity, and gating for channel proteins. Even though the MD field has experienced a vast increase in protein sizes as well as timescales that were modeled over the past 10 – 15 years (FIG. 4), it remains challenging to fold proteins of a ‘reasonable’ size (say, 150 residues) from an unstructured state in solution. The currently largest proteins folded are about 80 residues in size199 – one example is the soluble protein acylCoA whose simulation pushed several limits at once with an incredibly long simulation time of 30 ms. These timescales were reached by using specialized software that utilizes Graphics Processing Units (GPUs) on a distributed computing grid (Folding@home), while at the same time improving the sampling efficiency by representing the solvent implicity using the GBSA method200.
Figure 4.
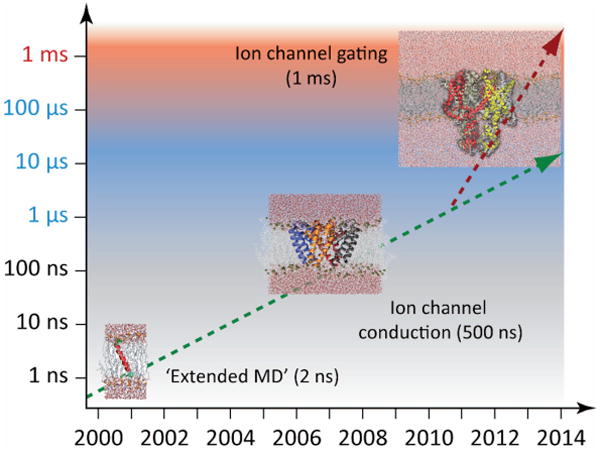
Growth of computational power for all-atom MD simulations as seen by simulation times and protein sizes to be modeled.
The first attempts at atomistic folding of MPs in explicit lipid bilayers proved challenging and exposed fundamental problems with simulation methods and force fields201,202. Recent studies, employing new lipid force fields203,204, have shown that atomic detail partitioning and folding of individual TM helices into explicit lipid bilayer membranes can be achieved via unbiased long-timescale molecular dynamics simulations on the multi-microsecond timescale205,206. However, folding of larger MP fragments and full multi-span membrane proteins remains difficult. Key challenges are: (1) the use of current protein force fields on the microsecond timescale has exposed flaws that need updating to reflect longer simulation timescales and new parameterizations might have to include solvent transfer free energies; (2) simulation timescales for protein folding are in the ms range and are now just starting to become accessible with modern computers, supercomputing clusters, and latest technologies such as GPUs207; (3) the inherent complexity of the membrane environment means that multi-span MPs need to be threaded correctly into the bilayer and prevented from getting trapped in non-native states; (4) the number of physical interactions (and the conformational search space) to be sampled increases dramatically for large proteins, such as MPs, and the associated lipids in the simulated unit cell. A review highlighting these challenges has recently been published208.
Coarse grained or all-atom simulations?
In 2003, Marrink and coworkers developed the coarse grained MARTINI force field to model the lipid bilayer explicitly209. MARTINI has since been extended to proteins185, and was used extensively to study events of the membrane bilayer such as raft formation210 (reviewed by Bennett & Tieleman211), membrane fusion212, and supramolecular assemblies in the membrane (like oligomerization of GPCRs213). Coarse-grained simulations are very useful for investigating larger-scale systems with MD, especially in the lipid bilayer, such as drug delivery using nanotubes214 or fullerenes215. However, studying the molecular mechanism of protein folding206,216, membrane partitioning217, or specific protein interactions (e. g. drug-protein218,219 or protein-lipid220) requires atomic detail representation of hydrogen bonds. For instance, atomic detail interactions were crucial to uncover the mechanisms of selectivity, ion conduction, and gating of potassium221,188 and sodium channels223, the voltage-dependent anion channel224, as well as an outer membrane protein225,190.
Which force field and software to use?
Several commonly used force fields were developed over the years and their accuracy was tested and compared to experimental data227. The most commonly used are AMBER179 and OPLS180, which have been used extensively for drug docking, and CHARMM228. Over the last few years highly accurate lipid parameterizations have been developed for these force fields, allowing simulation of proteins in a variety of atomic detail lipid bilayer membranes203,229. In addition, these all-atom force fields are supplemented with the previously mentioned MARTINI force field, which has coarse-grained representations of lipids and proteins. These force fields are implemented in several different software suites, i.e. most of the software can use most of the force fields. GROMACS230 is one of the fastest and most widely used packages, which support AMBER, OPLS, and CHARMM. Another popular package is NAMD231 and more recently Desmond232, which is fast, but not highly used at present.
Emerging techniques in Molecular Dynamics
Several methods have been introduced to accelerate MD simulations. Even though the methods discussed here are general approaches also applicable to soluble proteins, their impact on MP modeling is widespread because (1) of the larger size of MPs compared to most soluble proteins, and (2) of the large number of lipid molecules that need to be modeled in addition to the protein.
One of the most widely used methods is replica-exchange sampling233 (REMD, or parallel tempering). This method creates identical replicas of a system, which are simulated in parallel on an exponential temperature ladder. A Monte Carlo scheme is used to swap replicas regularly, melting kinetically trapped non-native conformations, accelerating sampling of phase space, and speeding up the transition towards the native state. The downside is that the kinetic information is lost when replicas are swapped.
Another related technique for increased sampling efficiency is MD at elevated temperatures. This approach was used to study the unbiased partitioning of peptides into the membrane and can speed up sampling of phase space by 2-3 orders of magnitude205,206, sufficient to accurately reproduce experimental membrane insertion free energies234. A requirement is a high thermostability of the peptide or protein that must be confirmed experimentally[2].
Whereas equilibrium MD is good at sampling low energy conformations, sampling of high-energy transition states (see reference 235 for a recent review about pathway sampling methods) is typically poor. Dynamic Importance Sampling (DIMS) solves this problem by sampling transition states between two known protein conformations. It relies on importance sampling, which assigns a higher ‘importance’ (a bias) to less frequently sampled transition states such that the distribution of computed states matches the experimentally observed one. In DIMS, however, the applied bias is constantly adjusted based on the current state that allows guiding the sampled protein conformation through high-energy states to a defined endpoint. DIMS-MD was applied to sample intermediate conformations in the transport cycle of LacY236 and to study gating transitions in voltage-gated potassium channels237.
While the above methods have allowed improvements of sampling and extension of simulation timescales, the largest improvements have come from the advancement in computer hardware. These developments were three-fold: (1) The rapid growth in speed and parallelization of Intel/AMD processors has extended the range of simulation timescales by 4 orders of magnitude over the last 15 years and shows no signs of abating; (2) the use of graphics cards (GPUs)207 for parallel scientific computations has resulted in a massive speedup. However, GPUs require a specific implementation of the code; (3) the development of distributed computing facilities such as Folding@home238, as well as dedicated massively parallel high-performance supercomputers, such as the Anton machine, which employs custom built hardware designed specifically for efficient MD simulation 204.
The future of MP modeling
Even though the MP modeling field has seen substantial progress in the past decade, many challenges lie ahead. Since we are now able to model MPs in a coarse-grained fashion, future research will most likely focus on MP function originating from different functional states of the proteins. At the same time, higher resolution representations of the protein (e.g. for design and for ligand binding) and the membrane itself (e.g. for specific lipid interactions) are important to understand MP function.
Membrane protein function and different conformational states
Even though knowledge of MP structure is beneficial to infer function, it is certainly insufficient. A single structure represents a protein trapped in a distinct conformational state (active or inactive, apo or holo, or an intermediate) and it does not reveal by itself how this protein functions, how it interacts with ligands or other proteins, or how it is regulated by other biomolecules or the membrane bilayer itself. Understanding MP function requires knowledge of structures in different conformational states that are often difficult to capture. The energy landscape of a protein depends on its sequence, ligand binding, protein-protein interactions, post-translational modifications, and membrane properties.240 Structural characterization of a particular state, such as a transition state, can be achieved by altering the system's variables and interactions to stabilize or trap the state of interest. As an example, GPCR functions span a range of conformational states, and capturing the active state requires stabilization to crystallize the protein (such as nanobodies for the muscarinic acetylcholine receptor241). Once stabilized, transient states can then be studied using different techniques such as NMR spectroscopy242, EPR, HD exchange experiments243, MD simulations244,245, normal mode analysis, and others. Even low-populated transient states can be captured with relaxation dispersion NMR spectroscopy246,247.
Protein function can be modulated by membrane properties. For instance, changes in membrane potential are sensed by the gating charges in the voltage sensor of voltage-gated channels, leading to increased pore domain fluctuations that ultimately open or close the channel248. Likewise, the spring constant of the lipid bilayer can cause channel opening and closing, as shown for the gramicidin channel249. An interesting experiment demonstrated that membrane asymmetry can also lead to channel opening: phospholipase has been used to cleave fatty acid chains from DOPC:DOPG lipids in one leaflet of the bilayer, converting them into lysophospholipids. Since flip-flop of the latter is orders of magnitude slower than for fatty acid chains, this resulted in membrane asymmetry leading to opening of the Mechanosensitive Channel of Large conductance (MscL)250.
The previous examples and future research will show that close integration of computational modeling with experimental validation is necessary and valuable to study MP function.
Membrane protein design
Protein design is one of the holy grails of structural biology. De novo design of soluble proteins remains difficult251,252 but some breakthroughs in recent years have been reported. The challenge for MPs is more pronounced, since both over-expression of sufficient material and experimental structure determination remain challenging. Point mutations and interface design are easier goals, even though the latter requires an understanding of the drivers for association, where helix-helix interfaces have been studied most85. De novo design and re-design of protein function are most challenging and require an advanced understanding of the underlying mechanism for MP stability and folding. Computational methods facilitate MP design but require accurate energy functions to discriminate ‘good’ from ‘bad’ designs, which ultimately determines which constructs are tested experimentally.
To bypass challenges in MP expression and solubilization, a soluble variant of the μ-opioid receptor was designed based on a homology model253. Interestingly, the solubilized form of the receptor had nanomolar affinity for the antagonist naltrexone, an affinity similar to the native human receptor. DeGrado's lab designed CHAMP peptides (computed helical anti-membrane protein) that disrupt association of, and therefore activate, integrin dimers (αIIbβ3 and αvβ3) in micelles, bacterial membranes, and mammalian cells254. Sapay et al. engineered a chimera of the muscarinic acetylcholine receptor M2 and the Kir6.2 potassium channel that can be used as a biosensor255. Ligand binding to the M2 receptor modulates Kir6.2 channel gating allowing the construct to be used for drug screening, biosensing, and diagnostics. Exciting developments are also the high-throughout methods such as the combined use of protein display and peptide libraries that has been used to identify a novel regulator for the GIRK2 potassium channel256. High-throughput screening of combinatorial synthetic peptide libraries has recently been successfully employed in the design and optimization of membrane active peptides257,258.
These examples demonstrate that MP design, even though a challenging endeavor, has made great initial progress with enormous potential for future research and that computational modeling is an integral part of the design process. Designs so far have concentrated on homology models and small functional changes whereas de novo design of novel function within MPs has yet to be realized.
Membrane asymmetry and lipid composition
Lipid composition asymmetry and distribution seem crucial for MP function, as some MPs function in certain membranes, but remain inactive in others. Lipid asymmetry results in different biophysical properties of the membrane leaflets and influences vesicle fusion, cell division, cell stability, apoptosis, and other processes. For example, phosphatidylserine and phosphatidylethanolamine occur chiefly in the cytosolic leaflet and contribute to membrane fluidity required for vesicle fusion, whereas the tighter packing of sphingolipids and sterols in the exoplasmic leaflet leads to enhanced barrier function and cell stability for instance in red blood cells259. Proteins responsible for flipping the phospholipids in the membrane are P-type ATPases246, lipid translocases, scramblases, and others261. Cholesterol asymmetry has been associated with amyloid processing and may explain the onset of Alzheimer's disease262. Membrane asymmetry can also explain differential sequestering of integrin complexes. The lipid asymmetry further affects the charge symmetry (or lack thereof) in MPs: the Dunbrack lab has found that the external side of outer membrane β-barrels has three times more charged residues than the internal side206.
Specific lipid interactions
An emerging direction is the investigation of lipid compositions and specific MP-lipid interactions264. Since MPs are usually stable in a variety of lipid or detergent compositions, the structural changes due to different lipid compositions and/or cofactors are likely small265. One example is the orientation of the voltage-sensor domain of KvAP that can be tuned between the resting and activated conformation by varying the charge in the lipid head group using lipids with or without phosphate groups266. In β-barrels, lipid binding can stabilize the protein in weakly stable regions267 using for instance sterol-binding motifs.
Modeling both membrane asymmetry as well as various lipid compositions and explicit MP-lipid interactions will require better energy functions that are able to distinguish native-like from non-native conformations. A current obstacle for parameterization of such energy functions is the lack of experimental data, i.e. MP structures in various membrane mimetic and/or bound to explicit lipids.
Thinking bigger: interaction networks
Interactions of MPs with ligands are studied regularly268, especially GPCRs269, as they are important drug targets. Inter-MP interactions are increasingly studied270 such that whole interaction networks can slowly be derived271, which in turn drive the development of new computational methods272, such as the prediction of interacting surface residues. Modeling MP association in MD simulations, however, is still on the horizon since it requires timescales to be modeled that are currently barely accessible, even with coarse-grained simulations273.
Conclusions
The past decade saw vast improvements in MP modeling, mainly due to an increasing availability of determined structures and newly developed algorithms. Secondary structure prediction and trans-membrane span prediction can generally be regarded as a solved problem, though improvements are needed in the prediction of more subtle phenomena such as kinks, re-entrant loops and half-helices, and precise beginnings and ends of secondary structure elements. Homology modeling has become more and more useful and important, mainly due to an increasing number of template structures combined with advances in modeling methods. The de novo prediction of both α-helical bundles as well as β-barrels has improved considerably in recent years, primarily because transitive correlations in evolutionary constraints can now be successfully filtered out such that modeling MPs up to 350 residues can yield models under 5 Å RMSD using information from multiple sequence alignments only. MD simulations have also made enormous progress through the improvement of energy functions and an increase in computational power that allows longer simulation times. Developments like these now make it possible to start explaining specific functions underlying MPs, that are modulated by asymmetric membranes and distinct lipid compositions of membrane bilayers. Recent progress that describes interactions between MPs/lipids and MPs/MPs up to MP interaction networks inspires the community with exciting developments still to come.
Acknowledgments
The authors would like to acknowledge the F. Stuart Hodgson Faculty Scholar Fund (to JJG) and NIH R01-GM078221 to JJG. We are also very grateful for the valuable comments from Kalina Hristova and Oliver Beckstein after proofreading the manuscript.
Footnotes
Terminology: A potential is a mathematical function describing the potential energy of a system, for instance the Lennard-Jones potential. A force field is the sum of individual potential energy terms. A scoring function describes the “correctness” of a prediction. The term scoring function is most often used when derived from a knowledge-base, the term force field is often used for physics-based potentials.
The method is generally not applicable to soluble proteins, which typically unfold even at slightly elevated temperatures of 40-50 °C.
Conflict of interest: The authors declare no conflict of interest.
References
- 1.Jones DT. Do transmembrane protein superfolds exist? FEBS Lett. 1998;423:281–285. doi: 10.1016/s0014-5793(98)00095-7. [DOI] [PubMed] [Google Scholar]
- 2.Wallin E, von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci Publ Protein Soc. 1998;7:1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yildirim MA, Goh KI, Cusick ME, Barabási AL, Vidal M. Drug-target network. Nat Biotechnol. 2007;25:1119–1126. doi: 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]
- 4.Grisshammer R, Tate CG. Overexpression of integral membrane proteins for structural studies. Q Rev Biophys. 1995;28:315–422. doi: 10.1017/s0033583500003504. [DOI] [PubMed] [Google Scholar]
- 5.Popot JL. Amphipols, nanodiscs, and fluorinated surfactants: three nonconventional approaches to studying membrane proteins in aqueous solutions. Annu Rev Biochem. 2010;79:737–75. doi: 10.1146/annurev.biochem.052208.114057. [DOI] [PubMed] [Google Scholar]
- 6.Bolla JR, Su CC, Yu EW. Biomolecular membrane protein crystallization. Philos Mag. 2012;92:2648–2661. doi: 10.1080/14786435.2012.670734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenbaum DM, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–73. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 8.Van Horn WD, et al. Solution nuclear magnetic resonance structure of membrane-integral diacylglycerol kinase. Science. 2009;324:1726–9. doi: 10.1126/science.1171716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cherezov V, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–65. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li X, et al. Structure of a presenilin family intramembrane aspartate protease. Nature. 2013;493:56–61. doi: 10.1038/nature11801. [DOI] [PubMed] [Google Scholar]
- 11.Baradaran R, Berrisford JM, Minhas GS, Sazanov LA. Crystal structure of the entire respiratory complex I. Nature. 2013;494:443–8. doi: 10.1038/nature11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kosinska Eriksson U, et al. Subangstrom resolution X-ray structure details aquaporin-water interactions. Science. 2013;340:1346–1349. doi: 10.1126/science.1234306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geibel S, Procko E, Hultgren SJ, Baker D, Waksman G. Structural and energetic basis of folded-protein transport by the FimD usher. Nature. 2013;496:243–246. doi: 10.1038/nature12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pervushin K, Riek R, Wider G, Wuthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci U A. 1997;94:12366–71. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanders CR, Sonnichsen F. Solution NMR of membrane proteins: practice and challenges. Magn Reson Chem. 2006;44 Spec No:S24–40. doi: 10.1002/mrc.1816. [DOI] [PubMed] [Google Scholar]
- 16.Otomo T, Teruya K, Uegaki K, Yamazaki T, Kyogoku Y. Improved segmental isotope labeling of proteins and application to a larger protein. J Biomol NMR. 1999;14:105–114. doi: 10.1023/a:1008308128050. [DOI] [PubMed] [Google Scholar]
- 17.Kainosho M, et al. Optimal isotope labelling for NMR protein structure determinations. Nature. 2006;440:52–57. doi: 10.1038/nature04525. [DOI] [PubMed] [Google Scholar]
- 18.Hyberts SG, Arthanari H, Robson SA, Wagner G. Perspectives in magnetic resonance: NMR in the post-FFT era. J Magn Reson San Diego Calif 1997. 2014;241:60–73. doi: 10.1016/j.jmr.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Voehler MW, Collier G, Young JK, Stone MP, Germann MW. Performance of cryogenic probes as a function of ionic strength and sample tube geometry. J Magn Reson. 2006;183:102–9. doi: 10.1016/j.jmr.2006.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koehler J, Meiler J. Expanding the utility of NMR restraints with paramagnetic compounds: Background and practical aspects. Prog Nucl Magn Reson Spectrosc. 2011;59:360–389. doi: 10.1016/j.pnmrs.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gautier A, Mott HR, Bostock MJ, Kirkpatrick JP, Nietlispach D. Structure determination of the seven-helix transmembrane receptor sensory rhodopsin II by solution NMR spectroscopy. Nat Struct Mol Biol. 2010;17:768–774. doi: 10.1038/nsmb.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reckel S, et al. Solution NMR structure of proteorhodopsin. Angew Chem Int Ed Engl. 2011;50:11942–6. doi: 10.1002/anie.201105648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patzelt H, et al. The structures of the active center in dark-adapted bacteriorhodopsin by solution-state NMR spectroscopy. Proc Natl Acad Sci U S A. 2002;99:9765–9770. doi: 10.1073/pnas.132253899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu L, et al. Nuclear magnetic resonance structural studies of a potassium channel-charybdotoxin complex. Biochemistry (Mosc) 2005;44:15834–41. doi: 10.1021/bi051656d. [DOI] [PubMed] [Google Scholar]
- 25.Berardi MJ, Shih WM, Harrison SC, Chou JJ. Mitochondrial uncoupling protein 2 structure determined by NMR molecular fragment searching. Nature. 2011;476:109–13. doi: 10.1038/nature10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bayrhuber M, et al. Structure of the human voltage-dependent anion channel. Proc Natl Acad Sci U S A. 2008;105:15370–5. doi: 10.1073/pnas.0808115105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shahid SA, et al. Membrane-protein structure determination by solid-state NMR spectroscopy of microcrystals. Nat Methods. 2012;9:1212–7. doi: 10.1038/nmeth.2248. [DOI] [PubMed] [Google Scholar]
- 28.Wang S, et al. Solid-state NMR spectroscopy structure determination of a lipid-embedded heptahelical membrane protein. Nat Methods. 2013;10:1007–1012. doi: 10.1038/nmeth.2635. [DOI] [PubMed] [Google Scholar]
- 29.Liao M, Cao E, Julius D, Cheng Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 2013;504:107–112. doi: 10.1038/nature12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gogala M, et al. Structures of the Sec61 complex engaged in nascent peptide translocation or membrane insertion. Nature. 2014;506:107–10. doi: 10.1038/nature12950. [DOI] [PubMed] [Google Scholar]
- 31.Kloppmann E, Punta M, Rost B. Structural genomics plucks high-hanging membrane proteins. Curr Opin Struct Biol. 2012;22:326–32. doi: 10.1016/j.sbi.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klammt C, et al. Facile backbone structure determination of human membrane proteins by NMR spectroscopy. Nat Methods. 2012;9:834–839. doi: 10.1038/nmeth.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pieper U, et al. Coordinating the impact of structural genomics on the human alpha-helical transmembrane proteome. Nat Struct Mol Biol. 2013;20:135–8. doi: 10.1038/nsmb.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hedin LE, Illergård K, Elofsson A. An introduction to membrane proteins. J Proteome Res. 2011;10:3324–3331. doi: 10.1021/pr200145a. [DOI] [PubMed] [Google Scholar]
- 35.Otzen DE, Andersen KK. Folding of outer membrane proteins. Arch Biochem Biophys. 2013;531:34–43. doi: 10.1016/j.abb.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 36.Rost B, Sander C. Third generation prediction of secondary structures. Methods Mol Biol Clifton NJ. 2000;143:71–95. doi: 10.1385/1-59259-368-2:71. [DOI] [PubMed] [Google Scholar]
- 37.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 38.Meiler J, Baker D. Coupled prediction of protein secondary and tertiary structure. Proc Natl Acad Sci U A. 2003;100:12105–10. doi: 10.1073/pnas.1831973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leman JK, Mueller R, Karakas M, Woetzel N, Meiler J. Simultaneous prediction of protein secondary structure and transmembrane spans. Proteins. 2013;81:1127–40. doi: 10.1002/prot.24258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rost B, Liu J. The Predict Protein server. Nucleic Acids Res. 2003;31:3300–4. doi: 10.1093/nar/gkg508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:W197–201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bull HB, Breese K. Surface tension of amino acid solutions: a hydrophobicity scale of the amino acid residues. Arch Biochem Biophys. 1974;161:665–70. doi: 10.1016/0003-9861(74)90352-x. [DOI] [PubMed] [Google Scholar]
- 43.Wimley WC, Creamer TP, White SH. Solvation energies of amino acid side chains and backbone in a family of host-guest pentapeptides. Biochemistry (Mosc) 1996;35:5109–24. doi: 10.1021/bi9600153. [DOI] [PubMed] [Google Scholar]
- 44.Engelman DM, Steitz TA, Goldman A. Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. Annu Rev Biophys Biophys Chem. 1986;15:321–53. doi: 10.1146/annurev.bb.15.060186.001541. [DOI] [PubMed] [Google Scholar]
- 45.Koehler J, Woetzel N, Staritzbichler R, Sanders CR, Meiler J. A unified hydrophobicity scale for multispan membrane proteins. Proteins. 2009;76:13–29. doi: 10.1002/prot.22315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–32. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 47.Eisenberg D, Weiss RM, Terwilliger TC. The Helical Hydrophobic Moment - a Measure of the Amphiphilicity of a Helix. Nature. 1982;299:371–374. doi: 10.1038/299371a0. [DOI] [PubMed] [Google Scholar]
- 48.Guy HR. Amino acid side-chain partition energies and distribution of residues in soluble proteins. Biophys J. 1985;47:61–70. doi: 10.1016/S0006-3495(85)83877-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Janin J. Surface and inside volumes in globular proteins. Nature. 1979;277:491–2. doi: 10.1038/277491a0. [DOI] [PubMed] [Google Scholar]
- 50.Beuming T, Weinstein H. Development and application of a knowledge-based scale for the analysis and prediction of interior facing surfaces in transmembrane protein domains. Biophys J. 2004;86:256a–256a. doi: 10.1093/bioinformatics/bth143. [DOI] [PubMed] [Google Scholar]
- 51.Hessa T, et al. Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature. 2007;450:1026–U2. doi: 10.1038/nature06387. [DOI] [PubMed] [Google Scholar]
- 52.Moon CP, Fleming KG. Side-chain hydrophobicity scale derived from transmembrane protein folding into lipid bilayers. Proc Natl Acad Sci U A. 2011;108:10174–7. doi: 10.1073/pnas.1103979108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peters C, Elofsson A. Why is the biological hydrophobicity scale more accurate than earlier experimental hydrophobicity scales? Proteins. 2014 doi: 10.1002/prot.24582. [DOI] [PubMed] [Google Scholar]
- 54.Viklund H, Elofsson A. OCTOPUS: improving topology prediction by two-track ANN-based preference scores and an extended topological grammar. Bioinformatics. 2008;24:1662–8. doi: 10.1093/bioinformatics/btn221. [DOI] [PubMed] [Google Scholar]
- 55.Kahsay RY, Gao G, Liao L. An improved hidden Markov model for transmembrane protein detection and topology prediction and its applications to complete genomes. Bioinforma Oxf Engl. 2005;21:1853–1858. doi: 10.1093/bioinformatics/bti303. [DOI] [PubMed] [Google Scholar]
- 56.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 57.Nugent T, Jones DT. Transmembrane protein topology prediction using support vector machines. BMC Bioinformatics. 2009;10 doi: 10.1186/1471-2105-10-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Von Heijne G. Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J Mol Biol. 1992;225:487–94. doi: 10.1016/0022-2836(92)90934-c. [DOI] [PubMed] [Google Scholar]
- 59.Tusnády GE, Kalmár L, Simon I. TOPDB: topology data bank of transmembrane proteins. Nucleic Acids Res. 2008;36:D234–239. doi: 10.1093/nar/gkm751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tsaousis GN, et al. ExTopoDB: a database of experimentally derived topological models of transmembrane proteins. Bioinformatics. 2010;26:2490–2. doi: 10.1093/bioinformatics/btq362. [DOI] [PubMed] [Google Scholar]
- 61.Tusnady GE, Kalmar L, Hegyi H, Tompa P, Simon I. TOPDOM: database of domains and motifs with conservative location in transmembrane proteins. Bioinformatics. 2008;24:1469–70. doi: 10.1093/bioinformatics/btn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hayat S, Elofsson A. BOCTOPUS: improved topology prediction of transmembrane beta barrel proteins. Bioinformatics. 2012;28:516–22. doi: 10.1093/bioinformatics/btr710. [DOI] [PubMed] [Google Scholar]
- 63.Savojardo C, Fariselli P, Casadio R. BETAWARE: a machine-learning tool to detect and predict transmembrane beta-barrel proteins in prokaryotes. Bioinforma Oxf Engl. 2013;29:504–505. doi: 10.1093/bioinformatics/bts728. [DOI] [PubMed] [Google Scholar]
- 64.Gromiha MM, Ahmad S, Suwa M. TMBETA-NET: discrimination and prediction of membrane spanning beta-strands in outer membrane proteins. Nucleic Acids Res. 2005;33:W164–167. doi: 10.1093/nar/gki367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Singh NK, Goodman A, Walter P, Helms V, Hayat S. TMBHMM: a frequency profile based HMM for predicting the topology of transmembrane beta barrel proteins and the exposure status of transmembrane residues. Biochim Biophys Acta. 2011;1814:664–70. doi: 10.1016/j.bbapap.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 66.Bigelow H, Rost B. PROFtmb: a web server for predicting bacterial transmembrane beta barrel proteins. Nucleic Acids Res. 2006;34:W186–8. doi: 10.1093/nar/gkl262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhai Y, Saier MH. A web-based program (WHAT) for the simultaneous prediction of hydropathy, amphipathicity, secondary structure and transmembrane topology for a single protein sequence. J Mol Microbiol Biotechnol. 2001;3:501–2. [PubMed] [Google Scholar]
- 68.Chou KC, Zhang CT, Maggiora GM. Disposition of amphiphilic helices in heteropolar environments. Proteins. 1997;28:99–108. [PubMed] [Google Scholar]
- 69.Sapay N, Guermeur Y, Deleage G. Prediction of amphipathic in-plane membrane anchors in monotopic proteins using a SVM classifier. BMC Bioinformatics. 2006;7:255. doi: 10.1186/1471-2105-7-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gautier R, Douguet D, Antonny B, Drin G. HELIQUEST: a web server to screen sequences with specific alpha-helical properties. Bioinformatics. 2008;24:2101–2. doi: 10.1093/bioinformatics/btn392. [DOI] [PubMed] [Google Scholar]
- 71.Meruelo AD, Samish I, Bowie JU. TMKink: a method to predict transmembrane helix kinks. Protein Sci Publ Protein Soc. 2011;20:1256–1264. doi: 10.1002/pro.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yohannan S, Faham S, Yang D, Whitelegge JP, Bowie JU. The evolution of transmembrane helix kinks and the structural diversity of G protein-coupled receptors. Proc Natl Acad Sci U S A. 2004;101:959–63. doi: 10.1073/pnas.0306077101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yuan Z, Zhang F, Davis MJ, Boden M, Teasdale RD. Predicting the solvent accessibility of transmembrane residues from protein sequence. J Proteome Res. 2006;5:1063–70. doi: 10.1021/pr050397b. [DOI] [PubMed] [Google Scholar]
- 74.Park Y, Hayat S, Helms V. Prediction of the burial status of transmembrane residues of helical membrane proteins. BMC Bioinformatics. 2007;8:302. doi: 10.1186/1471-2105-8-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nguyen D, Helms V, Lee PH. PRIMSIPLR: Prediction of inner-membrane situated pore-lining residues for alpha-helical transmembrane proteins. Proteins. 2014 doi: 10.1002/prot.24520. [DOI] [PubMed] [Google Scholar]
- 76.Adamian L, Nanda V, DeGrado WF, Liang J. Empirical lipid propensities of amino acid residues in multispan alpha helical membrane proteins. Proteins. 2005;59:496–509. doi: 10.1002/prot.20456. [DOI] [PubMed] [Google Scholar]
- 77.Adamian L, Liang J. Prediction of transmembrane helix orientation in polytopic membrane proteins. BMC Struct Biol. 2006;6:13. doi: 10.1186/1472-6807-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Petersen B, Petersen TN, Andersen P, Nielsen M, Lundegaard C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct Biol. 2009;9:51. doi: 10.1186/1472-6807-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Adamczak R, Porollo A, Meller J. Combining prediction of secondary structure and solvent accessibility in proteins. Proteins. 2005;59:467–75. doi: 10.1002/prot.20441. [DOI] [PubMed] [Google Scholar]
- 80.Lemmon MA, Flanagan JM, Treutlein HR, Zhang J, Engelman DM. Sequence specificity in the dimerization of transmembrane alpha-helices. Biochemistry (Mosc) 1992;31:12719–25. doi: 10.1021/bi00166a002. [DOI] [PubMed] [Google Scholar]
- 81.Senes A, Gerstein M, Engelman DM. Statistical analysis of amino acid patterns in transmembrane helices: the GxxxG motif occurs frequently and in association with beta-branched residues at neighboring positions. J Mol Biol. 2000;296:921–36. doi: 10.1006/jmbi.1999.3488. [DOI] [PubMed] [Google Scholar]
- 82.Curran AR, Engelman DM. Sequence motifs, polar interactions and conformational changes in helical membrane proteins. Curr Opin Struct Biol. 2003;13:412–7. doi: 10.1016/s0959-440x(03)00102-7. [DOI] [PubMed] [Google Scholar]
- 83.Langosch D, Arkin IT. Interaction and conformational dynamics of membrane-spanning protein helices. Protein Sci. 2009;18:1343–58. doi: 10.1002/pro.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Marsico A, et al. MeMotif: a database of linear motifs in alpha-helical transmembrane proteins. Nucleic Acids Res. 2010;38:D181–9. doi: 10.1093/nar/gkp1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Walters RF, DeGrado WF. Helix-packing motifs in membrane proteins. Proc Natl Acad Sci U S A. 2006;103:13658–63. doi: 10.1073/pnas.0605878103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yang J, Jang R, Zhang Y, Shen HB. High-accuracy prediction of transmembrane inter-helix contacts and application to GPCR 3D structure modeling. Bioinforma Oxf Engl. 2013;29:2579–2587. doi: 10.1093/bioinformatics/btt440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lee SY, Skolnick J. Benchmarking of TASSER_2.0: an improved protein structure prediction algorithm with more accurate predicted contact restraints. Biophys J. 2008;95:1956–64. doi: 10.1529/biophysj.108.129759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Polyansky AA, et al. PREDDIMER: a web server for prediction of transmembrane helical dimers. Bioinforma Oxf Engl. 2014;30:889–890. doi: 10.1093/bioinformatics/btt645. [DOI] [PubMed] [Google Scholar]
- 89.Li E, Wimley WC, Hristova K. Transmembrane helix dimerization: Beyond the search for sequence motifs. Biochim Biophys Acta BBA - Biomembr. 2012;1818:183–193. doi: 10.1016/j.bbamem.2011.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Baker D, Sali A. Protein structure prediction and structural genomics. Science. 2001;294:93–6. doi: 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- 91.Pirovano W, Feenstra KA, Heringa J. PRALINETM: a strategy for improved multiple alignment of transmembrane proteins. Bioinformatics. 2008;24:492–7. doi: 10.1093/bioinformatics/btm636. [DOI] [PubMed] [Google Scholar]
- 92.Ng PC, Henikoff JG, Henikoff S. PHAT: a transmembrane-specific substitution matrix. Predicted hydrophobic and transmembrane. Bioinformatics. 2000;16:760–6. doi: 10.1093/bioinformatics/16.9.760. [DOI] [PubMed] [Google Scholar]
- 93.Hill JR, Deane CM. MP-T: improving membrane protein alignment for structure prediction. Bioinforma Oxf Engl. 2013;29:54–61. doi: 10.1093/bioinformatics/bts640. [DOI] [PubMed] [Google Scholar]
- 94.Shi J, Blundell TL, Mizuguchi K. FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J Mol Biol. 2001;310:243–57. doi: 10.1006/jmbi.2001.4762. [DOI] [PubMed] [Google Scholar]
- 95.Chetwynd AP, Scott KA, Mokrab Y, Sansom MS. CGDB: a database of membrane protein/lipid interactions by coarse-grained molecular dynamics simulations. Mol Membr Biol. 2008;25:662–9. doi: 10.1080/09687680802446534. [DOI] [PubMed] [Google Scholar]
- 96.Yarov-Yarovoy V, Schonbrun J, Baker D. Multipass membrane protein structure prediction using Rosetta. Proteins-Struct Funct Bioinforma. 2006;62:1010–1025. doi: 10.1002/prot.20817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Barth P, Schonbrun J, Baker D. Toward high-resolution prediction and design of transmembrane helical protein structures. Proc Natl Acad Sci. 2007;104:15682–15687. doi: 10.1073/pnas.0702515104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yarov-Yarovoy V, Baker D, Catterall WA. Voltage sensor conformations in the open and closed states in ROSETTA structural models of K(+) channels. Proc Natl Acad Sci U A. 2006;103:7292–7. doi: 10.1073/pnas.0602350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Subbotina J, et al. Structural refinement of the hERG1 pore and voltage-sensing domains with ROSETTA-membrane and molecular dynamics simulations. Proteins. 2010;78:2922–34. doi: 10.1002/prot.22815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Eswar N, et al. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2007;Chapter 2, Unit 2 9 doi: 10.1002/0471140864.ps0209s50. [DOI] [PubMed] [Google Scholar]
- 101.Tabassum A, et al. Structural characterization and mutational assessment of podocin - A novel drug target to nephrotic syndrome - An in silico approach. Interdiscip Sci. 2014;6:32–9. doi: 10.1007/s12539-014-0190-4. [DOI] [PubMed] [Google Scholar]
- 102.Kelm S, Shi J, Deane CM. MEDELLER: homology-based coordinate generation for membrane proteins. Bioinformatics. 2010;26:2833–40. doi: 10.1093/bioinformatics/btq554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ebejer JP, Hill JR, Kelm S, Shi J, Deane CM. Memoir: template-based structure prediction for membrane proteins. Nucleic Acids Res. 2013;41:W379–383. doi: 10.1093/nar/gkt331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hildebrand A, Remmert M, Biegert A, Soding J. Fast and accurate automatic structure prediction with HHpred. Proteins. 2009;77 Suppl 9:128–32. doi: 10.1002/prot.22499. [DOI] [PubMed] [Google Scholar]
- 105.Guex N, Peitsch MC, Schwede T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis. 2009;30 Suppl 1:S162–173. doi: 10.1002/elps.200900140. [DOI] [PubMed] [Google Scholar]
- 106.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–38. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang H, He Z, Zhang C, Zhang L, Xu D. Transmembrane protein alignment and fold recognition based on predicted topology. PLoS One. 2013;8:e69744. doi: 10.1371/journal.pone.0069744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–60. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 109.Fagerberg L, Jonasson K, von Heijne G, Uhlén M, Berglund L. Prediction of the human membrane proteome. Proteomics. 2010;10:1141–1149. doi: 10.1002/pmic.200900258. [DOI] [PubMed] [Google Scholar]
- 110.Liu J, Rost B. Comparing function and structure between entire proteomes. Protein Sci. 2001;10:1970–9. doi: 10.1110/ps.10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lazaridis T. Effective energy function for proteins in lipid membranes. Proteins. 2003;52:176–92. doi: 10.1002/prot.10410. [DOI] [PubMed] [Google Scholar]
- 112.Karakaş M, et al. BCL∷Fold--de novo prediction of complex and large protein topologies by assembly of secondary structure elements. PloS One. 2012;7:e49240. doi: 10.1371/journal.pone.0049240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Woetzel N, et al. BCL∷Score--knowledge based energy potentials for ranking protein models represented by idealized secondary structure elements. PLoS One. 2012;7:e49242. doi: 10.1371/journal.pone.0049242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Weiner BE, Woetzel N, Karakaş M, Alexander N, Meiler J. BCL∷MP-fold: folding membrane proteins through assembly of transmembrane helices. Struct Lond Engl 1993. 2013;21:1107–1117. doi: 10.1016/j.str.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hopf TA, et al. Three-Dimensional Structures of Membrane Proteins from Genomic Sequencing. Cell. 2012;149:1607–1621. doi: 10.1016/j.cell.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nugent T, Jones DT. Accurate de novo structure prediction of large transmembrane protein domains using fragment-assembly and correlated mutation analysis. Proc Natl Acad Sci. 2012;109:E1540–E1547. doi: 10.1073/pnas.1120036109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Stevens RC, et al. The GPCR Network: a large-scale collaboration to determine human GPCR structure and function. Nat Rev Drug Discov. 2012;12:25–34. doi: 10.1038/nrd3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shacham S, et al. Modeling the 3D structure of GPCRs from sequence. Med Res Rev. 2001;21:472–83. doi: 10.1002/med.1019. [DOI] [PubMed] [Google Scholar]
- 119.Vaidehi N, et al. Prediction of structure and function of G protein-coupled receptors. Proc Natl Acad Sci U S A. 2002;99:12622–12627. doi: 10.1073/pnas.122357199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Michino M, Chen J, Stevens RC, Brooks CL., 3rd FoldGPCR: structure prediction protocol for the transmembrane domain of G protein-coupled receptors from class A. Proteins. 2010;78:2189–2201. doi: 10.1002/prot.22731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang Y, Devries ME, Skolnick J. Structure modeling of all identified G protein-coupled receptors in the human genome. PLoS Comput Biol. 2006;2:e13. doi: 10.1371/journal.pcbi.0020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Oberai A, Ihm Y, Kim S, Bowie JU. A limited universe of membrane protein families and folds. Protein Sci Publ Protein Soc. 2006;15:1723–1734. doi: 10.1110/ps.062109706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nishida M, Cadene M, Chait BT, MacKinnon R. Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. Embo J. 2007;26:4005–15. doi: 10.1038/sj.emboj.7601828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Whorton MR, MacKinnon R. Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell. 2011;147:199–208. doi: 10.1016/j.cell.2011.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gutman GA, et al. International Union of Pharmacology. XLI. Compendium of voltage-gated ion channels: potassium channels. Pharmacol Rev. 2003;55:583–586. doi: 10.1124/pr.55.4.9. [DOI] [PubMed] [Google Scholar]
- 127.Doyle DA, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 128.Jiang Y, et al. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 129.Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 130.Kuo A, et al. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 2003;300:1922–6. doi: 10.1126/science.1085028. [DOI] [PubMed] [Google Scholar]
- 131.Jiang Y, et al. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 2002;417:515–522. doi: 10.1038/417515a. [DOI] [PubMed] [Google Scholar]
- 132.Berneche S, Roux B. Molecular dynamics of the KcsA K(+) channel in a bilayer membrane. Biophys J. 2000;78:2900–17. doi: 10.1016/S0006-3495(00)76831-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Allen TW, Chung SH. Brownian dynamics study of an open-state KcsA potassium channel. Biochim Biophys Acta. 2001;1515:83–91. doi: 10.1016/s0005-2736(01)00395-9. [DOI] [PubMed] [Google Scholar]
- 134.Berneche S, Roux B. A microscopic view of ion conduction through the K+ channel. Proc Natl Acad Sci U A. 2003;100:8644–8. doi: 10.1073/pnas.1431750100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fernandez-Ballester G, Fernandez-Carvajal A, Gonzalez-Ros JM, Ferrer-Montiel A. Ionic channels as targets for drug design: a review on computational methods. Pharmaceutics. 2011;3:932–53. doi: 10.3390/pharmaceutics3040932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Saini L, Gupta SP, Kumar Satluri VS. A QSAR study on some series of sodium and potassium channel blockers. Med Chem. 2009;5:570–6. doi: 10.2174/157340609790170524. [DOI] [PubMed] [Google Scholar]
- 137.Peukert S, et al. Pharmacophore-based search, synthesis, and biological evaluation of anthranilic amides as novel blockers of the Kv1.5 channel. Bioorg Med Chem Lett. 2004;14:2823–7. doi: 10.1016/j.bmcl.2004.03.057. [DOI] [PubMed] [Google Scholar]
- 138.Du L, Li M, You Q, Xia L. A novel structure-based virtual screening model for the hERG channel blockers. Biochem Biophys Res Commun. 2007;355:889–894. doi: 10.1016/j.bbrc.2007.02.068. [DOI] [PubMed] [Google Scholar]
- 139.Butkiewicz M, et al. Benchmarking ligand-based virtual High-Throughput Screening with the PubChem database. Molecules. 2013;18:735–56. doi: 10.3390/molecules18010735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- 141.Durdagi S, Deshpande S, Duff HJ, Noskov SY. Modeling of open, closed, and open-inactivated states of the hERG1 channel: structural mechanisms of the state-dependent drug binding. J Chem Inf Model. 2012;52:2760–2774. doi: 10.1021/ci300353u. [DOI] [PubMed] [Google Scholar]
- 142.Ma J, et al. hERG Potassium Channel Blockage by Scorpion Toxin BmKKx2 Enhances Erythroid Differentiation of Human Leukemia Cells K562. PLoS One. 2013;8:e84903. doi: 10.1371/journal.pone.0084903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jimenez-Vargas JM, Restano-Cassulini R, Possani LD. Interacting sites of scorpion toxin ErgTx1 with hERG1 K+ channels. Toxicon Off J Int Soc Toxinology. 2012;59:633–641. doi: 10.1016/j.toxicon.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 144.Redaelli E, et al. Target promiscuity and heterogeneous effects of tarantula venom peptides affecting Na+ and K+ ion channels. J Biol Chem. 2010;285:4130–42. doi: 10.1074/jbc.M109.054718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Huang PT, Shiau YS, Lou KL. The interaction of spider gating modifier peptides with voltage-gated potassium channels. Toxicon. 2007;49:285–92. doi: 10.1016/j.toxicon.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 146.Wimley WC. The versatile beta-barrel membrane protein. Curr Opin Struct Biol. 2003;13:404–11. doi: 10.1016/s0959-440x(03)00099-x. [DOI] [PubMed] [Google Scholar]
- 147.Clote P, Waldispuhl J, Berger B, Steyaert JM. transFold: a web server for predicting the structure and residue contacts of transmembrane beta-barrels. Nucleic Acids Res. 2006;34:W189–W193. doi: 10.1093/nar/gkl205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Waldispuhl J, O'Donnell CW, Devadas S, Clote P, Berger B. Modeling ensembles of transmembrane beta-barrel proteins. Proteins-Struct Funct Bioinforma. 2008;71:1097–112. doi: 10.1002/prot.21788. [DOI] [PubMed] [Google Scholar]
- 149.Tran VDT, Chassignet P, Sheikh S, Steyaert JM. A graph-theoretic approach for classification and structure prediction of transmembrane beta-barrel proteins. Bmc Genomics. 2012;13 doi: 10.1186/1471-2164-13-S2-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Randall A, Cheng J, Sweredoski M, Baldi P. TMBpro: secondary structure, beta-contact and tertiary structure prediction of transmembrane beta-barrel proteins. Bioinformatics. 2008;24:513–20. doi: 10.1093/bioinformatics/btm548. [DOI] [PubMed] [Google Scholar]
- 151.Hayat S, Elofsson A. Ranking models of transmembrane beta-barrel proteins using Z-coordinate predictions. Bioinformatics. 2012;28:i90–6. doi: 10.1093/bioinformatics/bts233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Naveed H, Xu Y, Jackups R, Liang J. Predicting three-dimensional structures of transmembrane domains of beta-barrel membrane proteins. J Am Chem Soc. 2012;134:1775–81. doi: 10.1021/ja209895m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Defay T, Cohen FE. Evaluation of current techniques for Ab initio protein structure prediction. Proteins Struct Funct Bioinforma. 1995;23:431–445. doi: 10.1002/prot.340230317. [DOI] [PubMed] [Google Scholar]
- 154.Skolnick J, Kolinski A, Ortiz AR. MONSSTER: a method for folding globular proteins with a small number of distance restraints. J Mol Biol. 1997;265:217–41. doi: 10.1006/jmbi.1996.0720. [DOI] [PubMed] [Google Scholar]
- 155.Bowers PM, Strauss CE, Baker D. De novo protein structure determination using sparse NMR data. J Biomol NMR. 2000;18:311–318. doi: 10.1023/a:1026744431105. [DOI] [PubMed] [Google Scholar]
- 156.Meiler J, Baker D. Rapid protein fold determination using unassigned NMR data. Proc Natl Acad Sci U A. 2003;100:15404–9. doi: 10.1073/pnas.2434121100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Latek D, Ekonomiuk D, Kolinski A. Protein structure prediction: combining de novo modeling with sparse experimental data. J Comput Chem. 2007;28:1668–76. doi: 10.1002/jcc.20657. [DOI] [PubMed] [Google Scholar]
- 158.Raman S, et al. NMR structure determination for larger proteins using backbone-only data. Science. Feb 19;327:1014–8. doi: 10.1126/science.1183649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Thompson JM, et al. Accurate protein structure modeling using sparse NMR data and homologous structure information. Proc Natl Acad Sci U S A. 2012;109:9875–80. doi: 10.1073/pnas.1202485109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lange OF, Baker D. Resolution-adapted recombination of structural features significantly improves sampling in restraint-guided structure calculation. Proteins. 2012;80:884–895. doi: 10.1002/prot.23245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Berjanskii M, et al. GeNMR: a web server for rapid NMR-based protein structure determination. Nucleic Acids Res. 2009;37:W670–677. doi: 10.1093/nar/gkp280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Barth P, Wallner B, Baker D. Prediction of membrane protein structures with complex topologies using limited constraints. Proc Natl Acad Sci U A. 2009;106:1409–14. doi: 10.1073/pnas.0808323106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lange OF, et al. Determination of solution structures of proteins up to 40 kDa using CS-Rosetta with sparse NMR data from deuterated samples. Proc Natl Acad Sci U S A. 2012;109:10873–10878. doi: 10.1073/pnas.1203013109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Weiner BE, et al. BCL∷Fold-Protein topology determination from limited NMR restraints. Proteins. 2014;82:587–95. doi: 10.1002/prot.24427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Chen H, et al. Optimal mutation sites for PRE data collection and membrane protein structure prediction. Structure. 2011;19:484–95. doi: 10.1016/j.str.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kazmier K, Alexander NS, Meiler J, McHaourab HS. Algorithm for selection of optimized EPR distance restraints for de novo protein structure determination. J Struct Biol. 2011;173:549–557. doi: 10.1016/j.jsb.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hirst SJ, Alexander N, McHaourab HS, Meiler J. RosettaEPR: an integrated tool for protein structure determination from sparse EPR data. J Struct Biol. 2011;173:506–514. doi: 10.1016/j.jsb.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Alexander N, Bortolus M, Al-Mestarihi A, McHaourab H, Meiler J. De novo high-resolution protein structure determination from sparse spin-labeling EPR data. Structure. 2008;16:181–95. doi: 10.1016/j.str.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Islam SM, Stein RA, Mchaourab HS, Roux B. Structural Refinement from Restrained-Ensemble Simulations Based on EPR/DEER Data: Application to T4 Lysozyme. J Phys Chem B. 2013;117:4740–4754. doi: 10.1021/jp311723a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lindert S, et al. Ab initio protein modeling into CryoEM density maps using EM-Fold. Biopolymers. 2012;97:669–77. doi: 10.1002/bip.22027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.DiMaio F, Tyka MD, Baker ML, Chiu W, Baker D. Refinement of protein structures into low-resolution density maps using rosetta. J Mol Biol. 2009;392:181–90. doi: 10.1016/j.jmb.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Forster F, et al. Integration of small-angle X-ray scattering data into structural modeling of proteins and their assemblies. J Mol Biol. 2008;382:1089–106. doi: 10.1016/j.jmb.2008.07.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Pons C, et al. Structural characterization of protein-protein complexes by integrating computational docking with small-angle scattering data. J Mol Biol. 2010;403:217–30. doi: 10.1016/j.jmb.2010.08.029. [DOI] [PubMed] [Google Scholar]
- 174.Politis A, et al. A mass spectrometry-based hybrid method for structural modeling of protein complexes. Nat Methods. 2014 doi: 10.1038/nmeth.2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kahraman A, et al. Cross-link guided molecular modeling with ROSETTA. PLoS One. 2013;8:e73411. doi: 10.1371/journal.pone.0073411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Park E, et al. Structure of the SecY channel during initiation of protein translocation. Nature. 2014;506:102–106. doi: 10.1038/nature12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Greber BJ, et al. Architecture of the large subunit of the mammalian mitochondrial ribosome. Nature. 2014;505:515–9. doi: 10.1038/nature12890. [DOI] [PubMed] [Google Scholar]
- 178.Brooks BR, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 180.Jorgensen WL, Tirado-Rives J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J Am Chem Soc. 1988;110:1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 181.Lazaridis T, Karplus M. Effective energy function for proteins in solution. Proteins. 1999;35:133–52. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 182.Spassov VZ, Yan L, Szalma S. Introducing an Implicit Membrane in Generalized Born/Solvent Accessibility Continuum Solvent Models. J Phys Chem B. 2002;106:8726–8738. [Google Scholar]
- 183.Im W, Brooks CL., 3rd Interfacial folding and membrane insertion of designed peptides studied by molecular dynamics simulations. Proc Natl Acad Sci U S A. 2005;102:6771–6776. doi: 10.1073/pnas.0408135102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ulmschneider JP, Ulmschneider MB. Folding Simulations of the Transmembrane Helix of Virus Protein U in an Implicit Membrane Model. J Chem Theory Comput. 2007;3:2335–2346. doi: 10.1021/ct700103k. [DOI] [PubMed] [Google Scholar]
- 185.Monticelli L, et al. The MARTINI coarse-grained force field: Extension to proteins. J Chem Theory Comput. 2008;4:819–834. doi: 10.1021/ct700324x. [DOI] [PubMed] [Google Scholar]
- 186.Wassenaar TA, Ingolfsson HI, Priess M, Marrink SJ, Schafer LV. Mixing MARTINI: electrostatic coupling in hybrid atomistic-coarse-grained biomolecular simulations. J Phys Chem B. 2013;117:3516–30. doi: 10.1021/jp311533p. [DOI] [PubMed] [Google Scholar]
- 187.Adamian L, Liang J. Helix-helix packing and interfacial pairwise interactions of residues in membrane proteins. J Mol Biol. 2001;311:891–907. doi: 10.1006/jmbi.2001.4908. [DOI] [PubMed] [Google Scholar]
- 188.Fleishman SJ, Ben-Tal N. A novel scoring function for predicting the conformations of tightly packed pairs of transmembrane alpha-helices. J Mol Biol. 2002;321:363–378. doi: 10.1016/s0022-2836(02)00590-9. [DOI] [PubMed] [Google Scholar]
- 189.Wendel C, Gohlke H. Predicting transmembrane helix pair configurations with knowledge-based distance-dependent pair potentials. Proteins. 2008;70:984–999. doi: 10.1002/prot.21574. [DOI] [PubMed] [Google Scholar]
- 190.Park Y, Elsner M, Staritzbichler R, Helms V. Novel scoring function for modeling structures of oligomers of transmembrane alpha-helices. Proteins. 2004;57:577–585. doi: 10.1002/prot.20229. [DOI] [PubMed] [Google Scholar]
- 191.Illergard K, Callegari S, Elofsson A. MPRAP: an accessibility predictor for a-helical transmembrane proteins that performs well inside and outside the membrane. BMC Bioinformatics. 2010;11:333. doi: 10.1186/1471-2105-11-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Mihajlovic M, Lazaridis T. Calculations of pH-dependent binding of proteins to biological membranes. J Phys Chem B. 2006;110:3375–84. doi: 10.1021/jp055906b. [DOI] [PubMed] [Google Scholar]
- 193.Yuzlenko O, Lazaridis T. Interactions between ionizable amino acid side chains at a lipid bilayer-water interface. J Phys Chem B. 2011;115:13674–84. doi: 10.1021/jp2052213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Forrest LR, Woolf TB. Discrimination of native loop conformations in membrane proteins: decoy library design and evaluation of effective energy scoring functions. Proteins. 2003;52:492–509. doi: 10.1002/prot.10404. [DOI] [PubMed] [Google Scholar]
- 195.Gao C, Stern HA. Scoring function accuracy for membrane protein structure prediction. Proteins. 2007;68:67–75. doi: 10.1002/prot.21421. [DOI] [PubMed] [Google Scholar]
- 196.Yuzlenko O, Lazaridis T. Membrane protein native state discrimination by implicit membrane models. J Comput Chem. 2013;34:731–8. doi: 10.1002/jcc.23189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ray A, Lindahl E, Wallner B. Model quality assessment for membrane proteins. Bioinforma Oxf Engl. 2010;26:3067–3074. doi: 10.1093/bioinformatics/btq581. [DOI] [PubMed] [Google Scholar]
- 198.Heim AJ, Li Z. Developing a high-quality scoring function for membrane protein structures based on specific inter-residue interactions. J Comput Aided Mol Des. 2012;26:301–9. doi: 10.1007/s10822-012-9556-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Jiang P, Yasar F, Hansmann UH. Sampling of Protein Folding Transitions: Multicanonical Versus Replica Exchange Molecular Dynamics. J Chem Theory Comput. 2013;9 doi: 10.1021/ct400312d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Voelz VA, et al. Slow unfolded-state structuring in Acyl-CoA binding protein folding revealed by simulation and experiment. J Am Chem Soc. 2012;134:12565–77. doi: 10.1021/ja302528z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Nymeyer H, Woolf TB, Garcia AE. Folding is not required for bilayer insertion: replica exchange simulations of an alpha-helical peptide with an explicit lipid bilayer. Proteins. 2005;59:783–790. doi: 10.1002/prot.20460. [DOI] [PubMed] [Google Scholar]
- 202.Ulmschneider JP, Ulmschneider MB. Sampling efficiency in explicit and implicit membrane environments studied by peptide folding simulations. Proteins. 2009;75:586–597. doi: 10.1002/prot.22270. [DOI] [PubMed] [Google Scholar]
- 203.Ulmschneider JP, Ulmschneider MB. United Atom Lipid Parameters for Combination with the Optimized Potentials for Liquid Simulations All-Atom Force Field. J Chem Theory Comput. 2009;5:1803–1813. doi: 10.1021/ct900086b. [DOI] [PubMed] [Google Scholar]
- 204.Klauda JB, et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ulmschneider MB, Doux JPF, Killian JA, Smith JC, Ulmschneider JP. Mechanism and Kinetics of Peptide Partitioning into Membranes from All-Atom Simulations of Thermostable Peptides. J Am Chem Soc. 2010;132:3452–3460. doi: 10.1021/ja909347x. [DOI] [PubMed] [Google Scholar]
- 206.Ulmschneider JP, Smith JC, White SH, Ulmschneider MB. In silico partitioning and transmembrane insertion of hydrophobic peptides under equilibrium conditions. J Am Chem Soc. 2011;133:15487–95. doi: 10.1021/ja204042f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Stone JE, Hardy DJ, Ufimtsev IS, Schulten K. GPU-accelerated molecular modeling coming of age. J Mol Graph Model. 2010;29:116–125. doi: 10.1016/j.jmgm.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Freddolino PL, Harrison CB, Liu Y, Schulten K. Challenges in protein folding simulations: Timescale, representation, and analysis. Nat Phys. 2010;6:751–758. doi: 10.1038/nphys1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Marrink SJ, de Vries AH, Mark AE. Coarse grained model for semiquantitative lipid simulations. J Phys Chem B. 2004;108:750–760. [Google Scholar]
- 210.Parton DL, Tek A, Baaden M, Sansom MS. Formation of raft-like assemblies within clusters of influenza hemagglutinin observed by MD simulations. PLoS Comput Biol. 2013;9:e1003034. doi: 10.1371/journal.pcbi.1003034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Bennett WF, Tieleman DP. Computer simulations of lipid membrane domains. Biochim Biophys Acta. 2013;1828:1765–76. doi: 10.1016/j.bbamem.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 212.Pannuzzo M, De Jong DH, Raudino A, Marrink SJ. Simulation of polyethylene glycol and calcium-mediated membrane fusion. J Chem Phys. 2014;140:124905. doi: 10.1063/1.4869176. [DOI] [PubMed] [Google Scholar]
- 213.Periole X, Knepp AM, Sakmar TP, Marrink SJ, Huber T. Structural determinants of the supramolecular organization of G protein-coupled receptors in bilayers. J Am Chem Soc. 2012;134:10959–65. doi: 10.1021/ja303286e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Lelimousin M, Sansom MSP. Membrane perturbation by carbon nanotube insertion: pathways to internalization. Small Weinh Bergstr Ger. 2013;9:3639–3646. doi: 10.1002/smll.201202640. [DOI] [PubMed] [Google Scholar]
- 215.Wong-Ekkabut J, et al. Computer simulation study of fullerene translocation through lipid membranes. Nat Nanotechnol. 2008;3:363–8. doi: 10.1038/nnano.2008.130. [DOI] [PubMed] [Google Scholar]
- 216.Andersson M, Ulmschneider JP, Ulmschneider MB, White SH. Conformational states of melittin at a bilayer interface. Biophys J. 2013;104:L12–14. doi: 10.1016/j.bpj.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Gumbart J, Chipot C, Schulten K. Free-energy cost for translocon-assisted insertion of membrane proteins. Proc Natl Acad Sci U S A. 2011;108:3596–3601. doi: 10.1073/pnas.1012758108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Ziervogel BK, Roux B. The binding of antibiotics in OmpF porin. Struct Lond Engl 1993. 2013;21:76–87. doi: 10.1016/j.str.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Lin YL, Meng Y, Jiang W, Roux B. Explaining why Gleevec is a specific and potent inhibitor of Abl kinase. Proc Natl Acad Sci U S A. 2013;110:1664–9. doi: 10.1073/pnas.1214330110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Stansfeld PJ, Jefferys EE, Sansom MSP. Multiscale simulations reveal conserved patterns of lipid interactions with aquaporins. Struct Lond Engl 1993. 2013;21:810–819. doi: 10.1016/j.str.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Jensen MØ, et al. Principles of conduction and hydrophobic gating in K+ channels. Proc Natl Acad Sci U S A. 2010;107:5833–5838. doi: 10.1073/pnas.0911691107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Jensen MØ, et al. Mechanism of voltage gating in potassium channels. Science. 2012;336:229–233. doi: 10.1126/science.1216533. [DOI] [PubMed] [Google Scholar]
- 223.Ulmschneider MB, et al. Molecular dynamics of ion transport through the open conformation of a bacterial voltage-gated sodium channel. Proc Natl Acad Sci U A. 2013;110:6364–9. doi: 10.1073/pnas.1214667110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Choudhary OP, et al. Structure-guided simulations illuminate the mechanism of ATP transport through VDAC1. Nat Struct Mol Biol. 2014 doi: 10.1038/nsmb.2841. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Khalili-Araghi F, Ziervogel B, Gumbart JC, Roux B. Molecular dynamics simulations of membrane proteins under asymmetric ionic concentrations. J Gen Physiol. 2013;142:465–75. doi: 10.1085/jgp.201311014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Dhakshnamoorthy B, Ziervogel BK, Blachowicz L, Roux B. A structural study of ion permeation in OmpF porin from anomalous X-ray diffraction and molecular dynamics simulations. J Am Chem Soc. 2013;135:16561–16568. doi: 10.1021/ja407783a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Lindorff-Larsen K, et al. Systematic validation of protein force fields against experimental data. PloS One. 2012;7:e32131. doi: 10.1371/journal.pone.0032131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Brooks BR, et al. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 229.Lim JB, Rogaski B, Klauda JB. Update of the cholesterol force field parameters in CHARMM. J Phys Chem B. 2012;116:203–210. doi: 10.1021/jp207925m. [DOI] [PubMed] [Google Scholar]
- 230.Van Der Spoel D, et al. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 231.Phillips JC, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Shaw DE. A fast, scalable method for the parallel evaluation of distance-limited pairwise particle interactions. J Comput Chem. 2005;26:1318–1328. doi: 10.1002/jcc.20267. [DOI] [PubMed] [Google Scholar]
- 233.Sugita Y, Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett. 1999;314:141–151. [Google Scholar]
- 234.Ulmschneider MB, et al. Spontaneous transmembrane helix insertion thermodynamically mimics translocon-guided insertion. Nat Commun. 2014;5:4863. doi: 10.1038/ncomms5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Seyler S, Beckstein O. Sampling large conformational transitions: Adenylate kinase as a testing ground. Mol Simul. 2014 accepted. [Google Scholar]
- 236.Stelzl LS, Fowler PW, Sansom MSP, Beckstein O. Flexible Gates Generate Occluded Intermediates in the Transport Cycle of LacY. J Mol Biol. 2014;426:735–751. doi: 10.1016/j.jmb.2013.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Denning EJ, Woolf TB. Cooperative Nature of Gating Transitions in K+ Channels as seen from Dynamic Importance Sampling Calculations. Proteins. 2010;78:1105–1119. doi: 10.1002/prot.22632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Kohlhoff KJ, et al. Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways. Nat Chem. 2014;6:15–21. doi: 10.1038/nchem.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Shaw DE, et al. Anton, a Special-purpose Machine for Molecular Dynamics Simulation. Commun ACM. 2008;51:91–97. [Google Scholar]
- 240.Deupi X, Kobilka BK. Energy landscapes as a tool to integrate GPCR structure, dynamics, and function. Physiol Bethesda Md. 2010;25:293–303. doi: 10.1152/physiol.00002.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Kruse AC, et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504:101–106. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Nygaard R, et al. The dynamic process of β(2)-adrenergic receptor activation. Cell. 2013;152:532–542. doi: 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Kacprzyk-Stokowiec A, et al. Crucial Role of Perfringolysin O D1 Domain in Orchestrating Structural Transitions Leading to Membrane-Perforating Pores: A Hydrogen-Deuterium Exchange Study. J Biol Chem. 2014 doi: 10.1074/jbc.M114.577981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Rahman KS, Cui G, Harvey SC, McCarty NA. Modeling the conformational changes underlying channel opening in CFTR. PloS One. 2013;8:e74574. doi: 10.1371/journal.pone.0074574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Stolzenberg S, Khelashvili G, Weinstein H. Structural intermediates in a model of the substrate translocation path of the bacterial glutamate transporter homologue GltPh. J Phys Chem B. 2012;116:5372–5383. doi: 10.1021/jp301726s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Korzhnev DM, Kay LE. Probing invisible, low-populated States of protein molecules by relaxation dispersion NMR spectroscopy: an application to protein folding. Acc Chem Res. 2008;41:442–451. doi: 10.1021/ar700189y. [DOI] [PubMed] [Google Scholar]
- 247.Korzhnev DM, Religa TL, Banachewicz W, Fersht AR, Kay LE. A transient and low-populated protein-folding intermediate at atomic resolution. Science. 2010;329:1312–1316. doi: 10.1126/science.1191723. [DOI] [PubMed] [Google Scholar]
- 248.Denning EJ, Crozier PS, Sachs JN, Woolf TB. From the gating charge response to pore domain movement: initial motions of Kv1.2 dynamics under physiological voltage changes. Mol Membr Biol. 2009;26:397–421. doi: 10.3109/09687680903278539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Lundbaek JA, Koeppe RE, Andersen OS. Amphiphile regulation of ion channel function by changes in the bilayer spring constant. Proc Natl Acad Sci U S A. 2010;107:15427–15430. doi: 10.1073/pnas.1007455107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Charalambous K, et al. Engineering de novo membrane-mediated protein-protein communication networks. J Am Chem Soc. 2012;134:5746–5749. doi: 10.1021/ja300523q. [DOI] [PubMed] [Google Scholar]
- 251.Kuhlman B, et al. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 252.Koga N, et al. Principles for designing ideal protein structures. Nature. 2012;491:222–227. doi: 10.1038/nature11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Perez-Aguilar JM, et al. A Computationally Designed Water-Soluble Variant of a G-Protein-Coupled Receptor: The Human Mu Opioid Receptor. PLoS ONE. 2013;8:e66009. doi: 10.1371/journal.pone.0066009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Yin H, et al. Computational design of peptides that target transmembrane helices. Science. 2007;315:1817–1822. doi: 10.1126/science.1136782. [DOI] [PubMed] [Google Scholar]
- 255.Sapay N, Estrada-Mondragon A, Moreau C, Vivaudou M, Crouzy S. Rebuilding a macromolecular membrane complex at the atomic scale: Case of the Kir6.2 potassium channel coupled to the muscarinic acetylcholine receptor M2. Proteins. 2014;82:1694–1707. doi: 10.1002/prot.24521. [DOI] [PubMed] [Google Scholar]
- 256.Bagriantsev SN, et al. Tethered Protein Display Identifies a Novel Kir3.2 (GIRK2) Regulator from Protein Scaffold Libraries. ACS Chem Neurosci. 2014 doi: 10.1021/cn5000698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Rathinakumar R, Walkenhorst WF, Wimley WC. Broad-spectrum antimicrobial peptides by rational combinatorial design and high-throughput screening: the importance of interfacial activity. J Am Chem Soc. 2009;131:7609–7617. doi: 10.1021/ja8093247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Krauson AJ, He J, Wimley WC. Gain-of-function analogues of the pore-forming peptide melittin selected by orthogonal high-throughput screening. J Am Chem Soc. 2012;134:12732–12741. doi: 10.1021/ja3042004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Antia R, Schlegel RA, Williamson P. Binding of perforin to membranes is sensitive to lipid spacing and not headgroup. Immunol Lett. 1992;32:153–157. doi: 10.1016/0165-2478(92)90108-z. [DOI] [PubMed] [Google Scholar]
- 260.Lenoir G, Williamson P, Holthuis JC. On the origin of lipid asymmetry: the flip side of ion transport. Curr Opin Chem Biol. 2007;11:654–61. doi: 10.1016/j.cbpa.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 261.Ikeda M, Kihara A, Igarashi Y. Lipid asymmetry of the eukaryotic plasma membrane: functions and related enzymes. Biol Pharm Bull. 2006;29:1542–6. doi: 10.1248/bpb.29.1542. [DOI] [PubMed] [Google Scholar]
- 262.Liguori N, Nerenberg PS, Head-Gordon T. Embedding Aβ42 in heterogeneous membranes depends on cholesterol asymmetries. Biophys J. 2013;105:899–910. doi: 10.1016/j.bpj.2013.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Slusky JSG, Dunbrack RL., Jr Charge asymmetry in the proteins of the outer membrane. Bioinforma Oxf Engl. 2013;29:2122–2128. doi: 10.1093/bioinformatics/btt355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Denning EJ, Beckstein O. Influence of lipids on protein-mediated transmembrane transport. Chem Phys Lipids. 2013;169:57–71. doi: 10.1016/j.chemphyslip.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 265.Sanders CR, Mittendorf KF. Tolerance to changes in membrane lipid composition as a selected trait of membrane proteins. Biochemistry (Mosc) 2011;50:7858–67. doi: 10.1021/bi2011527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Li Q, Wanderling S, Sompornpisut P, Perozo E. Structural basis of lipid-driven conformational transitions in the KvAP voltage-sensing domain. Nat Struct Mol Biol. 2014;21:160–166. doi: 10.1038/nsmb.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Adamian L, Naveed H, Liang J. Lipid-binding surfaces of membrane proteins: evidence from evolutionary and structural analysis. Biochim Biophys Acta. 2011;1808:1092–1102. doi: 10.1016/j.bbamem.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Ong YS, Lakatos A, Becker-Baldus J, Pos KM, Glaubitz C. Detecting substrates bound to the secondary multidrug efflux pump EmrE by DNP-enhanced solid-state NMR. J Am Chem Soc. 2013;135:15754–15762. doi: 10.1021/ja402605s. [DOI] [PubMed] [Google Scholar]
- 269.Gregory KJ, et al. Identification of Specific Ligand-Receptor Interactions That Govern Binding and Cooperativity of Diverse Modulators to a Common Metabotropic Glutamate Receptor 5 Allosteric Site. ACS Chem Neurosci. 2014 doi: 10.1021/cn400225x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Moore DT, Berger BW, DeGrado WF. Protein-protein interactions in the membrane: sequence, structural, and biological motifs. Struct Lond Engl 1993. 2008;16:991–1001. doi: 10.1016/j.str.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Babu M, et al. Interaction landscape of membrane-protein complexes in Saccharomyces cerevisiae. Nature. 2012;489:585–589. doi: 10.1038/nature11354. [DOI] [PubMed] [Google Scholar]
- 272.Bordner AJ. Predicting protein-protein binding sites in membrane proteins. BMC Bioinformatics. 2009;10:312. doi: 10.1186/1471-2105-10-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Baaden M, Marrink SJ. Coarse-grain modelling of protein-protein interactions. Curr Opin Struct Biol. 2013;23:878–886. doi: 10.1016/j.sbi.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 274.Tusnady GE, Dosztanyi Z, Simon I. TMDET: web server for detecting transmembrane regions of proteins by using their 3D coordinates. Bioinformatics. 2005;21:1276–7. doi: 10.1093/bioinformatics/bti121. [DOI] [PubMed] [Google Scholar]
- 275.Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 2012;40:D370–376. doi: 10.1093/nar/gkr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Kozma D, Simon I, Tusnády GE. PDBTM: Protein Data Bank of transmembrane proteins after 8 years. Nucleic Acids Res. 2013;41:D524–529. doi: 10.1093/nar/gks1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Forrest LR, Tang CL, Honig B. On the accuracy of homology modeling and sequence alignment methods applied to membrane proteins. Biophys J. 2006;91:508–17. doi: 10.1529/biophysj.106.082313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Sandal M, et al. GOMoDo: A GPCRs online modeling and docking webserver. PloS One. 2013;8:e74092. doi: 10.1371/journal.pone.0074092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Kelm S, et al. Fragment-based modeling of membrane protein loops: successes, failures, and prospects for the future. Proteins. 2013 doi: 10.1002/prot.24299. [DOI] [PubMed] [Google Scholar]
- 280.Reif MM, Winger M, Oostenbrink C. Testing of the GROMOS Force-Field Parameter Set 54A8: Structural Properties of Electrolyte Solutions, Lipid Bilayers, and Proteins. J Chem Theory Comput. 2013;9:1247–1264. doi: 10.1021/ct300874c. [DOI] [PMC free article] [PubMed] [Google Scholar]