

Abstract
Objective
Vascular calcification is a characteristic feature of atherosclerosis, diabetes and end-stage renal disease. We have demonstrated that activation of AKT upregulates Runx2, a key osteogenic transcription factor that is crucial for calcification of vascular smooth muscle cells (VSMC). Using mice with SMC-specific deletion of PTEN, a major negative regulator of AKT, the present studies uncovered a novel molecular mechanism underlying PTEN/AKT/FOXO-mediated Runx2 upregulation and VSMC calcification.
Approaches and Results
SMC-specific PTEN deletion mice were generated by crossing PTEN floxed mice with SM22α-Cre transgenic mice. The PTEN deletion resulted in sustained activation of AKT that upregulated Runx2 and promoted VSMC calcification in vitro and arterial calcification ex vivo. Runx2 knockdown did not affect proliferation but blocked calcification of the PTEN deficient VSMC, suggesting that PTEN deletion promotes Runx2-depedent VSMC calcification that is independent of proliferation. At the molecular level, PTEN deficiency increased the amount of Runx2 post-transcriptionally by inhibiting Runx2 ubiquitination. AKT activation increased phosphorylation of FOXO1/3 that led to nuclear exclusion of FOXO1/3. FOXO1/3 knockdown in VSMC phenocopied the PTEN deficiency, demonstrating a novel function of FOXO1/3, as a downstream signaling of PTEN/AKT, in regulating Runx2 ubiquitination and VSMC calcification. Using heterozygous SMC-specific PTEN deficient mice and atherogenic ApoE−/− mice, we further demonstrated AKT activation, FOXO phosphorylation and Runx2 ubiquitination in vascular calcification in vivo.
Conclusions
Our studies have determined a new causative effect of SMC-specific PTEN deficiency on vascular calcification, and demonstrated that FOXO1/3 plays a crucial role in PTEN/AKT-modulated Runx2 ubiquitination and VSMC calcification.
Keywords: FOXO1/3, AKT, PTEN, Vascular calcification, Runx2, ubiquitination
INTRODUCTION
Vascular calcification, namely aberrant calcium deposition in the vessel wall, reduces elasticity and compliance of the vessel wall. It is a well-known predictive risk factor of subsequent cardiovascular mortality1–3. Vascular calcification has now been recognized as an active cell-regulated process resembling bone modeling, rather than simply passive calcium deposition4–10. Several cell types are involved in this process, including vascular smooth muscle cells (VSMC), which undergo osteogenic differentiation and calcification4–10.
We have demonstrated that runt-related transcription factor 2 (Runx2), the key osteogenic regulator for osteoblast differentiation and chondrocyte maturation11, 12, plays an essential role in regulating osteogenic differentiation of VSMC in vitro13 and vascular calcification in atherosclerosis in vivo14. Furthermore, we have determined that activation of AKT is crucial for oxidative stress-induced VSMC calcification through upregulation of Runx213, and enforced expression of constitutively activated AKT promotes VSMC calcification15. However, the molecular mechanisms underlying AKT-regulated upregulation of Runx2 and VSMC calcification are unknown. Activation of AKT is regulated by two major upstream signals: phosphatidylinositol 3-kinase (PI3K) and phosphatase and tensin homolog (PTEN). PTEN is a protein/lipid phosphatase, which was original discovered as a tumor repressor16, 17. PTEN inactivates AKT by hydrolyzing phosphatidylinositol-3,4,5-trisphosphate18. PTEN has been implicated in regulating neointimal smooth muscle cell proliferation and migration19, 20. PTEN deficiency in smooth muscle cells activates AKT in the mouse vasculature21–23, which contributes to increased smooth muscle cell proliferation that leads to intimal hyperplasia in mouse vascular development21, 23 and in response to injury22. In atherosclerosis studies, partial inactivation of PTEN does not appear to affect high-fat diet-induced atherosclerosis of the atherogenic ApoE−/− mice24, but chemical-induced expression of PTEN was found to be associated with inhibition of high-cholesterol diet-induced atherosclerosis in a rabbit model25. It is unknown whether PTEN plays a role in regulating vascular calcification. Using the SMC-specific PTEN deletion mice, we determined a causative effect of the PTEN deficiency on vascular calcification, independent of cell proliferation, and elucidated the underlying molecular mechanisms.
One of the known downstream signals regulated by activated AKT is the family of forkhead box O (FOXO) proteins. The FOXO family includes FOXO1, FOXO3, FOXO4 and FOXO6 in mammalian cells. FOXO1, FOXO3 and FOXO4 are ubiquitously expressed, while FOXO6 is specifically expressed in brain and liver26–29. Activated AKT phosphorylates FOXOs and leads to exclusion of the FOXOs from the nucleus, which blocks the transcriptional activity of FOXOs30. The role of the FOXOs in VSMC calcification is entirely unknown. Previous studies have suggested a potential link between FOXO and Runx2 expression in other cell types31–34. In osteoblasts and human embryonic stem cells, FOXO1 or FOXO3 increases Runx2 expression; whereas other studies suggest that FOXO1 inhibits Runx2 activity in osteoblasts or prostate cancer cells31–34. Therefore, the function of FOXOs in regulating Runx2 expression may be cell type-dependent. In this study, we have determined, for the first time, the function of FOXOs in regulating VSMC calcification and elucidated the role of FOXOs in AKT-regulated upregulation of Runx2 and VSMC calcification.
We have determined that SMC-specific PTEN deletion promoted VSMC calcification in vitro, aortic calcification ex vivo and in vivo, via increased activation of AKT that upregulates Runx2. VSMC calcification induced by the PTEN deficiency is independent of VSMC proliferation. Mechanistically activation of AKT by PTEN deficiency induces inhibition of FOXO1/3 that led to the upregulation of Runx2. The upregulation of Runx2 occurred post-transcriptionally through inhibiting Runx2 ubiquitination. Using heterozygous SMC-specific PTEN deficient mice and atherogenic ApoE−/− mice, we have also demonstrated AKT activation, FOXO phosphorylation and Runx2 ubiquitination in vascular calcification in vivo. Altogether, our studies have provided the first evidence demonstrating a causative effect of SMC-specific PTEN deficiency on vascular calcification; and identified a novel function of FOXO1/3 in the regulation of the Runx2 ubiquitination and vascular calcification.
METHODS
The smooth muscle specific-PTEN deficient mice were generated by crossing PTEN exon 5 floxed mice (PTENf/f)35 with the SM22α-Cre transgenic mice14, 36. Details of materials and experimental procedures are in the Methods section in the Online Data Supplement.
RESULTS
Activation of AKT by PTEN deficiency promotes Runx2 upregulation and VSMC calcification
We have demonstrated that activation of AKT is associated with VSMC calcification in vitro13, 15. Using mice with SMC-specific ablation of PTEN18, the key upstream negative regulator for AKT activation, we aimed to determine a direct effect of endogenous AKT activation on VSMC calcification. The SMC-specific PTEN deletion mice (PTENΔ/Δ) were generated by crossing the PTEN exon 5 floxed mice with SM22Cre transgenic mice. Similar to previous observation37, the SM22Cre-mediated PTEN deletion resulted in early death of the PTENΔ/Δ mice. We and others have reported that osteogenic differentiation of VSMC determines vascular calcification5, 6, 14, therefore, the effect of the PTEN deletion on osteogenic differentiation of VSMC was first characterized using primary VSMC from the PTENΔ/Δ mice and their control PTENf/f littermates.
The deletion of PTEN in VSMC was demonstrated by Western blot analysis (Fig. 1Aa). AKT1 was found to be the predominant isoform of AKT in VSMC. The PTEN deletion did not affect the expression of AKT mRNA in VSMC (Fig 1Ab), however, it increased activation of AKT1, as indicated by AKT phosphorylation at both serine 473 (S473) and threonine 308 (T308) residues in the PTENΔ/Δ VSMC cultured in growth media (Fig 1Aa). Marked increase in calcification was also observed in PTENΔ/Δ VSMC cultured in osteogenic media for 3 weeks compared with the PTENf/f VSMC (Fig 1Ba), which was further confirmed by quantitative calcium measurement (Fig 1Bb). Concurrently, the expression of osteogenic transcription factor Runx2 and Runx2-regulated osteogenic marker genes, including osteocalcin (OC) and collagen type I (Col Ia), was increased in the PTENΔ/Δ VSMC (Fig 1Bc). Notably, a dramatic increase in the Runx2 protein level was evident in the PTENΔ/Δ VSMC (Fig 1Bd). Accordingly, we conclude the PTEN deletion in VSMC constitutively activates AKT and promotes upregulation of Runx2 and osteogenic differentiation of VSMC.
Fig. 1. Increased AKT by PTEN deficiency promotes Runx2 upregulation and VSMC calcification.

A) Increased activation of AKT in PTEN deficient VSMC. VSMC were isolated from PTENf/f and SMC-specific PTEN deficient mice (PTENΔ/Δ) and cultured in growth media. a) Western blot analysis of the expression of AKT and its phosphorylated (activated) forms; and b) Real-time PCR analysis of AKT1, AKT2 and AKT3 in VSMC, normalized by the expression of GAPDH (n=5 mice per group, NS=not significant). The expression of AKT3 in PTENf/f VSMC is defined as 1. B) Calcification of VSMC from PTENf/f and PTENΔ/Δ mice. a) VSMC were cultured in osteogenic medium for 3 weeks, and calcification was determined by Alizarin Red staining. Results from three independent experiments performed in duplicate are shown. b) Calcification of VSMC in parallel experiments as in a), calcium content was quantified by Arsenazo III method. Results shown are normalized to total protein (n=3, *p = 0.017). c) Real-time PCR analysis of the expression of Runx2 and osteogenic marker genes, OC and ColIa in parallel experiments as in a). The expression of each gene in PTENf/f VSMC is defined as 1 (n=3, *p<0.05). d) Western blot analysis of the expression of PTEN and Runx2 in parallel experiments as in a). Representative blots from three independent experiments are shown. C) Inhibition of AKT attenuates PTEN deficiency-promoted calcification. PTENΔ/Δ and control PTENf/f VSMC were exposed to H2O2-containing osteogenic media with/without an AKT inhibitor (AKTIV, 1.0uM). a) Calcification was determined by Alizarin red staining; and b) Western blot analysis was performed to determine the AKT phosphorylation and Runx2 expression in the cells in a) and b). Results from 3 independent experiments are shown. D) Runx2 knockdown abolished PTEN deletion-promoted VSMC calcification. VSMC were stably infected with lentivirus carrying control scrambled shRNA (Control) or shRNA specific for Runx2 and selected by puromycin. Stably infected VSMC were exposed to H2O2-containing osteogenic medium. a) Calcification was determined by Alizarin red staining; and b) Western blot analysis was performed to determine the expression of pAKT and Runx2. Results from 3 independent experiments are shown.
Inhibition of AKT and Runx2 attenuates calcification of the PTEN deficient VSMC
The definitive role of AKT/Runx2 signaling axis in mediating the PTEN deficiency-induced VSMC calcification was further determined by loss of function studies. Inhibition of AKT activation by an AKT inhibitor abolished calcification of the PTENΔ/Δ VSMC (Fig 1Ca), which was associated with inhibition of Runx2 upregulation (Fig 1Cb). Furthermore, we found that knockdown of Runx2 by specific shRNA blocked calcification of PTENΔ/Δ VSMC (Fig 1Da). The Runx2 knockdown did not affect AKT phosphorylation (Fig 1Db). These data demonstrated that upregulation of Runx2 by activation of AKT is essential for the PTEN deficiency-induced VSMC calcification. Of note, the Runx2 knockdown blocked calcification but did not affect proliferation of the PTENΔ/Δ VSMC (Online Supplemental Fig I), suggesting that the Runx2 upregulation-dependent calcification in the PTEN deficient VSMC is independent of its effect on VSMC proliferation.
SMC-specific PTEN deletion promotes ex vivo aortic calcification
The effect of SMC-specific PTEN deletion on calcification of VSMC in their natural milieu was characterized in an ex vivo aortic ring culture system using descending aortas from PTENΔ/Δ mice and their control littermates. Immunohistochemical staining with a PTEN specific antibody demonstrated specific deletion of PTEN in the media, but not in the endothelium (Fig 2A, PTEN arrows). Histological analysis demonstrated increased medial thickness and decreased smooth muscle α-action (SMA), a SMC marker in the PTENΔ/Δ aortas (Fig 2A), which is consistent with the previous findings that PTEN regulates proliferation of SMCs19–23, 25. On the other hand, sustained increased phosphorylation of AKT was demonstrated in the PTENΔ/Δ aorta (Fig 2A). Similar to the observation with isolated VSMC, aortas from PTENΔ/Δ mice exhibited intensive calcification after cultured in osteogenic media for 2 weeks (Fig 2B, Alizarin red and Von Kossa), which was associated with decreased SMA and elevated Runx2 (Fig 2B). By contrast, vascular calcification was not evident in aortas from the control PTENf/f littermates under the same conditions (Fig 2B). These results demonstrated a direct effect of endogenous activation of AKT by the PTEN deletion in VSMC on upregulation of Runx2 and VSMC calcification.
Fig. 2. SMC-specific PTEN deletion promotes ex vivo aortic calcification.
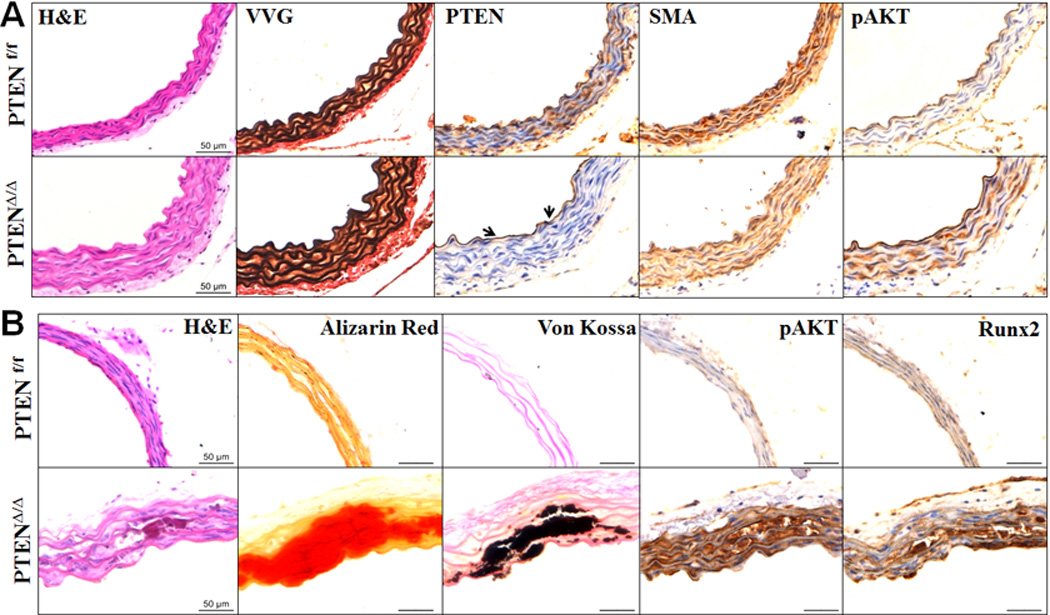
A) SMC-specific PTEN deficiency increased phosphorylated AKT in the media. Consecutive sections from descending aortas from PTENf/f and PTENΔ/Δ mice were stained with H&E (histology), VVG (elastin) or specific antibodies for PTEN, SMA, and phosphorylated AKT (pAKT). Representative images from 5 pairs of littermates are shown. B) SMC-specific PTEN deficiency promoted aortic calcification. Aorta rings from PTENf/f and PTENΔ/Δ mice were cultured in osteogenic medium containing H2O2 (0.3mM) for 2 weeks. Consecutive sections were stained by H&E, Alizarin Red (calcium), Von Kossa (calcium phosphate), and specific antibodies for pAKT and Runx2. Representative images from 5 independent experiments are shown.
The PTEN deficiency upregulates Runx2 by inhibiting Runx2 ubiquitination
To determine the molecular mechanism underlying activation of AKT in promoting upregulation of Runx2 and VSMC calcification, we first characterized Runx2 expression in the PTENΔ/Δ VSMC. The amount of the Runx2 protein was increased during calcification of the PTENΔ/Δ VSMC (Fig 1Bd), which was associated with a modest increase in expression of the Runx2 mRNA (Fig 1Bc). These observations led us to examine the amount of both Runx2 mRNA and protein in PTENΔ/Δ VSMC cultured in normal growth media, a basal condition. Under the basal condition, marked increase in the Runx2 protein level was also evident in the PTENΔ/Δ VSMC (Fig 3Aa), while expression of the Runx2 mRNA is not altered significantly (Fig 3Ab). Inhibition of de novo protein synthesis by cycloheximide decreased the amount of Runx2 in the control PTENf/f VSMC (Fig 3B) in a time dependent manner. In contrast, the amount of the Runx2 protein was sustained in the PTENΔ/Δ VSMC after exposure to cycloheximide for 12 hours, indicating that the PTEN deletion stabilized Runx2 by preventing Runx2 protein from degradation. As ubiquitination mediates degradation of Runx238, 39, we determined the effect of the PTEN deficiency on the Runx2 ubiquitination. The PTEN deletion did not affect the overall expression profile of poly-ubiquitin in VSMC (Fig 3C, input). However, modification of Runx2 by poly-ubiquitin was markedly decreased in the PTENΔ/Δ VSMC (Fig 3C, IP:Runx2), indicating that ubiquitination-mediated degradation of Runx2 was inhibited, which rendered Runx2 stable in the PTENΔ/Δ VSMC.
Fig. 3. PTEN deficiency increases Runx2 protein level in VSMC by inhibiting Runx2 ubiquitination.

A) Expression of Runx2 in VSMC from PTENf/f and PTENΔ/Δ mice cultured in growth media (basal conditions), determined by a) Western blot analysis of Runx2 protein; and b) Real-time PCR analysis of Runx2 mRNA. Representative results from VSMC from 3 pairs of littermates are shown (n=3, #NS, not significant). B) Effects of PTEN deletion on Runx2 stability. PTENf/f and PTENΔ/Δ VSMC were cultured in growth media with cycloheximide (50 µM) for up to 12 hours. The expression of PTEN and Runx2 protein was determined by Western blot. Representative blots from 3 independent experiments are shown. C) Runx2 ubiquitination determined by Western blot analysis. PTENf/f and PTENΔ/Δ VSMC were pretreated with MG132 (50 µM). Proteins were extracted, and the expression of Runx2 and poly-ubiquitin was determined by Western blot analysis (Input). Immunoprecipitation was performed with Runx2 antibody; Runx2-bound poly-ubiquitin was determined by Western blot analysis (IP: Runx2). Representative images from three independent experiments are shown.
AKT activation promotes cytosolic translocation of FOXO1/FOXO3
The FOXO protein family, one of several known downstream targets of activated AKT, has been associated with expression of Runx2 in osteoblasts, human embryonic stem cells and prostate cancer cells, although data from these studies are not compelling31–34. We found that FOXO1 and FOXO3a are highly expressed in VSMC compared with FOXO4 (Online Supplemental Fig II). The PTEN deficiency in VSMC did not affect the expression of FOXOs mRNA (Online Supplemental Fig II) or production of FOXO1/3 proteins (Fig 4A). In contrast, phosphorylation of FOXO1 and 3 was dramatically increased in the PTENΔ/Δ VSMC (Fig 4A). As phosphorylation of FOXO has been shown to be associated with nuclear exclusion40, we examined the cellular localization of FOXO1/3 in PTENΔ/Δ VSMC and control PTENf/f VSMC. The PTEN deletion in VSMC increased cytosolic translocation and decreased nuclear localization of FOXO1/3, which was associated with increased Runx2 in the nuclear (Fig 4B). Furthermore, activation of AKT using a lentivirus carrying a constitutively activated AKT similarly promoted cytosolic translocation of FOXO1/3 and abolished their nuclear localization (Fig 4C), which was again associated with increased amount of Runx2. These data demonstrate that activation of AKT, either by PTEN deficiency or by overexpression of constitutively activated AKT, leads to nuclear exclusion of FOXO1/FOXO3 in VSMC, which was associated with upregulation of Runx2.
Fig. 4. AKT activation promotes nuclear exclusion of FOXO1/3.

A) Increased phosphorylation of FOXO1/3 in PTEN deficient VSMC. Western blot analysis of the expression of FOXO1/3 and their phosphorylated forms in PTENf/f and PTENΔ/Δ VSMC cultured in growth media. B) Increased Runx2 expression and nuclear exclusion of FOXO1/3 in PTEN deficient VSMC. Western blot analysis of Runx2 and FOXO1/3 in the cytosolic and nuclear fractions of the PTENf/f and PTENΔ/Δ VSMC. C) Constitutively activated AKT promoted nuclear exclusion of FOXOX1/3 and upregulated Runx2. Wild type VSMC overexpressing constitutively active AKT (AKT) or control GFP protein (Con) were cultured in growth media. The expression of AKT, FOXO1/3 and Runx2 in cytosolic and nuclear fractions was determined by Western blot analysis. Representative blots from three independent experiments are shown.
FOXO1/3 knockdown inhibits Runx2 ubiquitination and promotes VSMC calcification
We further determined whether inhibition of FOXO1/3 directly affect Runx2 and VSMC calcification, by knocking down FOXO1 or FOXO3 in VSMC using specific shRNA. Knockdown of FOXO1 decreased the expression of FOXO3 in VSMC, and vice versa (Fig 5A). Such an observation is consistent with a previous report that FOXO1 and FOXO3 regulate each other in human fibroblasts41. Knockdown of FOXO1 or FOXO3 markedly increased the amount of Runx2 detected, without affecting expression of the Runx2 mRNA (Fig 5A & B). The effects of FOXO1/3 knockdown on ubiquitination of Runx2 were further determined. Similar to the PTEN deletion, FOXO1 or FOXO3 knockdown in VSMC did not affect the general expression profile of poly-ubiquitin, but inhibited Runx2-bound poly-ubiquitin (Fig 5C). Furthermore, knockdown of FOXO1 or FOXO3 promoted VSMC calcification, as determined by Alizarin red staining (Fig 5D) and quantitative calcium measurement (Fig 5E). These data demonstrate that FOXO1/3 negatively regulates the Runx2 stability in VSMC, and knockdown of FOXO1/3 phenocopies the PTEN deficiency.
Fig. 5. FOXO1/3 knockdown inhibits Runx2 ubiquitination and promotes VSMC calcification.

Wild type VSMC stably infected with lentivirus carrying control scrambled shRNA (shScr) or shRNA specific for FOXO1 (shFOXO1) or FOXO3 (shFOXO3) were generated and characterized. A) FOXO1/3 knockdown in VSMC increased Runx2 protein expression. Stably infected VSMC were cultured in growth media, and the expression of FOXO1/3 and Runx2 proteins was determined by Western blot analysis. Representative blots from 3 independent experiments are shown. B) FOXO1/3 knockdown in VSMC does not affect Runx2 mRNA expression. Real-time PCR analysis was performed to determine the expression of Runx2 mRNA in FOXO1 or 3 knockdown VSMC (n=3, #NS indicating not significant). C) Effects of FOXO1/3 knockdown on Runx2 ubiquitination. VSMCs with FOXO1 or FOXO3 knockdown and control VSMC were pretreated with MG132 (50 µM). Proteins were extracted, and the expression of Runx2 and poly-ubiquitin was determined by Western blot analysis (Input). Immunoprecipitation was performed with Runx2 antibody, and Runx2-bound poly-ubiquitin was determined by Western blot analysis (IP: Runx2). Representative blots from three independent experiments are shown. D) and E) FOXO1/3 knockdown promotes VSMC calcification. Stably infected VSMC from A were cultured in osteogenic medium with H2O2 (0.3 mM) for 3 weeks. Calcification was determined by D) Alizarin red staining; and E) calcium quantification with Arsenazo III, in parallel experiments. Results from 3 independent experiments are shown (n=3, * p≤ 0.002).
SMC-specific PTEN reduction promotes vascular calcification in mice
The role of AKT/FOXO/Runx2 signaling axis in vascular calcification was further determined in vivo. As the early death of the homozygous SMC-specific PTENΔ/Δ mice prevented us from carrying out long-term in vivo vascular calcification studies, the heterozygous SMC-specific PTEN deficient mice (PTENΔ/+) were utilized. The PTENΔ/+ mice appeared normal as their control littermates (PTENf/+). Decreased PTEN expression was evident in the vasculature of the heterozygous mice (Fig 6A). Importantly, the heterozygous SMC-specific PTEN mice fed on a normal chow diet spontaneously developed vascular calcification at 6 months of age (Fig 6A&B). Alizarin red staining revealed apparent calcium deposition in aortas from the PTENΔ/+ mice, whereas no calcification was detected in the aortas from control littermates (Fig 6A). Quantitative measurement of total calcium content in the aortic tissues further demonstrated increased vascular calcification in the PTENΔ/+ mice (Fig 6B). Moreover, increased vascular calcification in the PTENΔ/+ mice was associated with increased AKT activation, FOXO phosphorylation and Runx2 upregulation in the vasculature, as determined by immunostaining (Fig 6C) as well as Western blot analyses (Fig 6D).
Fig. 6.

Decreased PTEN in SMC promotes vascular calcification. Descending aortas were isolated from 6 month-old heterozygous SMC-specific PTEN deficient mice (PTEN Δ/+) and their control littermates (PTEN f/+). A) Analysis of vascular calcification in aortic sections. Consecutive sections were stained by H&E, antibody for PTEN and Alizarin Red. Higher magnification images of the boxed areas are shown to the right. Arrows indicate calcium deposition. B) Calcium content measured in descending aortas by Arsenazo III. The calcium content in aortas from the control mice was defined as 1 (n=3 mice/group, *p=0.03). C) Decreased PTEN in SMC increases AKT activation, FOXO phosphorylation and Runx2. Consecutive aortic sections from the heterozygous SMC-specific PTEN deficient mice and their control littermates were stained with specific antibodies for pAKT, pFOXO1, pFOXO1/3/4 and Runx2. Representative images from 3 mice/group are shown. D) Western blot analysis of the expression of PTEN, pAKT, pFOXO1, pFOXO3 and Runx2 in aortic lysates from PTEN Δ/+ and PTEN f/+ mice.
In addition, we determined the AKT/FOXO/Runx2 signaling axis in the atherogenic ApoE−/− mice, which developed atherosclerotic vascular calcification as we demonstrated previously14, 36. Consistently, HFD induced upregulation of Runx2 and vascular calcification (supplemental Figure IV). Similar to the observation with the PTENΔ/+ mice (Fig 6), Runx2 upregulation was associated with increased AKT activation and FOXO phosphorylation in calcified vasculature of the ApoE−/− mice (supplemental Figure IV, A&Ba). Furthermore, decreased Runx2 ubiquitination was also evident in aortas from the HFD-fed ApoE−/− mice (supplemental Figure IV, Bb). Taken together, results from these two animal models supported the important role of AKT/FOXO/Runx2 signaling axis in vascular calcification in vivo.
DISCUSSION
Runx2 is a key transcriptional regulator required for vascular calcification in vitro and in vivo. Activation of AKT also promotes calcification of VSMC. To uncover underlying mechanisms connecting activation of AKT and upregulation of Runx2 in the development of vascular calcification, we generated a mouse model with sustained AKT activation in VSMC by selective deletion of PTEN, an upstream phosphatase that negatively regulates AKT activation. Our studies reveal a novel causative effect of the SMC-specific PTEN deficiency in promoting vascular calcification. Using VSMC from the SMC-specific PTEN deletion mice, we have also elucidated a new function of PTEN/AKT/FOXO1/3 signaling axis in regulating ubiquitination of Runx2 and vascular calcification. The important role of the AKT/FOXO/Runx2 signaling axis in vascular calcification in vivo has been further demonstrated with the heterozygous SMC-specific PTEN deletion mice and the atherogenic ApoE−/− mice.
As a protein/lipid phosphatase, PTEN has diverse functions in different type of cells, most notable one is its tumor repression activity. In the vasculature, PTEN has been implicated in proliferation, differentiation and migration of smooth muscle cell19–23, 37, however, the role of PTEN in vascular calcification is entirely unknown. Our studies with comprehensive approaches using the SMC-specific PTEN deletion VSMC in vitro, aortas ex vivo and the heterozygous SMC-specific PTEN deletion mice in vivo have strongly supported a causative effect of the PTEN inhibition on vascular calcification. Using an AKT inhibitor, we confirmed that activation of AKT is crucial for calcification of the PTEN deficient VSMC. Furthermore, we demonstrated that upregulation of Runx2 was required for the calcification of VSMC from the PTEN deficient mice. Importantly, the Runx2 knockdown was found to only block calcification but not proliferation of the PTEN deficient VSMC, suggesting that PTEN deficiency-induced Runx2 upregulation promotes osteogenic differentiation of VSMC, which may be independent of its previously reported function in regulating VSMC proliferation, differentiation and migration19–23, 37. In normal VSMC, the amount of Runx2 detected is very low42, 43. In the PTEN deficient VSMC, however, markedly increased Runx2 was evident even under the basal condition. Since we have shown that upregulation of Runx2 is essential and sufficient to induce VSMC calcification13, 14, AKT activation-induced elevation of the Runx2 protein in the PTEN deficient VSMC may predispose the cells to undergo osteogenic differentiation. As a result, the PTEN deficiency promoted calcification of the PTENΔ/Δ VSMC cultured in osteogenic medium without any additional stimuli. Runx2-regulated osteogenic marker genes, including OC and Col1a13, 14, 44 increased concurrently. This finding is also supported by the data from the in vivo studies with SMC-specific reduction of PTEN. Apparently, increased AKT activation by the PTEN reduction contributes to Runx2 upregulation and the spontaneous development of vascular calcification in these mice.
The elevated amount of Runx2 protein in the PTEN deficient VSMC under basal conditions was not due to increased expression of Runx2 at the transcriptional level. Instead, the PTEN deficiency in VSMC stabilizes the Runx2 protein by inhibiting Runx2 degradation, an important mechanism that has been demonstrated to stabilize the Runx2 protein in bone cells38. In bone cells, post-translational modulation by the ubiquitin-proteasome pathway regulates stability and degradation of Runx2 protein39. Our studies have provided the first evidence that the PTEN deficiency increases the Runx2 stability by inhibiting ubiquitination of Runx2 in the PTENΔ/Δ VSMC. Studies in osteoblasts have identified several molecules that contribute to Runx2 degradation, including Smad ubiquitin regulatory factor 1 (Smurf1) and cyclin-D145, 46. Smurf1 is an E3-ligase that binds to Runx2 and mediates ubiquitin binding to Runx2 that leads to Runx2 degradation45. Cyclin-D1, on the other hand, contributes to proteasome-dependent degradation of Runx2 by cyclin D1/Cdk4-induced Runx2 phosphorylation in the C-terminus of Runx2, which is critical for Runx2 protein stability46. Additionally, other factors have been implicated in mediating Runx2 degradation by recruiting E3-ligases, such as Smad 6, which binds to Runx2 and serves as an adaptor for Smurf1-induced Runx2 degradation47, 48. However, the PTEN deficiency in VSMC did not affect the expression of the previously characterized regulators of Runx2 ubiquitination in the bone cells, such as Smurf1 and cyclin-D1. Therefore, the molecular regulators that modulate Runx2 degradation and ubiquitination in VSMC may be novel, which warrant further exploration.
Our studies have also demonstrated that the FOXO1/3 signaling axis is responsible for the inhibition of the Runx2 ubiquitination, thereby upregulates Runx2 which then promotes VSMC calcification. Specifically, AKT-regulated phosphorylation and cytosolic translocation of FOXO1/3 uncreased Runx2 in VSMC. The phosphorylation and subsequent nuclear exclusion of the FOXO family of proteins has been associated with neointimal hyperplasia in a rat balloon injury model40. Upregulation of AKT/FOXO signaling has been found in the vasculature in an aging rat atherosclerosis model49. However, the function of SMC-expressed FOXO family proteins in regulating vascular calcification is unknown. Using FOXO1/3 specific shRNA, we determined that FOXO1/3 regulates upregulation of Runx2 and subsequent calcification of VSMC. FOXO1 and/or 3 have been found to increase Runx2 in osteoblasts and human embryonic stem cells; however, other studies indicate that FOXO1 inhibits the Runx2 activity in osteoblasts or prostate cancer cells31–34. Our studies have delineated a novel function of FOXO1/3 in regulating the Runx2 protein stability in VSMC. Consistent with the cross-regulation of FOXO1/3 in human fibroblasts41, knockdown of FOXO1 decreases the expression of FOXO3 in VSMC and vice versa, implying a positive feedback loop controlling the expression of FOXO1/3. Knockdown of FOXO1/3 phenocopied the PTEN deficiency in terms of Runx2 ubiquitination and VSMC calcification, which support a novel function of FOXO1/3 in the process. More importantly, increased FOXO phosphorylation and Runx2 upregulation are associated with AKT activation in vascular calcification in the heterozygous SMC-specific PTEN deletion mice as well as the atherogenic ApoE−/− mice in vivo. Decreased Runx2 ubiquitination has been found in the vasculature of the HFD-fed ApoE−/− mice, which may lead to decreased Runx2 degradation and thus increase Runx2 and promote vascular calcification. These animal studies have further supported an important role of AKT activation, FOXO phosphorylation and Runx2 ubiquitination in vascular calcification in vivo.
In summary, the current studies have demonstrated a causative effect SMC-specific PTEN deficiency on vascular calcification via activation of AKT and upregulation of Runx2 in vitro and in vivo. We have further determined a novel function of the AKT/FOXO1/3 signaling axis in regulating Runx2 ubiquitination and stability. These studies have provided the first evidence demonstrating the role of FOXO1/3 in mediating activation of AKT-induced VSMC calcification, and identified that FOXO1/3-modulated ubiquitination of Runx2 as a novel mechanism in regulating vascular calcification. The new molecular insights gained in the present studies in the regulation of Runx2 in the development of VSMC calcification may lead to identification of novel therapeutic targets.
MATERIALS AND METHODS
The smooth muscle specific-PTEN deficient mice were generated by crossing PTEN exon 5 floxed mice (PTENf/f)1 with the SM22α-Cre transgenic mice2, 3. Details of materials and experimental procedures are in the Methods section in the Online Data Supplement.
Online Supplemental Methods
Generation of SMC-specific PTEN deletion mice
PTEN exon5 floxed mice (PTENf/f) mice1 and SM22α-Cre transgenic mice2, 3 were obtained from The Jackson Laboratory. The SM22α-Cre mice were bred with PTENf/f to generate smooth muscle-specific PTEN deletion mice (PTENΔ/Δ). Exon 5 encodes the phosphatase domain of PTEN1, which is the functional domain. As most of the smooth muscle-specific PTEN deletion mice died around 4–5 weeks of age, all experiments were performed with aortas or smooth muscle cells isolated from 25–30 days old littermates. Primer sets for genotyping are: Cre: F-5’-GCGGTCTGGCAGTAAAAACTATC-3’ and R-5’-GTGAAACAGCATTGCTGTCACTT-3’; PTEN: F-5’ - CAAGCACTCTGCGAACTGAG- 3’ and R-5’-AAGTTTTTGAAGGCAAGATGC- 3’.
In vivo calcification of the heterozygous SMC-specific PTEN deletion mice
The effects of smooth muscle-expressed PTEN on vascular calcification was determined in 6-months old heterozygous SMC-specific PTEN deletion mice and their control littermates on normal chow diet. Aortic calcification was determined by Alizarin red staining as well as calcium content measurement Arsenazo III method as described before3, 4.
Immunohistochemistry
Frozen aortic sections were processed for histology and immunohistochemistry as we described3, 4. In brief, Hematoxylin & Eosin (H&E) and Verhoeff-Van Gieson (VVG) was used for histological analysis. Antibody for PTEN, SMA, Runx2 and phosphorylated-AKT was applied to acetone-fixed cryosections. The sections were washed and exposed to a secondary antibody (horseradish peroxidase-conjugated antibodies), and antibody binding was visualized with diaminobenzidine. Sections were counterstained with hematoxylin.
Aortic ring culture
Descending aortas were cut into 2–3 mm rings, which were cultured in osteogenic medium containing 1% FBS with 0.3mM H2O2 for 2 weeks with medium changed every 3 days. At the end of experiments, aortic rings were harvested, fixed and embedded in paraffin.
Aortic calcification
Consecutive sections from the cultured aortic rings were stained with H&E for histology. Alizarin Red staining or Von Kossa staining (Sigma Aldrich) were used to detect calcification as we previously described3, 4
VSMC culture
Primary VSMC were isolated from mouse aorta and cultured in growth medium as we described previously5. All experiments were performed with VSMC at passages 3 to 5.
In vitro VSMC calcification5
VSMC calcification was induced as we previously described in osteogenic media containing 0.25 mmol/L L-ascorbic acid and 10 mmol/L β-glycerophosphate (Sigma Aldrich) with or without H2O2 (0.3 mmol/L). Calcification was determined by Alizarin Red staining or quantified by measuring total calcium in the cell lysates by the Arsenazo III method4.
In vitro VSMC proliferation6
Proliferation was assayed by the incorporation of 5-bromo-2-deoxyuridine (BrdU Proliferation Assay Kit, Calbiochem) as we reported6. VSMC were cultured in growth media in 96 well plates. BrdU incorporation in 24 hours was identified with a fluorescein-labeled anti-BrdU antibody (Calbiochem) and fluorogenic peroxidase secondary antibody using the Synergy 2 plate reader (BioTek).
Lentivirus transduction of VSMC
Lentiviral constructs carrying short hairpin RNA (shRNA) for Runx2, FOXO1 or FOXO3 were purchased from Open Biosystems and packaged into lentiviral particles as previously described5. Lentiviral vector expressing constitutively active AKT protein was obtained from Dr. Hongju Wu (Tulane University). Viral transductions were performed by incubating VSMC with recombinant lentiviruses in growth media supplemented with 10µg/mL Polybrene (Sigma). After 12 hours, virus-containing medium was changed to fresh medium and cultured for another 36 to 48 hours. 5µg/ml puromycin was used to select stably infected cells.
Real-time polymerase chain reaction (PCR)4
Total RNA was isolated using Trizol (Invitrogen) and reverse transcribed into cDNA. SYBR Green-based PCR was performed using specific primers for mouse Runx2, osteocalcin (OC), type I collagen (Col Ia), AKT1/2/3 and FOXO1/3 or 4 (Primer sequences are listed in table 1), using iQ SYBR Green Supermix on an iCycler Thermal Cycler (Bio-Rad).
Table 1.
Primer sequences (mouse) for real-time PCR
| Name | Chain | Sequence | Gene ID |
|---|---|---|---|
| Runx2 | Forward | 5’- CGGCCCTCCCTGAACTCT-3’ | NM_001145920.1 |
| Reverse | 5’- TGCCTGCCTGGGATCTGTA-3’ | ||
| OC | Forward | 5'-TGCTTGTGACGAGCTATCAG-3' | NM_007541.2 |
| Reverse | 5'-GAGGACAGGGAGGATCAAGT-3' | ||
| Col1 | Forward | 5'-CTGGCGGTTCAGGTCCAAT-3' | NM_007742.3 |
| Reverse | 5'-TTCCAGGCAATCCACGAGC-3' | ||
| AKT1 | Forward | 5'-GACCCACGACCGCCTCTG-3' | NM_009652.3 |
| Reverse | 5'-GACACAATCTCCGCACCATAGAAG-3' | ||
| AKT2 | Forward | 5'-GAGGACGCCATGGATTACAAG-3' | NM_007434.3 |
| Reverse | 5'-GACAGCTACCTCCATCATCTCAGA-3' | ||
| AKT3 | Forward | 5'-GAGTACCTGGCACCAGAGGT-3' | NM_011785.3 |
| Reverse | 5'-AGAAAGGCAACCTTCCACAC-3' | ||
| FOXO1 | Forward | 5'-TACGCCGACCTCATCACCAAG-3' | NM_019739.3 |
| Reverse | 5'-GCACGCTCTTCACCATCCACT-3' | ||
| FOXO3 | Forward | 5'-TCACTGAGGAAAGGGGAAATGG-3' | NM_019740.2 |
| Reverse | 5'-AAAGGTGTCAAGCTGTAAACGGA-3' | ||
| FOXO4 | Forward | 5'-CAAGAAGAAGCCGTCTGTCC-3' | NM_018789.2 |
| Reverse | 5'-CTGACGGTGCTAGCATTTGA-3' | ||
| Reverse | 5'-GCAGGAGAGGAAGTTGTTGG-3' | ||
| Smurf1 | Forward | 5'-AGCATCAAGATCCGTCTGACA-3' | NM_001038627.1 |
| Reverse | 5'-CCAGAGCCGTCCACAACAAT-3' | ||
| Cyclin-D1 | Forward | 5'-TCCTCTCCAAAATGCCAGAG-3' | NM_007631.2 |
| Reverse | 5'-GCAGGAGAGGAAGTTGTTGG-3' | ||
| GAPDH | Forward | 5'-AACACGGAAGGCCATGCCAG-3' | NM_008084.2 |
| Reverse | 5'-TGCATCCTGCACCACCAACT-3' |
Western blot analysis
Cytosolic and nuclear extracts were prepared and protein concentration was measured as we described3,5. Western blot analyses were performed with specific antibody for Runx2 (MBL, D130-3), PTEN (Cell Signaling Technology, CST, 9552), Total AKT (CST, 4685), pAKT(Ser473, CST 9271 and Thr308, CST 9275), FOXO1 (CST, 2880), pFOXO1(CST, 9461), FOXO3 (CST, 2497), pFOXO3 (CST, 2599), poly-ubiquitin (Abcam, ab7780), lamin B (Santa Cruz) and GAPDH (Fitzgerald, 10R-G109a), and detected with a chemiluminescence detection kit (Millipore).
Determination of Runx2 stability
VSMC at 90% confluence were treated with cycloheximide (50 µM) for 0, 4, 6 and 12 hours. Western blot analysis was performed to detect the expression of Runx2. The expression of PTEN was verified in these cells, and the expression of GAPDH was used as a loading control.
Determination of Runx2 ubiquitination
VSMC were pretreated with 50 µM MG132 (Calbiochem) for 6 hours to inhibit proteasomal degradation. Then protein extracts were collected with non-denaturing lysis buffer (20mM Tris.HCL pH 8, 150mM NaCl, 10% Glycerol and 1% Triton X-100). 1000 µg lysates were incubated with 2 µg Runx2 antibody for at least 1h. Immune complexes were recovered from the supernatant by incubation with 50 µl of 1:1 slurry of protein G-agarose beads (Invitrogen) overnight at 4 °C. Beads were washed by cold 1×PBS for 5 times, and resuspended in 20µl 2×loading buffer. Then, the mixture was boiled at 100°C for 10min. After brief centrifugation, proteins in the supernatant were analyzed by Western blot with Runx2 and poly-ubiquitin antibodies.
Characterization of FOXO phosphorylation and Runx2 ubiqutination in atherogenic ApoE−/− mice
8-week old ApoE−/− mice were fed a normal chow or a high-fat diet for 30 weeks7, 8. All experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham. Consecutive aortic sections were stained with Alizarin Red and antibodies for pAKT, pFOXO1/3/4 and Runx2. Western blot analysis was performed in aortic protein extracts using specific antibodies Aortic Runx2 ubiqutination was determined as described above by immunoprecipitation with Runx2 antibody, followed by Western blot using poly-ubiqutin antibody.
Statistical analysis
All the data are expressed as means ± SD. Differences between two groups were compared with Student’s paired 2-tailed t test. A p value less than 0.05 was considered statistically significant.
Supplementary Material
SIGNIFICANCE.
Vascular calcification is emerging as an important risk factor that predicts outcome of cardiovascular diseases. Clinical and experimental studies from our group and others have demonstrated a critical role of the osteogenic transcription factor Runx2 in regulating vascular calcification. The present studies have determined a new causative effect of SMC-specific PTEN deficiency on Runx2 upregulation and vascular calcification. The PTEN deficiency upregulates Runx2 through inhibiting Runx2 ubiquitination. Our studies have revealed that FOXO1/3 is the link between AKT activation and Runx2 upregulation since FOXO1/3 knockdown phenocopies the PTEN deficiency, including Runx2 ubiquitination and VSMC calcification. Using heterozygous SMC-specific PTEN deletion mice and the atherogenic ApoE−/− mice, we have demonstrated the important role of AKT/FOXO/Runx2 signaling axis in vascular calcification in vivo. Altogether, our studies have provided the first evidence demonstrating a new role of PTEN in regulating vascular calcification, and identified FOXO1/3-regulated Runx2 ubiquitination as a novel underlying mechanism.
ACKNOWLEDGEMENTS
The authors thank Jay M McDonald, MD (University of Alabama at Birmingham, UAB) for critical review and Kaiyu Yuan, MD (UAB) for technical assistance.
SOURCES OF FUNDING
This work was supported in part by grants from the National Institutes of Health HL092215 and DK100847 and Veterans Administration BX000369 and BX001591 (project 2) to YC. JH was supported by American Heart Association Grant 12PRE11840009.
NON-STANDARD ABBREVIATIONS AND ACRONYMS
- AKT
Protein kinase B
- ALP
alkaline phosphatase
- BMP2
bone morphogenetic protein 2
- Col I
type I collagen
- FOXO
Forkhead box O
- OC
osteocalcin
- PTEN
phosphatase and tensin homolog
- PI3K
phosphatidylinositol 3-kinase
- PIP3
phosphatidylinositol-3,4,5-trisphosphate
- Runx2
runt-related transcription factor 2
- shRNA
small hairpin RNA
- VSMC
vascular smooth muscle cells
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Wilson PW, Kauppila LI, O'Donnell CJ, Kiel DP, Hannan M, Polak JM, Cupples LA. Abdominal aortic calcific deposits are an important predictor of vascular morbidity and mortality. Circulation. 2001;103:1529–1534. doi: 10.1161/01.cir.103.11.1529. [DOI] [PubMed] [Google Scholar]
- 2.Towler DA, Demer LL. Thematic series on the pathobiology of vascular calcification: an introduction. Circ Res. 2011;108:1378–1380. doi: 10.1161/CIRCRESAHA.110.234419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bastos GF, Voute MT, Hoeks SE, Chonchol MB, Boersma EE, Stolker RJ, Verhagen HJ. Calcification of the abdominal aorta as an independent predictor of cardiovascular events: a meta-analysis. Heart. 2012;98:988–994. doi: 10.1136/heartjnl-2011-301464. [DOI] [PubMed] [Google Scholar]
- 4.Witztum JL. The oxidation hypothesis of atherosclerosis. Lancet. 1994;344:793–795. doi: 10.1016/s0140-6736(94)92346-9. [DOI] [PubMed] [Google Scholar]
- 5.Mody N, Parhami F, Sarafian TA, Demer LL. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radical Biology and Medicine. 2001;31:509–519. doi: 10.1016/s0891-5849(01)00610-4. [DOI] [PubMed] [Google Scholar]
- 6.Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, Schinke T, Karsenty G, Giachelli CM. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. 2001;89:1147–1154. doi: 10.1161/hh2401.101070. [DOI] [PubMed] [Google Scholar]
- 7.Boström KI. Cell differentiation in vascular calcification. Z Kardiol. 2000;89(Suppl 2):69–74. doi: 10.1007/s003920070102. [DOI] [PubMed] [Google Scholar]
- 8.Giachelli CM. Vascular calcification mechanisms. J Am Soc Nephrol. 2004;15:2959–2964. doi: 10.1097/01.ASN.0000145894.57533.C4. [DOI] [PubMed] [Google Scholar]
- 9.Shanahan CM, Cary NR, Salisbury JR, Proudfoot D, Weissberg PL, Edmonds ME. Medial localization of mineralization-regulating proteins in association with Mönckeberg's sclerosis: evidence for smooth muscle cell-mediated vascular calcification. Circulation. 1999;100:2168–2176. doi: 10.1161/01.cir.100.21.2168. [DOI] [PubMed] [Google Scholar]
- 10.Yao Y, Jumabay M, Ly A, Radparvar M, Cubberly MR, Boström KI. A role for the endothelium in vascular calcification. Circ Res. 2013;113:495–504. doi: 10.1161/CIRCRESAHA.113.301792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 12.Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB, Owen MJ. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 13.Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, McDonald JM, Chen Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. 2008;283:15319–15327. doi: 10.1074/jbc.M800021200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun Y, Byon CH, Yuan K, Chen J, Mao X, Heath JM, Javed A, Zhang K, Anderson PG, Chen Y. Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ Res. 2012;111:543–552. doi: 10.1161/CIRCRESAHA.112.267237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heath J, Sun Y, Yuan K, Bradley WE, Litovsky S, Dell'italia LJ, Chatham JC, Wu H, Chen Y. Activation of AKT by O-linked N-acetyglucosamine Induces Vascular Calcification in Diabetes. Circ Res. 2014;114:1094–1102. doi: 10.1161/CIRCRESAHA.114.302968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 17.Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 18.Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008;27:5527–5541. doi: 10.1038/onc.2008.247. [DOI] [PubMed] [Google Scholar]
- 19.Huang J, Kontos CD. Inhibition of vascular smooth muscle cell proliferation, migration, and survival by the tumor suppressor protein PTEN. Arterioscler Thromb Vasc Biol. 2002;22:745–751. doi: 10.1161/01.atv.0000016358.05294.8d. [DOI] [PubMed] [Google Scholar]
- 20.Mourani PM, Garl PJ, Wenzlau JM, Carpenter TC, Stenmark KR, Weiser-Evans MC. Unique, highly proliferative growth phenotype expressed by embryonic and neointimal smooth muscle cells is driven by constitutive Akt, mTOR, and p70S6K signaling and is actively repressed by PTEN. Circulation. 2004;109:1299–1306. doi: 10.1161/01.CIR.0000118462.22970.BE. [DOI] [PubMed] [Google Scholar]
- 21.Nemenoff RA, Simpson PA, Furgeson SB, Kaplan-Albuquerque N, Crossno J, Garl PJ, Cooper J, Weiser-Evans MC. Targeted deletion of PTEN in smooth muscle cells results in vascular remodeling and recruitment of progenitor cells through induction of stromal cell-derived factor-1alpha. Circ Res. 2008;102:1036–1045. doi: 10.1161/CIRCRESAHA.107.169896. [DOI] [PubMed] [Google Scholar]
- 22.Nemenoff RA, Horita H, Ostriker AC, Furgeson SB, Simpson PA, Vanputten V, Crossno J, Offermanns S, Weiser-Evans MC. SDF-1alpha induction in mature smooth muscle cells by inactivation of PTEN is a critical mediator of exacerbated injury-induced neointima formation. Arterioscler Thromb Vasc Biol. 2011;31:1300–1308. doi: 10.1161/ATVBAHA.111.223701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hernando E, Charytonowicz E, Dudas ME, Menendez S, Matushansky I, Mills J, Socci ND, Behrendt N, Ma L, Maki RG, Pandolfi PP, Cordon-Cardo C. The AKT-mTOR pathway plays a critical role in the development of leiomyosarcomas. Nat Med. 2007;13:748–753. doi: 10.1038/nm1560. [DOI] [PubMed] [Google Scholar]
- 24.Andres V, Gascon-Irun M, Pandolfi PP, Gonzalez-Navarro H. Atheroma development in apolipoprotein E-null mice is not affected by partial inactivation of PTEN. Front Biosci. 2006;11:2739–2745. doi: 10.2741/2003. [DOI] [PubMed] [Google Scholar]
- 25.Chen WJ, Lin KH, Lai YJ, Yang SH, Pang JH. Protective effect of propylthiouracil independent of its hypothyroid effect on atherogenesis in cholesterol-fed rabbits: PTEN induction and inhibition of vascular smooth muscle cell proliferation and migration. Circulation. 2004;110:1313–1319. doi: 10.1161/01.CIR.0000140764.15398.F3. [DOI] [PubMed] [Google Scholar]
- 26.Anderson MJ, Viars CS, Czekay S, Cavenee WK, Arden KC. Cloning and characterization of three human forkhead genes that comprise an FKHR-like gene subfamily. Genomics. 1998;47:187–199. doi: 10.1006/geno.1997.5122. [DOI] [PubMed] [Google Scholar]
- 27.Biggs WH, 3rd, Cavenee WK, Arden KC. Identification and characterization of members of the FKHRFOXO) subclass of winged-helix transcription factors in the mouse. Mamm Genome. 2001;12:416–425. doi: 10.1007/s003350020002. [DOI] [PubMed] [Google Scholar]
- 28.Jacobs FM, van der Heide LP, Wijchers PJ, Burbach JP, Hoekman MF, Smidt MP. FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J Biol Chem. 2003;278:35959–35967. doi: 10.1074/jbc.M302804200. [DOI] [PubMed] [Google Scholar]
- 29.Kim DH, Zhang T, Lee S, Dong HH. FoxO6 in Glucose Metabolism(FoxO6) J Diabetes. 2013;5:233–240. doi: 10.1111/1753-0407.12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Biggs WH, 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci U S A. 1999;96:7421–7426. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siqueira MF, Flowers S, Bhattacharya R, Faibish D, Behl Y, Kotton DN, Gerstenfeld L, Moran E, Graves DT. FOXO1 modulates osteoblast differentiation. Bone. 2011;48:1043–1051. doi: 10.1016/j.bone.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tseng PC, Hou SM, Chen RJ, Peng HW, Hsieh CF, Kuo ML, Yen ML. Resveratrol promotes osteogenesis of human mesenchymal stem cells by upregulating RUNX2 gene expression via the SIRT1/FOXO3A axis. J Bone Miner Res. 2011;26:2552–2563. doi: 10.1002/jbmr.460. [DOI] [PubMed] [Google Scholar]
- 33.Rached MT, Kode A, Silva BC, Jung DY, Gray S, Ong H, Paik JH, Depinho RA, Kim JK, Karsenty G, Kousteni S. FoxO1 expression in osteoblasts regulates glucose homeostasis through regulation of osteocalcin in mice. J Clin Invest. 2010;120:357–368. doi: 10.1172/JCI39901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang H, Pan Y, Zheng L, Choe C, Lindgren B, Jensen ED, Westendorf JJ, Cheng L, Huang H. FOXO1 inhibits Runx2 transcriptional activity and prostate cancer cell migration and invasion. Cancer Res. 2011;71:3257–3267. doi: 10.1158/0008-5472.CAN-10-2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H, Liu X, Wu H. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis. 2002;32:148–149. doi: 10.1002/gene.10036. [DOI] [PubMed] [Google Scholar]
- 36.Mao X, Debenedittis P, Sun Y, Chen J, Yuan K, Jiao K, Chen Y. Vascular smooth muscle cell Smad4 gene is important for mouse vascular development. Arterioscler Thromb Vasc Biol. 2012;32:2171–2177. doi: 10.1161/ATVBAHA.112.253872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Furgeson SB, Simpson PA, Park I, Vanputten V, Horita H, Kontos CD, Nemenoff RA, Weiser-Evans MC. Inactivation of the tumour suppressor, PTEN, in smooth muscle promotes a pro-inflammatory phenotype and enhances neointima formation. Cardiovasc Res. 2010;86:274–282. doi: 10.1093/cvr/cvp425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jonason JH, Xiao G, Zhang M, Xing L, Chen D. Post-translational Regulation of Runx2 in Bone and Cartilage. J Dent Res. 2009;88:693–703. doi: 10.1177/0022034509341629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bae SC, Lee YH. Phosphorylation, acetylation and ubiquitination: the molecular basis of RUNX regulation. Gene. 2006;366:58–66. doi: 10.1016/j.gene.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 40.Abid MR, Yano K, Guo S, Patel VI, Shrikhande G, Spokes KC, Ferran C, Aird WC. Forkhead transcription factors inhibit vascular smooth muscle cell proliferation and neointimal hyperplasia. J Biol Chem. 2005;280:29864–29873. doi: 10.1074/jbc.M502149200. [DOI] [PubMed] [Google Scholar]
- 41.Essaghir A, Dif N, Marbehant CY, Coffer PJ, Demoulin JB. The transcription of FOXO genes is stimulated by FOXO3 and repressed by growth factors. J Biol Chem. 2009;284:10334–10342. doi: 10.1074/jbc.M808848200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boström KI, Jumabay M, Matveyenko A, Nicholas SB, Yao Y. Activation of vascular bone morphogenetic protein signaling in diabetes mellitus. Circ Res. 2011;108:446–457. doi: 10.1161/CIRCRESAHA.110.236596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bialek P, Kern B, Yang X, Schrock M, Sosic D, Hong N, Wu H, Yu K, Ornitz DM, Olson EN, Justice MJ, Karsenty G. A twist code determines the onset of osteoblast differentiation. Dev Cell. 2004;6:423–435. doi: 10.1016/s1534-5807(04)00058-9. [DOI] [PubMed] [Google Scholar]
- 44.Karsenty G. Transcriptional control of skeletogenesis. Annu Rev Genomics Hum Genet. 2008;9:183–196. doi: 10.1146/annurev.genom.9.081307.164437. [DOI] [PubMed] [Google Scholar]
- 45.Zhao M, Qiao M, Oyajobi BO, Mundy GR, Chen D. E3 ubiquitin ligase Smurf1 mediates core-binding factor alpha1/Runx2 degradation and plays a specific role in osteoblast differentiation. J Biol Chem. 2003;278:27939–27944. doi: 10.1074/jbc.M304132200. [DOI] [PubMed] [Google Scholar]
- 46.Shen R, Wang X, Drissi H, Liu F, O'Keefe RJ, Chen D. Cyclin D1-cdk4 induce runx2 ubiquitination and degradation. J Biol Chem. 2006;281:16347–16353. doi: 10.1074/jbc.M603439200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones DC, Wein MN, Oukka M, Hofstaetter JG, Glimcher MJ, Glimcher LH. Regulation of adult bone mass by the zinc finger adapter protein Schnurri-3. Science. 2006;312:1223–1227. doi: 10.1126/science.1126313. [DOI] [PubMed] [Google Scholar]
- 48.Shen R, Chen M, Wang YJ, Kaneki H, Xing L, O'Keefe RJ, Chen D. Smad6 interacts with Runx2 and mediates Smad ubiquitin regulatory factor 1-induced Runx2 degradation. J Biol Chem. 2006;281:3569–3576. doi: 10.1074/jbc.M506761200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li M, Chiu JF, Gagne J, Fukagawa NK. Age-related differences in insulin-like growth factor-1 receptor signaling regulates Akt/FOXO3a and ERK/Fos pathways in vascular smooth muscle cells. J Cell Physiol. 2008;217:377–387. doi: 10.1002/jcp.21507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1.Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H, Liu X, Wu H. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis. 2002;32:148–149. doi: 10.1002/gene.10036. [DOI] [PubMed] [Google Scholar]
- 2.Mao X, Debenedittis P, Sun Y, Chen J, Yuan K, Jiao K, Chen Y. Vascular smooth muscle cell Smad4 gene is important for mouse vascular development. Arterioscler Thromb Vasc Biol. 2012;32:2171–2177. doi: 10.1161/ATVBAHA.112.253872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun Y, Byon CH, Yuan K, Chen J, Mao X, Heath JM, Javed A, Zhang K, Anderson PG, Chen Y. Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ Res. 2012;111(5):543–552. doi: 10.1161/CIRCRESAHA.112.267237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byon CH, Sun Y, Chen J, Yuan K, Mao X, Heath JM, Anderson PG, Tintut Y, Demer LL, Wang D, Chen Y. Runx2-Upregulated Receptor Activator of Nuclear Factor {kappa}B Ligand in Calcifying Smooth Muscle Cells Promotes Migration and Osteoclastic Differentiation of Macrophages. Arterioscler Thromb Vasc Biol. 2011;31:1387–1396. doi: 10.1161/ATVBAHA.110.222547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, McDonald JM, Chen Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. 2008;283:15319–15327. doi: 10.1074/jbc.M800021200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Y, Budd RC, Kelm RJ, Jr, Sobel BE, Schneider DJ. Augmentation of proliferation of vascular smooth muscle cells by plasminogen activator inhibitor type 1. Arterioscler Thromb Vasc Biol. 2006;26:1777–1783. doi: 10.1161/01.ATV.0000227514.50065.2a. [DOI] [PubMed] [Google Scholar]
- 7.Byon CH, Sun Y, Chen J, Yuan K, Mao X, Heath JM, Anderson PG, Tintut Y, Demer LL, Wang D, Chen Y. Runx2-Upregulated Receptor Activator of Nuclear Factor {kappa}B Ligand in Calcifying Smooth Muscle Cells Promotes Migration and Osteoclastic Differentiation of Macrophages. Arterioscler Thromb Vasc Biol. 2011;31:1387–1396. doi: 10.1161/ATVBAHA.110.222547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun Y, Byon C, Yuan K, Chen J, Mao X, Heath JM, Javed A, Zhang K, Anderson PG, Chen Y. Smooth Muscle Cell-Specific Runx2 Deficiency InhibitsVascular Calcification. Circ Res. 2012;111:543–552. doi: 10.1161/CIRCRESAHA.112.267237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.