

Abstract
Epidermal fatty acid-binding protein (E-FABP/FABP5/DA11) binds and transport long-chain fatty acids in the cytoplasm and may play a protecting role during neuronal injury. We examined whether E-FABP protects nerve growth factor-differentiated PC12 cells (NGFDPC12 cells) from lipotoxic injury observed after palmitic acid (C16:0; PAM) overload. NGFDPC12 cells cultures treated with PAM/bovine serum albumin at 0.3 mM/0.15 mM show PAM-induced lipotoxicity (PAM-LTx) and apoptosis. The apoptosis was preceded by a cellular accumulation of reactive oxygen species (ROS) and higher levels of E-FABP. Antioxidants MCI-186 and N-acetyl cysteine prevented E-FABP's induction in expression by PAM-LTx, while tert-butyl hydroperoxide increased ROS and E-FABP expression. Non-metabolized methyl ester of PAM, methyl palmitic acid (mPAM), failed to increase cellular ROS, E-FABP gene expression, or trigger apoptosis. Treatment of NGFDPC12 cultures with siE-FABP showed reduced E-FABP levels correlating with higher accumulation of ROS and cell death after exposure to PAM. In contrast, increasing E-FABP cellular levels by pre-loading the cells with recombinant E-FABP diminished the PAM-induced ROS and cell death. Finally, agonists for PPARβ (GW0742) or PPARγ (GW1929) increased E-FABP expression and enhanced the resistance of NGFDPC12 cells to PAM-LTx. We conclude that E-FABP protects NGFDPC12 cells from lipotoxic injury through mechanisms that involve reduction of ROS.
Epidermal fatty acid-binding protein (E-FABP) may protect nerve cells from the damaging exposure to high levels of free fatty acids (FA). We show that E-FABP can neutralize the effects of reactive oxygen species (ROS) generated by the high levels of FA in the cell and protect PC12 cells from lipotoxic injuries common in Type 2 diabetes neuropathy. Potentially, E-FABP gene up-regulation may be mediated through the NFkB pathway and future studies are needed to further evaluate this proposition.
Keywords: antioxidants, FABP, lipotoxicity, PPAR, reactive oxygen species
Epidermal fatty acid-binding protein (E-FABP/FABP5/DA11) belongs to a family of low-molecular-weight (14–15 kDa) cytosolic proteins that binds long-chain free fatty acids. The members of the FABP family share similar tertiary structure which consists of two short α-helical segments and a fatty acid binding cavity surrounded by 2 five-stranded anti-parallel β-sheets (Gutierrez-Gonzalez et al. 2002). FABPs are thought to play a prominent role in trafficking of long-chain free fatty acids to respective final metabolic or regulatory sites within the cell. Compared to other members of FABPs in the family, E-FABP has particularly wide tissue distribution, suggesting its diversified functions. E-FABP was first isolated from human epidermal cells (Siegenthaler et al. 1994), and the rat version (DA11) was later cloned from injured dorsal root ganglion cells (De Leon et al. 1996). E-FABP is also found expressed in tissues such as vasculature endothelial cells (Masouye et al. 1997), mammary tissue (Buhlmann et al. 1999), adipose tissue (Coe et al. 1999), pancreatic islets (Hyder et al. 2010), macrophages (Owada et al. 2001) in addition to all nerve cells studied. An analysis of the temporal and spatial expression profiles in the developing brain shows that E-FABP exhibits a robust expression during embryonic neurogenesis, axonal growth, neuronal migration, and terminal differentiation (Liu et al. 1997, 2000). It is highly expressed (more than 100-fold compared to adult) as early as E14 in the rat, and the high level of expression is sustained during the first 3 weeks post-natal followed by a lower expression in the adult brain and spinal cord (Owada et al. 1996b; Liu et al. 2000). E-FABP is up-regulated by nerve growth factor (NGF) and is required for NGF-induced and polyunsaturated fatty acid-potentiated neurite outgrowth in PC12 cells (Allen et al. 2000; Liu et al. 2008). E-FABP colocalizes with growth-associated protein 43 in differentiating PC12 cells and in retinal ganglion cells (Allen et al. 2000, 2001). Furthermore, we and others have reported that E-FABP is induced in motor neurons after peripheral nerve axonal injury (De Leon et al. 1996; Owada et al. 1997). Similarly, E-FABP expression is up-regulated in the hippocampus in response to kainic acid-induced seizure (Owada et al. 1996a) and in the cerebellum and hippocampus after experimental brain ischemia (Boneva et al. 2010; Ma et al. 2010). In addition, hypoxia causes elevated expression of E-FABP in aortic endothelial cells (Han et al. 2010) and PC12 cells (unpublished data). These results suggest a role of E-FABP during stress injury in the nerve cells, but its neuroprotective characteristics in the nervous system have not been addressed in the literature.
Neuronal tissue holds a larger amount of membrane lipids compared to other tissues and contains a high concentration of polyunsaturated fatty acids that are susceptible to lipid peroxidation, leading to oxidative damage. Modulation lipid metabolism plays an important role during neuronal injuries and neuronal disorders. Several studies have shown that traumatic injury and hypoxic–ischemic brain injury lead to activation of phospholipase A2 and sphingomyelinsse that release fatty acids and ceramide from membrane phospholipids and sphingolipids, respectively (Dhillon et al. 1997; Yu et al. 2000; Liu et al. 2006). The increase of arachidonic acid and ceramide initiates the cascade of inflammatory response (Farooqui et al. 2007; Nixon 2009), and the accumulation of free fatty acids, especially saturated fatty acids, can cause lipotoxicity (Scheff and Dhillon 2004). All of these events contribute to a great extent to the prolonged phase of secondary degenerative damage by various pathways, including generation of free radicals and lipid peroxidation, eventually leading to cell death (Hall and Braughler 1989; Lok et al. 2011). Furthermore, there is growing evidence suggesting that sustained elevation of saturated fatty acids in the circulation because of high calorie intake results in lipotoxicity, inflammation, and increased oxidative stress, which are believed to take part in the development of neurodegenerative diseases such as Alzheimer's disease and Parkinson's disease (Adibhatla and Hatcher 2008; Middleton and Yaffe 2009; Gupta et al. 2012), and metabolic disorders such as type 2 diabetes and non-alcoholic fatty liver disease (Unger and Orci 2001; Wang et al. 2006).
Our laboratory has been investigating the lipotoxic effect of palmitic acid (C16:0; PAM) in the nervous system. Pathophysiological concentration of PAM causes caspase-dependent and caspase-independent apoptotic cell death in NGF-differentiated PC12 (NGFDPC12) cells and Schwann cells (Ulloth et al. 2003; Padilla et al. 2011). The injury cascade starts with increasing intracellular calcium concentration and reactive oxygen species (ROS) generation (Almaguel et al. 2009; Padilla et al. 2011). Meanwhile, ER stress genes (CHOP, Xbp1, and GRP78) and apoptosis genes (BNIP3, Bax) have been shown to be involved in the process (Ulloth et al. 2003; Almaguel et al. 2009; Padilla et al. 2011), and Fas receptor and Fas ligand are dramatically increased (Ulloth et al. 2003; Almaguel et al. 2009). In NGFDPC12 cells, lysosomal membrane permeabilization precedes mitochondrial membrane permeabilization, and cathepsin L seems to be a key player of this lysosomal-mitochondrial crosstalk in PAM-induced lipotoxicity (PAM-LTx) (Almaguel et al. 2010). This study uses an NGFDPC12 cell model to evaluate whether E-FABP is a stress response protein that plays a protective role counteracting the ROS accumulation observed during PAM-LTx.
Material and Methods
Material
Rat pheochromocytoma (PC12) cells were obtained from American Type Culture Collection (Manassas, VA, USA) and all experiments were performed within seven passages. Cell culture medium was acquired from Mediatech (Manassas, VA, USA), while horse serum and fetal bovine serum (FBS) were purchased from Atlantic Biological (Lawrenceville, GA, USA). NGF is a product from Alomone Labs (Jerusalem, Israel). Fatty acid-free bovine serum albumin (BSA) was from EMD Millipore Corp. (Billerica, MA, USA) and palmitic acid (PAM), methyl palmitic acid (mPAM), N-acetyl cysteine (NAC), and tert-butyl hydroperoxide were from Sigma-Aldrich (St. Louis, MO, USA). Antioxidant MCI-186 was purchased from Biomol Research Laboratories (Plymouth Meeting, PA, USA) and nuclear factor kappa B (NF-ĸB) SN50 cell permeable inhibitory peptide and control peptide SN50M were from Enzo Life Sciences (Farmingdale, NY, USA). All of the peroxisome proliferator-activated receptor (PPAR) agonists were bought from R&D Systems (Minneapolis, MN, USA).
Cell culture and PAM treatment
PC12 cells were propagated with F-12K medium (Ham's F-12 Nutrient Mixture, Kaighn's Modification with glutamine) containing 15% horse serum, 2.5% FBS, and penicillin (100 units/mL)/streptomycin (100 μg/mL) (Full serum-medium). The cells were then reseeded in 6-well plates at the density of 70 000 cells/well and differentiated with 50 ng/mL of NGF in F-12K medium with 1% FBS and penicillin/streptomycin (1% FBS-NGF medium). The medium was replaced every 3 days and PC12 cells were differentiated for 7–10 days before treatments.
PAM was delivered to the cell culture in complex with fatty acid-free BSA at PAM to BSA molar ratio of 2 : 1, 1 : 1, or 0.5 : 1. Following previously described procedures (Ulloth et al. 2003; Almaguel et al. 2009), PAM was first dissolved in 100% ethanol to make 300 mM stock solution and then diluted into warm 1% FBS-NGF medium containing 150 μM fatty acid-free BSA to make 300 μM (2 : 1), 150 μM (1 : 1), and 75 μM (0.5 : 1) working solution. The final concentration of ethanol was less than 0.1%. With this method, the concentration of unbound free PAM in the medium was about 11 nM in the 300 μM PAM medium throughout the course of experiment. For control, the cells were treated with 1% FBS-NGF medium containing 150 μM fatty acid-free BSA and 0.1% ethanol, designated as ‘control medium.’
Cell morphology analysis
Cell morphology changes were monitored by phase contrast microscopy at different time points, while nuclear morphology of control and PAM-treated NGFDPC12 cells were assessed by staining with fluorescent dye Hoechst 33342 (Life Technologies, Carlsbad, CA, USA). Hoechst dye was added to the culture medium at 2.5 μg/mL and incubated for 10 min at 37°C. Nuclear morphology was visualized under fluorescent microscopy. Apoptotic cells were identified by the presence of highly condensed chromatin or fragmented nuclei.
Cell viability assay
At the end of experimental treatments, test medium was replaced by 1 mL/well of plain F-12K medium containing 50 μL of cell proliferation/viability reagent WST-1 (Roche Applied Science, Indianapolis, IN, USA). After 30-min incubation in the cell culture incubator, 100-μL aliquots were transferred to 96-well plate and optical density at 450 nm was determined by μQuant plate reader (Bio-Teck Instruments, Winooski, VT, USA). WST-1 solution without incubating with cells was served as the blank. Cell viability was expressed as relative optical density of experimental group opposed to that of control group. For comparison, crystal violet cell viability assay was also performed using the same procedure described previously (Padilla et al. 2011).
Reactive oxygen species analysis
The ROS levels in NGFDPC12 cells were evaluated by a florescent indicator 2′,7′ dichlorodihydrofluorescein diacetate (H2DCFDA, Life Technologies). After treatments, the cells were changed to plain F-12K medium with 10 μM H2DCFDA and incubated for 25 min in cell culture incubator. Cells were then detached and analyzed using a FACSCalibur flow cytometer and Flow-Jo software as described before (Almaguel et al. 2009; Padilla et al. 2011).
siRNA transfection
To knock down E-FABP in NGFDPC12 cells, we selected Ambion in vivo siRNA system (Ambion, Austin, TX, USA). Rat FABP5 siRNA (s139047) and negative control siRNA with scrambled sequences were acquired (Life Technologies) and introduced into cells by X-tremeGENE siRNA transfection reagent (Roche Applied Science). The siRNA transfection, following the manufacturer's instruction, was performed as below: siE-FABP or siControl stock was prepared at 100 μM in water and 2 μL of stock was diluted with 250 μL of Opti-MEM. Transfection reagent (10 μL) was also diluted with 250 μL of Opti-MEM. Diluted siRNA and transfection reagent were combined and incubated for 20 min. Three-day-differentiated PC12 cells in 6-well plates were change to antibiotics-free medium (1.5 mL/well) and then transfection solution (0.5 mL) was added to the well. After 24 h, the medium was changed to 1% FBS–NGF medium for continual differentiation for another 3–5 days. The transfected NGFDPC12 cells were treated with PAM accordingly.
Deliver recombinant E-FABP to NGFDPC12 cells
Recombinant rat E-FABP protein was produced using IMPACT kit (New England Biolabs, Beverly, MA, USA) and delipidated by Lipidex 1000 method as reported before (Liu et al. 2008). To increase the level of E-FABP in NGFDPC12 cells, recombinant E-FABP protein was delivered to the cells by BioPORTER Quik Ease kit (Gene Therapy Systems, San Diego, CA, USA). Dried BioPORTER reagent in the vials was hydrated with phosphate-buffered saline (PBS) and then incubated with recombinant E-FABP at 25°C for 5 min. Recombinant E-FABP/BioPORTER complex solution was diluted with plain F-12 medium before added to NGFDPC12 cells in 6-well plates (10 μg protein/well). BioPORTER reagent alone and BioPORTER complexed with a non-related protein, β-galactosidase, were used as controls. After 3- to 4-h incubation, full serum medium was added to the wells to let cells recover for 4 h and then the medium was changed to 1% FBS-NGF medium. The cells were treated with PAM accordingly on the following day.
Real-time RT-PCR analysis
Total cellular RNA was extracted using TRI reagent (Molecular Research Center, Cincinnati, OH, USA) and quantified by measuring the OD at 260 nm. RNA samples (800 ng) were first reversed transcribed to cDNA using iSCRIPT cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA, USA). E-FABP gene as well as another five FABP genes: intestinal type FABP (I-FABP), heart type FABP (H-FABP), adipocyte FABP (A-FABP), brain type FABP (B-FABP), and myelin FABP (M-FABP) were quantified by real-time PCR using CFX96 system (Bio-Rad Laboratories). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the reference gene. Table1 lists primer sequences used in real-time PCR. Reactions were performed in three replicates with a 25-μL mixture containing cDNA samples, primers, and iQ Sybr Green supermix (Bio-Rad Laboratories). The relative amount of mRNA in experimental cells was calculated using 2−ΔΔCT method. In addition, the sizes of final PCR products were verified with a 4% agarose gel followed by ethidium bromide staining.
Table 1.
Primer sequences for RT-qPCR
| Gene | Primer sequences (5′–3′) | Amplicon | |
|---|---|---|---|
| I-FABP | Forward | ATGCAGAAGGGCTAGCTTGG | 70 bp |
| Reverse | CACAGTGAGTGAGCCTGCAT | ||
| H-FABP | Forward | CATGGCGGACGCCTTTGTCG | 91 bp |
| Reverse | GGTGGCAAAGCCCACACCGA | ||
| A-FABP | Forward | AGAAGTGGGAGTTGGCTTCG | 103 bp |
| Reverse | ACTCTCTGACCGGATGACGA | ||
| E-FABP | Forward | TTACCCTCGACGGCAACAA | 106 bp |
| Reverse | CCATCAGCTGTGGTTTCATCA | ||
| B-FABP | Forward | GGGCGTGGGCTTTGCCACTA | 89 bp |
| Reverse | TCCGGATCACCACTTTGCCGC | ||
| M-FABP | Forward | TCCGGGGGCCTGGGCAGTTA | 117 bp |
| Reverse | GGAGGCTGCTCCTGTTGCCTG | ||
| GAPDH | Forward | GGGGCTCTCTGCTCCTCCCTG | 119 bp |
| Reverse | AGGCGTCCGATACGGCCAAA |
Western Blots
The polyclonal antibodies against E-FABP were generated in rabbits against recombinant E-FABP produced in the laboratory. After treatment, cells were pelleted and extracted with lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton X-100, 5% glycerol, 1 mM EDTA, 100 μM phenylmethylsulfonyl fluoride, 1 mM dithiothreitol, and protease inhibitor cocktail from Roche Applied Science). Protein extracts of NGFDPC12 cells (10 μg) were resolved on a NuPAGE Bis-Tris gel (Life Technologies) and transferred to a nitrocellulose membrane. After blocking with 7.5% milk in Tris-buffered saline with 0.05% Tween 20, pH 7.4 (TTBS), the membrane was incubated with E-FABP antiserum and anti-β-actin (clone AC-15; Sigma-Aldrich) in 5% milk TTBS at 4°C overnight. Subsequently, the membrane was washed with TTBS, incubated with horseradish peroxidase-goat anti-rabbit IgG and goat anti-mouse IgG for 1 h, and washed again. The signal was then detected by SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, Rockford, IL, USA). The relative amount of protein was quantified by densitometry analysis of the autoradiographs using Alpha Innotech (Protein Simple, Santa Clara, CA, USA).
Immunofluorescent staining
PC12 cells were seeded in collagen-coated 4-well culture slides (BD Biosciences, Bedford, MA, USA) and differentiated with NGF. After PA-LTx treatments, cells were fixed with 4% paraformaldehyde. After washed with PBS, the cells were incubated with blocking solution that consists of 20% normal donkey serum in PBST (PBS with 0.1% Tween 20) for 2 h. Primary antibody, anti-E-FABP antiserum, was prepared in 3% normal donkey serum with PBST and incubated with the cells overnight at 4°C. Next day, the slides were washed with PBST and incubated with secondary antibody, Alexa Fluor488 anti-rabbit (Life Technologies), for 2 h. Afterward, cells were counter-stained with Texas red-phalloidin (Liu et al. 2008) and examined with fluorescent microscopy.
Statistical analysis
All the experiments were repeated independently at least three times. Statistical comparisons were made using Student's t-test. Significance was accepted at p < 0.05.
Results
Lipotoxicity caused by palmitic acid (PAM) induces apoptosis in NGFDPC12 cells
The first series of experiments confirmed the effect of PAM-LTx in NGFDPC12 cells. Figure1(a–c) shows the morphological appearances of NGFDPC12 cells after PAM treatment for up to 48 h. The extensive neurite network seen in control non-treated cells started to disintegrate after 24 h and was largely absent after 36 and 48 h of treatment (Fig.1a). The loss of neurites coincided with roundup cell bodies (Fig.1a) and typical apoptotic nuclei in PAM-treated cells (Fig.1b). Immunocytochemistry analysis of E-FABP revealed that there is an accumulation of E-FABP in the neurite region after PAM treatment in NGFDPC12 cells (Fig.1c). Quantitative cell viability assays indicated a time-dependent decrease loss of NGFDPC12 cells following PAM treatment as shown in WST-1 assay and crystal violet assay (Fig.1d and e).
Figure 1.
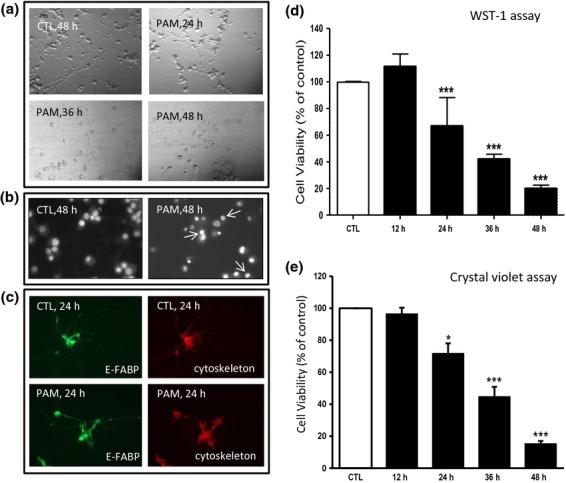
Palmitic acid (PAM) induces apoptotic cell death in NGFDPC12 cells. Cells were treated with control medium (CTL) or 300 μM PAM for different time periods (12–48 h). (a) Representative cellular morphological images were acquired by phase contrast microscopy. (b) Representative nuclear morphological images were analyzed with Hoechst staining and fluorescent microscopy. Arrows indicate chromatin condensation and fragmentation. (c) Representative immunofluorescent images are shown to exhibit intracellular epidermal fatty acid-binding protein (E-FABP) in green and cytoskeleton in red. (d) Cell viability of NGFDPC12 cells was determined by WST-1 assay. (e) Cell viability of NGFDPC12 cells was determined by crystal violet assay. The data represent mean ± SEM of three independent experiments. Significance symbol: *p < 0.05, ***p < 0.001 (compared to control group).
E-FABP is up-regulated under PAM-LTx in NGFDPC12 cells
In the next series of experiments, we determined the E-FABP mRNA levels in NGFDPC12 cells at various time points after cells were treated with 75, 150, and 300 μM PAM (Fig.2a). When near physiological concentration of PAM was used (75 μM), mRNA level of E-FABP did not change for up to 48 h. However, at pathophysiological concentration (150 μM), E-FABP mRNA showed a maximum increase of 2.5-fold at 18 h and dropped back to control level at 24 and 48 h. When NGFDPC12 cells were cultured with even higher PAM concentration of 300 μM, an earlier and a stronger response of E-FABP mRNA up-regulation was observed. Under such conditions, E-FABP mRNA level reached a maximum 3.5-fold at 12 h, followed by a lower stimulation at 18 h (threefold) and 24 h (twofold). Western blot analysis showed that E-FABP protein level in NGFDPC12 cells was increased continuously up to 24 h by 300 μM PAM, while there are no significant changes observed up to 24 h when treated with 150 μM PAM (Fig.2b and c). In addition to E-FABP, mRNA levels of the other five FABPs (I-FABP, H-FABP, A-FABP, B-FABP, and M-FABP) in NGFDPC12 cells were also analyzed and the results are summarized in Table2. The data show that E-FABP is the most abundant FABP in NGFDPC12 cells by two to three orders of magnitude higher and it was significantly increased by PAM exposure. PAM appears to also stimulate A-FABP expression, but it exhibits only very low expression in NGFDPC12 cells.
Figure 2.
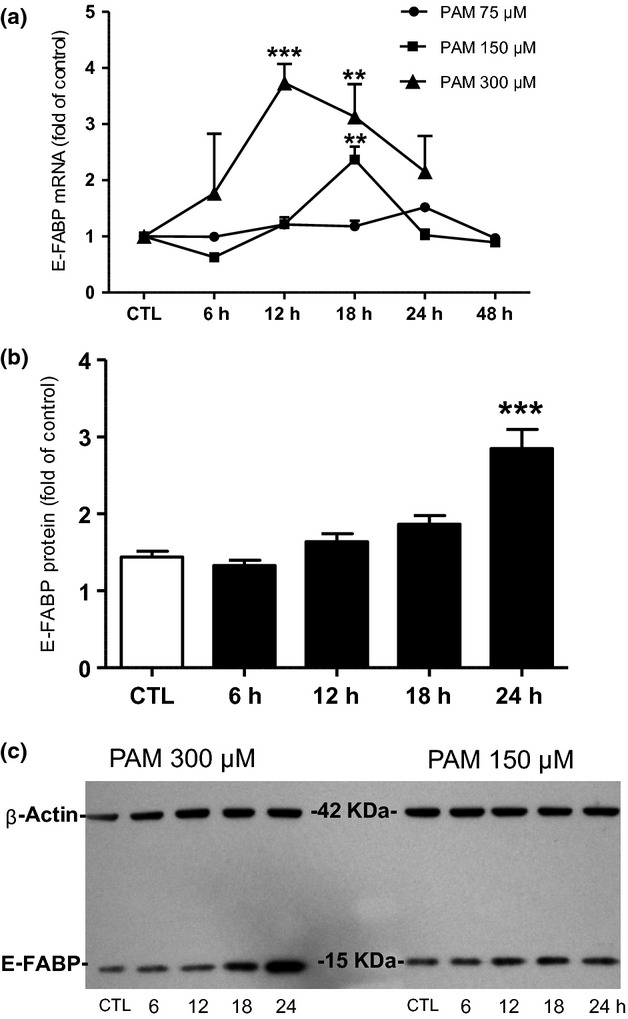
Epidermal fatty acid binding protein (E-FABP) is up-regulated under palmitic acid-induced lipotoxicity (PAM-LTx) in NGFDPC12 cells. (a) Cells were treated with control medium (CTL) or PAM at the concentrations of 75, 150, or 300 μM for 6–48 h. Quantification of E-FABP mRNA levels was performed by real-time RT-PCR. (b) Cells were treated with CTL or 300 μM PAM for 6–24 h. Quantification of E-FABP protein levels was done by Western blots followed by densitometry analysis, using β-actin as the loading control. (c) A representative Western blot for E-FABP (15 kDa) and β-actin (42 kDa) of NGFDPC12 cells treated with 300 or 150 μM PAM for 6–24 h. The data represent mean ± SEM of three independent experiments. Significance symbols: **p < 0.01 and ***p < 0.001 (compared to control group).
Table 2.
Relative level of FABP mRNAs in NGFDPC12 cells with and without PAM treatment
| E-FABP | I-FABP | H-FABP | A-FABP | B-FABP | M-FABP | |
|---|---|---|---|---|---|---|
| Ct(FABP)-Ct(GAPDH) | 0.00 ± 0.27 | 8.72 ± 2.15 | 9.93 ± 0.56 | 11.69 ± 2.20 | 11.76 ± 4.73 | 10.10 ± 2.88 |
| Relative Abundance | 1042.81 ± 196.69 | 5.51 ± 4.95 | 1.11 ± 0.32 | 0.73 ± 0.67 | 3.95 ± 4.00 | 3.45 ± 3.48 |
| PAM/CTL (fold) | 3.06 ± 0.90 | 1.18 ± 0.66 | 1.77 ± 1.40 | 4.71 ± 2.18 | 1.03 ± 0.57 | 1.48 ± 0.80 |
Relative abundance of selected FABPs in NGFDPC12 cells (without PAM-LTx) was determined by real-time RT-PCR, using GAPDH as the reference gene. Ct, threshold cycle. The amount of mRNA in experimental cells was calculated using 2−ΔΔCT method. Their mRNA changes upon PAM treatment are represented as mRNA level of PAM-treated cells/mRNA level of control-treated cells.
E-FABP up-regulation by PAM-LTx in NGFDPC12 cells is mediated through reactive oxygen species accumulation
The accumulation of ROS in NGFDPC12 cells after PAM treatment was analyzed using H2DCFDA assay. The results indicate that PAM caused a dose- and time-dependent increase of ROS levels in NGFDPC12 cells (Fig.3a). The increase of ROS by 300 and 150 μM PAM occurred as early as 6 h after treatment, which precedes the up-regulation of E-FABP mRNA seen in the same conditions (18 h, Fig.2a). Their ROS accumulation reached the highest level at 12 h and returned to control level at 24 h. Furthermore, ROS levels were not increased when NGFDPC12 cells were treated with 75 μM PAM, in which condition did not affect E-FABP mRNA levels (Fig.2a). Therefore, we hypothesized that the stimulation of E-FABP could be mediated through ROS generation. To test this hypothesis, antioxidants MCI-186 (100 μM) or N-acetyl cysteine (NAC, 100 μM) were added to NGFDPC12 cultures during PAM-LTx treatment. The results are shown in Fig.3(b). MCI-186 and NAC by themselves did not affect E-FABP expression but were able to partially (MCI-186) or completely (NAC) abolish the up-regulation of E-FABP caused by PAM. In addition, NGFDPC12 cells were treated with free radical tert-butyl hydroperoxide (100 μM), and the result showed that NGFDPC12 cells elicited a 2.5- to 3-fold induction of E-FABP mRNA expression after 12–24 h (Fig.3c), which is comparable to the stimulation seen by 300 μM PAM (Fig.2a). These results confirmed that E-FABP is induced via ROS generated by PAM-LTx.
Figure 3.
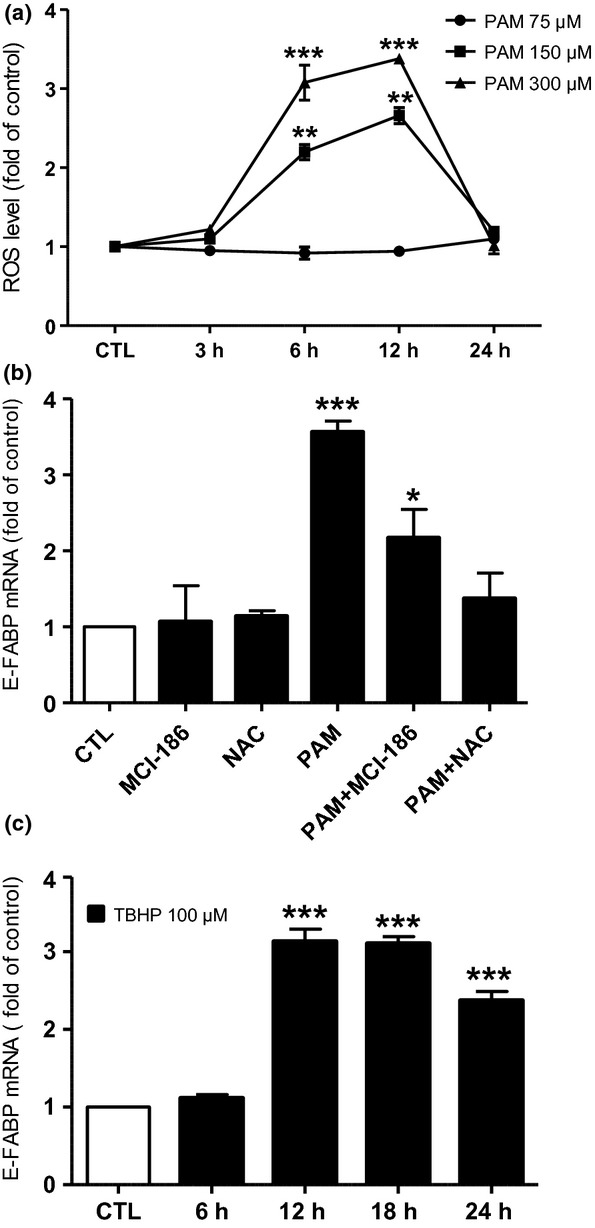
Cellular accumulation of reactive oxygen species (ROS) mediates palmitic acid-induced lipotoxicity (PAM-LTx)-triggered epidermal fatty acid-binding protein (E-FABP) up-regulation in NGFDPC12 cells. (a) Cells were treated with control medium (CTL) or PAM at the concentrations of 75, 150, or 300 μM for 3, 6, 12, and 24 h and cellular ROS level was measured with H2DCFDA method. (b) Cells were pre-treated with antioxidants MCI-186 (100 μM in control medium) or N-acetyl-cysteine (NAC, 100 μM in control medium) for 12 h and then incubated with 300 μM PAM in the presence of the same antioxidant for another 12 h. For comparison, cells were treated with CTL, 300 μM PAM or antioxidants alone for 12 h. Relative E-FABP mRNA levels were determined by real-time RT-PCR. (c) Cells were treated with CTL or tert-butyl hydroperoxide (TBHP, 100 μM in control medium) for 6, 12, 18, and 24 h and relative E-FABP mRNA levels were determined. The data represent mean ± SEM of three independent experiments. Significance symbols: *p < 0.05, **p < 0.01 and ***p < 0.001 (compared to control group).
ROS have been shown to activate a number of cellular signal transduction pathways that could mediate the increase of E-FABP gene expression observed during lipotoxic injury. Considering that E-FABP gene has two putative NF-ĸB binding sites in the promoter region (Fig.4a) that mediate the up-regulation of E-FABP in MCF-7 cells (Kannan-Thulasiraman et al. 2010), we proceeded to further explore the interactions of ROS and E-FABP regulation in cultures treated with SN50, which blocks nucleus translocation of NF-ĸB. Figure4(b) shows that the increase of E-FABP mRNA by PAM was significantly lower in cells cotreated with SN50. Determination of cellular ROS levels at a later time showed that cells cotreated with SN50 and PAM exhibited higher ROS level than cells treated with PAM alone, while the control peptide, SN50M, did not affect the ROS levels in NGFDPC12 cells (Fig.4c). In an additional experiment, we also examined if MAP kinase mediates the up-regulation of E-FABP by PAM-LTx. Figure4(d) indicates that in the presence or absence of the MAP kinase inhibitor, U0126, E-FABP mRNA was stimulated to similar level upon PAM treatment.
Figure 4.
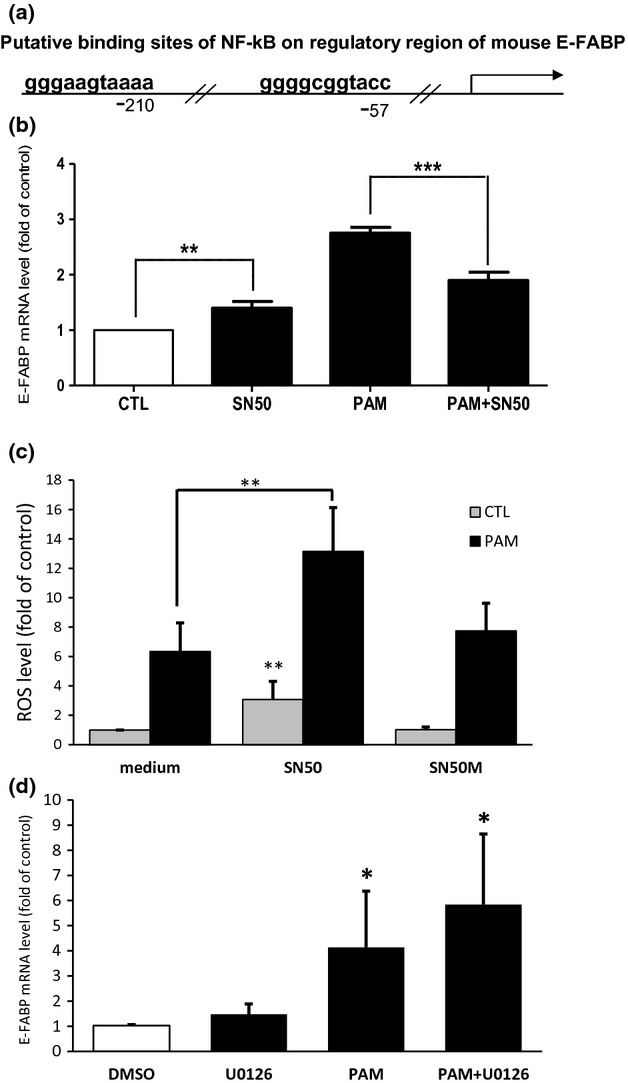
NF-ĸB inhibitor affects palmitic acid-induced lipotoxicity (PAM-LTx)-induced epidermal fatty acid-binding protein (E-FABP) expression and reactive oxygen species (ROS) accumulation in NGFDPC12 cells. (a) The putative binding sites of NF-kB on regulatory region of mouse E-FABP predicted by PROMO program (Farre et al. 2003). (b) Cells were treated with control medium (CTL), NF-kB inhibitor SN50 at 50 μg/mL in control medium (SN50), 300 μM PAM (PAM) or PAM together with SN50 (PAM+SN50) for 12 h and E-FABP mRNA levels were determined by real-time RT-PCR. (c) Cells were treated with control medium, SN50, or control peptide SN50M with or without 300 μM PAM for 18 h and ROS level was measured with H2DCFDA method. (d) Cells were treated with 20 μM of U0126 or control medium with 0.1% DMSO with or without 300 μM PAM for 12 h and E-FABP mRNA levels were determined by real-time RT-PCR. The data represent mean ± SEM of three independent experiments. Significance symbols: *p < 0.05, **p < 0.01 and ***p < 0.001. Significance symbols are shown above bars when compared to control group and are shown above lines when compared between two linked groups.
E-FABP up-regulation by PAM-LTx in NGFDPC12 cells may involve PAM metabolism
To determine whether metabolized PAM is essential for producing lipotoxicity, a non-metabolized methyl ester of PAM, methyl palmitic acid (mPAM), was tested (Parker et al. 2003). Our results revealed that mPAM does not alter either the E-FABP mRNA (Fig.5a) or ROS level (Fig.5b) and NGFDPC12 cells did not lose their morphological integrity as they did with PAM treatment (Fig.5c). Hence, PAM activation and metabolism are prerequisite in the lipotoxic process.
Figure 5.
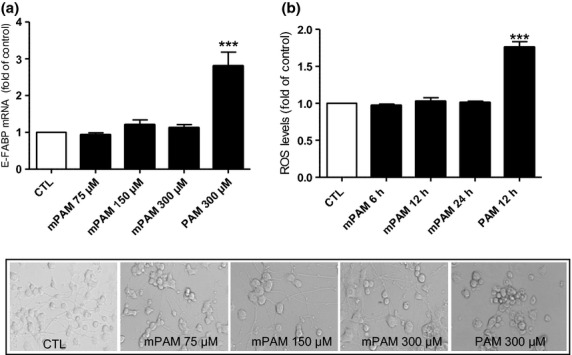
Non-metabolized PAM, methyl palmitic acid (mPAM), does not cause lipotoxicity in NGFDPC12 cells. (a) Cells were treated with control medium (CTL), 300 μM PAM or mPAM at the concentrations of 75, 150, or 300 μM for 12 h. Relative epidermal fatty acid-binding protein (E-FABP) mRNA levels were determined by real-time RT-PCR. (b) Cells were treated with control medium (CTL), 300 μM PAM for 12 h or 300 μM mPAM for 6, 12, and 24 h and reactive oxygen species (ROS) level was measured with H2DCFDA method. The data represent mean ± SEM of three independent experiments. Significance symbols: ***p < 0.001 (compared to control group). (c) Representative morphological photographs of NGFDPC12 cells 48 h after being treated as in (a).
Cellular levels of E-FABP affect ROS concentration and NGFDPC12 cells viability during PAM-LTx
To investigate the significance of E-FABP up-regulation during lipotoxicity, we tested lipotoxic effects on NGFDPC12 cells having decreased or increased intracellular E-FABP protein levels.
By transfecting siRNA, both the E-FABP protein level and mRNA level in NGFDPC12 cells were significantly reduced when compared to siControl-treated cells (Fig.6a). PAM treatment resulted in elevated E-FABP mRNA in siE-FABP cells and siControl cells, but E-FABP in siE-FABP cells were significant lower than that in siControl cells 12 and 18 h after PAM-LTx (Fig.6a). The effect of decreased E-FABP in NGFDPC12 cells on intracellular ROS is shown in Fig.6(b). In the absence of PAM, siE-FABP cells showed higher basal level of ROS than siControl cells, presumably because of lacking ROS-buffering effect from E-FABP. PAM-LTx caused time-dependent increase of ROS accumulation at 12 and 18 h. As expected, siE-FABP cells exhibited higher ROS than siControl did (Fig.6b). Cell viability assay showed no difference between siControl and siE-FABP cells without PAM (Fig.6c). However, a significant higher loss of cell viability in siE-FABP-transfected cells during lipotoxicity was found when compared to that of siControl (Fig.6c). These results suggest that the E-FABP can reduce ROS accumulation and protect NGFDPC12 cells from apoptosis during PAM-LTx.
Figure 6.
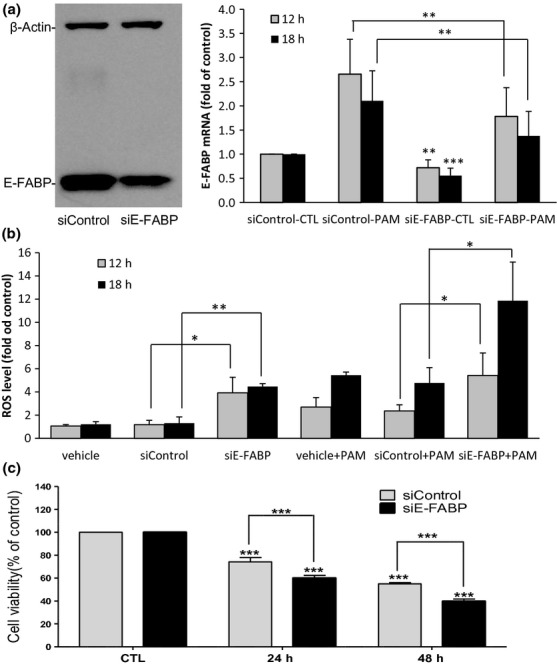
Lowering epidermal fatty acid-binding protein (E-FABP) levels in NGFDPC12 cells by siRNA increases reactive oxygen species (ROS) accumulation and PAM-LTx-induced cell death. (a) Left: Three-day-differentiated PC12 cells were transfected with siControl or siE-FABP for 5 days. A representative E-FABP Western blot is shown. Right: siControl- and siE-FABP-transfected cells were treated with control medium or 300 μM PAM for 12 and 18 h. Relative E-FABP mRNA levels were determined by real-time RT-PCR. (b) Cellular ROS levels of transfection reagent (vehicle), siControl and siE-FABP treated cells were measured by H2DCFDA method at 12 and 18 h after PAM (or control medium) treatment. (c) Cell viability of siControl and siE-FABP cells were measured by WST-1 assay at 24 and 48 h after PAM treatment. CTL: control medium at 48 h. The data represent mean ± SEM of three independent experiments. Significance symbols: *p < 0.05, **p < 0.01 and ***p < 0.001. Significance symbols are shown above bars when compared to control group and are shown above lines when compared between two linked groups.
In a previous report, we showed that increasing cellular levels of E-FABP significantly potentiated neurite extension in differentiating PC12 cells (Liu et al. 2008). Using a similar approach, we delivered recombinant E-FABP protein to NGFDPC12 cells before exposure to PAM overload. Figure7(a) indicates that NGFDPC12 cells receiving recombinant E-FABP protein showed higher E-FABP levels at both basal and lipotoxic condition. Upon PAM treatment, the cells loaded with recombinant E-FABP protein did not show significant elevation of ROS as seen in vehicle-treated cells (Fig.7b). Furthermore, this ROS-reducing effect appears to be specific to E-FABP because cells loaded with vehicle (BioPORTER) or unrelated protein (β-galactosidase) still displayed increased ROS level after PAM treatment (Fig.7b). Figure7(c) shows that the PAM+E-FABP group, which exhibits lower ROS levels, had more viable cells than the other three groups at the end of PAM treatment.
Figure 7.
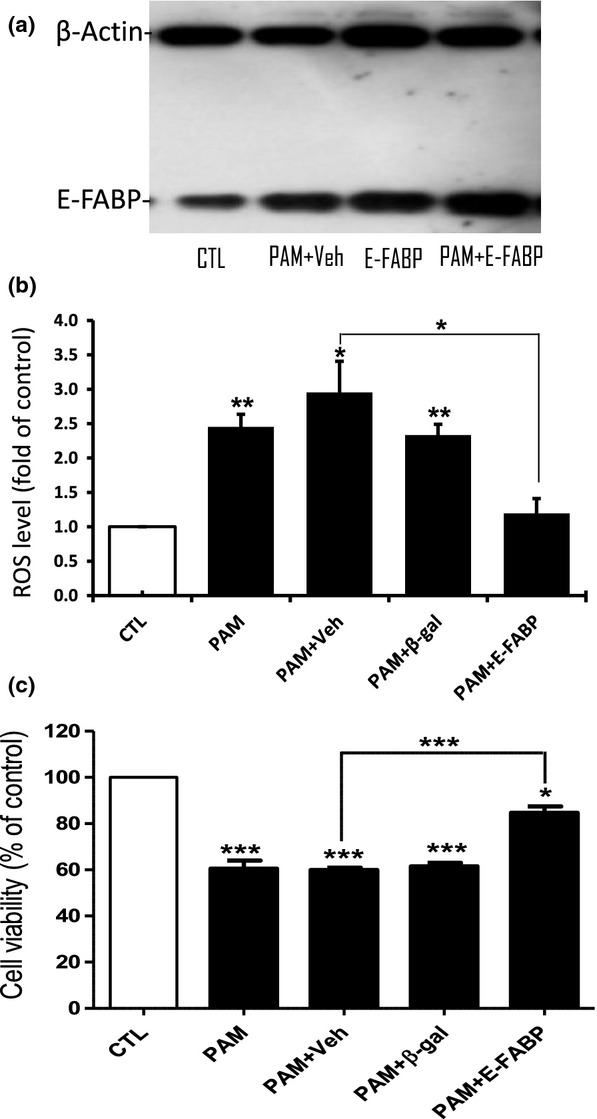
Preloading NGFDPC12 cells with recombinant epidermal fatty acid-binding protein (E-FABP) decreases palmitic acid-induced cellular reactive oxygen species (ROS) accumulation and cell death. Recombinant proteins were delivered to NGFDPC12 cells by BioPORTER Quik Ease kit 1 day before the addition of 300 μM PAM to the cell culture. BioPORTER alone was used as the vehicle control. (a) E-FABP western blot was performed 24 h after treatment and a representative blot is shown. Veh, control medium added to vehicle-loaded cells; PAM+Veh, PAM added to vehicle-loaded cells; E-FABP, control medium added to E-FABP-loaded cells; PAM+E-FABP, PAM added to E-FABP-loaded cells. (b) Cellular ROS level was evaluated by H2DCFDA method at 12 h after PAM treatment. CTL, control medium added to vehicle-loaded cells; PAM, PAM added to untreated cells; PAM+Veh, PAM added to vehicle-loaded cells; PAM+β-gal, PAM added to β-galactosidase-loaded cells; PAM+E-FABP, PAM added to E-FABP-loaded cells. (c) Cell viability was determined by WST-1 assay at 48 h after PAM treatment. Significance symbols: *p < 0.05, **p < 0.01 and ***p < 0.001. Significance symbols are shown above bars when compared to control group and are shown above lines when compared between two linked groups.
E-FABP has been shown to be regulated by PPARs (Hyder et al. 2010). We used specific PPAR agonists WY14643 (for PPARα), GW0742 (for PPARβ/δ), and GW1929 (for PPARγ) to further examine the effect of increased levels of E-FAPB during PAM-LTx. Among the PPAR agonists tested (Fig.8a), PPARβ/δ agonist, GW0742, elicited 6-fold stimulation, and PPARγ agonist, GW1929, produced 2-fold stimulation of E-FABP expression at 24 h but not at 12 h. Based on this result, we pre-treated NGFDPC12 cells with PPARβ/δ agonist and PPARγ agonist to increase E-FABP levels before PAM-LTx experiment. Consistent with our hypothesis, elevated cellular levels of E-FABP in NGFDPC12 culture resulting from treatment with PPARβ/δ and PPARγ agonists increased cell resistance to lipotoxic insult (Fig.8b). Furthermore, the protecting effect provided by PPARβ/δ and PPARγ agonists was abolished in siE-FABP-transfected cells (Fig.8c), confirming the important role of E-FABP in the PPAR agonists-initiated resistance during PAM-LTx.
Figure 8.
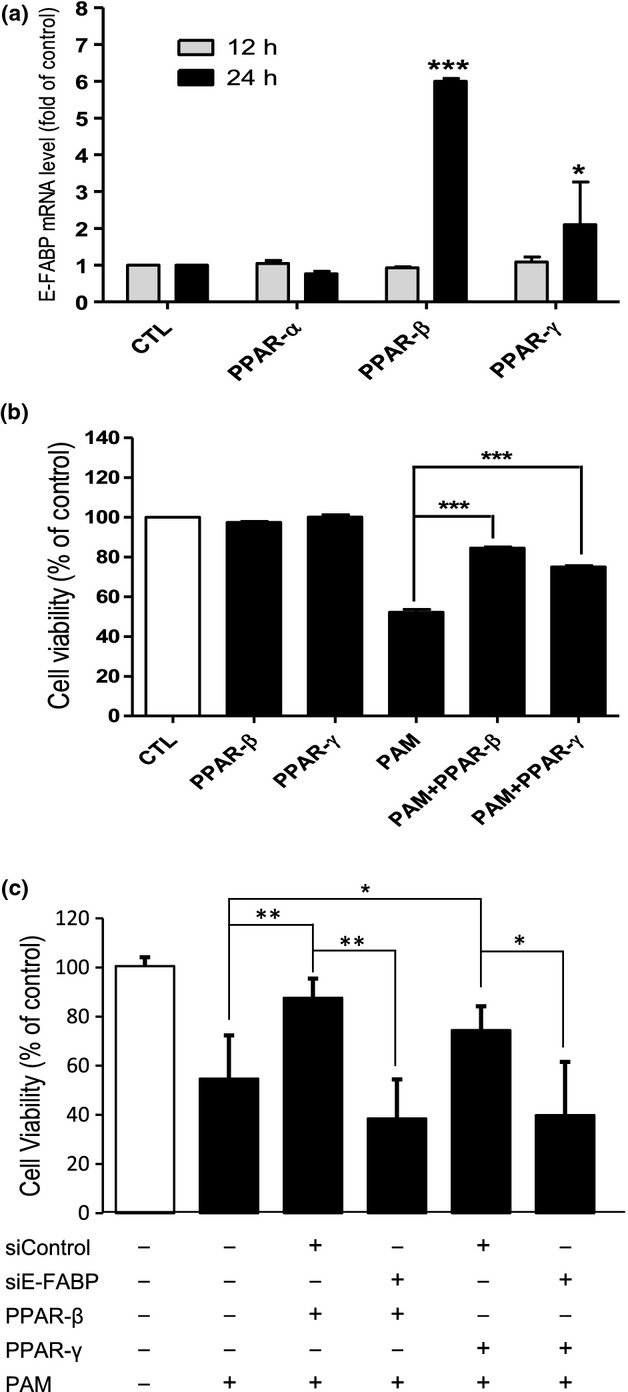
Peroxisome proliferator-activated receptors (PPAR) agonists stimulate epidermal fatty acid-binding protein (E-FABP) expression and protect NGFDPC12 cells from PAM-LTx. (a) Cells were treated with 10 μM of PPARα agonist (WY14643), PPARβ agonist (GW0742), and PPARγ agonist (GW1929) in control medium for 12 and 24 h. The stock solution of chemicals was made at 20 mM in DMSO. CTL, control medium with 0.05% DMSO. Relative E-FABP mRNA levels of cells upon treatments were determined at 12 and 24 h by real-time RT-PCR. (b) NGFDPC12 cells were treated with 300 μM PAM without PPAR agonists pre-treatment (PAM), with GW0742 pre-treatment (PAM+ PPAR-β), or with GW1929 pre-treatment (PAM+ PPAR-γ) for 2 days. Each group was also treated with control medium without PAM. Cell viability was determined by WST-1 assay at 48 h. (c) siControl and siE-FABP-transfected cells were treated with 10 μM GW0742 or GW0742 for 2 days before PAM-LTx experiment. Cell viability was determined by WST-1 assay at 48 h. The data represent mean ± SEM of three independent experiments. Significance symbols: *p < 0.05, **p < 0.01 and ***p < 0.001. Significance symbols are shown above bars when compared to control group and are shown above lines when compared between two linked groups.
Discussion
This study provides new experimental evidence that E-FABP is an oxidative stress response protein that plays a protective role as an antioxidant in NGFDPC12 cells exposed to PAM-LTx injury. We demonstrate that experimental conditions that increase cellular levels of E-FABP (administration of E-FABP recombinant protein and treatment with PPAR agonists) lower ROS levels and improve cell survival in PAM-injured NGFDPC12 cells. Inhibition of E-FABP gene expression using E-FABP siRNA magnifies the detrimental effects of PAM-LTx by increasing ROS accumulation and cell death. Thus, E-FABP levels in the cell can regulate the outcome of PAM-LTx.
Previous work from our laboratory showed that the detrimental effects of PAM-LTx can be inhibited by antioxidant treatment (Almaguel et al. 2009; Padilla et al. 2011), suggesting that cellular ROS accumulation is an integral part of this PAM-LTx process. The mitochondrial electron transport chain (ETC) has been recognized as one of the major sites to generate ROS in the cell, mainly from enzyme complexes, Complex I and Complex III. Long-chain non-esterified fatty acids modulate mitochondrial ROS production by interacting or uncoupling of the ETC (Schonfeld and Wojtczak 2008). For PAM, in particular, has been shown to increase mitochondrial superoxide generation in cardiomyocytes and INS-1 beta cells (Fauconnier et al. 2007; Lin et al. 2012). Palmitoyl-carnitine that serves as the substrate for β-oxidation releases significant amount of H2O2 (St-Pierre et al. 2002). Furthermore, the mitochondrial H2O2 emission rates in permeabilized muscle fibers are higher during respiration by palmitoyl-carnitine oxidation (fatty acid metabolism) than by pyruvate oxidation (glucose metabolism) (Anderson et al. 2007). Palmitoyl-CoA was shown to inhibit directly (without β-oxidation) adenine nucleotide translocator and results in reduction in the extramitochondrial ATP/ADP ratio and increased ROS production in liver mitochondria (Ciapaite et al. 2006). On the other hand, succinate-dependent ROS release from Complex I is decreased by palmitoyl-CoA in heart mitochondria (Bortolami et al. 2008). PAM can also be metabolized in peroxisome and the balance between H2O2 production (by acyl-CoA oxidases) and H2O2 reduction (by catalase) controls the net H2O2 output from peroxisome. Recently, peroxisomal ROS generation has also been suggested to mediate the PAM-LTx in insulin-producing cells (Elsner et al. 2011). While this study demonstrates that PAM activation is required for generating ROS and producing lipotoxicity, further studies are required to assess the respective contributions of metabolic oxidation and the generation of lipid-derived signals in the cytosol.
ROS can be toxic to the cells, because of their strong reactivity with proteins, lipids and nucleic acids that causes deleterious modification of these macromolecules (Aruoma et al. 1991; Radi et al. 1991; Kramer et al. 1994). High intracellular accumulation of ROS can also regulate gene expression as seen in the redox-sensitive NF-ĸB signaling pathway (Morgan and Liu 2011). NF-ĸB proteins regulate the expression of many genes involved in inflammation, immune response, cell growth/differentiation, and apoptosis. Our findings showing SN50 reduces the increase of E-FABP gene expression in NGFDPC12 cells during lipotoxic injury points to a potential contribution of the NF-ĸB pathway in this process, which leads to future studies to examine the role of this important signaling pathway. The NF-ĸB proteins are composed of dimers of the Rel family that bind 10-base pair ĸB sites. NF-ĸB dimers are sequestered in the cytosol by inhibitor proteins IĸBs. The activity of IĸBs is controlled through phosphorylation by IĸB kinases (IKKs). Upon phosphorylation, IĸBs become ubiquitinated and degraded in proteosomes that allow NF-ĸB dimers to translocate to the nucleus, bind to ĸB motif and activate gene transcription. ROS have both stimulatory and inhibitory effects in NF-ĸB signaling pathway (Byun et al. 2002). Mitochondrial ROS generated during hypoxia/reoxygenation activate NF-ĸB through phosphorylation of IĸBα at alternative phosphorylation site tyrosine 42 (Fan et al. 2003). Tyrosine 42-phosphorylated IĸBα is bound by regulatory subunit of PI3-kinase, which consequently unmasks NF-ĸB and allows it to translocate to the nucleus (Beraud et al. 1999). On the other hand, H2O2 reduces NF-ĸB signaling by inhibiting inflammatory-stimulated IKK (Reynaert et al. 2006). Furthermore, IKK is known to be phosphorylated and activated by upstream kinase NIK (NF-ĸB-inducing kinase), and this activity is also modulated by H2O2 (Li and Engelhardt 2006). Along these lines, PAM can regulate genes such as interleukin-6 in the skeletal muscle (Weigert et al. 2004) and adipocytes (Ajuwon and Spurlock 2005) through NF-ĸB activation. PAM has been shown to stimulate DNA binding of NF-ĸB, decrease cytosolic IĸBα, and increase nuclear levels of p65/RalA in L6 myotubes, which is the pathway implicated in fatty acid-induced insulin resistance in skeletal muscle (Sinha et al. 2004).
The fact that treating NGFDPC12 cultures with PPAR agonists is followed by induction of E-FABP suggests an important role of these proteins in protecting cells during PAM-LTx. PPARs, comprising three isotypes PPARα, PPARβ/δ, and PPARγ, are transcription factors activated by a wide range of naturally occurring or metabolized lipids derived from the diet or from intracellular signaling pathways and subsequently modulate lipid-related gene expression (Feige et al. 2006). PPARs serve as a lipid signal sensor in the cell and play a crucial role to integrate various pathways involved in lipid metabolism, inflammation, cellular proliferation, and differentiation (Feige et al. 2006). Elevated plasma saturated fatty acid because of over nutrition as well as traumatic/ischemic CNS injury lead to the activation of inflammatory cascades, as NF-ĸB the key regulator of inflammation (Fleming et al. 2006; Cai 2013). PPARs are shown to suppress NF-ĸB through various mechanisms (Wahli and Michalik 2012) and PPARγ is considered a promising neuroprotective strategy mainly through its anti-neuroinflammatory activity (Munhoz et al. 2008; Heneka et al. 2011). FABPs are able to facilitate this neuroprotective pathway through channeling lipid ligands to respective PPAR ligand binding domain (Velkov 2013). Members of the FABP family are regulated by PPARs. Adipocyte FABP and liver FABP are among the best characterized PPAR target genes (Tontonoz et al. 1994; Poirier et al. 1997; Mochizuki et al. 2001). The impressive up-regulation of E-FABP by PPARβ/δ agonist observed in this study is similar to the effect of PPARβ/δ in prostate cancer (Morgan et al. 2010). In addition, the neuronal differentiating activity of PPARβ/δ can occur through modulating MAP kinase pathway (D'Angelo et al. 2011), which is consistent with our previous study showing that MAP kinase mediates the NGF-induced up-regulation of E-FABP gene in PC12 cells (Liu et al. 2008). However, data presented in this study suggest that PAM-stimulated E-FABP gene expression is dependent on NF-ĸB but not MAP kinase. Our data agree with the notion that the expression of E-FABP may be at the center of neuronal differentiating and injury processes (De Leon et al. 1996; Liu et al. 1997, 2000, 2008).
Multiple lines of evidence support an antioxidant role of FABPs as function of their demonstrated fatty acid/lipid binding properties (Terrasa et al. 1998; Wang et al. 2005, 2007). For instance, Ek et al. (1997) showed that FABPs can serve as an intracellular fatty acids buffer that can alter their availability for cell metabolic functions. Therefore, the increase in the level of E-FABP by PAM-LTx observed in our study may reduce the effective amount of PAM to generate additional ROS. Furthermore, FABPs can also bind to long-chain fatty acid oxidation derivatives (Raza et al. 1989) and, thus, may neutralize their potential toxic effect. In addition, FABPs are known to directly involve PPAR-mediated gene control by transporting PPAR ligands to the nucleus (Wolfrum et al. 2001; Kaczocha et al. 2009), and PPARs regulate the balance between cell survival and apoptosis (Feige et al. 2006). Along these lines, Tan et al. (2002) reported that E-FABP and adipocyte FABP can transport selective ligands to PPARβ/δ and PPARγ, respectively, to enhance their transcriptional activity. For instance, in breast cancer, retinoic acid either inhibits or promotes cell growth depending on how FABPs, i.e., cellular retinoic acid binding protein II and E-FABP, channel it to either the nuclear retinoic acid receptor or PPARβ/δ, respectively (Schug et al. 2008). In another series of studies, FABPs have been shown to serve as antioxidant proteins through modification of their amino acid residues not through their ligand binding. For example, E-FABP protein has the highest number of cysteine residue among all FABPs (Odani et al. 2000). Disulfide bonds may be formed between spatially close cysteine pairs: Cys-120/Cys-127 and Cys-67/Cys-87 in the case of rat E-FABP, and they seem not to be directly involved in fatty acid binding. That the cysteine pairs form disulfide bonds or remain free thiols is affected by the cellular redox state suggests that E-FABP can serve as an intracellular free radical scavenger (Odani et al. 2000). The protective role of E-FABP was also evident in the study by Bennaars-Eiden et al. (2002) showing that Cys-120 and Cys-127, to a lesser extent, are covalently modified by 4-hydroxynonenal (4-HNE), a toxic lipid peroxidation product, and the 4-HNE modification is potentiated by fatty acid binding. The 4-HNE modification of cysteine side chains was later reported for adipocyte FABP and liver FABP (Grimsrud et al. 2007; Smathers et al. 2012). Besides cysteine residue, oxidative modification of methionine residues was also identified in liver FABP, but the antioxidant effect was reduced after fatty acid binding (Yan et al. 2009). The mechanism by which E-FABP diminishes ROS produced by PAM-LTx demonstrated in this study needs further investigation. However, it is reasonable to suggest that the protective role of E-FABP in PAM-LTx may have important clinical implications in neuronal injuries that involve oxidative stress or lipid peroxidation.
Acknowledgments and conflict of interest disclosure
We would like to acknowledge Dr Johnny Figueroa and Lorena Salto for their valuable input in the preparation of the manuscript. This work was supported, in part, by NIH grants 5R25GM060507 and 5P2MD006988.
All experiments were conducted in compliance with the ARRIVE guidelines. The authors have no conflict of interest to declare.
Glossary
- 4-HNE
4-hydroxynonenal
- BSA
bovine serum albumin
- E-FABP
epidermal fatty acid-binding protein
- FBS
fetal bovine serum
- PBS
phosphate-buffered saline
- ROS
reactive oxygen species
References
- Adibhatla RM, Hatcher JF. Altered lipid metabolism in brain injury and disorders. Subcell. Biochem. 2008;49:241–268. doi: 10.1007/978-1-4020-8831-5_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajuwon KM, Spurlock ME. Palmitate activates the NF-kappaB transcription factor and induces IL-6 and TNFalpha expression in 3T3-L1 adipocytes. J. Nutr. 2005;135:1841–1846. doi: 10.1093/jn/135.8.1841. [DOI] [PubMed] [Google Scholar]
- Allen GW, Liu JW, De Leon M. Depletion of a fatty acid-binding protein impairs neurite outgrowth in PC12 cells. Brain Res. Mol. Brain Res. 2000;76:315–324. doi: 10.1016/s0169-328x(00)00014-0. [DOI] [PubMed] [Google Scholar]
- Allen GW, Liu J, Kirby MA, De Leon M. Induction and axonal localization of epithelial/epidermal fatty acid-binding protein in retinal ganglion cells are associated with axon development and regeneration. J. Neurosci. Res. 2001;66:396–405. doi: 10.1002/jnr.1232. [DOI] [PubMed] [Google Scholar]
- Almaguel FG, Liu JW, Pacheco FJ, Casiano CA, De Leon M. Activation and reversal of lipotoxicity in PC12 and rat cortical cells following exposure to palmitic acid. J. Neurosci. Res. 2009;87:1207–1218. doi: 10.1002/jnr.21918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almaguel FG, Liu JW, Pacheco FJ, De Leon D, Casiano CA, De Leon M. Lipotoxicity-mediated cell dysfunction and death involve lysosomal membrane permeabilization and cathepsin L activity. Brain Res. 2010;1318:133–143. doi: 10.1016/j.brainres.2009.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson EJ, Yamazaki H, Neufer PD. Induction of endogenous uncoupling protein 3 suppresses mitochondrial oxidant emission during fatty acid-supported respiration. J. Biol. Chem. 2007;282:31257–31266. doi: 10.1074/jbc.M706129200. [DOI] [PubMed] [Google Scholar]
- Aruoma OI, Halliwell B, Gajewski E, Dizdaroglu M. Copper-ion-dependent damage to the bases in DNA in the presence of hydrogen peroxide. Biochem. J. 1991;273(Pt 3):601–604. doi: 10.1042/bj2730601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennaars-Eiden A, Higgins L, Hertzel AV, Kapphahn RJ, Ferrington DA, Bernlohr DA. Covalent modification of epithelial fatty acid-binding protein by 4-hydroxynonenal in vitro and in vivo. Evidence for a role in antioxidant biology. J. Biol. Chem. 2002;277:50693–50702. doi: 10.1074/jbc.M209493200. [DOI] [PubMed] [Google Scholar]
- Beraud C, Henzel WJ, Baeuerle PA. Involvement of regulatory and catalytic subunits of phosphoinositide 3-kinase in NF-kappaB activation. Proc. Natl Acad. Sci. USA. 1999;96:429–434. doi: 10.1073/pnas.96.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boneva NB, Mori Y, Kaplamadzhiev DB, Kikuchi H, Zhu H, Kikuchi M, Tonchev AB, Yamashima T. Differential expression of FABP 3, 5, 7 in infantile and adult monkey cerebellum. Neurosci. Res. 2010;68:94–102. doi: 10.1016/j.neures.2010.07.2028. [DOI] [PubMed] [Google Scholar]
- Bortolami S, Comelato E, Zoccarato F, Alexandre A, Cavallini L. Long chain fatty acyl-CoA modulation of H(2)O (2) release at mitochondrial complex I. J. Bioenerg. Biomembr. 2008;40:9–18. doi: 10.1007/s10863-008-9126-1. [DOI] [PubMed] [Google Scholar]
- Buhlmann C, Borchers T, Pollak M, Spener F. Fatty acid metabolism in human breast cancer cells (MCF7) transfected with heart-type fatty acid binding protein. Mol. Cell. Biochem. 1999;199:41–48. doi: 10.1023/a:1006986629206. [DOI] [PubMed] [Google Scholar]
- Byun MS, Jeon KI, Choi JW, Shim JY, Jue DM. Dual effect of oxidative stress on NF-kappakB activation in HeLa cells. Exp. Mol. Med. 2002;34:332–339. doi: 10.1038/emm.2002.47. [DOI] [PubMed] [Google Scholar]
- Cai D. Neuroinflammation and neurodegeneration in overnutrition-induced diseases. Trends Endocrinol. Metab. 2013;24:40–47. doi: 10.1016/j.tem.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciapaite J, Bakker SJ, Diamant M, van Eikenhorst G, Heine RJ, Westerhoff HV, Krab K. Metabolic control of mitochondrial properties by adenine nucleotide translocator determines palmitoyl-CoA effects. Implications for a mechanism linking obesity and type 2 diabetes. FEBS J. 2006;273:5288–5302. doi: 10.1111/j.1742-4658.2006.05523.x. [DOI] [PubMed] [Google Scholar]
- Coe NR, Simpson MA, Bernlohr DA. Targeted disruption of the adipocyte lipid-binding protein (aP2 protein) gene impairs fat cell lipolysis and increases cellular fatty acid levels. J. Lipid Res. 1999;40:967–972. [PubMed] [Google Scholar]
- D'Angelo B, Benedetti E, Di Loreto S, Cristiano L, Laurenti G, Ceru MP, Cimini A. Signal transduction pathways involved in PPARbeta/delta-induced neuronal differentiation. J. Cell. Physiol. 2011;226:2170–2180. doi: 10.1002/jcp.22552. [DOI] [PubMed] [Google Scholar]
- De Leon M, Welcher AA, Nahin RH, Liu Y, Ruda MA, Shooter EM, Molina CA. Fatty acid binding protein is induced in neurons of the dorsal root ganglia after peripheral nerve injury. J. Neurosci. Res. 1996;44:283–292. doi: 10.1002/(SICI)1097-4547(19960501)44:3<283::AID-JNR9>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Dhillon HS, Dose JM, Scheff SW, Prasad MR. Time course of changes in lactate and free fatty acids after experimental brain injury and relationship to morphologic damage. Exp. Neurol. 1997;146:240–249. doi: 10.1006/exnr.1997.6524. [DOI] [PubMed] [Google Scholar]
- Ek BA, Cistola DP, Hamilton JA, Kaduce TL, Spector AA. Fatty acid binding proteins reduce 15-lipoxygenase-induced oxygenation of linoleic acid and arachidonic acid. Biochim. Biophys. Acta. 1997;1346:75–85. doi: 10.1016/s0005-2760(97)00021-0. [DOI] [PubMed] [Google Scholar]
- Elsner M, Gehrmann W, Lenzen S. Peroxisome-generated hydrogen peroxide as important mediator of lipotoxicity in insulin-producing cells. Diabetes. 2011;60:200–208. doi: 10.2337/db09-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan C, Li Q, Ross D, Engelhardt JF. Tyrosine phosphorylation of I kappa B alpha activates NF kappa B through a redox-regulated and c-Src-dependent mechanism following hypoxia/reoxygenation. J. Biol. Chem. 2003;278:2072–2080. doi: 10.1074/jbc.M206718200. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Horrocks LA, Farooqui T. Modulation of inflammation in brain: a matter of fat. J. Neurochem. 2007;101:577–599. doi: 10.1111/j.1471-4159.2006.04371.x. [DOI] [PubMed] [Google Scholar]
- Farre D, Roset R, Huerta M, Adsuara JE, Rosello L, Alba MM, Messeguer X. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 2003;31:3651–3653. doi: 10.1093/nar/gkg605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauconnier J, Andersson DC, Zhang SJ, Lanner JT, Wibom R, Katz A, Bruton JD, Westerblad H. Effects of palmitate on Ca(2 + ) handling in adult control and ob/ob cardiomyocytes: impact of mitochondrial reactive oxygen species. Diabetes. 2007;56:1136–1142. doi: 10.2337/db06-0739. [DOI] [PubMed] [Google Scholar]
- Feige JN, Gelman L, Michalik L, Desvergne B, Wahli W. From molecular action to physiological outputs: peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006;45:120–159. doi: 10.1016/j.plipres.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Fleming JC, Norenberg MD, Ramsay DA, Dekaban GA, Marcillo AE, Saenz AD, Pasquale-Styles M, Dietrich WD, Weaver LC. The cellular inflammatory response in human spinal cords after injury. Brain. 2006;129:3249–3269. doi: 10.1093/brain/awl296. [DOI] [PubMed] [Google Scholar]
- Grimsrud PA, Picklo MJ, Sr, Griffin TJ, Bernlohr DA. Carbonylation of adipose proteins in obesity and insulin resistance: identification of adipocyte fatty acid-binding protein as a cellular target of 4-hydroxynonenal. Mol. Cell. Proteomics. 2007;6:624–637. doi: 10.1074/mcp.M600120-MCP200. [DOI] [PubMed] [Google Scholar]
- Gupta S, Knight AG, Gupta S, Keller JN, Bruce-Keller AJ. Saturated long-chain fatty acids activate inflammatory signaling in astrocytes. J. Neurochem. 2012;120:1060–1071. doi: 10.1111/j.1471-4159.2012.07660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Gonzalez LH, Ludwig C, Hohoff C, Rademacher M, Hanhoff T, Ruterjans H, Spener F, Lucke C. Solution structure and backbone dynamics of human epidermal-type fatty acid-binding protein (E-FABP) Biochem. J. 2002;364:725–737. doi: 10.1042/BJ20020039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ED, Braughler JM. Central nervous system trauma and stroke. II. Physiological and pharmacological evidence for involvement of oxygen radicals and lipid peroxidation. Free Radical Biol. Med. 1989;6:303–313. doi: 10.1016/0891-5849(89)90057-9. [DOI] [PubMed] [Google Scholar]
- Han Q, Yeung SC, Ip MS, Mak JC. Effects of intermittent hypoxia on A-/E-FABP expression in human aortic endothelial cells. Int. J. Cardiol. 2010;145:396–398. doi: 10.1016/j.ijcard.2010.04.027. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Reyes-Irisarri E, Hull M, Kummer MP. Impact and Therapeutic Potential of PPARs in Alzheimer's Disease. Curr. Neuropharmacol. 2011;9:643–650. doi: 10.2174/157015911798376325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyder A, Zenhom M, Klapper M, Herrmann J, Schrezenmeir J. Expression of fatty acid binding proteins 3 and 5 genes in rat pancreatic islets and INS-1E cells: regulation by fatty acids and glucose. Islets. 2010;2:174–184. doi: 10.4161/isl.2.3.11454. [DOI] [PubMed] [Google Scholar]
- Kaczocha M, Glaser ST, Deutsch DG. Identification of intracellular carriers for the endocannabinoid anandamide. Proc. Natl Acad. Sci. USA. 2009;106:6375–6380. doi: 10.1073/pnas.0901515106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan-Thulasiraman P, Seachrist DD, Mahabeleshwar GH, Jain MK, Noy N. Fatty acid-binding protein 5 and PPARβ/γ are critical mediators of epidermal growth factor receptor-induced carcinoma cell growth. J. Biol. Chem. 2010;285:19106–191155. doi: 10.1074/jbc.M109.099770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer JH, Misik V, Weglicki WB. Lipid peroxidation-derived free radical production and postischemic myocardial reperfusion injury. Ann. N. Y. Acad. Sci. 1994;723:180–196. [PubMed] [Google Scholar]
- Li Q, Engelhardt JF. Interleukin-1beta induction of NFkappaB is partially regulated by H2O2-mediated activation of NFkappaB-inducing kinase. J. Biol. Chem. 2006;281:1495–1505. doi: 10.1074/jbc.M511153200. [DOI] [PubMed] [Google Scholar]
- Lin N, Chen H, Zhang H, Wan X, Su Q. Mitochondrial reactive oxygen species (ROS) inhibition ameliorates palmitate-induced INS-1 beta cell death. Endocrine. 2012;42:107–117. doi: 10.1007/s12020-012-9633-z. [DOI] [PubMed] [Google Scholar]
- Liu Y, Molina CA, Welcher AA, Longo LD, De Leon M. Expression of DA11, a neuronal-injury-induced fatty acid binding protein, coincides with axon growth and neuronal differentiation during central nervous system development. J. Neurosci. Res. 1997;48:551–562. doi: 10.1002/(sici)1097-4547(19970615)48:6<551::aid-jnr8>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Liu Y, Longo LD, De Leon M. In situ and immunocytochemical localization of E-FABP mRNA and protein during neuronal migration and differentiation in the rat brain. Brain Res. 2000;852:16–27. doi: 10.1016/s0006-8993(99)02158-7. [DOI] [PubMed] [Google Scholar]
- Liu NK, Zhang YP, Titsworth WL, Jiang X, Han S, Lu PH, Shields CB, Xu XM. A novel role of phospholipase A2 in mediating spinal cord secondary injury. Ann. Neurol. 2006;59:606–619. doi: 10.1002/ana.20798. [DOI] [PubMed] [Google Scholar]
- Liu JW, Almaguel FG, Bu L, De Leon DD, De Leon M. Expression of E-FABP in PC12 cells increases neurite extension during differentiation: involvement of n-3 and n-6 fatty acids. J. Neurochem. 2008;106:2015–2029. doi: 10.1111/j.1471-4159.2008.05507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok J, Leung W, Murphy S, Butler W, Noviski N, Lo EH. Intracranial hemorrhage: mechanisms of secondary brain injury. Acta Neurochir. Suppl. 2011;111:63–69. doi: 10.1007/978-3-7091-0693-8_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Zhang M, Mori Y, Yao C, Larsen CP, Yamashima T, Zhou L. Cellular localization of epidermal-type and brain-type fatty acid-binding proteins in adult hippocampus and their response to cerebral ischemia. Hippocampus. 2010;20:811–819. doi: 10.1002/hipo.20682. [DOI] [PubMed] [Google Scholar]
- Masouye I, Hagens G, Van Kuppevelt TH, Madsen P, Saurat JH, Veerkamp JH, Pepper MS, Siegenthaler G. Endothelial cells of the human microvasculature express epidermal fatty acid-binding protein. Circ. Res. 1997;81:297–303. doi: 10.1161/01.res.81.3.297. [DOI] [PubMed] [Google Scholar]
- Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch. Neurol. 2009;66:1210–1215. doi: 10.1001/archneurol.2009.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki K, Suruga K, Kitagawa M, Takase S, Goda T. Modulation of the expression of peroxisome proliferator-activated receptor-dependent genes through disproportional expression of two subtypes in the small intestine. Arch. Biochem. Biophys. 2001;389:41–48. doi: 10.1006/abbi.2001.2305. [DOI] [PubMed] [Google Scholar]
- Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan E, Kannan-Thulasiraman P, Noy N. Involvement of fatty acid binding protein 5 and PPARbeta/delta in prostate cancer cell growth. PPAR Res. 2010;2010:1–9. doi: 10.1155/2010/234629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munhoz CD, Garcia-Bueno B, Madrigal JL, Lepsch LB, Scavone C, Leza JC. Stress-induced neuroinflammation: mechanisms and new pharmacological targets. Braz. J. Med. Biol. Res. 2008;41:1037–1046. doi: 10.1590/s0100-879x2008001200001. [DOI] [PubMed] [Google Scholar]
- Nixon GF. Sphingolipids in inflammation: pathological implications and potential therapeutic targets. Br. J. Pharmacol. 2009;158:982–993. doi: 10.1111/j.1476-5381.2009.00281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odani S, Namba Y, Ishii A, Ono T, Fujii H. Disulfide bonds in rat cutaneous fatty acid-binding protein. J. Biochem. 2000;128:355–361. doi: 10.1093/oxfordjournals.jbchem.a022761. [DOI] [PubMed] [Google Scholar]
- Owada Y, Yoshimoto T, Kondo H. Increased expression of the mRNA for brain- and skin-type but not heart-type fatty acid binding proteins following kainic acid systemic administration in the hippocampal glia of adult rats. Brain Res. Mol. Brain Res. 1996a;42:156–160. doi: 10.1016/s0169-328x(96)00182-9. [DOI] [PubMed] [Google Scholar]
- Owada Y, Yoshimoto T, Kondo H. Spatio-temporally differential expression of genes for three members of fatty acid binding proteins in developing and mature rat brains. J. Chem. Neuroanat. 1996b;12:113–122. doi: 10.1016/s0891-0618(96)00192-5. [DOI] [PubMed] [Google Scholar]
- Owada Y, Utsunomiya A, Yoshimoto T, Kondo H. Changes in gene expression for skin-type fatty acid binding protein in hypoglossal motor neurons following nerve crush. Neurosci. Lett. 1997;223:25–28. doi: 10.1016/s0304-3940(97)13390-0. [DOI] [PubMed] [Google Scholar]
- Owada Y, Abdelwahab SA, Suzuki R, Iwasa H, Sakagami H, Spener F, Kondo H. Localization of epidermal-type fatty acid binding protein in alveolar macrophages and some alveolar type II epithelial cells in mouse lung. Histochem. J. 2001;33:453–457. doi: 10.1023/a:1014420330284. [DOI] [PubMed] [Google Scholar]
- Padilla A, Descorbeth M, Almeyda AL, Payne K, De Leon M. Hyperglycemia magnifies Schwann cell dysfunction and cell death triggered by PA-induced lipotoxicity. Brain Res. 2011;1370:64–79. doi: 10.1016/j.brainres.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker SM, Moore PC, Johnson LM, Poitout V. Palmitate potentiation of glucose-induced insulin release: a study using 2-bromopalmitate. Metab. Clin. Exp. 2003;52:1367–1371. doi: 10.1016/s0026-0495(03)00279-8. [DOI] [PubMed] [Google Scholar]
- Poirier H, Braissant O, Niot I, Wahli W, Besnard P. 9-cis-retinoic acid enhances fatty acid-induced expression of the liver fatty acid-binding protein gene. FEBS Lett. 1997;412:480–484. doi: 10.1016/s0014-5793(97)00830-2. [DOI] [PubMed] [Google Scholar]
- Radi R, Bush KM, Cosgrove TP, Freeman BA. Reaction of xanthine oxidase-derived oxidants with lipid and protein of human plasma. Arch. Biochem. Biophys. 1991;286:117–125. doi: 10.1016/0003-9861(91)90016-c. [DOI] [PubMed] [Google Scholar]
- Raza H, Pongubala JR, Sorof S. Specific high affinity binding of lipoxygenase metabolites of arachidonic acid by liver fatty acid binding protein. Biochem. Biophys. Res. Commun. 1989;161:448–455. doi: 10.1016/0006-291x(89)92619-3. [DOI] [PubMed] [Google Scholar]
- Reynaert NL, van der Vliet A, Guala AS, et al. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc. Natl Acad. Sci. USA. 2006;103:13086–13091. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Dhillon HS. Creatine-enhanced diet alters levels of lactate and free fatty acids after experimental brain injury. Neurochem. Res. 2004;29:469–479. doi: 10.1023/b:nere.0000013753.22615.59. [DOI] [PubMed] [Google Scholar]
- Schonfeld P, Wojtczak L. Fatty acids as modulators of the cellular production of reactive oxygen species. Free Radical Biol. Med. 2008;45:231–241. doi: 10.1016/j.freeradbiomed.2008.04.029. [DOI] [PubMed] [Google Scholar]
- Schug TT, Berry DC, Toshkov IA, Cheng L, Nikitin AY, Noy N. Overcoming retinoic acid-resistance of mammary carcinomas by diverting retinoic acid from PPARbeta/delta to RAR. Proc. Natl Acad. Sci. USA. 2008;105:7546–7551. doi: 10.1073/pnas.0709981105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegenthaler G, Hotz R, Chatellard-Gruaz D, Didierjean L, Hellman U, Saurat JH. Purification and characterization of the human epidermal fatty acid-binding protein: localization during epidermal cell differentiation in vivo and in vitro. Biochem. J. 1994;302(Pt 2):363–371. doi: 10.1042/bj3020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Perdomo G, Brown NF, O'Doherty RM. Fatty acid-induced insulin resistance in L6 myotubes is prevented by inhibition of activation and nuclear localization of nuclear factor kappa B. J. Biol. Chem. 2004;279:41294–41301. doi: 10.1074/jbc.M406514200. [DOI] [PubMed] [Google Scholar]
- Smathers RL, Fritz KS, Galligan JJ, Shearn CT, Reigan P, Marks MJ, Petersen DR. Characterization of 4-HNE modified L-FABP reveals alterations in structural and functional dynamics. PLoS ONE. 2012;7:e38459. doi: 10.1371/journal.pone.0038459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- Tan NS, Shaw NS, Vinckenbosch N, Liu P, Yasmin R, Desvergne B, Wahli W, Noy N. Selective cooperation between fatty acid binding proteins and peroxisome proliferator-activated receptors in regulating transcription. Mol. Cell. Biol. 2002;22:5114–5127. doi: 10.1128/MCB.22.14.5114-5127.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrasa A, Guajardo M, Catala A. Lipoperoxidation of rod outer segments of bovine retina is inhibited by soluble binding proteins for fatty acids. Mol. Cell. Biochem. 1998;178:181–186. doi: 10.1023/a:1006839413757. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994;8:1224–1234. doi: 10.1101/gad.8.10.1224. [DOI] [PubMed] [Google Scholar]
- Ulloth JE, Casiano CA, De Leon M. Palmitic and stearic fatty acids induce caspase-dependent and -independent cell death in nerve growth factor differentiated PC12 cells. J. Neurochem. 2003;84:655–668. doi: 10.1046/j.1471-4159.2003.01571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger RH, Orci L. Diseases of liporegulation: new perspective on obesity and related disorders. FASEB J. 2001;15:312–321. doi: 10.1096/fj.00-0590. [DOI] [PubMed] [Google Scholar]
- Velkov T. Interactions between human liver fatty acid binding protein and peroxisome proliferator activated receptor selective drugs. PPAR Res. 2013;2013:938401. doi: 10.1155/2013/938401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahli W, Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012;23:351–363. doi: 10.1016/j.tem.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Wang G, Gong Y, Anderson J, Sun D, Minuk G, Roberts MS, Burczynski FJ. Antioxidative function of L-FABP in L-FABP stably transfected Chang liver cells. Hepatology. 2005;42:871–879. doi: 10.1002/hep.20857. [DOI] [PubMed] [Google Scholar]
- Wang D, Wei Y, Pagliassotti MJ. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology. 2006;147:943–951. doi: 10.1210/en.2005-0570. [DOI] [PubMed] [Google Scholar]
- Wang G, Shen H, Rajaraman G, Roberts MS, Gong Y, Jiang P, Burczynski F. Expression and antioxidant function of liver fatty acid binding protein in normal and bile-duct ligated rats. Eur. J. Pharmacol. 2007;560:61–68. doi: 10.1016/j.ejphar.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Weigert C, Brodbeck K, Staiger H, Kausch C, Machicao F, Haring HU, Schleicher ED. Palmitate, but not unsaturated fatty acids, induces the expression of interleukin-6 in human myotubes through proteasome-dependent activation of nuclear factor-kappaB. J. Biol. Chem. 2004;279:23942–23952. doi: 10.1074/jbc.M312692200. [DOI] [PubMed] [Google Scholar]
- Wolfrum C, Borrmann CM, Borchers T, Spener F. Fatty acids and hypolipidemic drugs regulate peroxisome proliferator-activated receptors alpha - and gamma-mediated gene expression via liver fatty acid binding protein: a signaling path to the nucleus. Proc. Natl Acad. Sci. USA. 2001;98:2323–2328. doi: 10.1073/pnas.051619898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Gong Y, She YM, Wang G, Roberts MS, Burczynski FJ. Molecular mechanism of recombinant liver fatty acid binding protein's antioxidant activity. J. Lipid Res. 2009;50:2445–2454. doi: 10.1194/jlr.M900177-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu ZF, Nikolova-Karakashian M, Zhou D, Cheng G, Schuchman EH, Mattson MP. Pivotal role for acidic sphingomyelinase in cerebral ischemia-induced ceramide and cytokine production, and neuronal apoptosis. J. Mol. Neurosci. 2000;15:85–97. doi: 10.1385/JMN:15:2:85. [DOI] [PubMed] [Google Scholar]